

Abstract
Translation initiation site usage on the human rhinovirus 2 internal ribosome entry site (IRES) has been examined in a mixed reticulocyte lysate/HeLa cell extract system. There are two relevant AUG triplets, both in a base-paired hairpin structure (domain VI), with one on the 5′ side at nucleotide (nt) 576, base paired with the other at nt 611, which is the initiation site for polyprotein synthesis. A single residue was inserted in the apical loop to put AUG-576 in frame with AUG-611, and in addition another in-frame AUG was introduced at nt 593. When most of the IRES was deleted to generate a monocistronic mRNA, the use of these AUGs conformed to the scanning ribosome model: improving the AUG-576 context increased initiation at this site and decreased initiation at downstream sites, whereas the converse was seen when AUG-576 was mutated to GUA; and AUG-593, when present, took complete precedence over AUG-611. Under IRES-dependent conditions, by contrast, much less initiation occurred at AUG-576 than in a monocistronic mRNA with the same AUG-576 context, mutation of AUG-576 decreased initiation at downstream sites by ∼70%, and introduction of AUG-593 did not completely abrogate initiation at AUG-611, unless the apical base pairing in domain VI was destroyed by point mutations. These results indicate that ribosomes first bind at the AUG-576 site, but instead of initiating there, most of them are transferred to AUG-611, the majority by strictly linear scanning and a substantial minority by direct transfer, which is possibly facilitated by the occasional persistence of base pairing in the apical part of the domain VI stem.
Until the recent discovery of animal picornaviruses with internal ribosome entry sites (IRESs) resembling that of hepatitis C virus, most picornavirus IRESs have been classified into two groups (1, 17): type 1 (exemplified by entero- and rhinoviruses) and type 2 (cardio- and aphthoviruses). Primary sequences and especially secondary structures are strongly conserved within each group but there is very little similarity between the two groups apart from an AUG triplet at the 3′ end of the IRES (as defined by deletion analysis), which is preceded by a ∼25 nucleotide (nt) pyrimidine-rich tract (17). In type 2 IRESs, notably encephalomyocarditis virus (EMCV), this AUG triplet is the authentic initiation codon for viral polyprotein synthesis, and the totality of the evidence indicates that all ribosomes bind at, or very close to, this AUG and that all initiate translation at this site (18, 19). The foot-and-mouth disease virus (FMDV), although a type 2 IRES, is not quite so straightforward in that a minority of initiation events occur at the AUG immediately downstream of the oligopyrimidine tract, and the rest occur at the next AUG, 84 nt downstream (3, 45).
In contrast, initiation on type 1 IRESs seems much more complicated and rather puzzling. The first puzzling feature is that there is very little, if any, initiation at the AUG just downstream of the oligopyrimidine tract, at nt 586 in poliovirus type 1 (PV-1) (39), and the initiation site for polyprotein synthesis is the next AUG further downstream, at a distance of ∼160 nt in enteroviruses and ∼35 nt in rhinoviruses (17). Nevertheless, AUG-586 is important for efficient initiation at the authentic polyprotein initiation site. Mutation of AUG-586 in a PV-1 infectious clone was found to be quasi-infectious (42), while mutation of the equivalent site in PV-2 conferred a small-plaque phenotype and reduced initiation at the polyprotein initiation site by ∼70% in both in vitro assays and in transfection assays (32, 33, 37).
This observation has led to the idea that ribosomes first bind at AUG-586, but instead of initiating at this site, virtually all of them get transferred to the polyprotein initiation site (17). This raises questions as to the nature of the transfer process. Because insertion of an AUG codon between PV-1 nt 586 and the authentic initiation site conferred a small-plaque phenotype and because all large-plaque pseudo-revertants had lost the inserted AUG either by deletion or point mutation (25, 26), linear scanning is likely to be important. However, as the insertion resulted in a small-plaque phenotype rather than lethality, there remains the possibility that some ribosomes were transferred directly without scanning the whole distance. This has also been suggested on the grounds that insertion of AUGs or a hairpin loop between nt 586 and the authentic initiation site of PV-1 did not seem to reduce polyprotein synthesis in vitro as much as might be expected if the authentic initiation site is accessed by strictly linear scanning (8).
The final puzzle is that AUG-586 is located in a stem-loop structure, domain VI (Fig. 1A), which is conserved in all entero- and rhinoviruses apart from bovine enterovirus. If the initiating 40S subunits do inspect AUG-586 in some way, albeit an unproductive way, this stem-loop would need to open at least partly, if not completely. This need for domain VI to be opened might be considered an impediment to efficient initiation, and yet its strong conservation suggests the opposite, namely, that it might have a positive effect. Precise deletion of the spacer downstream of AUG-586 in PV-1(Mahoney), so that polyprotein synthesis now started at 586, reduced virus yield by ∼10-fold (39), and in an independent study a deletion that brought the polyprotein initiation site to nt 586 or 580 caused a very similar growth defect in PV-1(Sabin) although the defect was considerably less in a Mahoney background (13, 27). On the other hand, two smaller deletions in PV-1(Sabin) that retained just the whole base-paired domain VI or only its 5′ side, placing the polyprotein initiation site 52 or 31 nt, respectively, downstream of AUG-586, did not confer any significant negative phenotype (13, 27). Taken together, these results would seem to imply that the base pairing in domain VI is neutral to initiation efficiency, but the primary sequence of its 5′ side may confer a moderate positive effect. In this respect it is interesting that bovine enterovirus retains most of the sequence of the 5′ side of domain VI but lacks the complementary sequence of the 3′ side.
FIG. 1.

(A) Sequence and base pairing of IRES domain VI of HRV-2 and PV-1(Mahoney), numbered with respect to the viral genome sequence. (B) Hypothetical model for the opening of HRV-2 domain VI in two stages, showing that in the intermediate state AUG-576 and AUG-611 are both exposed.
We have reexamined these issues but in the context of human rhinovirus 2 (HRV-2), mainly because the close proximity of the polyprotein initiation site (at nt 611) to the AUG (at nt 576) just downstream of the oligopyrimidine tract makes the interpretation of results less ambiguous than is the case with enteroviruses. A recent comprehensive sequence comparison of 106 different HRV strains plus 10 field isolates shows that HRV-2 domain VI is typical of the 106 serotypes and the one field isolate that differs in domain VI from its parent strain (35). In 95% of these sequences, the number of residues between the two AUG codons is in the range of 28 to 34 nt (median, 31 nt), with five outliers at 20 or 22 nt. The two AUGs are invariably base paired in a back-to-back configuration (Fig. 1A), and the intervening residues fold into a base-paired structure, usually with a single mismatch (Fig. 1A) or at least one G-U codon at around the mid-point and an apical loop of 3 to 6 residues (depending on the strain). The base-paired stem of enteroviruses is considerably shorter (usually without a mismatch), and the extra length in HRV domain VI generally consists of A-U and U-A pairs (often alternating) in the apical part (Fig. 1A). In 23% of these 107 HRV domain VI sequences, the two AUGs are in the same reading frame, and in 17 (approximately two-thirds) of these there is no in-frame stop codon between them so that any initiation at the upstream AUG would result in synthesis of a VP0 protein (and, hence, also VP4) with an N-terminal extension.
We first asked whether AUG-576 in HRV-2 is similar to AUG-586 in PV-1 in that there is very little initiation at this site, and yet AUG-576 is important for efficient initiation at the downstream polyprotein initiation site. We then looked for evidence that the domain VI stem-loop opens and whether all ribosomes access the authentic initiation site (AUG-611) by strictly linear scanning from some upstream site. We conclude that most ribosomes do access AUG-611 in this way, but a significant minority may take a shortcut, which could be facilitated if the apical part of this domain remains closed and base paired, with the single mismatch in the domain VI stem possibly causing the opening of this domain to occur in two stages (Fig. 1B).
MATERIALS AND METHODS
Plasmid constructs.
The parent construct was pXLJHRV10-611, which has been described previously (4, 5). It encodes a dicistronic mRNA under the control of the T7 promoter in pGEM2 (Promega) and consists of the Xenopus laevis cyclin B2 open reading frame (ORF) followed by nucleotides 10 to 611 of the HRV-2 5′ untranslated region (UTR) and a slightly truncated derivative of the influenza virus NS1 protein coding sequence (NS), followed by the complete 3′ UTR of NS. The initiation codon of the NS reading frame occupies the same position relative to the viral 5′ UTR sequences as does the viral polyprotein initiation codon. pXLJHRV10-611 and all mutants derived from it were linearized with EcoRI prior to transcription to generate the dicistronic mRNAs.
Mutations were introduced into the domain VI region of the HRV 5′ UTR in the dicistronic construct, pXLJHRV10-611, using specific oligonucleotides and PCR-based techniques, and the sequences of the mutants are shown in Fig. 2. The monocistronic mutants (with IRESs deleted) were made by PCR amplification of nucleotides 525 to 611 of the HRV-2 5′ UTR and include the NS coding sequence followed by the NS 3′ UTR, with a unique HindIII site introduced at the 5′ end. The resultant PCR product was ligated into pGEM2 in a positive orientation with respect to the T7 promoter. All mutants were fully sequenced to verify their authenticity, and all plasmids were propagated according to standard procedures (44).
FIG. 2.

(A) A schematic diagram of the dicistronic mRNAs, showing the upstream cistron, the Xenopus laevis cyclin B2 gene, and the influenza virus nonstructural protein, NS, downstream of the HRV-2 5′ UTR. The HRV 5′ UTR is shown as a series of stem structures which are designated I to VI. Also shown is the position of the unique EcoRI site at which the plasmid is linearized prior to transcription. (B) The nucleotide sequences of domain VI of the wild-type and mutant RNAs. AUG-611 is shown in bold. The underlined AUGs are in frame with the NS cistron while those not underlined (the upstream AUG at nt 576 in wt and mutant wt.1) are not in frame. Only changes from the wild-type sequence are shown for the mutants. The dash in the top (wild-type) sequence and in mutants wt.1 and wt.2 signifies a gap introduced to facilitate alignments with the other sequences which have an A inserted in this position. The positions of annealing of oligodeoxynucleotides used in RNase H cleavage assays are shown as solid lines, with S1 annealing to wt domain VI and S2 annealing to mutant 3.1d domain VI. LHS, left-hand side; RHS, right-hand side.
In vitro transcription reactions.
Uncapped and capped RNAs for subsequent translation were synthesized using bacteriophage T7 RNA polymerase and trace labeled using [α-32P]UTP to allow accurate quantitation. The RNA products were isolated and quantitated exactly as described previously (6). RNAs for oligodeoxynucleotide/RNase H assays were prepared similarly, except that the RNAs were labeled to a slightly higher specific activity (∼0.16 μCi/μg of RNA).
In vitro translation reactions.
All in vitro translation reactions were carried out using rabbit reticulocyte lysate (RRL; Promega) that was treated with micrococcal nuclease as described previously (16). All reactions were carried out in 10-μl volumes comprising 5.5 μl of RRL and 2 μl of HeLa cell high-salt S100 (HS-S100) prepared as described previously (10), plus the following additions at the final concentrations stated: 70 mM KCl (100 mM for capped monocistronic IRES-deleted mRNA), 0.5 mM MgCl2, 10 mM creatine phosphate, 50 μg/ml creatine kinase, 10 μM hemin, 0.1 mM each amino acid (except methionine), and 5 μCi/ml [35S]methionine. Each RNA preparation was initially assayed at 40, 20, 10, 5, and 2.5 μg of RNA/ml of reaction mixture. On the basis of these assays a concentration of 5 μg/ml was chosen for the assays shown here as the best compromise between a good signal and one that would saturate the translation capacity of the system. Assays were incubated at 30°C for 1.5 h. The translation products were separated on 20% polyacrylamide-SDS gels. Quantitation of the autoradiographs was carried out by densitometric analysis using Phoretix software. The preparation of eukaryotic initiation factor 4G (eIF4G)-depleted reticulocyte lysate and the purification of recombinant the His-tagged dominant negative mutant eIF4A R362Q were carried out exactly as described previously (2, 41).
Oligodeoxynucleotide/RNase H assays.
Oligodeoxynucleotide/RNase H assays were carried out in buffer and also under in vitro translation conditions. The assays in buffer contained 70 mM KCl, 20 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 11.6 nM 32P-labeled dicistronic RNA, 232 nM specific oligodeoxynucleotide, and 10 units of RNase H in a total volume of 20 μl. Assays were incubated at 30°C for 10 min, and then the RNA was precipitated and resuspended in formamide solution. The assays were also conducted under in vitro translation conditions using essentially the same conditions as for in vitro translation reactions, except that reaction mixtures also contained 4 mM 2-aminopurine, with the same concentrations of 32P-labeled dicistronic RNA, oligodeoxynucleotide, and RNase H as specified above. To some assay mixtures inhibitors of protein synthesis were also added at concentrations sufficient for complete inhibition: edeine at 1 μM, cycloheximide at 100 μM, or dominant negative eIF4A R362Q at 350 μg/ml. RRL-only and HeLa-only conditions were exactly as those described above but lacked HeLa cell HS-S100 extract and RRL, respectively. The HeLa-only assay was also supplemented with 0.5 mM ATP, 0.1 mM GTP, and 50 μg/ml creatine kinase. Assays were incubated at 30°C for 10 min, and then 5 μl was removed and analyzed for products of translation by 20% polyacrylamide-SDS gel electrophoresis. To the remaining 15 μl was added 32 mg of proteinase K, and the incubation was continued at 37°C for 30 min. The sample was then phenol extracted and precipitated before being resuspended in formamide solution. All samples were counted in a Beckman liquid scintillation counter, and equal counts were loaded in each lane of a 3.5% polyacrylamide-urea gel. Control reaction mixtures lacking added RNase H were not feasible because the RRL had significant endogenous RNase H activity.
The sequence of the control oligodeoxynucleotide (C) complementary to a 5′-proximal part of the NS coding sequence was 5′-AACTCGTTTGCGGACATGCC-3′. Two different domain VI stem-specific oligodeoxynucleotides were used: S1, 5′-ATATATTTTATATATTGTCACCATAA-3′, which is complementary to the domain VI wild-type sequence; and S2, 5′-TGGCCATTTTTAATATTGTCACCATGG-3′, which is complementary to mutant 3.1d domain VI. The positions where these oligonucleotides anneal to domain VI are shown in Fig. 2.
RESULTS
Constructs and mutants used in this study.
For the purposes of this study, the HRV-2 IRES from nt 10 to 611 was inserted between the Xenopus laevis cyclin B gene and the influenza virus NS cistron (4, 5). The influenza virus NS coding sequence plus 3′ UTR is fused directly to the authentic HRV initiation codon at position 611 and essentially substitutes for the viral polyprotein coding sequences. In the wild-type HRV-2 sequence, AUG-576, which is located just downstream of the pyrimidine-rich tract and at the base of the 5′ side of the domain VI stem-loop, is out of frame with the viral polyprotein coding sequence. Initiation at AUG-576 would yield a product of only 18 amino acids in length that could not be easily detected or quantified using conventional SDS-PAGE. To overcome this problem and to allow us to monitor the use of AUG-576 as an initiation codon, AUG-576 was made in frame with the NS reporter by introducing a single A residue into the apical loop sequence of stem VI, with the result that the product of initiation at AUG-576 is NS with a 12-amino-acid extension at the N terminus. (The larger apical loop occurs naturally in some HRV strains [35].) This construct is designated mutant 2 (Fig. 2), which also contains further sequence modifications to allow optimal expression from AUG-593 (to be introduced into mutant 3) while maintaining the stability of stem VI. To avoid potential confusion, we have not renumbered the residues downstream of this insertion, and so the authentic initiation codon in this mutant and its derivatives will still be designated AUG-611. As a means of monitoring whether ribosomes can scan through the apical loop sequence in an initiation-competent state, an additional AUG was introduced by site-directed point mutagenesis into the modified domain VI loop sequence at nt 593, where it is in frame with both AUG-576 and -611 (Fig. 2, mutant 3).
We generated a further set of three mutants in the background of mutant 2, mutant 3, and the wild type, where the context of AUG-576 was optimized by point mutations converting the wild-type context from CUUAUGGUG to ACCAUGGUG; these constructs are designated mutant 2.1, 3.1, and wt.1, respectively (Fig. 2). Another set of three mutants (designated wt.2, mutant 2.2, and mutant 3.2) destroyed AUG-576 by mutating it to GUA in its wild-type context. Monocistronic derivatives of all these constructs were prepared in which the entire cyclin cistron and all HRV IRES sequences up to and including nt 524 had been deleted.
Uncapped IRES-containing dicistronic mRNA transcripts were translated in a mixed RRL/HeLa cell-free system, where the reaction mixture was 55% (vol/vol) nuclease-treated RRL and 20% (vol/vol) HeLa cell high-salt S100 (HS-S100), which is the high-speed supernatant prepared from HeLa cell cytoplasmic (S10) extract treated with 0.5 M KCl (12). HS-S100 thus represents total HeLa cell cytoplasm except for salt-washed ribosomes and is used as a source of the trans-acting factors necessary for HRV IRES function (11, 12).
The translation products were separated by SDS-PAGE and visualized by autoradiography, and their yield was quantitated by densitometry using exposures within the linear range of film response. We show a representative example of the autoradiographs from each type of experiment, with the band intensity data given below each lane. After adjusting for the different methionine contents of the different products, these data were processed to give the relative proportion (as percentages) of domain VI initiation events occurring at each in-frame AUG. These values are summarized for all the experiments shown here in Table 1, which also serves to illustrate the reproducibility between different experiments (compare the three sets of values for each of mutants 2, 3, and 3.1).
TABLE 1.
Relative initiation frequency at each AUG in domain VI
| Figure no. | Lane no. | Mutanta | Relative initiation frequency (%) at:b |
|||
|---|---|---|---|---|---|---|
| AUG-576 | AUG-593 | AUG-611 | AUG-626 | |||
| 3A | 2 | 2 | 43 | − | 57 | − |
| 3 | 3 | 36 | 64 | ND | − | |
| 5 | 2.1 | 86 | − | 14 | − | |
| 6 | 3.1 | 92 | 8 | ND | − | |
| 7 | wt.2 | − | − | 100 | − | |
| 8 | 2.2 | − | − | 100 | − | |
| 9 | 3.2 | − | 100 | ND | − | |
| 3B | 2 | 2 | 7 | − | 93 | − |
| 3 | 3 | 6 | 66 | 28 | − | |
| 5 | 2.1 | 48 | − | 52 | − | |
| 6 | 3.1 | 45 | 41 | 14 | − | |
| 7 | wt.2 | − | − | 100 | − | |
| 8 | 2.2 | − | − | 100 | − | |
| 9 | 3.2 | − | 60 | 40 | − | |
| 5 | 1 | 2 | 7 | − | 93 | − |
| 2 | 2d | 12 | − | 88 | − | |
| 3 | 3 | 7 | 70 | 23 | − | |
| 4 | 3d | 7 | 88 | 5 | − | |
| 5 | 3.1 | 47 | 37 | 16 | − | |
| 6 | 3.1d | 50 | 46 | 4 | − | |
| 7 | 1 | 2 | 7 | − | 93 | − |
| 2 | 3 | 7 | 68 | 25 | − | |
| 3 | 3m | 10 | 82 | − | 8 | |
| 4 | 3.1 | 46 | 37 | 17 | − | |
| 5 | 3.1m | 55 | 39 | − | 6 | |
Data for the wild-type and mutant wt.1 are not included because the usage of AUG-576 (which is out of frame in these mRNAs) is unknown.
ND, no product initiated at this AUG was detected; −, the AUG was not present in this mutant.
Initiation site selection on the monocistronic IRES-deleted constructs.
Before examining initiation site selection directed by the HRV IRES, it is instructive to first see what happens with the IRES-deleted monocistronic mRNAs which will be translated by the cap-dependent scanning mechanism. In general, the results (Fig. 3A) conformed with the expectations of cap-dependent initiation via the scanning model. For example, when AUG-576 was removed by point mutation, all initiation took place at the next AUG downstream, i.e., at AUG-593 when present and otherwise at AUG-611, with no significant change in the total number of initiation events occurring in domain VI (Fig. 3A, lanes 7 to 9). Conversely, when the context of AUG-576 was optimized, the large increase in initiation frequency at this site was accompanied by a corresponding decrease in initiation at downstream sites (Fig. 3A, compare lane 2 with 5, and lane 3 with 6). In addition, when AUG-593 was introduced, it took complete precedence over AUG-611 (Fig. 3A, lanes 3, 6, and 9).
FIG. 3.

Products of in vitro translation of wild-type and mutant HRV IRES-derived mRNAs. The data are shown for the mutants in which the local sequence context of AUG-576 had been improved and also for mutants in which AUG-576 had been mutated to GUA. The translation products were separated by SDS-PAGE, and the autoradiographs of the dried gels are shown. The positions of cyclin (upstream cistron) and NS derivatives (downstream cistron) are shown. The intensity of each of the NS-derived products was quantitated by densitometry. The intensity of each band, relative to the intensity of the wild-type AUG-611 product (set at 100), is shown in the table below each autoradiograph. of, the AUG codon was out of frame with the reporter cistron; −, the AUG was absent from the mRNA under test; nd, the band was nondetectable. (A) Translation products of capped monocistronic mRNAs (lacking the IRES and cyclin cistron) at 5 μg/ml. (B) Translation products of uncapped dicistronic mRNAs at 5 μg/ml. Lane M was loaded with marker proteins (56, 43, and 36 kDa).
The slight surprise is that the 5′-proximal AUG-576 was not as efficient an initiation site as might be expected, even taking context effects into account (21). In the wild-type context only ∼40% of all initiation events in domain VI occurred at AUG-576 (Fig. 3A, lane 2), despite the presence of a favorable G residue in the +4 position. When the context was optimized (ACCAUGG), this AUG still failed to capture all of the scanning ribosomes despite its 5′-proximal position (Fig. 3A, lanes 5 and 6); in contrast, when AUG-593 was inserted as the most 5′-proximal AUG with a similarly favorable context (AAAAUGG), it exerted complete precedence over AUG-611 (Fig. 3A, lanes 3, 6, and 9). This suggests that the discriminatory mechanism described in the Discussion senses something unfavorable and negative about the 576 site outside its local context (positions −3 to +4).
Another surprise was that the total frequency of domain VI initiation events was much lower with mutant 3.1 than with any other mRNA, a result which was consistently observed even with different mRNA preparations. There is no obvious explanation for this apparent anomaly, but it needs emphasizing that a similar outcome was not seen with the IRES-dependent mRNAs, as shown in Fig. 3B, 5, and 7.
FIG. 5.

Products of in vitro translation of uncapped dicistronic mRNAs with mutant HRV IRESs, in which the base pairing of the apical part of domain VI has been disrupted (mutants 2d, 3d, and 3.1d), and their corresponding controls. Each mRNA was translated at a final concentration of 5 μg/ml, and the translation products were separated by SDS-PAGE. The autoradiograph of the dried gel is shown. The positions of cyclin (upstream cistron) and NS derivatives (IRES-dependent cistron) are shown on the left. The intensity of each of the NS-derived products was quantitated by densitometry and is expressed relative to the intensity of the AUG-611 product of mutant 2 (set at 100) in the table below the autoradiograph. −, the AUG concerned was absent from the mRNA under test.
FIG. 7.
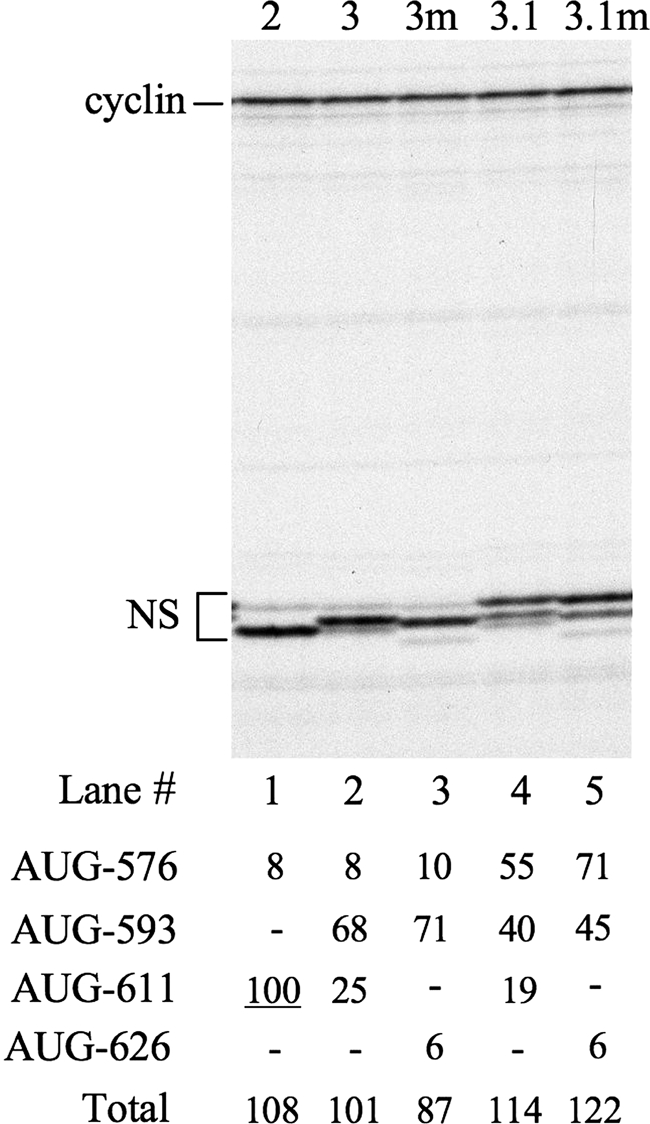
Products of in vitro translation of uncapped dicistronic mRNAs with mutant HRV IRESs, in which AUG-611 had been destroyed and a new AUG was introduced at nt 626 (mutants 3m and 3.1m), and their corresponding controls. Each mRNA was translated at a final concentration of 5 μg/ml, and the translation products were separated by SDS-PAGE. The autoradiograph of the dried gel is shown. The positions of cyclin (upstream cistron) and NS derivatives (IRES dependent) are shown on the left. The intensity of each of the NS-derived products was quantitated by densitometry and expressed relative to the intensity of the AUG-611 product of mutant 2, which was set at 100. The values are shown in the table below the autoradiograph. −, the AUG concerned was absent from the mRNA under test.
Initiation site selection under IRES-dependent conditions.
When the same mutants were examined in the background of dicistronic mRNAs with the HRV IRES (Fig. 2A), three striking differences were evident. First, regardless of the actual local sequence context, the initiation frequency at AUG-576 was much less than in a cap-dependent monocistronic mRNA with the same context (Table 1). Second, mutation of AUG-576 to GUA drastically reduced the frequency of initiation occurring at downstream sites within domain VI. Third, when AUG-593 was inserted, it did not exert the complete precedence over AUG-611 seen with the scanning-dependent monocistronic constructs.
When AUG-576 was in its wild-type context, only 6 to 7% of all domain VI initiation events occurred at this site (Table 1; Fig. 3B, lanes 2 and 3). With the improved context there was a modest 15 to 20% increase in overall productive ribosome recruitment, as reflected in an increase in the total number of initiation events, but only 45 to 50% of all initiation occurred at the 576 site (Table 1; Fig. 3B, lanes 5 and 6). This unexpectedly low use of an AUG in a supposedly optimal context is reminiscent of what happens with scanning-dependent mRNAs when the AUG is positioned very close to the 5′ cap (22, 40), and in this case it is considered to be due to some imprecision or variability in the exact position where the 40S ribosomal subunit is loaded and starts scanning. Accordingly, we examined the effect of lengthening the oligopyrimidine tract by 10 residues but found that this made no significant difference to the total number of initiation events occurring within domain VI or to the proportion of such events occurring at AUG-576 (data not shown). Thus, we conclude that a low efficiency of AUG-576 is an intrinsic feature of IRES-dependent initiation.
When AUG-576 was mutated to GUA, there was a 3- to 4-fold decrease in the total number of IRES-driven initiation events (Fig. 3B, lanes 7 to 9), whereas the same mutation in the monocistronic scanning-dependent mRNA caused a reduction of no more than 20% (Fig. 3A, lanes 7 to 9). This result parallels what has been seen previously with poliovirus IRESs, as described in the introduction. We noted, however, that in one such previous study where the absence of AUG-586 in the PV-1 infectious clone was quasi-infectious, the presence of a GUG in this position resulted in a small-plaque phenotype (42). Given that in all entero- and rhinoviruses this AUG is invariably followed by GU, and usually GUG (Fig. 1), the ability of GUG to act as a weak substitute for AUG-586 in PV-1 may explain why mutation of HRV-2 AUG-576 does not completely abolish all initiation at downstream sites. However, we found that the effect of the double mutation (AUGGUG → GUAGUA) was indistinguishable from that of the AUG → GUA single mutation (data not shown). Thus, the residual initiation at downstream sites when just the AUG is destroyed does not seem to be dependent on the neighboring GUG.
The other peculiarity of IRES-dependent initiation is that the relative use of AUG-593 and AUG-611 is only in the range of 2:1 to 3:1 (Fig. 3B, lanes 3 and 6), whereas the scanning-dependent system showed complete preference for AUG-593 over AUG-611 (Fig. 3A, lanes 3, 6, and 9). This weaker precedence of AUG-593 over the downstream AUG is again similar to what is observed with cap-dependent initiation when the 5′-proximal AUG is very close to the 5′ cap (22, 40), and so a possible explanation for the weaker preference in IRES-dependent initiation could be that AUG-593 is too close to the actual 40S subunit entry point, or landing site. An alternative explanation is that in IRES-dependent initiation, the upper part of the base-paired stem fails to open completely 25 to 30% of the time (as depicted in Fig. 1B), preventing access to AUG-593 and allowing a ribosomal bypass to AUG-611. We will show later, in Fig. 5, results which favor the latter explanation, but first we describe the outcome of an alternative approach to examining the status of base pairing in the domain VI stem-loop.
Oligodeoxynucleotide/RNase H assays of stem opening.
In order to test whether the stem-loop of domain VI opens during translation, we developed an oligodeoxynucleotide/RNase H cleavage assay. Two oligodeoxynucleotides were used in separate reactions: a 26- or 27-mer oligonucleotide (designated S1 or S2, respectively), complementary to the 5′ side and apical loop of stem VI of different mutants, and a 20-mer control (designated C), complementary to a region in the NS reporter ORF. In the presence of the control oligodeoxynucleotide, RNase H is expected to cleave the dicistronic RNA into a fragment ∼2,040 nt long (composed of the cyclin gene plus the HRV IRES and the first ∼40 nt of the NS coding region) and an ∼810-nt fragment composed of the rest of the NS sequence. With the stem-specific oligodeoxynucleotides (S1 and S2), the dicistronic RNA should be cleaved into an ∼1,970-nt fragment composed of the cyclin gene plus the 5′ end of the HRV IRES and an ∼880-nt fragment composed of the entire NS sequence plus the 3′ side of domain VI (Fig. 4A). Unless otherwise stated, these assays were done under conditions suitable for in vitro translation reactions, and the translation products were examined in parallel (Fig. 4B).
FIG. 4.

Oligodeoxynucleotide/RNase H cleavage assays performed on uncapped dicistronic mRNA containing the wild-type HRV-2 IRES. The assays were performed under various different incubation conditions, including the combination of RRL and HeLa cell extract (RRL+HeLa) to which was also added the inhibitors of protein synthesis, edeine, cycloheximide (CHX), and the dominant negative eIF4A R362Q mutant. Both the control (C) and S1 test (S) oligodeoxynucleotides were added at a 20-fold molar excess with respect to the dicistronic mRNA. 4G-depl RRL, eIF4G-depleted RRL. (A) The RNA products of the RNase H assay. The positions of the intact dicistronic mRNA and the cleavage products are shown on the left, while the markers are shown on the right. (B) The in vitro translation products synthesized in the RNase H assay. The positions of cyclin (upstream cistron product) and NS derivatives (downstream cistron products) are shown on the left. Lane M, molecular weight marker.
Apart from the different methods, it is important to appreciate that there is another significant difference between this approach and the translation assays presented in Fig. 3. The cleavage assay reports the status of all the mRNA present in the system without differentiating between mRNA that is being actively translated and the pool of untranslated RNAs. In contrast, in showing the accessibility of AUGs-576, -593, and -611, the translation assays give information, albeit indirect, on the status of just those mRNAs that are actually being translated.
With oligonucleotide C, efficient cleavage of a dicistronic mRNA with the wild-type domain VI was seen under all conditions (Fig. 4A), even in the presence of buffer alone (Fig. 4A, lane 17). This is as expected because oligonucleotide C was targeted at a segment of the NS ORF that is predicted to be largely unstructured. The parallel translation reactions confirm that cleavage in the presence of oligonucleotide C was almost complete, with only a trace of full-length NS protein still produced (Fig. 4B, lane 2), together with a low yield of some smaller products which are likely to have arisen by leaky scanning of the uncapped NS reporter cistron fragment. In contrast, with oligonucleotide S1 only a trace of cleavage was seen in buffer alone (Fig. 4A, lane 16), and even under translation conditions cleavage was rather incomplete (Fig. 4A, lane 2). Maximum cleavage was seen in the mixed RRL/HeLa translation system (Fig. 4A, lane 2), and the parallel translation assays show that this was the only condition under which significant amounts of NS protein were produced in the presence of S1 (Fig. 4B, lane 1), which is likely to be due to a combination of IRES-dependent translation of residual uncleaved dicistronic mRNA and translation of the uncapped, but complete, NS reporter ORF fragment by scanning. Cleavage in the presence of S1 was rather inefficient in unsupplemented RRL (Fig. 4A, lane 10), which is unable to carry out IRES-dependent translation because it is deficient in the required trans-acting factors (11, 12). The HeLa cell HS-S100, which has these factors but is incapable of translation because it lacks ribosomes (Fig. 4B, lanes 11 and 12), shows significantly better cleavage in the presence of oligonucleotide S1 (Fig. 4A, lane 12).
Cleavage with oligonucleotide S1 in the mixed RRL/HeLa system was reduced by the presence of all three translation inhibitors tested (edeine, cycloheximide, and the dominant negative eIF4A R362Q mutant), with the eIF4A mutant showing the strongest inhibition and edeine the weakest (Fig. 4A, compare lane 2 with lanes 4, 6, and 8). Edeine inhibits recognition of the AUG initiation codon, and in cap-dependent translation the 40S subunits usually scan past such codons (24); with these IRES-dependent mRNAs, however, edeine is also likely to inhibit 40S ribosomal subunit recruitment for the reasons explained in the Discussion. Cycloheximide will stall ribosomes on the mRNA immediately following the ribosomal subunit joining step which leads to 80S initiation complex formation, and this would protect 28 nt of mRNA centered approximately on the AUG wobble position (see Discussion). With the wild-type IRES construct used here, most such 80S initiation complexes will be at AUG-611, with a few at AUG-576 (Fig. 3B, lanes 1 and 2). Initiation complexes formed at either site would almost certainly force the complete opening of domain VI, but with the difference that those at AUG-611 would most likely not prevent annealing of S1 to the 5′ side of domain VI and the consequent cleavage while those at AUG-576 would block annealing and cleavage. Dominant negative eIF4A R362Q will inhibit the activity of the eIF4F complex (36), resulting in inhibition of the mRNA unwinding dependent on this factor and also inhibition of productive 40S subunit loading onto the mRNA (Fig. 4B, lanes 7 and 8), which requires either the complete eIF4F complex or, at a minimum, the central domain of its eIF4G component plus eIF4A (31).
Taken as a whole, these oligodeoxynucleotide/RNase H cleavage assays show that the default status of domain VI under physiological conditions of salt and temperature is the closed base-paired conformation, but it can be opened in a process dependent on eIF4F (or eIF4G/eIF4A) and other factors which are present in higher abundance in HeLa cells than in RRL.
The effect of destabilizing the domain VI stem-loop.
Although the results described in the previous section show that the domain VI stem-loop can be opened, there remains a possibility that the apical part might sometimes remain closed and base paired (Fig. 1B), which could prevent ribosomes from accessing AUG-593 (if present) and encourage a ribosome bypass to AUG-611. To explore this possibility, we examined the effect of mutations which should open the upper part of this stem. These mutations (mutants 2d, 3d, and 3.1d in Fig. 2) were introduced into the 3′ side of the stem, leaving the sequence of the 5′ side intact since the conservation of the 5′ sequence in all rhino- and enteroviruses and even in bovine enterovirus, which lacks the 3′ complementary sequence and so lacks a base-paired stem, suggests that the primary sequence just downstream of HRV-2 AUG-576 could be important. The mutations were made in only the upper part of the stem, thus leaving the lower part still capable of base pairing in theory though likely destabilized by the larger apical loop. In an otherwise wild-type background, these mutations had no significant effect on the frequency of initiation at AUG-611, nor did destabilizing the stem stimulate productive ribosome recruitment since there was no significant increase in total initiation events (Fig. 5, compare lanes 1 and 2). However, when AUG-593 was present, the destabilizing mutations increased the relative frequency of initiation at this site (Table 1; Fig. 5, compare lanes 3 and 4 and lanes 5 and 6) at the expense of an ∼4-fold decrease in the proportion of initiation events occurring at AUG-611 (Table 1; Fig. 5, compare lanes 3 and 4 and lanes 5 and 6). (Note that the amino acid sequence changes resulting from the destabilizing mutations slightly reduced the mobility of the products initiated at AUG-576 and -593.) Thus, the effect of the mutations was to make AUG selection more processive, consistent with the view that destroying the base pairing at the top of the stem favored strictly linear scanning and therefore enhanced the precedence of AUG-593 over AUG-611.
Because the distance of AUG-593 from the putative 40S subunit entry point (landing site) at or around AUG-576 was unchanged in these destabilized mutants, these results indicate that the reason why some ribosomes initiate at AUG-611 rather than at AUG-593 in the parent construct is not likely to be due to the latter site being too close to this entry point. On the contrary, the findings favor the notion that, in the absence of the destabilizing mutants, a few ribosomes select AUG-611 over AUG-593 because the stem-loop sometimes fails to open completely (Fig. 1B), and when this happens, some ribosomes bypass AUG-593 by a type of skipping process.
When the oligodeoxynucleotide/RNase H cleavage assay was used to test whether this mutation had actually destabilized the domain VI stem, we found that cleavage in the presence of oligonucleotide S2, which is complementary to the mutant 3.1d sequence, could, indeed, now occur efficiently even in buffer alone (Fig. 6A, lane 6). Moreover, cleavage of mutant 3.1d was as efficient in unsupplemented RRL as in the mixed RRL/HeLa system (Fig. 6A, lanes 2 to 6), in contrast to results with the wild-type stem-loop (Fig. 4A, compare lane 2 with lane 10). The parallel translation assays of the destabilized mutant show that cleavage with oligonucleotide S2 decreases IRES-dependent initiation at AUG-576 and -593 in the mixed RRL/HeLa system, as expected, and results in the appearance of a product of the same size as NS, which likely arises from scanning-dependent translation of the uncapped NS cistron fragment (Fig. 6B, compare lanes 9 and 10) and which is also seen, but at lower yield, in the corresponding assay with unsupplemented RRL (lane 7).
FIG. 6.

Oligodeoxynucleotide/RNase H cleavage assays performed on uncapped dicistronic mRNA with HRV IRES mutant 3.1d in which the base pairing of the apical part of domain VI has been disrupted. The assays were performed under various different incubation conditions in the presence or absence of cycloheximide (CHX), as indicated. Both the control (C) and S2 test (S) oligodeoxynucleotides were added at 20-fold molar excess with respect to the dicistronic mRNA. (A) The RNA products of the RNase H assay. The positions of the intact dicistronic mRNA and the RNase H cleavage products are shown on the left. In RNase H lanes the reaction was performed in buffer. (B) The in vitro translation products synthesized in the RNase H assays (lacking cycloheximide) of dicistronic mRNAs with the wild-type or mutant 3.1d IRES. −, no oligodeoxynucleotide was added. The positions of cyclin (upstream cistron product) and NS derivatives (downstream cistron products) are shown on the left.
As a converse to disrupting the base pairing at the top of the stem, we tried increasing the stability of this pairing by introducing a number of G-C pairs near the top in mutants 2s, 3s, 2.1s, and 3.1s (Fig. 2). This had a strong negative effect on initiation at all sites, reducing it even more drastically than the mutation of AUG-576 to GUA (data not shown). A possible explanation is that the G-C pairing extended so far down the stem that it prevented sufficient opening of the base of the stem to allow ribosome recruitment to the putative entry site at AUG-576.
The effect of displacing AUG-611.
Because the above results suggest that the initiation seen at AUG-611 when the domain VI stem is fully base paired might arise from some ribosomes bypassing AUG-593, we asked whether 611 was in some way a privileged position for initiation by these ribosomes. In this respect it is noteworthy that the back-to-back configuration of AUG-576 and -611 (Fig. 1A) is conserved in all human rhinoviruses (35). Accordingly, we displaced this AUG further downstream by 5 codons in the background of a construct which included AUG-593 (mutants 3m and 3.1m in Fig. 2). The relative frequency of initiation at this new position (designated AUG-626) was 3- to 4-fold lower than when it was at position 611 in the background of a construct which likewise included AUG-593 (Table 1; Fig. 7, compare lane 3 with 2 and lane 5 with 4). This suggests that whatever the mechanism may be by which some ribosomes bypass AUG-593 and initiate at the next AUG further downstream, AUG-611 is in an especially favorable position for this to occur.
DISCUSSION
The results of this work need to be evaluated in the light of what is known about how ribosomes interact with mRNA. An 80S ribosome which is in elongation mode protects a length of 28 nt of mRNA from promiscuous ribonucleases that exhibit no sequence specificity, and 80S initiation complexes cover the same length (14, 48). In both cases, toe printing shows that the leading edge of the 80S ribosome is 16 nt downstream of the first residue of the P-site codon (40, 43), and so the 28-nt protected fragment is centered approximately on the wobble position of this P-site codon. In addition, ribosomes that are in elongation mode are capable of unwinding almost all RNA secondary structures that they encounter. So, for a ribosome to initiate at HRV-2 AUG-576, domain VI would likely have to unwind completely, and it would stay unwound until the trailing edge of the ribosome had completely cleared domain VI, whereupon this domain could snap back into its base-paired state unless another ribosome had initiated translation at AUG-576 in the meantime.
In contrast, 48S initiation complexes (a 40S subunit with associated initiation factors plus initiator tRNA and bound to mRNA at the initiation codon) protect a much greater length of mRNA against RNase A and RNase T1 than 80S complexes (20, 23, 28, 29, 30): at least 65 nt (provided the mRNA 5′ UTR is longer than ∼50 nt) yet with the leading edge 16 nt downstream of the P-site codon, exactly the same as in 80S initiation complexes (38, 40). The extra 35+ nt protected against nuclease attack by the 48S complex must therefore be mRNA sequences further upstream, i.e., on the 5′ side of the initiation complex. These are likely to be protected by eIF3, which interacts with the 40S subunit in an appropriate position for binding these upstream sequences, and by the eIF4F complex (consisting of eIF4A, -4E, and -4G), which interacts with eIF3 via its eIF4G subunit (46).
The 28-nt segment of mRNA that is in direct contact with an 80S ribosome in elongation mode is almost certainly fully unstructured as a result of unwinding by the advancing ribosome, but this need not necessarily be the case at initiation, as is shown (for eubacterial initiation) by two unusual bacteriophage T4 mRNAs, gene 25 and gene 38 mRNAs. In both of these cases the linear spacing between the Shine-Dalgarno (S-D) motif and the AUG initiation codon appears to be far too long to allow efficient initiation if it were completely unstructured, but in both cases there is the potential for a hairpin structure, located between the S-D motif and the AUG, to bring the S-D motif into an appropriate position to allow the 30S ribosomal subunit to form the S-D interaction and bind with the AUG in its P site. For gene 38 mRNA the hairpin looks very stable, due to a UNCG apical loop flanked by four contiguous G-C pairs, so that it must surely exist in reality (7). The putative hairpin in gene 25 mRNA appears less stable, but its existence is supported by the effects on gene 25 expression and T4 burst size (phage yield) of mutations that would destabilize the base pairing and of compensatory mutations that would restore it (34). Thus, it appears that in bacterial 30S initiation complexes, the ribosomal subunit does not bind the whole mRNA segment in a tightly closed channel but in something more akin to a open slot or “trough” that could accommodate an mRNA fragment with a small hairpin, at a distance of 4 to 8 nt upstream of the AUG, with the hairpin protruding out of the open slot (7).
If initiating mammalian 40S subunits engage with mRNA in a similar manner, then HRV domain VI need not necessarily open completely to allow initiation at AUG-611, and initiation could still occur even when the apical part of the stem remained paired, as depicted in Fig. 1B.
The results obtained with the IRES-deleted cap-dependent monocistronic constructs were largely as expected from the scanning ribosome model, assuming complete unwinding of domain VI. Thus, mutation of AUG-576 to GUA resulted in increased initiation at the next downstream AUG, whereas improving the context of AUG-576 had the opposite effect; moreover, if AUG-593 was present, it took complete precedence over AUG-611. The only surprises were that AUG-576 in its wild-type context was rather less efficient as an initiation site than might be expected (21), given that it has a G in the +4 position and that optimizing its context did not result in this AUG capturing all the scanning ribosomes. This suggests that this initiation site may have some (unidentified) negative features outside the local context (−3 to +4, inclusive).
A comparison of initiation site utilization under IRES-dependent conditions with the above results for cap-dependent (monocistronic) mRNA shows some striking differences. First, initiation at AUG-576 was even less efficient with the IRES than in a monocistronic mRNA with the same local sequence context. Even more striking is the fact that mutation of this AUG to a non-AUG codon strongly reduced initiation at all downstream AUGs. Last, the precedence of AUG-593 (when present) over initiation at AUG-611 was significantly weaker in IRES-dependent initiation than in cap-dependent scanning initiation.
Undoubtedly, the most puzzling feature of HRV-2 AUG-576 is that it is clearly important (though not absolutely essential) for productive recruitment of ribosomes, yet very little initiation occurs at this site. Its importance for ribosome recruitment suggests that the anticodon of Met-tRNAi associated with the incoming 40S subunit engages the AUG codon, resulting in either an increase in the on-rate of 40S subunit binding or a decrease in the off-rate. Such engagement would certainly require at least the lower half of domain VI to be melted. In the scanning mechanism of initiation, codon-anticodon pairing normally leads to commitment to initiate, especially if the context is highly favorable. This commitment step involves 40S-associated eIF1 moving its position on the 40S subunit in a way that allows eIF5, which has GAP function, to trigger hydrolysis of eIF2-associated GTP and phosphate release (15, 47). In the absence of eIF1, cap-dependent initiation occurs equally efficiently at any AUG (and even at some non-AUG codons), irrespective of local sequence context or of whether it is located within 8 nt of the 5′-cap; when eIF1 is present, however, cap-proximal AUGs and AUGs with a highly unfavorable context are ignored (40), presumably because there is no movement of the eIF1, and so the 40S subunit continues scanning. eIF1 therefore acts as a critical discriminatory factor in initiation site selection. Our results with the monocistronic IRES-deleted mRNAs show that AUG-576 in its native context is quite an inefficient initiation site, and even with an optimized context it is less than 100% efficient, suggesting that the eIF1-dependent discrimination mechanism senses some negative features other than immediate local sequence context (positions −3 to +4, inclusive). Moreover, the discrimination mechanism seems to sense that this site is even more unfavorable in the situation of IRES-dependent initiation because there is even less initiation at AUG-576 in the IRES-dependent mRNAs than in a cap-dependent monocistronic mRNA with the same local sequence context. One possible explanation might be that the additional negative feature is the persistence of some base pairing of the apical part of domain VI, as in the intermediate structure pictured in Fig. 1B, but this can likely be discounted because deliberately destroying this base pairing did not result in a really significant increase in initiation at AUG-576 (Table 1 and Fig. 5).
The properties of HRV-2 AUG-576 are very similar to those of the equivalent AUG in polioviruses. Mutation of this AUG in PV-2 IRES constructs with a reporter fused to the polyprotein initiation site reduced reporter expression in both transfection assays and in vitro translation to the same degree we observed (32, 33). Its mutation to UUG in a PV-2 infectious clone conferred a small-plaque phenotype (37), and the absence of the equivalent AUG (at nt 586) in the PV-1 infectious clone was quasi-infectious (42). In an independent study, no detectable initiation at PV-1 AUG-586 was observed in vitro unless its context was improved, which resulted in an ∼50% decrease in initiation at the polyprotein initiation site (39).
Although the FMDV IRES is often considered similar to the PV IRES in so far as a minority of initiation events occur at the Lab initiation site, i.e., the AUG immediately downstream of the oligopyrimidine tract (3, 45), and a majority occur at the next AUG (the Lb site), we will show elsewhere that initiation site selection on the FMDV IRES differs markedly from that of entero-/rhinovirus IRESs. In particular, mutation of the upstream Lab AUG usually results in increased initiation at the Lb site and in only a modest reduction in total initiation events rather than the strong decrease seen with mutation of the equivalent AUG in entero-/rhinoviruses.
With the native HRV IRES, almost all initiation occurs at AUG-611 (apart from the very low initiation at AUG-576), implying that following initial binding at AUG-576, the 40S initiation complex is transferred to nt 611. If this transfer occurred invariably by strictly linear scanning, we would expect that an AUG inserted at nt 593 would take virtually complete precedence over AUG-611, just as was the case with the monocistronic cap-dependent IRES-deleted mRNA (Fig. 3A). However, in the IRES background the precedence was significantly less extreme, with initiation frequency at 593 only 2- to 3-fold higher than that at AUG-611. It is unlikely that AUG-593 is underutilized because it is too close to AUG-576 (which is where such scanning probably starts), given that these two sites are 17 nt apart, yet in cap-dependent mRNAs, it is only AUGs within 8 nt of the cap that are bypassed if eIF1 is present (40). Moreover, we found that when the domain VI stem was destabilized, the precedence of AUG-593 over AUG-611 markedly increased even though there was no change in the distance between AUG-576 and AUG-593.
This outcome implies that destabilizing the domain VI stem has made the linearity of scanning more stringent, which in turn suggests that with the native domain VI base pairing, some 40S initiation complexes bypass AUG-593. This could occur if the apical part of domain VI sometimes remained base paired (as depicted in Fig. 1B) at the time of transfer of the 40S initiation complex from AUG-576 to downstream sites; as a result AUG-593 would be bypassed, and the initiation complex would engage AUG-611 with an mRNA configuration quite similar to that which is thought to be adopted at initiation on bacteriophage T4 gene 25 and gene 38 mRNAs, as described above.
We also found that displacing AUG-611 from its back-to-back position with respect to AUG-576 and moving it 15 nt downstream markedly reduced initiation at this site (when AUG-593 was present), indicating that nt 611 is an especially favorable position, presumably for those ribosomes which bypass AUG-593 rather than for those which access AUG-611 by strictly linear scanning. It is interesting that when an AUG codon was introduced into PV-1 at the equivalent back-to-back position (which coincidentally happens to be at PV-1 nt 611), this codon was used in vitro at a much higher frequency than if the AUG was placed, in what seemed to be an equally favorable local sequence context, just one codon further downstream, at nt 614 (8). Taken together with our results, this suggests that the back-to-back configuration of the two AUG triplets is strongly favorable for initiation at the downstream AUG in a way that cannot readily be explained by an effect on scanning from the upstream AUG but could be explained if some ribosomes access AUG-611 by a direct transfer process or bypass.
The recent sequence analysis of over 100 HRV strains (35), published after our experimental work had been completed, showed that this back-to-back configuration is conserved in all of them, and the authors suggested that this would favor ribosomes accessing the authentic initiation site by a mechanism of “switching” directly to it from the upstream AUG triplet without scanning the intervening sequence. Our results indicate that such switching can, indeed, occur, though in fact the data suggest that only 25 to 35% of the ribosomes accessed AUG-611 by this bypass mechanism in our experiments. However, the relative frequencies of bypass versus scanning could well be influenced by the particular conditions (K+ and Mg2+ concentrations and temperature, for example), and so we cannot exclude the possibility of a higher frequency of switching in the infected cell.
Although our proposed bypass may seem at first sight to be similar to the process known as ribosome shunting, we think there are significant differences, apart from the fact that the two processes can coexist on the same mRNA, with some ribosomes scanning the whole 5′ UTR and some shunting. Cauliflower mosaic virus (CaMV) 35S mRNA and the adenovirus late mRNAs transcribed from the major late promoter (so all share the same tripartite 5′ UTR) are by far the best understood examples of shunting. In both cases initiation is cap dependent and involves the 43S preinitiation complex scanning through the first section of the 5′ UTR, which is unstructured (10, 49). However, the 43S complex then seems to dissociate from the mRNA and reengage with it close to the initiation codon, which is ∼180 nt further downstream from the putative take-off point in the case of the adenovirus mRNAs and over 500 nt downstream in CaMV 35S mRNA. The intervening segment which is bypassed by this shunt consists of a very large (∼480 nt) irregular stem-loop in CaMV mRNA and three shorter irregular hairpins in the adenovirus mRNAs. In both cases these structures seem essential for shunting (10, 49, 50) although a shorter perfectly base-paired hairpin (stability of −48 kcal/mole) can substitute for the large native structure in CaMV 35S mRNA (9). Remarkably, shunting on the adenovirus mRNAs is unaffected by insertion, at a site just 25 nt upstream of the initiation codon, of a perfect hairpin that is sufficiently stable (−70 kcal/mole) to act as a complete barrier to scanning (49). Thus, there seems little doubt that for a very brief moment, between the disengagement of the 43S complex from the mRNA and reengagement at the downstream site, there is no mRNA in the 43S mRNA binding channel even though the 43S complex may well remain tethered to the mRNA in some other way. In contrast, we envisage that the mRNA channel is always occupied during transfer of the 43S complex over the short distance from HRV AUG-576 to AUG-611, but the mRNA may sometimes pass through this channel with the extreme apical part of the domain VI stem still base paired, which results in the bypassing of AUG-593 (when present).
Acknowledgments
We thank Jenny Reed and Rosemary Farrell for technical support and Ian Brierley for helpful advice.
This work was supported by a Wellcome Trust Programme grant (062348).
Footnotes
Published ahead of print on 28 April 2010.
REFERENCES
- 1.Alexander, L., H. H. Lu, and E. Wimmer. 1994. Polioviruses containing picornavirus type 1 and/or type 2 internal ribosomal entry site elements: genetic hybrids and the expression of a foreign gene. Proc. Natl. Acad. Sci. U. S. A. 91:1406-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali, I. K., L. McKendrick, S. J. Morley, and R. J. Jackson. 2001. Truncated initiation factor eIF4G lacking an eIF4E binding site can support capped mRNA translation. EMBO J. 20:4233-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belsham, G. J. 1992. Dual initiation sites of protein synthesis on foot-and-mouth disease virus RNA are selected following internal entry and scanning of ribosomes in vivo. EMBO J. 11:1105-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borman, A., M. T. Howell, J. G. Patton, and R. J. Jackson. 1993. The involvement of a spliceosome component in internal initiation of human rhinovirus RNA translation. J. Gen. Virol. 74:1775-1788. [DOI] [PubMed] [Google Scholar]
- 5.Borman, A., and R. J. Jackson. 1992. Initiation of translation of human rhinovirus RNA: Mapping the internal ribosome entry site. Virology 188:685-696. [DOI] [PubMed] [Google Scholar]
- 6.Dasso, M. C., and R. J. Jackson. 1989. On the fidelity of mRNA translation in the nuclease-treated rabbit reticulocyte lysate system. Nucleic Acids Res. 17:3129-3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gold, L. 1988. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu. Rev. Biochem. 57:199-233. [DOI] [PubMed] [Google Scholar]
- 8.Hellen, C. U. T., T. V. Pestova, and E. Wimmer. 1994. Effect of mutations downstream of the internal ribosome entry site on initiation of poliovirus protein synthesis. J. Virol. 68:6312-6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hemmings-Mieszczak, M., and T. Hohn. 1999. A stable hairpin preceded by a short open reading frame promotes nonlinear ribosome migration on a synthetic mRNA leader. RNA 5:1149-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hohn, T., S. Corsten, D. Dominguez, J. Fütterer, D. Kirk, M. Hemmings-Mieszczak, M. Pooggin, N. Schärer-Hernandez, and L. Ryabova. 2001. Shunting is a translation strategy used by plant pararetroviruses (Caulimoviridae). Micron 32:51-57. [DOI] [PubMed] [Google Scholar]
- 11.Hunt, S. L., J. J. Hsuan, N. Totty, and R. J. Jackson. 1999. Unr, a cellular cytoplasmic RNA-binding protein with five cold shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 13:437-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt, S. L., and R. J. Jackson. 1999. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA 5:344-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iizuka, N., M. Kohara, K. Hagino-Yamagishi, S. Abe, T. Komatsu, K. Tago, M. Arita, and A. Nomoto. 1989. Construction of less neurovirulent polioviruses by introducing deletions into the 5′ noncoding sequence of the genome. J. Virol. 63:5354-5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingolia, N. T., S. Ghaemmaghami, J. R. S. Newman, and J. J. Weissman. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324:218-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson, R. J., C. U. T. Hellen, and T. V. Pestova. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell. Biol. 10:113-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson, R. J., and T. Hunt. 1983. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. Methods Enzymol. 96:50-74. [DOI] [PubMed] [Google Scholar]
- 17.Jackson, R. J., and A. Kaminski. 1995. Internal initiation of translation in eukaryotes: the picornavirus paradigm and beyond. RNA 1:985-1000. [PMC free article] [PubMed] [Google Scholar]
- 18.Kaminski, A., G. J. Belsham, and R. J. Jackson. 1994. Translation of encephalomyocarditis virus RNA: parameters influencing the selection of the internal initiation site. EMBO J. 13:1673-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaminski, A., M. T. Howell, and R. J. Jackson. 1990. Initiation of encephalomyocarditis virus RNA translation: the authentic initiation site is not selected by a scanning mechanism. EMBO J. 9:3753-3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozak, M. 1977. Nucleotide sequences of 5′-terminal ribosome-protected initiation regions from two reovirus messages. Nature 269:391-394. [DOI] [PubMed] [Google Scholar]
- 21.Kozak, M. 1986. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44:283-292. [DOI] [PubMed] [Google Scholar]
- 22.Kozak, M. 1991. A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr. 1:111-115. [PMC free article] [PubMed] [Google Scholar]
- 23.Kozak, M., and A. Shatkin. 1976. Characterization of ribosome-protected fragments from reovirus messenger RNA. J. Biol. Chem. 251:4259-4266. [PubMed] [Google Scholar]
- 24.Kozak, M., and A. Shatkin. 1978. Migration of 40S ribosomal subunits on messenger RNA in the presence of edeine. J. Biol. Chem. 253:6568-6577. [PubMed] [Google Scholar]
- 25.Kuge, S., N. Kawamura, and A. Nomoto. 1989. Genetic variation occurring on the genome of an in vitro insertion mutant of poliovirus type 1. J. Virol. 63:1069-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuge, S., N. Kawamura, and A. Nomoto. 1989. Strong inclination towards transition mutation in nucleotide substitutions by poliovirus replicase. J. Mol. Biol. 207:175-182. [DOI] [PubMed] [Google Scholar]
- 27.Kuge, S., and A. Nomoto. 1987. Construction of viable deletion and insertion mutants of the Sabin strain of type 1 poliovirus: function of the 5′ noncoding sequence in viral replication. J. Virol. 61:1478-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazarowitz, S. G., and H. D. Robertson. 1977. Initiator regions from the small size class of reovirus messenger RNA protected by rabbit reticulocyte ribosomes. J. Biol. Chem. 252:7842-7849. [PubMed] [Google Scholar]
- 29.Legon, S. 1976. Characterization of the ribosome-protected regions of 125I-labelled rabbit globin messenger RNA. J. Mol. Biol. 106:37-53. [DOI] [PubMed] [Google Scholar]
- 30.Legon, S., P. Model, and H. D. Robertson. 1977. Interaction of rabbit reticulocyte ribosomes with bacteriophage f1 mRNA and of Escherichia coli ribosomes with rabbit globin mRNA. Proc. Natl. Acad. Sci. U. S. A. 74:2692-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomakin, I. V., C. U. T. Hellen, and T. V. Pestova. 2000. Physical association of eukaryotic initiation factor 4G (eIF4G) with eIF4A strongly enhances binding of eIF4G to the internal ribosomal entry site of encephalomyocarditis virus is required for internal initiation of translation. Mol. Cell. Biol. 20:6019-6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meerovitch, K., R. Nicholson, and N. Sonenberg. 1991. In vitro mutational analysis of cis-acting RNA translational elements within the poliovirus type 2 5′ untranslated region. J. Virol. 65:5895-5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholson, R., J. Pelletier, S.-Y. Le, and N. Sonenberg. 1991. Structural and functional analysis of the ribosome landing pad of poliovirus type 2: in vivo translation studies. J. Virol. 65:5886-5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nivinskas, R., N. Malys, V. Klausa, R. Vaiskunaite, and E. Gineikiene. 1999. Post-transcriptional control of bacteriophage T4 gene 25 expression: mRNA secondary structure that enhances translational initiation. J. Mol. Biol. 288:291-304. [DOI] [PubMed] [Google Scholar]
- 35.Palmenberg, A. C., D. Spiro, R. Kuzmickas, S. Wang, A. Djikeng, J. A. Rathe, C. M. Frase-Liggett, and S. B. Liggett. 2009. Sequencing and analysis of all known human rhinovirus genomes reveal structure and evolution. Science 324:55-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pause, A., N. Méthot, Y. Svitkin, W. C. Merrick, and N. Sonenberg. 1994. Dominant negative mutants of mammalian translation initiation factor eIF4A define a critical role for eIF4F in cap-dependent and cap-independent initiation of translation. EMBO J. 13:1205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelletier, J., M. E. Flynn, G. Kaplan, V. Racaniello, and N. Sonenberg. 1988. Mutational analysis of upstream AUG codons of poliovirus RNA. J. Virol. 62:4486-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pestova, T. V., S. I. Borukhov, and C. U. T. Hellen. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394:854-859. [DOI] [PubMed] [Google Scholar]
- 39.Pestova, T. V., C. U. T. Hellen, and E. Wimmer. 1994. A conserved AUG triplet in the 5′ nontranslated region of poliovirus can function as an initiation codon in vitro and in vivo. Virology 204:729-737. [DOI] [PubMed] [Google Scholar]
- 40.Pestova, T. V., and V. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pestova, T. V., I. N. Shatsky, S. P. Fletcher, R. J. Jackson, and C. U. T. Hellen. 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12:67-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pilipenko, E. V., A. P. Gmyl, S. V. Maslova, Y. V. Svitkin, A. N. Sinyakov, and V. I. Agol. 1992. Prokaryotic-like cis elements in the cap-independent internal initiation of translation on picornavirus RNA. Cell 68:119-131. [DOI] [PubMed] [Google Scholar]
- 43.Pisarev, A. V., C. U. T. Hellen, and T. V. Pestova. 2007. Recycling of eukaryotic posttermination ribosomal complexes. Cell 131:286-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 45.Sangar, D. V., S. E. Newton, D. J. Rowlands, and B. E. Clarke. 1987. All foot and mouth disease virus serotypes initiate protein synthesis at two separate AUGs. Nucleic Acids Res. 15:3305-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siridechadilok, B., C. S. Fraser, R. J. Hall, J. A. Doudna, and E. Nogales. 2005. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science 310:1513-1515. [DOI] [PubMed] [Google Scholar]
- 47.Unbehaun, A., S. I. Borukhov, C. U. T. Hellen, and T. V. Pestova. 2004. Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev. 18:3078-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolin, S. L., and P. Walter. 1988. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 7:3559-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yueh, A., and R. J. Schneider. 1996. Selective translation initiation by ribosome jumping in adenovirus-infected and heat-shocked cells. Genes Dev. 10:1557-1567. [DOI] [PubMed] [Google Scholar]
- 50.Yueh, A., and R. J. Schneider. 2000. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 14:414-421. [PMC free article] [PubMed] [Google Scholar]