

Abstract
Myeloid differentiation primary response protein 88 (MyD88), which can be induced by alpha interferon (IFN-α), has an antiviral activity against the hepatitis B virus (HBV). The mechanism of this antiviral activity remains poorly understood. Here, we report that MyD88 inhibited HBV replication in HepG2.2.15 cells and in a mouse model. The knockdown of MyD88 expression weakened the IFN-α-induced inhibition of HBV replication. Furthermore, MyD88 posttranscriptionally reduced the levels of viral RNA. Remarkably, MyD88 accelerated the decay of viral pregenomic RNA in the cytoplasm. Mapping analysis showed that the RNA sequence located in the 5′-proximal region of the pregenomic RNA was critical for the decay. In addition, MyD88 inhibited the nuclear export of pre-S/S RNAs via the posttranscriptional regulatory element (PRE). The retained pre-S/S RNAs were shown to degrade in the nucleus. Finally, we found that MyD88 inhibited the expression of polypyrimidine tract-binding protein (PTB), a key nuclear export factor for PRE-containing RNA. Taken together, our results define a novel antiviral mechanism against HBV mediated by MyD88.
Hepatitis B virus (HBV) is a noncytopathic, enveloped virus with a circular, double-stranded DNA genome. It causes both acute and chronic infection of the human liver. Although a highly effective preventive vaccine is now available, HBV infection remains a major health problem worldwide. It is estimated that chronic HBV infection affects 350 to 400 million people globally, about a quarter of whom will eventually develop severe liver diseases, including liver cirrhosis, liver failure, and hepatocellular carcinoma (HCC) (4).
Current antiviral therapies involve the use of nucleoside analogs and alpha interferon (IFN-α) (28). IFN-α, a type I interferon, engages the IFN-α receptor complex to activate the Jak/Stat pathway and trigger the transcription of a diverse set of genes, referred to as IFN-stimulated genes (ISGs) (2, 40). In total, the gene products of ISGs establish an antiviral response in target cells (2, 40). IFN-α inhibits HBV replication through a variety of mechanisms. It was reported previously that IFN-α can suppress viral gene expression, prevent the formation of viral RNA-containing core particles, and reduce the accumulation of viral replicative intermediates (11, 35, 37, 46-48). Importantly, the precise antiviral mechanism of IFN-α and the biological functions of many ISGs have not been fully elucidated.
Myeloid differentiation primary response protein 88 (MyD88) is a key adaptor in the signaling cascade of the innate immune response (22). We and others have shown that MyD88 expression can be induced by IFN-α and that MyD88 has an antiviral activity against HBV in hepatoma cells that is mediated by nuclear factor κB (NF-κB) activation (12, 25, 51, 52). To counteract its inhibition, the HBV polymerase dampens the activation of the MyD88 promoter by blocking the nuclear translocation of Stat1, thereby reducing IFN-α-inducible MyD88 expression (50), further suggesting a critical role for MyD88 in antiviral activity against HBV. The aim of the present study was to further investigate the antiviral activity of MyD88 and the mechanism of action. We show that MyD88 inhibited HBV replication in HepG2.2.15 cells and in a mouse model. Furthermore, MyD88 decreased the stability of HBV pregenomic RNA and inhibited the nuclear export of HBV pre-S/S RNAs mediated by the posttranscriptional regulatory element (PRE).
MATERIALS AND METHODS
Plasmids and viruses.
To construct HBV plasmid pHBV1.3, a terminally redundant (1.3× copy), replication-competent HBV genome (subtype adw; nucleotides 957 to 1952; GenBank accession number AF100309) was inserted into pUC19 (Promega). pCIdA-HBV expresses all the HBV proteins, but it lacks the 5′ ɛ RNA signal, which eliminates pregenomic RNA packaging and DNA replication (27). Both pCMV-HBV (6) and pTet-HBV (36) contain the wild-type HBV 1.1-mer overlength genomic sequence. For these plasmids, the synthesis of the pregenomic RNA is driven by the cytomegalovirus (CMV) promoter and the Tet promoter, respectively. pCMV-HBV M2 is a derivative of pCMV-HBV in which the La protein-binding sites have been mutated (5). pCMV-HBVΔPRE is a PRE deletion mutant of pCMV-HBV (15). The luciferase reporter constructs of HBV promoters/enhancers (ENII/Cp, Sp1, Sp2, and ENI/Xp) were described previously (33). The pRSV138PRE-CAT and pRSV-CAT constructs were described previously (39). To construct Myc-tagged MyD88, polypyrimidine tract-binding protein 1 (PTB1), and nuclear export signal (NES)(−)RanBP plasmids, the complete cDNAs of human MyD88 carried on pCDNA3-MyD88 (51), PTB1 carried on His-PTB1 (43), and NES(−)RanBP carried on green fluorescent protein (GFP)-NES(−)RanBP1 (57) were inserted into the EcoRI and NotI sites of pCMV/Myc vectors (Invitrogen). cDNA fragments of HBV pregenomic RNA from pCMV-HBV and the complete HBV cDNA from pCMV-HBVΔPRE were inserted into pcDNA3.1/Luc (9) at the 3′ end of the luciferase coding sequence before the bovine growth hormone (BGH) polyadenylation signal (see Fig. 6A). The sequence of HBV(1804-2454) was deleted from pCMV-HBV by inverse PCR using a KOD-Plus mutagenesis kit (Toyobo) according to the manufacturer's instructions, and the resultant plasmid was named pCMV-HBVΔ1804-2454. The complete cDNA of the pre-S2/S RNA was inserted into pCDNA3.1 (Invitrogen) and pTRE2hyg (Clontech), and the resultant plasmids were named pCDNA3.1-preS2/S and pTRE2hyg-preS2/S, respectively. All primer sequences are available upon request. All of the constructs were confirmed by DNA sequencing.
FIG. 6.

Mapping of the RNA sequence(s) in HBV pregenomic RNA responsible for HBV RNA decay. (A) Schematic diagram of the truncation and deletion mutants of HBV pregenomic RNA. The numbers indicate the HBV DNA sequence with 1 at the unique EcoRI site in the HBV genome. (B) Huh7 cells were transfected with 0.1 μg of luciferase fusion constructs containing the truncation mutants of HBV DNA and 0.2 μg of pCMV/Myc or pCMV/Myc-MyD88 for 48 h, and the luciferase activity was then assessed. For each transfection, 0.01 μg of pRL-tk was included as an internal control of transfection efficiency. Results represent the means of data from three independent experiments performed in duplicate. *, P < 0.05. (C) Huh7 cells were transfected with 1 μg of pCDNA3.1-Luc, Luc-HBV(1804-2454), or Luc-HBV(1151-1684) together with 2 μg of pCMV/Myc or pCMV/Myc-MyD88 for 48 h. Levels of luciferase mRNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control. (D) Huh7 cells were transfected with 1 μg of Luc-ΔHBV(1151-1684) or Luc-ΔHBV(1804-2454) together with 2 μg of pCMV/Myc or pCMV/Myc-MyD88 for 48 h. Levels of luciferase mRNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control. (E) Huh7 cells were transfected with 1 μg of pCMV-HBV, pCMV-HBVΔ1804-2454, or pCMV-HBVΔ1151-1684 (also named pCMV-HBVΔPRE in this study) together with 2 μg of pCMV/Myc or pCMV/Myc-MyD88 for 48 h. Levels of luciferase mRNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control.
Adenoviruses expressing enhanced green fluorescent protein (EGFP) (Ad-EGFP) or MyD88 (Ad-MyD88) were created by using the bacterial plasmid recombination system AdMax (VGTC, China). The viral titers were determined by a cytopathic effect assay that involved determining the median tissue culture infective dose (TCID50) for HEK293 cells.
Cell culture.
The hepatoma cell lines HepG2 and Huh7, the African green monkey kidney cell line Vero, and the human cervical carcinoma cell line HeLa were obtained from the ATCC; the HBV-expressing stable cell lines HepG2.2.15 (42) and HepAD38 (23) were kindly provided by Yumei Wen (Fudan University, China). All cell lines were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (Invitrogen), 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2.
Cell transfection and virus infection.
Transient transfection was performed by using FuGENE 6 reagent (Roche) according to the manufacturer's instructions. Confluent cell monolayers (60 to 80%) were transfected with a 3:1 ratio of liposomes to DNA. For adenovirus infection, HepG2.2.15 or HepAD38 cells were infected at a multiplicity of infection (MOI) of 100. Cells were harvested at 48 h posttransfection or postinfection. The levels of HBV RNA and core particle-associated DNA were determined by Northern blot and Southern blot analyses, respectively.
Northern blot and Southern blot analyses.
Total RNA was isolated by use of TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Cytoplasmic or nuclear RNA was extracted by using a Cytoplasmic & Nuclear RNA purification kit (Norgen) as recommended by the manufacturer. RNA (15 μg) was separated on a denaturing formaldehyde-1% agarose gel and transferred onto a positively charged nylon membrane (Roche) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer. HBV RNA was detected with a 32P-radiolabeled HBV DNA probe prepared by using the Random Priming labeling kit (Roche) in Ultrahyb ultrasensitive hybridization buffer (Ambion). Hybridization signals were visualized by phosphorimaging (Fujifilm) and quantified by use of MultiGauge V2.2 software. The blots were then stripped and rehybridized with a 32P-radiolabeled GAPDH (glyceraldehyde-3-phosphate dehydrogenase) DNA probe for loading normalization.
Intracellular HBV core particle-associated DNA was extracted as described previously (33). After extraction, viral DNA was separated on a 1% agarose gel. The gel was then subjected to denaturalization in a solution containing 0.5 M NaOH and 1.5 M NaCl, followed by neutralization in a buffer containing 1.5 M NaCl and 1 M Tris-HCl (pH 7.4). The DNA was blotted onto a positively charged nylon membrane and detected with an HBV probe via an approach identical to that described above. β-Galactosidase (β-Gal) activities were measured to normalize viral DNA loading.
Hydrodynamics-based transfection in mice.
Specific-pathogen-free female BALB/c mice were purchased from the Center of Experimental Animals of the Shanghai Institute of Biological Sciences (Shanghai, China). A total of 10 μg of pHBV1.3 and 20 μg of pCMV/Myc or pCMV/Myc-MyD88 were diluted in 2 ml of saline and injected into the tail vein of 6- to 8-week-old mice within 5 to 8 s. Four days after the injection, viral DNA and RNA were extracted and analyzed by Southern and Northern blotting, respectively. Briefly, a piece of liver tissue was digested overnight in 400 μl of proteinase K-SDS digestion buffer, containing 10 mM EDTA, 100 mM NaCl, 1 mg/ml proteinase K, 0.5% SDS, and 25 mM Tris-HCl (pH 7.9), at 55°C. The DNA was phenol-chloroform extracted from the lysate. Thirty micrograms of DNA was then digested with HindIII, which linearized input plasmid pHBV1.3. Total liver RNA was isolated by use of TRIzol reagent, and 20 μg of purified RNA was submitted to Northern blot analysis. The animal experiments were carried out according to the guide for the care and use of medical laboratory animals (Ministry of Health, People's Republic of China).
RNA interference.
Huh7 cells at 30 to 40% confluence were transfected with 40 nM small interfering RNA (siRNA) duplexes (Invitrogen) by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 24 h posttransfection, the cells were transfected with 20 nM the same siRNA duplexes and the indicated plasmids.
Viral RNA stability analysis.
To assess the half-life of viral pregenomic RNA in Huh7 cells, cells were transfected with 1.5 μg of pTet-HBV, 0.5 μg of pUHD-TA (which expresses tTA, the tetracycline-controlled transactivator), and 3 μg of pCMV/Myc or pCMV/Myc-MyD88. To assess the half-life of viral pregenomic RNA in HepAD38 cells, cells were infected with Ad-EGFP or Ad-MyD88 at an MOI of 100. The synthesis of viral pregenomic RNA was blocked by the addition of 1 μg/ml doxycycline to the culture medium 39 h after transfection or infection. Cells were harvested directly upon the addition of doxycycline or at 3, 6, or 9 h thereafter. RNA was prepared and analyzed by Northern blotting. Two independent experiments were performed in duplicate, and viral RNA levels were normalized against RNA loading. The half-life of viral pre-S2/S RNA was determined in a similar manner by using the pTRE2hyg-preS2/S construct.
Western blot analysis.
Cell lysates were separated by SDS-PAGE and electrophoretically blotted onto nitrocellulose membranes (Roche). The membranes were blocked for 1 h with phosphate-buffered saline (PBS) containing 0.05% Tween 20 and 5% nonfat dry milk, followed by overnight incubation at 4°C with primary antibodies against Myc (Sigma), MyD88 (Sigma), DCP2 (Santa Cruz), EXOSC5 (Novus Biologicals), PTB (Sigma), or β-actin (Sigma). Bound antibodies were revealed by horseradish peroxidase (HRP)-labeled secondary antibodies (Sigma) and visualized with enhanced chemiluminescence (ECL) detection reagents (Amersham).
Reporter assays.
Luciferase assays were performed as described previously (50). For chloramphenicol acetyltransferase (CAT) assays, Huh7 cells were transfected with the indicated plasmids. At 48 h posttransfection, cells were harvested for the quantification of CAT activity by using a CAT enzyme-linked immunosorbent assay (ELISA) kit (Roche) according to the manufacturer's protocol. An aliquot of each lysate was assayed for β-Gal activity to standardize the results according to transfection efficiency.
Statistics.
Results are reported as means ± standard deviations. t tests were applied for comparisons between groups; a P value of <0.05 was considered to indicate statistical significance.
RESULTS
MyD88 inhibits HBV replication in HepG2.2.15 cells and in a mouse model.
We and others have previously shown that MyD88 inhibits transient HBV replication in hepatoma cells (12, 25, 51). To determine the effect of MyD88 on established HBV replication, a cell line stably transformed with replicating HBV genomic DNA, HepG2.2.15, was mock infected or infected with adenovirus expressing EGFP (Ad-EGFP) or MyD88 (Ad-MyD88). As a control, one set of HepG2.2.15 cells was treated with IFN-α. The amounts of viral RNA and core particle-associated DNA were determined by Northern and Southern blot analyses, respectively. As shown in Fig. 1 A, the levels of HBV RNA and DNA were decreased in Ad-MyD88-infected cells compared to mock-infected cells, while Ad-EGFP infection did not reduce the levels of HBV RNA or DNA. As a control, IFN-α treatment resulted in a decrease in the levels of viral RNA and DNA to a greater extent (Fig. 1A).
FIG. 1.

MyD88 inhibits HBV replication in HepG2.2.15 cells and in a mouse model. (A) HepG2.2.15 cells were mock infected or infected with Ad-EGFP or Ad-MyD88 or treated with 5,000 IU/ml IFN-α. The levels of viral RNA and core particle-associated DNA were determined by Northern (top) and Southern (middle) blot analyses, respectively, using a 32P-radiolabeled HBV DNA probe. After the detection of HBV RNA, the blots were stripped and rehybridized with a 32P-radiolabeled GAPDH DNA probe to control for gel loading. The positions of HBV pregenomic RNA (3.5 kb), pre-S1/S RNA (2.4 kb), and pre-S2/S RNA (2.1 kb) and the positions of relaxed circular (RC), single-stranded (SS) DNAs are indicated. MyD88 expression was confirmed by Western blot analysis using an anti-MyD88 antibody. The levels of the β-actin protein were determined to normalize for protein loading (bottom). (B) BALB/c mice were hydrodynamically coinjected with pHBV1.3 and pCMV/Myc or pCMV/Myc-MyD88. At 4 days postinjection, total liver DNA was extracted and subjected to digestion with HindIII restriction endonuclease. Input pHBV1.3 and viral core particle-associated DNA were examined by Southern blot analysis (top). The intensity of input pHBV1.3 in each lane was used as an internal control indicating equivalent transfection efficiency. Total liver RNA was analyzed by Northern blotting, and GAPDH was analyzed as a loading control (middle). MyD88 expression was confirmed by Western blot analysis using an anti-Myc antibody (bottom). Eight comparable pairs in which each part showed similar amounts of input DNA were obtained from two independent injections, and two representative pairs are shown. (C) Huh7 (top) and HepG2 (bottom) cells were cotransfected with the indicated amounts of pCIdA-HBV and pCMV/Myc or pCMV/Myc-MyD88. Levels of HBV RNAs were determined by Northern blot analysis. GAPDH was analyzed as a loading control.
Recently, a mouse model of acute HBV infection was established by using hydrodynamics-based transfection (53). To examine the antiviral effect of MyD88 in vivo, BALB/c mice were hydrodynamically coinjected with plasmids expressing HBV (pHBV1.3) and MyD88 (pCMV/Myc-MyD88). Total liver DNA and RNA were analyzed by Southern and Northern blotting, respectively. Consistent with in vitro results, MyD88 significantly reduced the levels of viral core particle-associated DNA and RNA (Fig. 1B, top and middle). The expression of MyD88 in transfected mouse livers was confirmed by Western blot analysis (Fig. 1B, bottom). Furthermore, the possible toxicity of MyD88 to liver was assessed by determining the alanine aminotransferase (ALT) levels in the sera of mice. No difference in ALT levels was observed between mice injected with pCMV/Myc and those injected with pCMV/Myc-MyD88 (data not shown).
Interestingly, viral DNA levels were decreased to the same extent as viral RNA levels (Fig, 1A and B), which is in agreement with previous findings (12, 25, 51). According to the HBV life cycle, we reasoned that the main primary antiviral target of MyD88 was most likely the viral RNA. To evaluate this hypothesis, we investigated whether MyD88 overexpression decreased the levels of viral RNA by using the pCIdA-HBV construct, which is capable of viral gene expression and incapable of viral DNA replication. As shown in Fig. 1C (top), the expression of MyD88 greatly downregulated viral RNA levels. This inhibitory effect was not restricted to Huh7 cells; it was also seen for HepG2 cells (Fig. 1C, bottom). Collectively, these results suggest that MyD88 has a strong inhibitory effect on HBV replication both in vitro and in vivo and that it inhibits HBV replication primarily by downregulating viral RNA levels.
Knocking down MyD88 weakens IFN-α-induced inhibition of HBV replication.
The above-described data and previous results (25, 51) indicate that the ectopic expression of MyD88 inhibits HBV replication; however, an antiviral activity of MyD88 has not yet been shown at physiological levels. For this reason, we investigated whether the silencing of MyD88 expression could weaken the inhibitory effect of IFN-α against HBV. Huh7 cells were transfected with either control EGFP siRNA or siRNA targeting MyD88, and the antiviral activity of IFN-α was tested. As shown in Fig. 2 A (top and middle, lane 3 versus lane 1) and Fig. 2 B (bar 3 versus bar 1), HBV showed a high sensitivity to IFN-α treatment in the control cells, as expected. In contrast, when treated with IFN-α, MyD88 knockdown cells showed a slight increase in levels of viral RNA and DNA (Fig. 2A, top and middle, lane 4 versus lane 3, and 2B, bar 4 versus bar 3). The effectiveness of the siRNA targeting of MyD88 was confirmed by Western blot analysis (Fig. 2A, bottom). These results indicate that MyD88 plays an active antiviral role in the IFN-α-mediated inhibition of HBV replication.
FIG. 2.
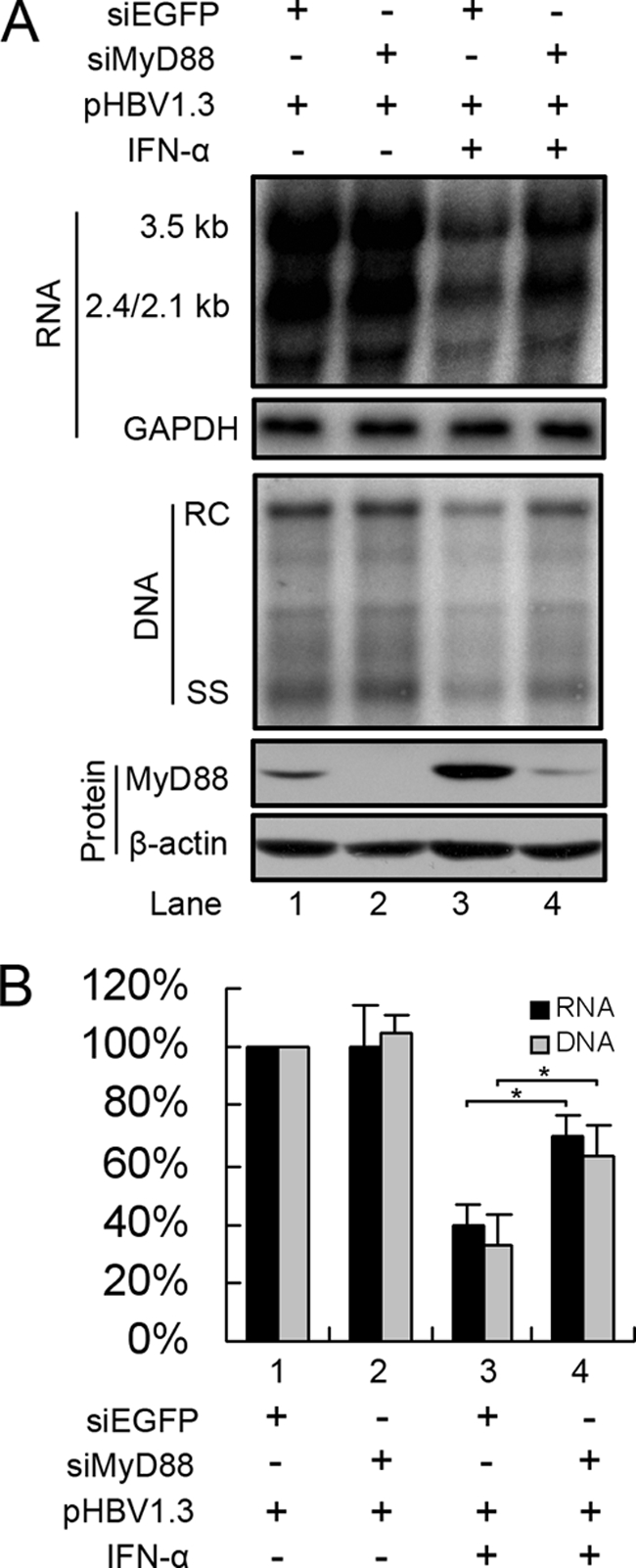
Knockdown of MyD88 expression by RNA interference partially abrogates IFN-α-induced inhibition of HBV replication. (A) Huh7 cells were transfected with 40 nM siRNAs targeting EGFP (siEGFP) or MyD88. After 24 h, the cells were transfected with 20 nM the same siRNAs together with pHBV1.3 and a β-Gal control plasmid, followed by treatment with or without 5,000 IU/ml IFN-α for 48 h. Viral RNA and DNA were analyzed by Northern blotting and Southern blotting, respectively. (B) Viral RNA and DNA levels from three independent experiments were quantified by phosphorimaging and normalized against GAPDH levels and β-Gal activity, respectively. *, P < 0.05.
MyD88 downregulates HBV RNAs by a posttranscriptional mechanism.
The above-described analysis suggested that MyD88 downregulates viral RNA levels. To determine whether this inhibition occurs transcriptionally or posttranscriptionally, we first employed reporter plasmids in which the luciferase reporter gene was under the control of HBV promoters/enhancers. At 48 h after the cotransfection of Huh7 or HepG2 cells with pCMV/Myc-MyD88, the cells were harvested, and the luciferase activity in the lysates was determined. The results showed that MyD88 had little inhibitory effect on the activity of the viral promoters/enhancers in both Huh7 (Fig. 3A) and HepG2 (data not shown) cells.
FIG. 3.

MyD88 downregulates HBV RNAs by a posttranscriptional mechanism. (A) Huh7 cells were cotransfected with 0.1 μg of each of four reporter plasmids for HBV promoters and enhancers (ENII/Cp, Sp1, Sp2, and ENI/Xp, respectively) and pCMV/Myc or pCMV/Myc-MyD88. The cells were harvested at 48 h posttransfection, and luciferase activity in the lysates was assessed. For each transfection, 0.01 μg of pRL-tk was included as an internal control of transfection efficiency. Results represent the means of data from three independent experiments performed in duplicate. (B). Huh7 cells were cotransfected with the indicated amounts of pCMV-HBV and pCMV/Myc or pCMV/Myc-MyD88. Levels of the pregenomic RNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control. (C) Huh7 cells were cotransfected with the indicated amounts of pCDNA3.1-Luc and pCMV/Myc or pCMV/Myc-MyD88. Levels of luciferase mRNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control. (D) Vero cells were transfected as described above (B). Levels of pregenomic RNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control.
We next examined whether HBV ENII/Cp was required for the downregulation of viral pregenomic RNA by MyD88. pCMV-HBV was cotransfected into Huh7 or HepG2 cells together with pCMV/Myc-MyD88, and the levels of pregenomic RNA were examined by Northern blot analysis. Our results showed that the expression of MyD88 significantly downregulated the pregenomic RNA levels in Huh7 (Fig. 3B, lanes 3 and 2 versus lane 1) and HepG2 (data not shown) cells. The inhibition was not due to a decreased transcriptional activity of the CMV promoter itself, as MyD88 could not inhibit CMV promoter-driven luciferase expression (Fig. 3C, lane 2 versus lane 1). Interestingly, we found that this inhibition could not be extended to Vero (Fig. 3D, lanes 3 and 2 versus lane 1) or HeLa (data not shown) cells, suggesting that the inhibitory effect may be hepatocyte specific. These results suggest that MyD88 posttranscriptionally reduces the levels of HBV RNA.
MyD88 accelerates the decay of HBV pregenomic RNA in cytoplasm.
Because the inhibition of pregenomic RNA expression is a posttranscriptional event, we investigated whether the decrease in RNA levels was due to an accelerated turnover rate of the pregenomic RNA. Huh7 cells were transfected with pTet-HBV, pUHD-TA, and pCMV/Myc-MyD88. At 39 h posttransfection, the cells were treated with doxycycline to turn off HBV pregenomic RNA transcription. The cells were harvested, and the levels of pregenomic RNA were measured by Northern blot analysis at different time points posttreatment. As shown in Fig. 4 A and B, the half-life of the pregenomic RNA in MyD88-overexpressing cells was shortened by about 2 h compared with that in control cells. A similar effect of MyD88 on pregenomic RNA decay was observed for HepAD38 cells (data not shown). In addition, cytoplasmic and nuclear fractionation analysis showed that a MyD88-induced decay of the pregenomic RNA occurred in the cytoplasm (Fig. 4C, left, and D) and not in the nucleus (Fig. 4C, right, and E).
FIG. 4.

MyD88 accelerates the decay of HBV pregenomic RNA in the cytoplasm. (A) Huh7 cells were cotransfected with pTet-HBV and pUHD-TA together with pCMV/Myc or pCMV/Myc-MyD88. At 39 h posttransfection, the cells were harvested directly upon the addition of doxycycline (Dox) or at 3, 6, or 9 h thereafter. The levels of pregenomic RNA were determined by Northern blot analysis. To normalize RNA loading, the same blots were hybridized with a GAPDH probe. (B) The half-life of the pregenomic RNA was determined by Northern blot analysis, and the bands were quantified by phosphorimaging. Two independent experiments performed in duplicate were analyzed, and the levels of pregenomic RNA were normalized against RNA loading. (C) Huh7 cells were transfected as described above (A). Cytoplasmic (left) and nuclear (right) RNAs were prepared and analyzed by Northern blotting. (D and E) The half-lives of pregenomic RNA in the cytoplasm (D) and nucleus (E) were determined by Northern blot analysis, and the bands were quantified by phosphorimaging. Two independent experiments performed in duplicate were analyzed and quantified by phosphorimaging, and the levels of pregenomic RNA were normalized against RNA loading. (F) Huh7 cells were transfected with 40 nM siRNA targeting EGFP, DCP2, or EXOSC5. At 24 h posttransfection, the cells were transfected with 20 nM the same siRNA together with pHBV1.3 and pCMV/Myc-MyD88. At 48 h after the second transfection, the cells were harvested, and viral pregenomic RNA was analyzed by Northern blot analysis (top). The expression of target proteins was evaluated by Western blot analysis using anti-DCP2 and anti-EXOSC5 antibodies (bottom).
In mammalian cells, mRNA decay occurs mainly in the cytoplasm, where mRNA degradation proceeds through two main pathways (8, 17). The 5′-to-3′ mRNA decay pathway is initiated by the removal of the 5′ cap by the decapping enzymes DCP1 and DCP2, whereas 3′-to-5′ mRNA decay is mediated by a large complex of 3′-to-5′ exonucleases known as the exosome, which includes exosome component 5 (EXOSC5). Considering that pregenomic RNA resembles cellular mRNA in structure, we determined whether one or both of these mRNA decay pathways are required for the MyD88-induced decay of pregenomic RNA. We knocked down the expression of DCP2 or EXOSC5 in Huh7 cells to block these two pathways independently. The results showed that siRNAs targeting DCP2 or EXOSC5 abrogated the MyD88-mediated inhibition of viral pregenomic RNA levels (Fig. 4F, top, lanes 3 and 4 versus lane 2). The effectiveness of siRNAs targeted against DCP2 or EXOSC5 was confirmed by Western blot analysis (Fig. 4F, bottom). Collectively, the above-described results suggest that MyD88 reduced the levels of HBV pregenomic RNA mainly through accelerating its decay in the cytoplasm and that RNA degradation proceeds through both the 5′-to-3′ and 3′-to-5′ mRNA decay pathways.
The RNA sequence in the 5′-proximal region of HBV pregenomic RNA mediates its decay.
It was reported previously that the La protein contributes to HBV pregenomic RNA stability through specific binding to the viral RNA, while cytotoxic T-lymphocyte (CTL) and interleukin-2 (IL-2) treatment results in the fragmentation of the La protein, which renders viral RNA vulnerable to degradation by cellular nucleases (5, 13, 14, 16, 44). To determine whether MyD88 induces the fragmentation of the La protein, we studied the expression of the La protein in MyD88-overexpressing cells by Western blot analysis. Our results showed that MyD88 overexpression did not result in a decrease in levels of the La protein in Huh7 cells in the absence or presence of HBV replication (Fig. 5 A). Importantly, MyD88 inhibited the La protein-binding-deficient pregenomic RNA from pCMV-HBV M2 to the same extent as wild-type pregenomic RNA (Fig. 5B).
FIG. 5.
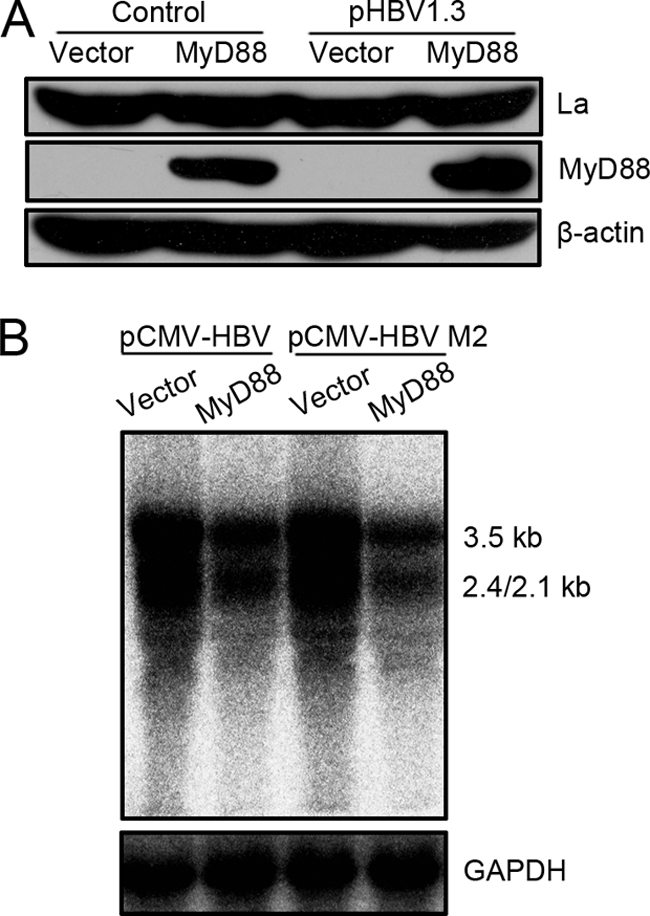
MyD88-induced decay of viral pregenomic RNA was independent of the interaction of La with viral pregenomic RNA. (A) Huh7 cells were transfected with 1 μg of pCMV/Myc or pCMV/Myc-MyD88, with or without 0.5 μg of pHBV1.3, for 48 h. Western blot analysis for La was performed as described in Materials and Methods. (B) Huh7 cells were transfected with 1 μg of pCMV-HBV or pCMV-HBV M2 together with 2 μg of pCMV/Myc or pCMV/Myc-MyD88. Levels of pregenomic RNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control.
Because the La protein-binding sequence is not required for MyD88-induced decay, we attempted to map the MyD88-responsive sequences in HBV pregenomic RNA. A series of HBV fragments was individually inserted into the multiple-cloning site of a CMV promoter-driven luciferase expression plasmid (pcDNA3.1/Luc) (Fig. 6 A). The resultant chimeric plasmids were transfected into Huh7 or HepG2 cells with pCMV/Myc-MyD88. Luciferase assays showed that MyD88 overexpression significantly decreased the luciferase activity derived from the Luc-HBV(1804-3170), Luc-HBV(3171-1986), Luc-HBV(1804-2454), Luc-HBV(837-1986), and Luc-HBV(1151-1684) constructs but not the Luc-HBV(2455-3170), Luc-HBV(3171-836), Luc-HBV(837-1308), and Luc-HBV(1309-1986) constructs in Huh7 cells (Fig. 6B). A similar result was observed for HepG2 cells (data not shown). We were able to demonstrate that the decreases in luciferase activity derived from the Luc-HBV(1804-2454) and Luc-HBV(1151-1684) constructs reflected the levels of luciferase mRNA (Fig. 6C, lane 4 versus lane 3 and lane 6 versus lane 5), suggesting that HBV(1804-2454) and HBV(1151-1684) are MyD88-responsive regions of the pregenomic RNA. To investigate the relative contribution of the two MyD88-responsive sequences to the MyD88-induced decay of viral pregenomic RNA, we constructed deletion mutants of these sequences in the context of Luc-HBV(1804-1986) and tested their response to MyD88. Our results showed that the HBV(1151-1684) deletion mutant retained sensitivity to MyD88 (Fig. 6D, lane 2 versus lane 1), while the HBV(1804-2454) deletion mutant was resistant to MyD88 (Fig. 6D, lane 4 versus lane 3). To exclude the influence of the luciferase RNA sequence on the response of the two deletion mutants to MyD88, we used the constructs pCMV-HBVΔ1804-2454 and pCMV-HBVΔ1151-1684, in which HBV(1804-2454) and HBV(1151-1684) were deleted in the context of pCMV-HBV, respectively, and found that the construct pCMV-HBVΔ1151-1684 showed a sensitivity to MyD88 similar to that of wild-type pCMV-HBV, while the construct pCMV-HBVΔ1804-2454 lost sensitivity to MyD88 (Fig. 6E). These results define the HBV(1804-2454) region as a crucial cis-regulatory sequence for the MyD88-induced decay of viral pregenomic RNA.
The RNA region of HBV(1151-1684) selectively mediates MyD88-induced decay of HBV pre-S/S RNAs in the nucleus.
Because the HBV(1151-1684) region, which is located in the 3′-overlapping region of the pregenomic RNA and pre-S/S RNAs, was not required for the MyD88-induced decay of pregenomic RNA, we determined whether it selectively conferred a sensitivity of pre-S/S RNAs to MyD88. We deleted this sequence in the context of pre-S2/S RNA and pre-S1/S RNA and found that the two deletion mutants lost responsiveness to MyD88 compared with the wild-type versions (Fig. 7 A and data not shown).
FIG. 7.

MyD88 accelerates the decay of HBV pre-S2/S RNA in the nucleus. (A) Huh7 cells were transfected with 1 μg of pCDNA3.1-pre-S2/S or its deletion mutant of HBV [HBV(1151-1684)] together with 2 μg of pCMV/Myc or pCMV/Myc-MyD88 for 48 h. Levels of viral RNA were determined by Northern blot analysis. GAPDH was analyzed as a loading control. (B) Huh7 cells were cotransfected with pTRE2hyg-preS2/S and pUHD-TA together with pCMV/Myc or pCMV/Myc-MyD88. Cells were harvested directly upon the addition of doxycycline or 3, 6, and 9 h thereafter. (B) Nuclear (bottom) and cytoplasmic (top) RNAs were prepared and analyzed by Northern blotting. (C and D) The half-life of pre-S2/S RNA in the nucleus (C) and cytoplasm (D) was determined by Northern blot analysis and quantified by phosphorimaging. Data from two independent experiments performed in duplicate were analyzed and quantified by phosphorimaging, and the levels of pre-S2/S RNA were normalized against RNA loading.
To confirm that MyD88 accelerated the decay of pre-S2/S RNA, the stability of cytoplasmic and nuclear pre-S2/S RNAs was determined. Our results showed that the overexpression of MyD88 accelerated the degradation of nuclear pre-S2/S RNA in Huh7 cells (Fig. 7B, top) and shortened the nuclear pre-S2/S RNA half-life by approximately 2.5 h (Fig. 7C). The stability of cytoplasmic pre-S2/S RNA was not significantly affected by MyD88 overexpression (Fig. 7B, bottom, and D). A similar result was also obtained with pre-S1/S RNA (data not shown). In summary, the above-described results suggest that the HBV(1151-1684) sequence mediates the MyD88-induced decay of HBV pre-S/S RNAs in the nucleus.
MyD88 inhibits the nuclear export of HBV pre-S/S RNAs mediated by PRE.
Interestingly, we found that the HBV(1151-1684) region overlaps with an RNA cis element termed the posttranscriptional regulatory element (PRE) (18-20). The PRE mediates the nuclear export of viral pre-S/S RNAs, but it does not affect the nuclear export of pregenomic RNA (15, 18-20). In addition, viral pre-S/S RNAs lacking the PRE fail to translocate to the cytoplasm and degrade in the nucleus via a mechanism that has remained elusive (54, 55).
We evaluated whether the decay of pre-S/S RNAs in the nucleus was associated with a deficiency in nuclear transport mediated by the PRE. We used the pRSV138PRE-CAT construct (39), which expresses a transcript that contains the PRE sequence and the coding sequence for CAT between a splicing donor and a splicing acceptor site. Because the sequence encoding the reporter enzyme is located within an intron, the reporter cannot be expressed after the transcript is spliced. However, the presence of the PRE within the same intron allows the efficient nuclear export of the nonspliced mRNA and results in CAT expression. CAT activity derived from the PRE-containing transcript was significantly decreased by MyD88 (Fig. 8A) compared to that derived from pRSV-CAT (Fig. 8A), suggesting that MyD88 impairs PRE-mediated nuclear export. To exclude the possibility that MyD88 directly induces the decay of the PRE-containing transcripts in the nucleus, we tested whether MyD88 inhibited CAT expression when PRE-mediated nuclear export was blocked by the expression of NES(−)RanBP1 (55), which is an inhibitor of PRE-mediated nuclear export. Our results showed that MyD88 did not further diminish CAT expression when coexpressed with NES(−)RanBP1 (Fig. 8B, bars 3 and 4 versus bar 2). We performed the converse experiments by determining whether the expression of polypyrimidine tract-binding protein (PTB) (including PTB1 and PTB4, encoded by the same gene [49]), which is an export factor for PRE-containing RNA (54), antagonized the inhibition of CAT expression. The results showed that the expression of PTB1 almost fully restored the function of the PRE (Fig. 8C, bars 4 and 5 versus bar 3).
FIG. 8.

The decay of HBV pre-S2/S RNA in the nucleus is associated with the deficiency in PRE-dependent nuclear transport. (A) Huh7 cells were transfected with 0.2 μg of pRSV-CAT or pRSV138PRE-CAT together with 0.4 μg of pCMV/Myc or pCMV/Myc-MyD88. At 48 h posttransfection, the cells were harvested, and CAT activity was measured. For each transfection, 0.1 μg of β-Gal was included as an internal control of transfection efficiency. Results represent the means of data from three independent experiments performed in duplicate. *, P < 0.05. (B and C) Huh7 cells were transfected with the indicated amounts of pRSV138PRE-CAT and pCMV/Myc-MyD88 together with pCMV/Myc-NES(−)RanBP or pCMV/Myc-PTB1. At 48 h posttransfection, the cells were harvested, and CAT activity was measured as described above (A). (D and E) Huh7 cells were transfected with the indicated amounts of pRSV138PRE-CAT and pCMV/Myc-MyD88 together with pCMV/Myc-NES(-)RanBP or pCMV/Myc-PTB1 for 48 h. Cytoplasmic and nuclear RNAs were prepared and analyzed by Northern blotting with a 32P-radiolabeled CAT DNA probe. To normalize RNA loading, the same blots were hybridized with a GAPDH probe.
Given that the CAT assays performed as described above represent only an indirect measure of RNA levels, we also performed Northern blot analysis for CAT RNA in the nucleus and cytoplasm. As expected from previous data (55), NES(−)RanBP1 expression resulted in decreases in both nuclear and cytoplasmic unspliced CAT RNA levels (Fig. 8D, lane 2 versus lane 1). The coexpression of MyD88 did not promote a further decay of nuclear unspliced CAT RNA (Fig. 8D, lanes 3 and 4 versus lane 2). In addition, the coexpression of PTB1 abrogated MyD88-induced decreases in both nuclear and cytoplasmic unspliced CAT RNA levels (Fig. 8E, lanes 3 and 4 versus lane 2). These changes in RNA levels are in good agreement with the observed changes in CAT activity. Therefore, from the results presented above, we conclude that MyD88 inhibits the nuclear export of HBV pre-S/S RNAs mediated by the PRE.
MyD88 transcriptionally inhibits the expression of PTB.
It was reported previously that IκBα, an NF-κB-responsive protein, can reduce HBV PRE-dependent nuclear export (39). As mentioned above, PTB, a PRE-interacting protein, is involved in the process of the nuclear transport of pre-S/S RNAs (54). To uncover the mechanism underlying the impaired PRE function in nuclear export, we evaluated the expression of IκBα and PTB in MyD88-overexpressing cells by Western blot analysis. The results showed that MyD88 did not change the expression levels of IκBα or PTB in Huh7 cells in the absence of HBV replication (Fig. 9A, lane 2 versus lane 1). In the presence of HBV replication, the expression of PTB was greatly downregulated by MyD88, in contrast to IκBα (Fig. 9A, lane 4 versus lane 3). A similar result was obtained for HepG2 cells (data not shown). Considering that the impaired function of the PRE was almost fully restored by PTB1 (Fig. 8C and E), we conclude that the reduction in levels of PTB expression may be the main cause of the impairment of HBV PRE function.
FIG. 9.

MyD88 transcriptionally inhibits the expression of PTB. (A) Huh7 cells were transfected with 1 μg of pCMV/Myc or pCMV/Myc-MyD88, with or without 0.5 μg of pHBV1.3, for 48 h. Western blot analysis for PTB and IκBα was performed as described in Materials and Methods. (B) Huh7 cells were transfected with 2 μg of pCMV/Myc or pCMV/Myc-MyD88 together with 1 μg of pHBV1.3 for 48 h. Total RNA was prepared and analyzed by Northern blotting with a 32P-radiolabeled PTB DNA probe. (C) Huh7 cells were transfected as described above (B). At 44 h posttransfection, the cells were treated with actinomycin D. The cells were harvested directly upon the addition of actinomycin D or at 2 or 4 h thereafter. Total RNA was prepared, and PTB mRNA levels were determined by Northern blot analysis. (D) Two independent experiments performed in duplicate were analyzed, data were quantified by phosphorimaging, and the levels of PTB mRNA were normalized against RNA loading.
We investigated the effect of MyD88 on PTB mRNA levels to determine whether changes in the levels of mRNA were responsible for the changes in protein expression levels. As shown in Fig. 9B, the overexpression of MyD88 significantly decreased the levels of PTB mRNA. As the decreased levels of PTB mRNA induced by MyD88 could result from an increased rate of mRNA degradation or a decreased rate of transcription, we treated cells with actinomycin, a general inhibitor of transcription, and monitored PTB mRNA levels by Northern blot analysis. Our results showed that the expression of MyD88 could not accelerate the degradation of PTB mRNA (Fig. 9C and D). Therefore, the inhibition of transcription, and not the acceleration of mRNA degradation, is responsible for the MyD88-induced decrease in PTB mRNA levels.
DISCUSSION
In this study, the effect of MyD88 on HBV replication and the mechanism of this effect were further investigated. Based on the data presented above, we propose the following model for the MyD88-mediated inhibition of HBV replication (illustrated in Fig. 10). After induction by IFN-α, MyD88 posttranscriptionally regulates HBV viral RNA expression. MyD88 accelerates the degradation of HBV pregenomic RNA in the cytoplasm through a process that requires the HBV(1804-2454) region. In addition, MyD88 inhibits the nuclear export of HBV pre-S/S RNAs mediated by the PRE by decreasing PTB expression. The retained pre-S/S RNAs are degraded in the nucleus.
FIG. 10.

Proposed model for MyD88-mediated inhibition of HBV replication in hepatoma cells. See the text for a more detailed discussion. cccDNA, covalently closed circular DNA.
Although IFN-α has been used for the treatment of HBV infection for 2 decades, the downstream effectors are still elusive. It was reported previously that the IFN-inducible protein MxA blocked HBV replication both in vitro and in vivo (11, 29). However, it was also reported that IFN-α induced the suppression of HBV replication in MxA-deficient cells (34). Members of the APOBEC3 family of cytidine deaminases, which have been shown to target a wide range of retroviruses, were reported to inhibit HBV replication (1, 26, 27, 38, 45). Whether these enzymes are the main mediators of the action of IFNs on HBV remains controversial (21, 32). Recently, TRIM22 was reported to be expressed in response to IFNs and displayed anti-HBV activity both in vitro and in vivo, but it is uncertain whether TRIM22 would displays such an activity at physiological levels (7). In this study, we showed that MyD88 inhibited HBV replication in HepG2.215 cells and in a mouse model (Fig. 1A and B). The knockdown of MyD88 expression weakened the IFN-α-induced inhibition of HBV replication in Huh7 cells (Fig. 2). In addition, we did not observe enhanced HBV replication when the basal level of MyD88 was knocked down (Fig. 2A, lane 2 versus lane 1, and B, bar 2 versus bar 1). This result might be due partially to a defect of the IFN induction pathway in Huh7 cells (24, 31). Nevertheless, from these data, we conclude that MyD88 partially accounts for the antiviral action of IFN-α in our system.
Previous studies demonstrated that IFN-α targets multiple steps of the HBV life cycle, including transcription, the export and degradation of viral RNAs, as well as the formation of the core particle and DNA replication (11, 35, 37, 46-48). Here, we showed that there was no marked inhibitory effect of MyD88 on the activity of three HBV regulatory elements (Sp1, Sp2, and ENI/Xp), except that a slight dose-dependent decrease in the activity of ENII/Cp was observed (Fig. 3A). As there was a significant inhibition of MyD88 on viral pregenomic RNA expression (Fig. 1), the changes in HBV pregenomic RNA transcription could not account for the large reduction in viral pregenomic RNA levels. In addition, we did not detect changes in the expression of the liver-enriched transcription factors HNF1 and HNF4 (data not shown), which were reported previously to regulate the activity of ENII/Cp (56). Besides, MyD88 reduced the levels of both HBV pregenomic RNA and pre-S2/S RNA transcribed from the CMV promoter (Fig. 3B and 7A). This reduction is likely not due to an inhibition of the CMV promoter itself, given that MyD88 did not inhibit luciferase expression from pcDNA3.1-Luc (Fig. 3C and 6B and C). Thus, it is reasonable to consider that MyD88 downregulates HBV RNA primarily through posttranscriptional regulation rather than through a modification of transcription.
In our effort to investigate the underlying mechanism of the posttranscriptional control of HBV RNA by MyD88, we found that MyD88 accelerated the decay of HBV pregenomic RNA in the cytoplasm (Fig. 4). It should be noted that, based on the presented data, we cannot exclude other mechanisms used by MyD88 to posttranscriptionally control viral pregenomic RNA. However, it appears certain that accelerated decay is responsible for the main reduction of viral pregenomic RNA levels. In fact, the promotion of viral RNA decay has been adopted by other ISGs as a strategy against virus replication. For example, it was reported previously that the activation of the 2′-5′(A) synthetase/RNase L pathway by IFNs inhibits a variety of RNA viruses by targeting viral RNAs for degradation (3, 10, 41).
Similar to transcription and translation, mRNA decay is a tightly controlled process that is determined by cis-acting elements within the mRNA and trans-acting factors in the host cell. In this study, we identified the HBV(1804-2454) region as a crucial cis-regulatory sequence for the MyD88-induced decay of HBV pregenomic RNA (Fig. 6). Notably, the binding sites for the La protein are not included in this region. Consistent with this fact, we found that the decay induced by MyD88 was independent of the interaction between La and viral pregenomic RNA (Fig. 5). Interestingly, a previous report identified a 65-kDa cellular protein that binds to the 5′ end of HBV pregenomic RNA and is probably involved in the posttranscriptional regulation of HBV RNA expression (30). One might therefore hypothesize that MyD88 acts by blocking this protein and thus results in the decay of HBV pregenomic RNA. In addition, one or more of the MyD88-induced trans-acting factors may be hepatocyte specific, given that the observed RNA decay could not be extended to Vero or HeLa cells (Fig. 3). Nevertheless, future studies are needed to more accurately delimitate the target sequence and identify the host factors that mediate the MyD88-induced decay of viral pregenomic RNA.
Guo and colleagues previously identified the RNA sequence of HBV(835-2327) as being responsive to MyD88 within the 3′-overlapping region of the pregenomic RNA and pre-S/S RNAs (12). The MyD88-responsive element HBV(1151-1684) that we identified is within this region (Fig. 6) and is completely included in the HBV(835-2327) region and almost completely overlaps the HBV PRE.
Similar to the HIV Rev response element (RRE), the HBV PRE mediates the nuclear export of unspliced viral RNAs (18-20). Specifically, the HBV PRE promotes the nuclear export of pre-S/S RNAs and not of the pregenomic RNA (15). It was reported previously that MxA inhibits the nuclear export of pre-S/S RNAs mediated by the HBV PRE (11). In this study, we showed that MyD88 also blocked PRE-dependent nuclear export (Fig. 8). It was previously shown that the IFN-inducible protein RBP9-27 inhibits Rev/RRE-mediated HIV expression by interfering with Rev function. In a manner similar to that of RBP9-27, MyD88 inhibits PRE-mediated HBV expression by targeting PTB (Fig. 9), an export factor for PRE-containing RNA (54). Interestingly, MyD88 exerted this effect only on HBV-infected cells (Fig. 9A). This might be due to the finding that MyD88 alone is not a strong activator of NF-κB, but it can strongly activate NF-κB through synergy with HBV (51).
Taken together, our results provide further insights into the mechanism of MyD88 antiviral activity. An elucidation of this antiviral pathway may ultimately lead to the development of new therapeutics for acute and chronic HBV infection.
Acknowledgments
We thank Jianming Hu for providing pCIdA-HBV and pCMV-HBV, Andreas Rang for providing pTet-HBV, Tilman Heise for providing pCMV-HBV M2 and pCMV-HBVΔPRE, Dina Kremsdorf for providing pRSV138PRE-CAT and pRSV-CAT, Chris Smith for providing His-PTB1, and Barbara Felber for providing GFP-NES(−)RanBP1.
This work was supported by a Chinese State Basic Research Foundation grant (grant 2005CB522902), the National Natural Science Fund for distinguished scholars (grant 30425041), National Megaprojects for Infectious Diseases (grant 2008ZX10203), and the Program for Outstanding Medical Academic Leader of Shanghai.
Footnotes
Published ahead of print on 21 April 2010.
REFERENCES
- 1.Bonvin, M., F. Achermann, I. Greeve, D. Stroka, A. Keogh, D. Inderbitzin, D. Candinas, P. Sommer, S. Wain-Hobson, J. P. Vartanian, and J. Greeve. 2006. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43:1364-1374. [DOI] [PubMed] [Google Scholar]
- 2.Borden, E. C., G. C. Sen, G. Uze, R. H. Silverman, R. M. Ransohoff, G. R. Foster, and G. R. Stark. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chebath, J., P. Benech, M. Revel, and M. Vigneron. 1987. Constitutive expression of (2′-5′) oligo A synthetase confers resistance to picornavirus infection. Nature 330:587-588. [DOI] [PubMed] [Google Scholar]
- 4.Dienstag, J. L. 2008. Hepatitis B virus infection. N. Engl. J. Med. 359:1486-1500. [DOI] [PubMed] [Google Scholar]
- 5.Ehlers, I., S. Horke, K. Reumann, A. Rang, F. Grosse, H. Will, and T. Heise. 2004. Functional characterization of the interaction between human La and hepatitis B virus RNA. J. Biol. Chem. 279:43437-43447. [DOI] [PubMed] [Google Scholar]
- 6.Fallows, D. A., and S. P. Goff. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao, B., Z. Duan, W. Xu, and S. Xiong. 2009. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50:424-433. [DOI] [PubMed] [Google Scholar]
- 8.Garneau, N. L., J. Wilusz, and C. J. Wilusz. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8:113-126. [DOI] [PubMed] [Google Scholar]
- 9.Georgantas, R. W., III, R. Hildreth, S. Morisot, J. Alder, C. G. Liu, S. Heimfeld, G. A. Calin, C. M. Croce, and C. I. Civin. 2007. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl. Acad. Sci. U. S. A. 104:2750-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh, A., S. N. Sarkar, and G. C. Sen. 2000. Cell growth regulatory and antiviral effects of the P69 isozyme of 2-5(A) synthetase. Virology 266:319-328. [DOI] [PubMed] [Google Scholar]
- 11.Gordien, E., O. Rosmorduc, C. Peltekian, F. Garreau, C. Brechot, and D. Kremsdorf. 2001. Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J. Virol. 75:2684-2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo, H., D. Jiang, D. Ma, J. Chang, A. M. Dougherty, A. Cuconati, T. M. Block, and J. T. Guo. 2009. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J. Virol. 83:847-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heise, T., L. G. Guidotti, V. J. Cavanaugh, and F. V. Chisari. 1999. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J. Virol. 73:474-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heise, T., L. G. Guidotti, and F. V. Chisari. 1999. La autoantigen specifically recognizes a predicted stem-loop in hepatitis B virus RNA. J. Virol. 73:5767-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heise, T., G. Sommer, K. Reumann, I. Meyer, H. Will, and H. Schaal. 2006. The hepatitis B virus PRE contains a splicing regulatory element. Nucleic Acids Res. 34:353-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horke, S., K. Reumann, A. Rang, and T. Heise. 2002. Molecular characterization of the human La protein.hepatitis B virus RNA.B interaction in vitro. J. Biol. Chem. 277:34949-34958. [DOI] [PubMed] [Google Scholar]
- 17.Houseley, J., and D. Tollervey. 2009. The many pathways of RNA degradation. Cell 136:763-776. [DOI] [PubMed] [Google Scholar]
- 18.Huang, J., and T. J. Liang. 1993. A novel hepatitis B virus (HBV) genetic element with Rev response element-like properties that is essential for expression of HBV gene products. Mol. Cell. Biol. 13:7476-7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang, Z. M., and T. S. Yen. 1994. Hepatitis B virus RNA element that facilitates accumulation of surface gene transcripts in the cytoplasm. J. Virol. 68:3193-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang, Z. M., and T. S. Yen. 1995. Role of the hepatitis B virus posttranscriptional regulatory element in export of intronless transcripts. Mol. Cell. Biol. 15:3864-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jost, S., P. Turelli, B. Mangeat, U. Protzer, and D. Trono. 2007. Induction of antiviral cytidine deaminases does not explain the inhibition of hepatitis B virus replication by interferons. J. Virol. 81:10588-10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawai, T., and S. Akira. 2007. TLR signaling. Semin. Immunol. 19:24-32. [DOI] [PubMed] [Google Scholar]
- 23.Ladner, S. K., M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger, and R. W. King. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, K., Z. Chen, N. Kato, M. Gale, Jr., and S. M. Lemon. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 280:16739-16747. [DOI] [PubMed] [Google Scholar]
- 25.Lin, S., M. Wu, Y. Xu, W. Xiong, Z. Yi, X. Zhang, and Y. Zhenghong. 2007. Inhibition of hepatitis B virus replication by MyD88 is mediated by nuclear factor-kappaB activation. Biochim. Biophys. Acta 1772:1150-1157. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen, D. H., S. Gummuluru, and J. Hu. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81:4465-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen, D. H., and J. Hu. 2008. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 82:6852-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papatheodoridis, G. V., S. Manolakopoulos, G. Dusheiko, and A. J. Archimandritis. 2008. Therapeutic strategies in the management of patients with chronic hepatitis B virus infection. Lancet Infect. Dis. 8:167-178. [DOI] [PubMed] [Google Scholar]
- 29.Peltekian, C., E. Gordien, F. Garreau, V. Meas-Yedid, P. Soussan, V. Willams, M. L. Chaix, J. C. Olivo-Marin, C. Brechot, and D. Kremsdorf. 2005. Human MxA protein participates to the interferon-related inhibition of hepatitis B virus replication in female transgenic mice. J. Hepatol. 43:965-972. [DOI] [PubMed] [Google Scholar]
- 30.Perri, S., and D. Ganem. 1996. A host factor that binds near the termini of hepatitis B virus pregenomic RNA. J. Virol. 70:6803-6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preiss, S., A. Thompson, X. Chen, S. Rodgers, V. Markovska, P. Desmond, K. Visvanathan, K. Li, S. Locarnini, and P. Revill. 2008. Characterization of the innate immune signalling pathways in hepatocyte cell lines. J. Viral Hepat. 15:888-900. [DOI] [PubMed] [Google Scholar]
- 32.Proto, S., J. A. Taylor, S. Chokshi, N. Navaratnam, and N. V. Naoumov. 2008. APOBEC and iNOS are not the main intracellular effectors of IFN-gamma-mediated inactivation of hepatitis B virus replication. Antiviral Res. 78:260-267. [DOI] [PubMed] [Google Scholar]
- 33.Qin, J., J. Zhai, R. Hong, S. Shan, Y. Kong, Y. Wen, Y. Wang, J. Liu, and Y. Xie. 2009. Prospero-related homeobox protein (Prox1) inhibits hepatitis B virus replication through repressing multiple cis regulatory elements. J. Gen. Virol. 90:1246-1255. [DOI] [PubMed] [Google Scholar]
- 34.Rang, A., M. Bruns, T. Heise, and H. Will. 2002. Antiviral activity of interferon-alpha against hepatitis B virus can be studied in non-hepatic cells and is independent of MxA. J. Biol. Chem. 277:7645-7647. [DOI] [PubMed] [Google Scholar]
- 35.Rang, A., S. Gunther, and H. Will. 1999. Effect of interferon alpha on hepatitis B virus replication and gene expression in transiently transfected human hepatoma cells. J. Hepatol. 31:791-799. [DOI] [PubMed] [Google Scholar]
- 36.Rang, A., and H. Will. 2000. The tetracycline-responsive promoter contains functional interferon-inducible response elements. Nucleic Acids Res. 28:1120-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romero, R., and J. E. Lavine. 1996. Cytokine inhibition of the hepatitis B virus core promoter. Hepatology 23:17-23. [DOI] [PubMed] [Google Scholar]
- 38.Rösler, C., J. Kock, M. Kann, M. H. Malim, H. E. Blum, T. F. Baumert, and F. von Weizsacker. 2005. APOBEC-mediated interference with hepadnavirus production. Hepatology 42:301-309. [DOI] [PubMed] [Google Scholar]
- 39.Roth, J., and M. Dobbelstein. 1997. Export of hepatitis B virus RNA on a Rev-like pathway: inhibition by the regenerating liver inhibitory factor IkappaB alpha. J. Virol. 71:8933-8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadler, A. J., and B. R. Williams. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scherbik, S. V., J. M. Paranjape, B. M. Stockman, R. H. Silverman, and M. A. Brinton. 2006. RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 80:2987-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sells, M. A., M. L. Chen, and G. Acs. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. U. S. A. 84:1005-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spellman, R., M. Llorian, and C. W. Smith. 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol. Cell 27:420-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsui, L. V., L. G. Guidotti, T. Ishikawa, and F. V. Chisari. 1995. Posttranscriptional clearance of hepatitis B virus RNA by cytotoxic T lymphocyte-activated hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 92:12398-12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turelli, P., B. Mangeat, S. Jost, S. Vianin, and D. Trono. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. [DOI] [PubMed] [Google Scholar]
- 46.Uprichard, S. L., S. F. Wieland, A. Althage, and F. V. Chisari. 2003. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc. Natl. Acad. Sci. U. S. A. 100:1310-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wieland, S. F., A. Eustaquio, C. Whitten-Bauer, B. Boyd, and F. V. Chisari. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 102:9913-9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wieland, S. F., L. G. Guidotti, and F. V. Chisari. 2000. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. 74:4165-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wollerton, M. C., C. Gooding, F. Robinson, E. C. Brown, R. J. Jackson, and C. W. Smith. 2001. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 7:819-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu, M., Y. Xu, S. Lin, X. Zhang, L. Xiang, and Z. Yuan. 2007. Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 88:3260-3269. [DOI] [PubMed] [Google Scholar]
- 51.Xiong, W., X. Wang, X. Liu, L. Xiang, L. Zheng, and Z. Yuan. 2004. Interferon-inducible MyD88 protein inhibits hepatitis B virus replication. Virology 319:306-314. [DOI] [PubMed] [Google Scholar]
- 52.Xiong, W., X. Wang, X. Y. Liu, L. Xiang, L. J. Zheng, J. X. Liu, and Z. H. Yuan. 2003. Analysis of gene expression in hepatitis B virus transfected cell line induced by interferon. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 35:1053-1060. (In Chinese.) [PubMed] [Google Scholar]
- 53.Yang, P. L., A. Althage, J. Chung, and F. V. Chisari. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:13825-13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zang, W. Q., B. Li, P. Y. Huang, M. M. Lai, and T. S. Yen. 2001. Role of polypyrimidine tract binding protein in the function of the hepatitis B virus posttranscriptional regulatory element. J. Virol. 75:10779-10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zang, W. Q., and T. S. Yen. 1999. Distinct export pathway utilized by the hepatitis B virus posttranscriptional regulatory element. Virology 259:299-304. [DOI] [PubMed] [Google Scholar]
- 56.Zheng, Y., J. Li, and J. H. Ou. 2004. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J. Virol. 78:6908-6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zolotukhin, A. S., and B. K. Felber. 1997. Mutations in the nuclear export signal of human ran-binding protein RanBP1 block the Rev-mediated posttranscriptional regulation of human immunodeficiency virus type 1. J. Biol. Chem. 272:11356-11360. [DOI] [PubMed] [Google Scholar]