

Abstract
H2S, the most recently discovered gasotransmitter, might in fact be the evolutionary matriarch of this family, being both ancient and highly reduced. Disruption of γ-cystathionase in mice leads to cardiovascular dysfunction and marked hypertension, suggesting a key role for this enzyme in H2S production in the vasculature. However, patients with inherited deficiency in γ-cystathionase apparently do not present vascular pathology. A mitochondrial pathway disposes sulfide and couples it to oxidative phosphorylation while also exposing cytochrome c oxidase to this metabolic poison. This report focuses on the biochemistry of H2S biogenesis and clearance, on the molecular mechanisms of its action, and on its varied biological effects.
Keywords: Metabolism, Metalloproteins, Signal Transduction, Sulfotransferase, Sulfur, Hydrogen Sulfide, Sulfur Metabolism, Transsulfuration
Introduction
Sulfur cycles through several biologically relevant oxidation states ranging from −2 as in hydrogen and metal sulfides to +6 in sulfate. H2S, a colorless gas with the odor of rotten eggs, is important in the biogeochemical sulfur cycle and is used as an energy source by microbes such as the purple and green sulfur bacteria. It is a weak acid with pKa1 and pKa2 of 6.9 and >12 (1) and an aqueous solubility of ∼80 mm at 37 °C. Hence, at the physiological pH of 7.4, the ratio of HS−:H2S is 3:1. For brevity, H2S is used to refer to the total free sulfide pool (i.e. H2S + HS− + S2−) in this report unless noted otherwise. The ready ionization of H2S at physiological pH suggests impeded permeation through the lipid bilayer when compared with other gases, viz. NO or CO. On the other hand, transport of the gas, H2S, across the membrane does not appear to be facilitated (2).
The toxicity of H2S is thought to have influenced evolution. The presence of a metastable H2S-enriched oceanic stratum is postulated to have limited early metazoan colonization of the continental shelf (3), and an increase in H2S has been implicated in the Permian-Triassic extinction >250 million years ago (4). However, the reputation of H2S as a toxic gas is enjoying a facelift, with increasing numbers of reports that it modulates a range of biological processes. Despite the rising interest in H2S biochemistry, fundamental questions regarding regulation of its production, its mechanism of action, and its destruction remain. In addition, perhaps most critical to the field is the issue of what constitutes biologically relevant levels of H2S with reports varying over a 105-fold concentration range. Using a gas chromatography-based chemiluminescent sulfur detection method, free H2S (H2S + HS−) in blood was estimated to be ∼100 pm, and in tissues, it was estimated to be ∼15 nm (5). These values are considerably lower than the ∼30–300 μm concentrations reported in a spate of recent studies (reviewed in Ref. 6). The high values can be ascribed to technical artifacts introduced by long processing times and harsh (either acidic or alkaline) conditions used to shift the equilibrium toward H2S or S2−, respectively. Under these conditions, sulfide leaches from iron-sulfur cluster-containing proteins or is eliminated via desulfuration, leading to overestimation. With very few exceptions (5, 7), most studies on H2S measurements in biological samples do not report on the sensitivity of the assay method nor include controls for background rates of cysteine (used as substrate at very high concentrations) desulfuration, which can be substantial (5), and hence, this body of data must be viewed with caution. Because most physiological effects of H2S appear to be mediated in the tens to hundreds of micromolar concentration range, it sets a reference point for the magnitude of increase in local H2S concentrations that must be realized to trigger biological signaling.
Biogenesis of H2S
H2S biogenesis in metazoans is the apparent byproduct of promiscuity of three PLP2-dependent enzymes (Fig. 1a). Thus, aspartate aminotransferase also deaminates cysteine to give mercaptopyruvate, which in a subsequent step catalyzed by MST liberates H2S and pyruvate. Similarly the PLP enzymes in the transsulfuration pathway, CBS and CSE, generate H2S by a multitude of reactions and substrate combinations. The ability to biosynthesize H2S is found in all three kingdoms of life, pointing to it being an ancient metabolic capability. H2S can also be formed non-enzymatically from polysulfides found in garlic (8).
FIGURE 1.

H2S biogenesis and catabolism. a, The enzymes in the transsulfuration pathway, CBS and CSE, catalyze multiple H2S-producing reactions, and the dominant reaction is shown in red. CAT and MST are components of the cysteine catabolism pathway. The preferred sulfur acceptor of MST is not known. b, components of the mitochondrial sulfide oxidation pathway that couple to the electron transfer chain. Q is ubiquinone. This figure was adapted from Ref. 30.
MST belongs to the family of sulfurtransferases, which catalyze the transfer of sulfane sulfur from persulfide or thiosulfate (in the rhodanese subfamily) or MP (in the MST subfamily) to an acceptor. MST catalyzes transsulfuration reactions (Reactions 1 and 2) to various donors (9, 10).
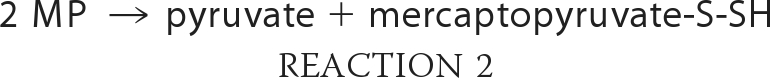 |
 |
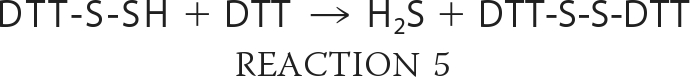 |
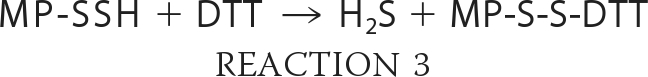
The enzymatic sulfur transfer reaction yields persulfide, not H2S. The release of H2S from mercaptopyruvate catalyzed by MST could be the product of a non-enzymatic reaction in the presence of dithiothreitol (i.e. Reactions 2 and 3 or 4 and 5) used in the reaction buffer (11) and needs to be investigated. Trypanosomal variants of MST have a C-terminal thioredoxin-like domain (12), which suggests that a sulfur acceptor for MST is thioredoxin. The activities of both MST and CAT have been characterized at alkaline pH (>9.5). Hence, direct comparison of the relative efficiency of the MST versus the transsulfuration pathway enzymes in H2S generation is currently not possible.
The relaxed substrate specificity of CBS and CSE combined with the catalytic versatility of CSE to act at both the β-carbons and the γ-carbons of substrates results in multiple H2S-generating reactions (Fig. 1a) (13, 14). Extensive kinetic characterization of these reactions at a physiologically relevant pH have led to the following conclusions. (i) H2S generation from cysteine is primarily catalyzed by CSE (13). (ii) H2S production by CBS preferentially occurs via condensation of cysteine and homocysteine (14, 15). (iii) H2S production by CSE but not CBS is responsive to the concentration of homocysteine and is increased in homocystinuric patients (13). (iv) H2S production by CBS is up-regulated by the allosteric activator, S-adenosylmethionine (14, 15). (v) Cystine, expected to be present at very low concentrations in the reducing intracellular milieu, is not a substrate for H2S production by human CSE (13). (vi) Novel thioether products of unknown function, lanthionine and homolanthionine, can be produced during H2S generation (13, 14). The substrate Km values for human CBS (6.8 mm for cysteine and 3.2 mm for homocysteine) and CSE (3.7 mm for cysteine and 2.7 mm for homocysteine), although high relative to the intracellular concentrations of these amino acids (13, 14), are substantially lower than for CAT (22 mm for cysteine) (16). The relatively high Km for CAT is consistent with the role of this enzyme together with MST in the cysteine catabolic pathway (discussed below).
MST is present in both the cytoplasmic and the mitochondrial compartments (17). CBS and CSE are cytoplasmic but might also be in the nucleus under some conditions because they are both substrates for sumoylation (18). Under oxidative stress conditions, when the cysteine demand increases to support glutathione replenishment, MST is inactive, whereas flux through the transsulfuration pathway is enhanced (19). Reversible redox regulation of MST results from the oxidative lability of a catalytic cysteine residue (20). Hence, different pathways for H2S generation might be operational under normoxic versus oxidative stress conditions and in different cell types reflecting the tissue distribution of the individual enzymes. Mechanisms for transient up-regulation of H2S production, as is expected for a signaling molecule, remain to be elucidated. Neither nitric oxide (21) nor calmodulin (22, 23), reported to modulate the activities of CSE and CBS, appears to do so (13, 15).3 Peroxynitrite (24) and carbon monoxide (25) inhibit human CBS.
Catabolism of H2S
Early indications of the metabolic lability of H2S came from labeling studies with [35S]sulfide, which demonstrated its rapid oxidation by tissues with the efficiency being organ-specific (26). Localization of sulfide oxidation activity to mitochondria was first reported more than two decades ago (27), and more recently, sulfide became the first inorganic substrate reported for human cells (28). H2S oxidation occurs in many, if not all, tissues including colonic mucosa (where it is responsible for detoxifying large quantities of H2S generated by colonic bacteria), liver, lungs, kidney, and brain. A series of oxidation steps converts H2S to persulfide, sulfite, thiosulfate (S2O32−), and sulfate (SO42−) (Fig. 1b). Sulfate comprises 77–92% of total urinary sulfur (29).
The first step in the sulfide oxidation pathway is catalyzed by SQR, a mitochondrial membrane flavoprotein that oxidizes H2S to protein-bound persulfide. The rat liver enzyme has a relatively low Km for H2S (2.9 ± 0.3 μm) (30), supporting efficient catabolism of a toxic sulfur substrate. The electron acceptor in this reaction is ubiquinone, which transfers electrons to complex III, thus linking H2S catabolism to the electron transfer chain. The sequential actions of a sulfur dioxygenase and a sulfur transferase (or rhodanese) are proposed to convert SQR-bound persulfide into sulfite and thiosulfate, respectively. Alternatively, low molecular weight thiols such as glutathione or dihydrolipoate might be involved in transferring the persulfide group from SQR to the sulfur dioxygenase, and indeed, in vitro, glutathione persulfide is a substrate for the dioxygenase (31). If CAT and MST are primarily involved in cysteine catabolism, they are likely to feed into the mitochondrial sulfide oxidation pathway described in Fig. 1b. Although the natural sulfide acceptor(s) for MST is not known, like rhodanese (32), MST can transfer the sulfide group to thioredoxin (33).
Inherited mutations in sulfur dioxygenase, the product of the ethe1 gene, result in ethylmalonic encephalopathy, an autosomal recessive disorder (34). ETHE1 is a non-heme iron-containing protein in the mitochondrial matrix. Genetic disruption of ETHE1 in mice results in accumulation of H2S above control levels and reduced levels and activity of cytochrome c oxidase in some (luminal colonocytes, muscle, brain) but not other (liver, kidney) tissues (31). The ethe1−/− mice exhibit an ∼3-fold lower sulfide-dependent oxygen consumption in liver extracts. Purified ETHE1 catalyzes glutathione persulfide-dependent O2 consumption. ETHE1 physically interacts with rhodanese, which together with the existence of fusions between these two genes in some bacterial genomes (31) suggests that the two enzymes work in a complex to convert the product of SQR, persulfide, to thiosulfate (Fig. 1b). Curiously, urinary thiosulfate was elevated, whereas sulfite was undetectable in the ethe1−/− mice. Inhibition of aerobic energy metabolism by sulfide accumulation due to ETHE1 deficiency explains some of the clinical manifestations of ethylmalonic encephalopathy (e.g. acrocyanosis and vascular damage and chronic diarrhea) (31).
The product of the sulfur dioxygenase reaction, sulfite, can be directly oxidized to sulfate by sulfite oxidase. Alternatively, sulfite can be converted by rhodanese to thiosulfate, which is presumably metabolized to sulfate via the actions of thiosulfate reductase (35) and sulfite oxidase (30). Sulfite oxidase is a molybdopterin-containing hemeprotein, and its deficiency, inherited as an autosomal recessive disorder, results in severe neurological problems (36).
Physiological Roles of H2S
The list of physiological effects mediated by H2S has been rapidly expanding and is the subject of several recent reviews (51–54). Hence, only a few salient biological effects are discussed here. Three major loci of H2S action are the cardiovascular and central nervous systems and energy metabolism. The efficacy of H2S in attenuating myocardial ischemia-reperfusion injury is mediated via protection of mitochondrial function (37). CSE−/− mice exhibit reduced endothelium-dependent vasorelaxation, are hypertensive, and have reduced H2S levels in the serum and lower H2S production rates in aorta and heart (23). H2S facilitates the induction of long term potentiation in hippocampal neurons, which requires activation of the N-methyl-d-aspartic acid receptor (38). Relaxation of smooth muscle cells is apparently mediated via opening of ATP-sensitive K+ channels (21). Remarkably, H2S induces a state of suspended animation by reversible inhibition of cytochrome c oxidase with consequent lowering of metabolic rate and body temperature (39).
In the relatively young field of H2S signaling, it is important to pay particular attention to the following issues: (i) the concentration of H2S used to elicit biological responses, (ii) the handling of this redox-active and air-sensitive compound, (iii) the substrates used to assess H2S production given the preferential utilization of cysteine versus cysteine + homocysteine by CSE and CBS, respectively (Fig. 1a), and (iv) the inhibitors of biosynthetic enzymes used to modulate its production. For instance, studies using iodoacetamide and hydroxylamine as “specific” inhibitors of CSE and CBS must be viewed with caution because these reactive compounds are completely bereft of specificity. Iodoacetamide is a cell-permeable thiol-alkylating agent that derivatizes all accessible and reactive thiols on proteins as well as small molecules e.g. glutathione, cysteine, and indeed, H2S itself. Hydroxylamine is widely used to release the cofactor from PLP-containing enzymes and inhibits not only all three PLP-dependent H2S-generating routes but also other PLP enzymes.
It is important to compare the clinical pathologies associated with deficiencies in enzymes involved in the three H2S-generating routes to the broad range of physiological effects attributed to H2S. Defects in CBS lead to a condition known as homocystinuria and affects four major organ systems: cardiovascular, ocular, skeletal, and the central nervous system (40). Deficiency of CSE gives rise to a relatively benign condition, cystathioninuria, which is not characteristically associated with a set of clinical abnormalities (40). Secondary association of cystathioninuria with a range of diseases including diabetes insipidus, Down syndrome, neuroblastoma, hepatoblastoma, and celiac disease has been reported (41). Deficiency of MST leads to mercaptolactate-cysteine disulfiduria, which may or many not be associated with mental retardation. The metabolite that accumulates in urine is formed by conversion of mercaptopyruvate to mercaptolactate by lactate dehydrogenase followed by its oxidation to form a mixed disulfide with cysteine (42).
Mechanisms of H2S Action
Despite the increasing number of reports on the biological effects of H2S, insights into its mechanisms of action and its molecular targets are largely lacking. Taking lessons from other gas sensors and gas-based signaling pathways, it is reasonable to expect that metalloproteins, particularly heme-containing ones, might mediate H2S signaling. The reversible inhibition of cytochrome c oxidase at moderately high H2S concentrations (∼80 ppm) is associated with induction of a suspended animation state (39). Although H2S is a potent inhibitor of purified cytochrome c oxidase (Ki = 0.2 μm), the situation is more complex in tissues where H2S is also a substrate for the respiratory chain (Fig. 1b). At low concentrations, it stimulates oxygen consumption (43). At higher tissue concentrations (>20 μm), the mitochondrial respiratory chain is inhibited (43). Thus, at the reported tissue concentrations of tens to hundreds of micromolar of free H2S, virtually complete inhibition of cytochrome c oxidase would be expected. H2S also reduces cytochrome c oxidase (44) and cytochrome c (45). The two-electron redox potential of H2S (Reaction 6)
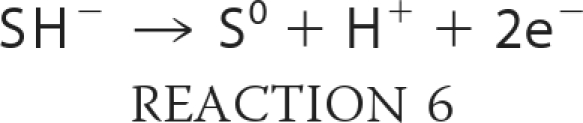 |
is +0.17 V at pH 7.0 (45), which makes it a significantly weaker reductant than the much more abundant intracellular thiols, glutathione and cysteine (E0 ∼−0.25 V). The considerably smaller size of H2S in comparison with other low molecular weight thiols would afford it preferential access to metal centers. However, based on its low concentration relative to glutathione and its high redox potential, a quantitatively significant role for H2S in cellular antioxidant function would appear unlikely.
The reactivity of H2S with globins is varied. Invertebrates living in sulfide-rich habitats encounter H2S concentrations of up to 1 mm and use hemoglobin to transport sulfide to symbiotic bacteria (46). The sulfide is apparently “carried” by zinc rather than the heme or protein thiols in the giant hydrothermal vent tubeworm, Riftia pachyptila (47). In contrast, hemoglobin I from the clam Lucina pectinata that dwells in sulfide-rich mangroves uses the ferric heme iron center to deliver sulfide to bacteria, and in turn, to protect itself from sulfide toxicity (46). The active site of hemoglobin I has evolved for H2S selectivity in contrast to the other two hemoglobins in this organism that do not bind H2S. It has been proposed that the polarity of the distal heme pocket influences the fate of H2S bound to ferric heme (48) (Fig. 2a). Active sites with a high dielectric constant stabilize ferric H2S and promote reduction by a second mole of H2S-generating ferrous heme and hydrogen persulfide (Reaction 7).
In contrast, in non-polar active sites, H2S dissociates from the heme-Fe(III)-H2S complex without redox chemistry. The affinity of sulfide for globins has been reported to range from 90 nm for the sulfide-avid L. pectinata hemoglobin I to 20 μm for sperm whale myoglobin (49).
FIGURE 2.

Molecular mechanisms for H2S targeting. a, H2S can ligate to ferric heme in globins, and depending on the stereoelectronic properties of the distal heme site, can remain bound to, dissociate from, or reduce the iron center. b, persulfidation of proteins requires attack of H2S to an oxidized side chain, e.g. a sulfenic acid or disulfide. c, once formed, persulfides need to be protected from small molecule thiols (e.g. glutathione) or thiol groups on the same or different proteins. Conversely, the same chemical reactions can be exploited to liberate H2S from the “stored sulfur” pool. GSH is glutathione.
In analogy with protein S-nitrosylation, protein S-sulfhydration has been proposed as a mechanism for H2S-mediated signaling (50). For this posttranslation modification to occur, the cysteine residue must exist in an oxidized state, e.g. sulfenic acid or a disulfide, which is subsequently attacked by the hydrosulfide anion to give a persulfide product (Fig. 2b). In oxidizing compartments, e.g. the endoplasmic reticulum or the extracellular milieu, oxidized cysteines on proteins are relatively common. In contrast, in the cytoplasmic compartment, which is reducing, oxidized cysteines on proteins have to be stabilized and sequestered. By the same token, cysteine persulfides, when formed, also have to be stabilized against (i) disulfide exchange and reduction by the abundant intracellular thiol, glutathione and (ii) intramolecular displacement by a vicinal cysteine forming a disulfide or by antioxidant enzymes like thioredoxin. In all cases, H2S is eliminated (Fig. 2c). Because the cysteine redox status of the protein substrate is a critical determinant of its ability to be modified by H2S, attention to sample handling conditions is essential for monitoring persulfide modifications. For instance, during treatment of cell lysates with NaHS to observe sulfhydration, cysteines are readily oxidized, thus artifactually enabling the modification chemistry shown in Fig. 2b. To avoid this, preparation and incubation of cells lysates must be conducted under strictly anaerobic conditions. Furthermore, treatment of cells with unphysiologically high concentrations of cysteine, which preferentially supports H2S production by CSE versus CBS (which requires homocysteine in addition to cysteine) or CAT and MST (which requires α-ketoglutarate in addition to cysteine or mercaptopyruvate) (Fig. 1a), biases conclusions about the relative importance of one versus the other pathways for H2S generation. It is presently unclear whether cellular sulfane sulfur stores present under steady-state conditions are mobilized or whether de novo H2S biogenesis is up-regulated in response to signals to trigger H2S-based signaling.
Future Perspectives
Despite the controversies in the field surrounding the physiologically relevant H2S concentrations and its biological effects (e.g. whether it is pro- or anti-inflammatory), or perhaps because of it, research on H2S biochemistry and cell biology is in the midst of a lively expansion. The field would benefit immensely from the development of chemical tools for H2S delivery, quantitation, and imaging and the design of specific inhibitors of H2S-producing and -consuming enzymes. Key biochemical questions that warrant elucidation include identification of the signals that turn on transient de novo production of H2S or its release from sulfane-sulfur stores and their tissue specificity, of the molecular targets and mechanisms of H2S action, and finally, of the pathway(s) for its rapid removal. Insights into the biochemistry of H2S metabolism would then allow the dots between the reception of an enabling (or disabling) signal and the network of biological responses that is elicited to be connected.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant HL58984 (to R. B.). This minireview will be reprinted in the 2010 Minireview Compendium, which will be available in January, 2011.
D. Padovani and R. Banerjee, unpublished observations.
- PLP
- pyridoxal 5′-phosphate
- CBS
- cystathionine β-synthase
- CSE
- γ-cystathionase
- MST
- mercaptopyruvate sulfur transferase
- MP
- mercaptopyruvate
- CAT
- cysteine aminotransferase
- SQR
- sulfide:quinone oxidoreductase
- DTT
- dithiothreitol.
REFERENCES
- 1.Vorobets V. S., Kovach S. K., Kolbasov G. Y. (2002) Russian J. Appl. Chem. 75, 229–234 [Google Scholar]
- 2.Mathai J. C., Missner A., Kügler P., Saparov S. M., Zeidel M. L., Lee J. K., Pohl P. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li C., Love G. D., Lyons T. W., Fike D. A., Sessions A. L., Chu X. (2010) Science 328, 80–83 [DOI] [PubMed] [Google Scholar]
- 4.Grice K., Cao C., Love G. D., Böttcher M. E., Twitchett R. J., Grosjean E., Summons R. E., Turgeon S. C., Dunning W., Jin Y. (2005) Science 307, 706–709 [DOI] [PubMed] [Google Scholar]
- 5.Furne J., Saeed A., Levitt M. D. (2008) Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1479–R1485 [DOI] [PubMed] [Google Scholar]
- 6.Olson K. R. (2009) Biochim. Biophys. Acta 1787, 856–863 [DOI] [PubMed] [Google Scholar]
- 7.Whitfield N. L., Kreimier E. L., Verdial F. C., Skovgaard N., Olson K. R. (2008) Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1930–R1937 [DOI] [PubMed] [Google Scholar]
- 8.Benavides G. A., Squadrito G. L., Mills R. W., Patel H. D., Isbell T. S., Patel R. P., Darley-Usmar V. M., Doeller J. E., Kraus D. W. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 17977–17982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarabak R., Westley J. (1978) Arch. Biochem. Biophys. 185, 458–465 [DOI] [PubMed] [Google Scholar]
- 10.Nagahara N., Okazaki T., Nishino T. (1995) J. Biol. Chem. 270, 16230–16235 [DOI] [PubMed] [Google Scholar]
- 11.Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., Kimura H. (2009) Antioxid. Redox Signal. 11, 703–714 [DOI] [PubMed] [Google Scholar]
- 12.Williams R. A., Kelly S. M., Mottram J. C., Coombs G. H. (2003) J. Biol. Chem. 278, 1480–1486 [DOI] [PubMed] [Google Scholar]
- 13.Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. (2009) J. Biol. Chem. 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X., Jhee K. H., Kruger W. D. (2004) J. Biol. Chem. 279, 52082–52086 [DOI] [PubMed] [Google Scholar]
- 16.Akagi R. (1982) Acta Med. Okayama 36, 187–197 [DOI] [PubMed] [Google Scholar]
- 17.Nagahara N., Ito T., Kitamura H., Nishino T. (1998) Histochem. Cell Biol. 110, 243–250 [DOI] [PubMed] [Google Scholar]
- 18.Agrawal N., Banerjee R. (2008) PLoS One 3, e4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosharov E., Cranford M. R., Banerjee R. (2000) Biochemistry 39, 13005–13011 [DOI] [PubMed] [Google Scholar]
- 20.Nagahara N., Katayama A. (2005) J. Biol. Chem. 280, 34569–34576 [DOI] [PubMed] [Google Scholar]
- 21.Zhao W., Zhang J., Lu Y., Wang R. (2001) EMBO J. 20, 6008–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eto K., Kimura H. (2002) J. Biol. Chem. 277, 42680–42685 [DOI] [PubMed] [Google Scholar]
- 23.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., Wang R. (2008) Science 322, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celano L., Gil M., Carballal S., Durán R., Denicola A., Banerjee R., Alvarez B. (2009) Arch. Biochem. Biophys. 491, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taoka S., Banerjee R. (2001) J. Inorg. Biochem. 87, 245–251 [DOI] [PubMed] [Google Scholar]
- 26.Bartholomew T. C., Powell G. M., Dodgson K. S., Curtis C. G. (1980) Biochem. Pharmacol. 29, 2431–2437 [DOI] [PubMed] [Google Scholar]
- 27.Powell M. A., Somero G. N. (1986) Science 233, 563–566 [DOI] [PubMed] [Google Scholar]
- 28.Goubern M., Andriamihaja M., Nübel T., Blachier F., Bouillaud F. (2007) FASEB J. 21, 1699–1706 [DOI] [PubMed] [Google Scholar]
- 29.Beauchamp R. O., Jr., Bus J. S., Popp J. A., Boreiko C. J., Andjelkovich D. A. (1984) Crit. Rev. Toxicol. 13, 25–97 [DOI] [PubMed] [Google Scholar]
- 30.Hildebrandt T. M., Grieshaber M. K. (2008) FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 31.Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., Zeviani M. (2009) Nat. Med. 15, 200–205 [DOI] [PubMed] [Google Scholar]
- 32.Nandi D. L., Westley J. (1998) Int. J. Biochem. Cell Biol. 30, 973–977 [DOI] [PubMed] [Google Scholar]
- 33.Westrop G. D., Georg I., Coombs G. H. (2009) J. Biol. Chem. 284, 33485–33494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiranti V., D'Adamo P., Briem E., Ferrari G., Mineri R., Lamantea E., Mandel H., Balestri P., Garcia-Silva M. T., Vollmer B., Rinaldo P., Hahn S. H., Leonard J., Rahman S., Dionisi-Vici C., Garavaglia B., Gasparini P., Zeviani M. (2004) Am. J. Hum. Genet. 74, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller F. H., Bandeiras T. M., Urich T., Teixeira M., Gomes C. M., Kletzin A. (2004) Mol. Microbiol. 53, 1147–1160 [DOI] [PubMed] [Google Scholar]
- 36.Mudd S. H., Irreverre F., Laster L. (1967) Science 156, 1599–1602 [DOI] [PubMed] [Google Scholar]
- 37.Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., Lefer D. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abe K., Kimura H. (1996) J. Neurosci. 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackstone E., Morrison M., Roth M. B. (2005) Science 308, 518. [DOI] [PubMed] [Google Scholar]
- 40.Mudd S. H., Levy H. L., Kraus J. P. (2001) in The Online Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Vogelstein B., Kinzler K. W., Childs B. eds) pp. 2007–2056, McGraw-Hill Co., New York [Google Scholar]
- 41.Wang J., Hegele R. A. (2003) Hum. Genet. 112, 404–408 [DOI] [PubMed] [Google Scholar]
- 42.Crawhall J. C., Parker R., Sneddon W., Young E. P., Ampola M. G., Efron M. L., Bixby E. M. (1968) Science 160, 419–420 [DOI] [PubMed] [Google Scholar]
- 43.Cooper C. E., Brown G. C. (2008) J. Bioenerg. Biomembr. 40, 533–539 [DOI] [PubMed] [Google Scholar]
- 44.Nicholls P., Kim J. K. (1982) Can. J. Biochem. 60, 613–623 [DOI] [PubMed] [Google Scholar]
- 45.Collman J. P., Ghosh S., Dey A., Decréau R. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 22090–22095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber R. E., Vinogradov S. N. (2001) Physiol. Rev. 81, 569–628 [DOI] [PubMed] [Google Scholar]
- 47.Flores J. F., Fisher C. R., Carney S. L., Green B. N., Freytag J. K., Schaeffer S. W., Royer W. E., Jr. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2713–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietri R., Lewis A., León R. G., Casabona G., Kiger L., Yeh S. R., Fernandez-Alberti S., Marden M. C., Cadilla C. L., López-Garriga J. (2009) Biochemistry 48, 4881–4894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicoletti F. P., Comandini A., Bonamore A., Boechi L., Boubeta F. M., Feis A., Smulevich G., Boffi A. (2010) Biochemistry 49, 2269–2278 [DOI] [PubMed] [Google Scholar]
- 50.Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. (2009) Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimura H. (2010) Antioxid. Redox Signal. 12, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 52.Gadalla M. M., Snyder S. H. (2010) J. Neurochem., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li L., Moore P. K. (2008) Trends Pharmacol. Sci. 29, 84–90 [DOI] [PubMed] [Google Scholar]
- 54.Calvert J. W., Coetzee W. A., Lefer D. J. (2010) Antioxid. Redox Signal. 12, 1203–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.