

Abstract
In thermogenic brown adipose tissue, uncoupling protein 1 (UCP1) catalyzes the dissipation of mitochondrial proton motive force as heat. In a cellular environment of high oxidative capacity such as brown adipose tissue (BAT), mitochondrial uncoupling could also reduce deleterious reactive oxygen species, but the specific involvement of UCP1 in this process is disputed. By comparing brown adipose tissue mitochondria of wild type mice and UCP1-ablated litter mates, we show that UCP1 potently reduces mitochondrial superoxide production after cold acclimation and during fatty acid oxidation. We address the sites of superoxide production and suggest diminished probability of “reverse electron transport” facilitated by uncoupled respiration as the underlying mechanism of reactive oxygen species suppression in BAT. Furthermore, ablation of UCP1 represses the cold-stimulated increase of substrate oxidation normally seen in active BAT, resulting in lower superoxide production, presumably avoiding deleterious oxidative damage. We conclude that UCP1 allows high oxidative capacity without promoting oxidative damage by simultaneously lowering superoxide production.
Keywords: Adipose Tissue, Bioenergetics, Electron Transport, Mitochondria, Reactive Oxygen Species (ROS), Brown Adipose Tissue, Cold Acclimation, Uncoupling Protein 1
Introduction
The physiological role of endogenous heat production by nonshivering thermogenesis in brown adipose tissue (BAT)2 of mammals, in particular small mammals, hibernators, and neonates, is the maintenance of a constant high body temperature (Tb) independent of daily and seasonal temperature fluctuations (1). Heat production by BAT is, however, also activated by high calorie diets (2), and because the presence of BAT has been recently shown in adult humans, it is in the focus as a potential therapeutic target for the treatment of human body weight disorders (3–5).
BAT is characterized by an exceptionally high presence of mitochondria that possess only a minor amount of ATP synthase to convert nutritional into cellular energy equivalents (6). Instead, brown adipose tissue mitochondria are specialized by a high content of mitochondrial uncoupling protein 1 (UCP1), up to 8% of total mitochondrial protein (7), which generates a proton leak in the mitochondrial inner membrane dissipating proton motive force as heat (8). UCP1 can be inhibited by physiological concentrations of purine nucleoside di- and triphosphates before free fatty acids can overcome inhibition (8).
Cold exposure stimulates lipolysis in BAT, thereby activating UCP1. Long term cold acclimation further increases the thermogenic capacity of BAT by enhanced UCP1 expression and recruitment of oxidative capacity (9). The importance of UCP1 for brown adipose tissue-derived heat production has been confirmed in UCP1-ablated mice, which are unable to defend their body temperature when acutely exposed to the cold (4 °C) (10). Successive acclimation improves the cold tolerance of UCP1-ablated mice, suggesting that other sources of heat production partially compensate for the loss of functional BAT, namely muscle shivering (11, 12) and accelerated metabolic flux in white adipose tissue (13, 14). These alternative thermogenic mechanisms appear to be less efficient and are disadvantageous because the median survival rate of UCP1-ablated mice decreases in the cold from >24 to ∼13 weeks as compared with wild type mice, raising the question about the advantages of UCP1-mediated thermogenesis.
The cold recruitment of oxidative capacity increases the chance of oxidative damage. According to the free radical theory of aging, an increase of radical production could potentially decrease life span (15, 16). Superoxide and reactive oxygen species are potently caused by mitochondria; escaping electrons from respiratory chain complexes are the major source of cellular superoxide and its derived reactive oxygen species (17, 18). Complex I (NADH:ubiquinone oxidoreductase) (19) and complex III (ubiquinol:cytochrome c oxidoreductase) (20) are postulated as the major producers of cellular superoxide, but also the mitochondrial glycerophosphate dehydrogenase, which is highly abundant in BAT (21, 22), appears to produce superoxide (23–25).
Functional studies on UCPs (UCP1, 2, and 3) showed activation of uncoupling activity by superoxide and peroxidation metabolites like 4-hydroxy-2-nonenal (26, 27), associating UCPs with a potential role in the prevention of superoxide production. These observations were highly disputed (28–30), and specifically the involvement of UCP1 in cellular reactive oxygen species regulation is questioned (29, 30). Studies comparing BAT from wild type and UCP1-ablated mice found no activation of UCP1 with 4-hydroxy-2-nonenal, unaltered uncoupling function with manipulated matrix superoxide levels, identical rates of superoxide release, and no difference in markers of oxidative damage (29, 30). Together with unchanged superoxide scavenging systems (29), these results led to the conclusion that conditions of high oxygen tension and high substrate supply in brown adipose tissue do not necessarily lead to increased oxidative stress.
Using mitochondria sampled from wild type and UCP1-ablated mice, we here show that the presence of UCP1 potently lowers mitochondrial superoxide production during cold acclimation and fatty acid oxidation. Our results therefore demonstrate that uncoupling is a physiologically relevant mechanism to reduce oxidative stress in a mammalian organ of high oxidative capacity.
EXPERIMENTAL PROCEDURES
Animals
Wild type and UCP1-ablated mice were provided by Dr. Leslie Kozak (Pennington Medical Research Center), and littermates (genetic background C57Bl/6J) were derived from heterozygous breeding pairs in the animal facility of the Philipps-Universität of Marburg. In these mice UCP1 was inactivated by homologous recombination with a deletion vector in which exon 2 and parts of exon 3 had been replaced with a neomycin resistance gene. In brown adipose tissue of these mice, no UCP1 could be detected with polyclonal antibodies (10). For experimental procedures, only homozygotes for UCP1 (wild type; UCP1+/+) and UCP1-ablated (UCP1−/−) mice were used. The mice were fed ad libitum (Sniff 1534) with free access to water and kept on a 12 h of light/12 h of dark cycle. For experiments, the animals were kept in single cages. Warm-acclimated mice (WA) were maintained at 30 °C at least 3 weeks prior to sacrifice, whereas wild type and UCP1-ablated adult mice were cold-acclimated (CA) by first transferring them to 18 °C for 3 weeks followed by 3 weeks at 5 °C (11). The final body mass of the mice did not differ between the experimental groups (WA, UCP1+/+, 23.5 ± 1.0 g, and UCP1−/−, 24.5 ± 1.0 g; CA, UCP1+/+, 24.7 ± 0.3 g, and UCP1−/−, 24.4 ± 0.4 g). All of the experimental procedures were approved by the German Animal Welfare Authorities.
Genotyping
The mice were genotyped by amplifying 201-bp (UCP1+/+) and 409-bp (UCP1−/−) fragments from the UCP1 gene using the primers 8265-5F (GGT AGT ATG CAA GAG AGG TGT), E2Rev (CCT AAT GGT ACT GGA AGC CTG), and NeoRev (CCT ACC CGC TTG CAT TGC TCA) according to a protocol kindly provided by L. Kozak. Additionally the presence or absence of UCP1 protein was validated post mortem by immunological detection in brown adipose tissue mitochondria (as published previously in Ref. 31). The membranes were probed with a rabbit anti-UCP1 polyclonal antibody (1:30,000 dilution; 3046; Chemicon), followed by the relevant peroxidase-conjugated secondary antibody (goat anti-rabbit-IgG at 1:10,000 dilution; Dako). The antigens were visualized on x-ray film (Super RX; Fuji) using an ECL Plus Western blotting detection system (SRX-101A; Konika Minolta).
Mitochondria Isolation
Mitochondria were prepared by homogenization and differential centrifugation as described previously (32). Mitochondria from one wild type and one UCP1-ablated mouse were always isolated in parallel to control for possible day-by-day variability of mitochondrial quality. All available brown adipose tissue depots were removed (interscapular, dorsal cervical, axillar, suprasternal, and subcostal) and immediately placed in ice-cold isolation medium. Protein concentration was determined photometrically using the Bradford method (33).
Measurement of Oxygen Consumption
Mitochondrial oxygen consumption was measured using a Clark-type oxygen electrode (Rank Brothers Ltd.) maintained at 37 °C and calibrated with air-saturated medium (50 mm KCl, 5 mm TES, 2 mm MgCl2 × 6H2O, 4 mm KH2PO4, 1 mm EGTA, 0.4% bovine serum albumin (w/v), pH 7.2 at room temperature) that was assumed to contain 406 nmol of O ml−1 (34). Mitochondrial respiration was measured in 500 μl of medium at a concentration of 0.1 mg ml−1 incubated with 4.8 μm rotenone (inhibiting complex I of the respiratory chain) and 2 μm oligomycin (inhibiting the ATP synthase). Mitochondria were energized by the addition of glycerol-3-phosphate (G3P, titrating up to 16 mm) or 4 mm succinate (state 4 respiration). Subsequently GDP was titrated to a final concentration of 5 mm. The ability of GDP to establish respiratory control in wild type mice also confirmed the integrity of the mitochondria after the isolation process. At the end of each respiration measurement, the artificial uncoupler FCCP was titrated in ∼1 μm steps to estimate the maximal substrate oxidation rate. State 4 respiration in some cases was directly translated into the proton leak rate, assuming that 6 protons were transported (and leaked back to the matrix) per atomic oxygen when respiring on succinate or glycerol-3-phosphate.
Measurements of Mitochondrial Hydrogen Peroxide Release
Measurements of hydrogen peroxide production of isolated mitochondria were performed similarly to the procedures of Lambert et al. (35). 10–20 μg of brown adipose tissue mitochondria were incubated in assay buffer (50 mm KCl, 5 mm TES, 2 mm MgCl2 × 6H2O, 4 mm KH2PO4, 1 mm EGTA, bovine serum albumin 0.4% (w/v), pH 7.2 at room temperature) containing a mixture of the fluorescent probe Amplex Red (50 μm; Invitrogen), 30 units ml−1 superoxide dismutase (to convert superoxide to hydrogen peroxide), 6 units ml−1 horseradish peroxidase (catalyzing the reaction of hydrogen peroxide with Amplex Red resulting in fluorescent resorufin), and 2 μm oligomycin (to inhibit ATP synthase). Amplex Red reacts with H2O2 at a 1:1 stoichiometry, whereas the stoichiometry of conversion from superoxide to H2O2 is assumed to be 1:2.
H2O2 formation was initiated by the addition of glycerol-3-phosphate (15 mm), succinate (5 mm), or a mixture of pyruvate (5 mm) and malate (3 mm). Experiments aimed to measure the H2O2 formation after palmitate addition were performed according to the protocol/substrates of the mitochondrial respiration measurements (see below). Fluorescence was detected at 37 °C in a microplate reader (BMG Labtech, FLUOstar Optima) in 96-well microplates (Greiner 96-Well μClear, F-Bottom, black). The excitation wavelength was set to 560–10 nm, and the fluorescence emission was detected at 590 nm. Fluorescence was calibrated using known amounts of H2O2 at each experimental day. Optionally, superoxide production was measured in the presence of rotenone (2 μm, inhibiting complex I-derived reactive oxygen species production), GDP (5 mm, to inhibit UCP1), and carboxyatractylate (2.5 nm) to distinguish from adenine nucleotide transporter-dependent effects.
Measurement of Oxygen Consumption and Hydrogen Peroxide Release during β-Oxidation of Palmitoyl-CoA
To measure the palmitate-dependent mitochondrial respiration and hydrogen peroxide release, we incubated the mitochondria with 5 μm coenzyme A and 2 mm l-carnitine in a measuring buffer without bovine serum albumin. The mitochondria were energized with 3 mm malate. After 7 min we added 1 mm ATP (to allow the activation of residual free fatty acids). After a further 15 min, 20 μm palmitate (equilibrated in a final concentration of 0.02% bovine serum albumin) was added that activated β-oxidation and UCP1 in parallel (36).
Statistics
In figures and text, the data are expressed as the means ± S.E. In some cases, normal distribution of data could not be assumed. Therefore, to reveal significant differences in respiration rates or reactive oxygen species production rates between genotypes, Student's t test or Mann Whitney U test was used, as indicated. To assess the effect of different substrates or inhibitors within genotypes, a paired t test or Wilcoxon test was used. All of the calculations were performed using SigmaStat 3.5. The overall level of significance was set to p < 0.05. To account for type one errors in multiple comparisons, the effective p values were adjusted according to Bonferroni.
RESULTS
In the following experiments, we investigated the impact of different substrates on mitochondrial respiration and superoxide production in isolated BAT mitochondria from warm-acclimated (30 °C) and cold-acclimated (5 °C) wild type and UCP1-ablated mice. Fig. 1 gives an overview on the respiratory chain complexes in the mitochondrial inner membrane, potential sites of substrate entry and electron leak, the latter resulting in superoxide production.
FIGURE 1.

Sites of substrate entry and superoxide production in the respiratory chain of BAT mitochondria. NADH-linked substrates donate their electrons to complex I. Further substrate entry sites, which are investigated in this study, are the mitochondrial glycerol-3-phosphate dehydrogenase (G3PDH), the succinate dehydrogenase (complex II), and the electron transfer flavoprotein (ETF), which is fueled by β-oxidation respectively. β-Oxidation also generates NADH, which enters complex I in parallel. The electrons from the entry complexes are transferred to ubiquinone (Q), forming QH2. In the normal electron forward transfer, the electrons are donated to complex III and then complex IV before finally reducing oxygen to water. Electrons may leak from the IF site and the IQ site of complex I. These sites can be distinguished by using the complex I inhibitor rotenone (ROT). A substantial amount of superoxide production can also derive from the glycerol-3-phosphate dehydrogenase and the electron transfer flavoprotein. The sites of superoxide production are indicated with stars in the figure. Proton motive force, generated by complexes I, III, and IV, is dissipated by either the ATP synthase (complex V) or UCP1. UCP1 is activated by free fatty acids (FFA) and inhibited by purine nucleoside di- and triphosphates (here, GDP).
UCP1 Reduces Superoxide Production in BAT Mitochondria of Warm-acclimated Mice
Initially, we investigated mitochondria respiring on G3P because BAT has substantial amounts of glycerol-3-phosphate dehydrogenase (Fig. 2a). Because the affinity of the G3P dehydrogenase to its substrate is comparably low, we titrated G3P to saturating amounts (supplemental Fig. S1). State 4 respiration in wild type and UCP1-ablated mice up to 7 mm G3P was similar. More than 7 mm G3P resulted in a further increase of respiration rates of wild type but not of UCP1-ablated mice, before saturating at concentrations above 13 mm (Fig. 2a). Then GDP was added to inhibit UCP1 and to induce respiratory control. The concentration of 5 mm GDP, which fully inhibited UCP1, was determined in the mitochondria of CA wild type animals (supplemental Fig. S1). By addition of GDP and recoupling of wild type mitochondria, we also assured mitochondrial integrity (because brown adipose tissue mitochondria possess very little ATP synthase, state 3 respiration is not applicable to show respiratory control, and oligomycin had minor or no effect on respiration). The addition of 5 mm GDP strongly recoupled wild type mitochondria but had only minor effects in UCP1-ablated mitochondria (Fig. 2a). At the end of each measurement, FCCP was added to induce maximal uncoupled respiration. FCCP-induced respiration was similar between wild type and UCP1-ablated mitochondria.
FIGURE 2.

Respiration and superoxide production rates of brown adipose tissue mitochondria from warm-acclimated wild type (UCP1+/+) and UCP1-ablated (UCP1−/−) mice energized with glycerol-3-phosphate (a and b) or with succinate (c and d). The effect of UCP1 on superoxide derived from different sites is illustrated in e. Mitochondrial respiration of 0.1 mg ml−1 mitochondrial protein was measured in a temperature-controlled chamber with a Clark-type electrode at 37 °C. 2 μm oligomycin, 4.8 μm rotenone, and 16 mm G3P (a) or 4 mm succinate (c) were added to establish state 4 respiration, 5 mm GDP was added to inhibit UCP1 and recouple mitochondria and at the end of each run, FCCP was added to artificially uncouple mitochondria and estimate maximum substrate oxidation. Comparing succinate with G3P respiration, succinate respiration was higher in every treatment. State 4 respiration with G3P tended to be higher in wild type as compared with UCP1-ablated mitochondria (p = 0.1, Student's t test, n = 8/group) but was significantly different during succinate respiration. (***, p < 0.001, Student's t test, n = 7/group). Superoxide production was calculated from hydrogen peroxide release assayed with Amplex Red in the presence of superoxide dismutase (30 units ml−1). Superoxide production was measured in mitochondria incubated with oligomycin (2 μm, state 4), 5 mm GDP was added to inhibit UCP1, and 4.8 μm rotenone was added to inhibit complex I. Genotype differences in superoxide production were found in state 4 and in the presence of rotenone (inhibiting complex I) (*, p < 0.05, Student's t test, n = 7/group). Superoxide production after the addition of succinate (d) was generally lower, but rotenone addition resulted in a greater decrease, demonstrating that the major proportion of superoxide was produced at complex I (#, p < 0.05, Mann Whitney U test, UCP1+/+, n = 11, and UCP1−/−, n = 15 (12 with GDP)). In e, rotenone inhibits superoxide production from the IQ site of complex I. The superoxide leaving from the IQ site (brackets a′ and a) when respiring on glycerol-3-phosphate is ∼40–50% lower in brown adipose tissue mitochondria of wild type mice as compared with UCP1-ablated mice. Respiring on succinate with rotenone, the residual superoxide production rate (brackets c′ and c) derives from other sites such as the Q radical and complex III. This residual superoxide production rate is ∼50% lower in wild type mice. Subtracting c and c′ from the rotenone-insensitive rate with glycerol-3-phosphate determines superoxide generated by the glycerol-3-phosphate dehydrogenase (brackets b and b′). This rate was ∼20% lower in UCP1-wild type mice. All of the experiments were performed from brown adipose tissue mitochondria of individual animals kept at 30 °C.
We then measured the hydrogen peroxide release rate from isolated brown adipose tissue mitochondria, which is established to report mitochondrial superoxide production rates (37, 38). Mitochondrial superoxide production rates were directly calculated from hydrogen peroxide concentrations. Superoxide production rates at state 4 (leak) respiration were lower in wild type mitochondria than in UCP1-ablated mitochondria (Fig. 2b). This reduction was attributable to UCP1 because the addition of the UCP1 inhibitor GDP abolished genotypic differences. The administration of rotenone allowed determination of the amount of superoxide production by the IQ site of complex I (Fig. 1). G3P-dependent superoxide production was only slightly sensitive to rotenone (Fig. 2b), indicating that the major proportion of superoxide was produced independently of complex I. Superoxide production rates and the associated effect of GDP were highly dependent on substrate concentration and only slightly sensitive to rotenone (supplemental Fig. S1), supporting the idea that the G3P dehydrogenase itself generates superoxide. In particular, when aiming to compare superoxide levels from warm- and cold-acclimated animals, membrane potential and UCP1-sensitive effects on superoxide production may be overwritten by differences in G3P dehydrogenase content and its self-generated superoxide (25). Therefore, we chose to further investigate mitochondria respiring on succinate (Fig. 2c), because the succinate dehydrogenase possesses high substrate affinity and is a weak producer of superoxide.
When respiring on succinate, leak respiration was ∼1.5-fold higher in wild type than in UCP1-ablated BAT mitochondria (Fig. 2c). In wild type mitochondria, leak respiration could be reduced to UCP1-ablated levels by administration of GDP. FCCP respiration was indistinguishable between wild type and UCP1-lacking mitochondria. Superoxide production rates of wild type mitochondria respiring on succinate were barely detectable but GDP-sensitive, whereas in UCP1-ablated mitochondria, the production rate was ∼5-fold higher and GDP-insensitive (Fig. 2d).
In the presence of GDP, superoxide production increased in wild type to levels as high as in UCP1-ablated mice that showed no GDP effect on superoxide production. Although superoxide production with G3P could only be slightly inhibited in the presence of rotenone, the potent inhibition in the presence of succinate demonstrated that the major proportion was produced by complex I (Fig. 2d).
The Sites of UCP1-dependent Superoxide Generation
The experimental setup measuring superoxide production with G3P and succinate, as well as the administration of rotenone, allowed for the determination of superoxide producing sites in BAT mitochondria. The rotenone-sensitive proportion of superoxide can be attributed to the IQ site of complex I. Superoxide generated by the G3P dehydrogenase can be calculated by subtracting the rotenone-insensitive superoxide production rate of succinate (residual superoxide) from the rate with G3P energization. Analyzing this (Fig. 2e), UCP1 decreases superoxide production at the IQ site by ∼40–50%, decreases superoxide production at the G3P dehydrogenase just ∼20%, and decreases superoxide production at residual sites such as the Q pool and complex III by 50–70%.
Mitochondrial Respiration and Superoxide Production during Cold Acclimation
Although brown adipose tissue should be inactive in the thermoneutral zone (30 °C) of the mouse, cold temperatures should lead to proliferation and activation of brown adipose tissue to produce heat. To examine this physiological condition, we acclimated the two genotypes (UCP1+/+/UCP1−/−) to 5 °C as described under “Experimental Procedures.” We then investigated thermoregulatory effects on respiration and superoxide production of isolated brown adipose tissue mitochondria.
Cold acclimation caused an increase in UCP1 protein levels (Fig. 3a) and in leak respiration in wild type mitochondria (compare Fig. 2c with Fig. 3b) but had no increasing effect in UCP1-ablated mice. The genotype difference in succinate respiration observed in WA animals was further increased in response to cold acclimation (Fig. 3b). Surprisingly, state 4 respiration in the CA UCP1-ablated mice (Fig. 3b) was decreased in contrast to WA UCP1-ablated mice (Fig. 2c), whereas in wild type mitochondria, cold acclimation led to an increase of state 4 respiration (Fig. 3b). The addition of 5 mm GDP inhibited UCP1 and decreased state 4 respiration of cold-acclimated wild type mice mitochondria (Fig. 3b). Similar to WA animals, GDP had only minor effects on UCP1-lacking mitochondria (Fig. 3b). The maximal substrate oxidation rate of CA UCP1-ablated mice determined with FCCP was not increased, suggesting that their mitochondria were unable to recruit oxidative capacity in the cold. The state 4 superoxide production rate of BAT mitochondria from CA animals was ∼7.5-fold lower in wild type than in UCP1-ablated mitochondria (Fig. 3d). The addition of GDP increased superoxide production of wild type mitochondria by 30-fold, exceeding values found for UCP1-ablated mice.
FIGURE 3.

Cold-induced adaptive changes in mitochondrial physiology. Succinate respiration rates/UCP1-dependent proton leak rate and superoxide production rates/UCP1-dependent superoxide production of brown adipose tissue mitochondria from cold-acclimated wild type (UCP1+/+) and UCP1-ablated (UCP1−/−) mice are shown. The ablation and cold-induced protein levels of UCP1 were confirmed by immunological detection of UCP1 in brown adipose tissue with an anti-hamster UCP1 antibody (a). Experiments on mitochondrial respiration were performed on 0.1 mg of mitochondrial protein/ml from wild type (n = 7) and UCP1-ablated mice mitochondria (n = 5) incubated in an air-saturated medium at 37 °C (b). Respiration was started in state 4 with 2 μm oligomycin, 4.8 μm rotenone (ROT), and 4 mm succinate. The addition of GDP (5 mm) acted as an inhibitor of UCP1, and a final addition of 2–4 μm FCCP was made to compare maximum substrate oxidation rates. State 4 respiration of CA wild type mitochondria was higher than of UCP1-lacking mitochondria, and the difference persisted after GDP addition (b) (#, p < 0.05, Mann Whitney U test). The UCP1-dependent proton leak rate (c) was calculated based on the assumption that 6 protons are transported per molecule of oxygen and increased by 1.6 μmol min−1 mg−1 in response to cold acclimation. The data for WA mice were taken from Fig. 1 (***, p < 0.001, Student's t test). Superoxide production was assessed by measurement of the hydrogen peroxide release rate, determined fluorometrically over the reduction of Amplex Red to resorufin in the presence of superoxide dismutase (30 units ml−1) and horseradish peroxidase (6 units ml−1). In state 4 conditions (with 2 μm oligomycin), superoxide production was higher in UCP1-ablated mitochondria (n = 5). Administration of GDP increased wild type (n = 7) mitochondrial superoxide production, exceeding the levels found for UCP1-ablated mitochondria as well as WA wild type mitochondria (see Fig. 1d) (Fig. 1; #, p < 0.05, Mann Whitney U test) (d). The UCP1-dependent superoxide production rate was calculated as superoxide production after GDP inhibition minus the respective state 4 superoxide production, resulting in a 2.7-fold increase of UCP1-dependent superoxide production of wild type mitochondria in response to cold (#, p < 0.05, Mann Whitney U test) (e).
UCP1-sensitive Changes in Proton Leak Rate
Mitochondrial oxygen consumption in state 4 can be directly translated to proton leak rate, assuming that with complex II substrates, 6 protons are transported out of the matrix (and leak back) per reduced oxygen atom. Calculated from Fig. 2c, GDP led to a decrease of proton leak rate by 1.00 μmol of protons min−1 mg−1 protein in WA wild type but only by 0.18 μmol of protons min−1 mg−1 mitochondrial protein in UCP1-ablated mice (UCP1-independent effects). The UCP1-dependent proportion of proton leak rate of mitochondria respiring on succinate in state 4 can therefore be given as 0.82 μmol of protons min−1 mg−1 (Fig. 3c). To investigate the physiological relevance of UCP1-mediated proton leak during cold acclimation, we also determined the UCP1-dependent proportion of proton leak from CA wild type brown adipose tissue mitochondria (Fig. 3b), which was ∼2.74 and ∼0.30 μmol of protons min−1 mg−1 for UCP1-lacking mitochondria. Consequentially 2.44 μmol of protons min−1 mg−1 of the proton leak was GDP-dependent. Taken together, the UCP1-dependent proton turnover rate in response to cold was increased by ∼1.6 μmol of protons min−1 mg−1 or 300% (Fig. 3c).
UCP1-dependent Changes in Superoxide Production
What is the contribution of UCP1 in reducing mitochondrial superoxide? Inhibiting UCP1 with GDP should give a differential value of superoxide production rate that reflects the production rate depending on UCP1. This value can be further corrected with the effect of GDP in the UCP1-ablated mice. The calculation of the UCP1-dependent superoxide production (Fig. 3d) showed that under warm conditions UCP1 blunts 83% of potential mitochondrial superoxide production. During cold stress, when the respiration rate and the flow of electrons through the respiratory chain are elevated, the protective function of UCP1 increased and mitigated more than 96% of potential superoxide production (calculated as the difference of values ± GDP).
Other Potential Sources of Uncoupling Affecting Mitochondrial Superoxide Production
The adenine nucleotide translocase contributes significantly to basal proton leak (39) and can also regulate uncoupling (40–42), albeit less than UCP(s). In BAT mitochondria of our experiments, the adenine nucleotide translocase, inhibited with carboxyatractylate, did not affect superoxide production rates (supplemental Fig. S2), although it has been shown previously that isoforms may contribute to uncoupling in BAT mitochondria (29).
Superoxide Production with the Complex I Substrates Pyruvate/Malate
Using pyruvate and malate as substrate of BAT mitochondria from both CA genotypes, we found barely detectable superoxide production rates (Fig. 4) that were not increased by GDP. Substantial superoxide production could only be provoked by adding rotenone (produced at the IF site, see Fig. 1) and was higher in the mitochondria of CA wild type compared with UCP1-ablated mice (Fig. 4).
FIGURE 4.
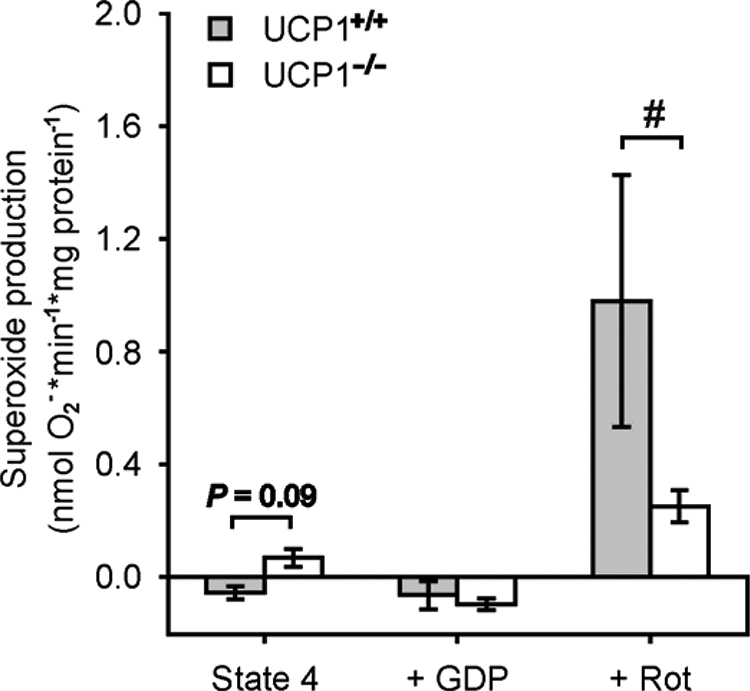
Superoxide production of brown adipose tissue mitochondria from cold-acclimated wild type (UCP1+/+) and UCP1-ablated (UCP1−/−) mice mitochondria by complex I (pyruvate/malate). A mixture of pyruvate (5 mm) and malate (3 mm) was used to provide complex I with NADH. By supplemental addition of rotenone (Rot), we showed that UCP1 provides superoxide production by reverse electron transfer. Independent experiments were performed with wild type mitochondria (n = 4) and UCP1-ablated mice mitochondria (n = 8) (#, p < 0.05 Mann Whitney U test).
UCP1 Mitigates Mitochondrial Superoxide during Fatty Acid Oxidation
During adaptive nonshivering thermogenesis, triglycerides are broken down by lipolytic activity, and fatty acids are delivered to brown adipose tissue mitochondria. Free fatty acids activate UCP1, but in the presence of CoA and ATP, they are subsequently converted to acyl-CoA clearing this physiological UCP1 activator. To highlight the relevance of UCP1 in preventing superoxide production in a physiological scenario by mimicking cold-stimulated lipolysis, we incubated brown adipose tissue mitochondria from cold-acclimated mice with compounds of the carnitine cycle (l-carnitine, CoA), let them respire on malate to provide C4-bodies for β-oxidation, and then added ATP to activate and oxidize residual free fatty acids as described previously (Ref. 43 and Fig. 5). The addition of ATP increased mitochondrial respiration in all mice (Fig. 5, a and b), clearly demonstrating that fatty acids were oxidized despite the absence or partial purine nucleotide-related inhibition of UCP1. After subsequent addition of a 20 μm pulse of equilibrated palmitate (containing 512 nm free fatty acids according to Ref. 44), the initial respiratory burst appeared to be larger in wild type mice. This increase of respiration is likely caused by a combination of palmitoyl-CoA oxidation and uncoupled respiration and was, however, depressed in UCP1-ablated mitochondria.
FIGURE 5.

Effects of UCP1 ablation on superoxide production during fatty acid oxidation in brown adipose tissue mitochondria of cold-acclimated animals. a and b, palmitatoyl-CoA oxygen consumption response curve of a wild type mouse (a) and of a UCP1-ablated mouse (b). The arrows indicate the time points when substrates were added. c and d, superoxide production of the same wild type mouse as shown above measured as fluorescence (c) and of the same UCP1-ablated mouse (d). At t = 0 the mitochondria were respiring on malate (3 mm) in all of the performed measurements. e and f, the mean values ± S.E. of wild type mice (n = 4) and UCP1-ablated mice (n = 8) for every treatment (***, p < 0.001, one-way RM analysis of variance (Bonferroni-adjusted)).
Although superoxide production rates of mitochondria from both genotypes energized with malate were similar, the activation of endogenous free fatty acids induced by adding 1 mm ATP resulted in a more pronounced increase of superoxide production of UCP1-ablated mitochondria as compared with wild type mitochondria (Fig. 5, c–f). Although the following palmitate pulse induced a stronger respiration rate of CA wild type mitochondria, the superoxide production rate remained on a constant level, whereas it further increased in UCP1-ablated mitochondria.
DISCUSSION
Cold stress causes the activation of brown adipose tissue mitochondria, which quantitatively increases the influx of electrons, the reduction state of the respiratory chain, and the amount of respiratory complexes. This is seen in our experiments by an increase of UCP1-dependent proton leak rate (Fig. 3c). The probability of electrons to leak from the respiratory chain to form superoxide should increase, but our results show that this increase is blunted by UCP1. Major sites of superoxide production that are affected by the presence of UCP1 are the IQ site (∼40–50% lower) and other sites in the respiratory chain like the Q pool or complex III (∼50–70% lower).
In contrast, a previous study (29) concluded no effect of UCP1 on superoxide production but measured rates in the presence of rotenone (inhibiting complex I) and antimycin A (inhibiting complex III). Under these conditions, however, there is reduced electron flow, and membrane potential is negligible (Fig. 1). Therefore, no effect of UCP1 under those conditions is expected.
We show that the glycerol-3-phosphate dehydrogenase, which is highly abundant in brown adipose tissue, produces a major proportion of superoxide that can only be mildly reduced (∼20%) by uncoupling. Previous reports confirm that the glycerol-3-phosphate dehydrogenase per se generates high levels of superoxide under in vitro conditions (24).
The oxidative capacity of BAT mitochondria from warm-acclimated animals, as measured by maximal uncoupling with FCCP, shows no genotype differences. However, during cold acclimation, it appears that only BAT of wild type mice can derive the typical cold-induced oxidative capacity requiring the presence of UCP1.
In fact, superoxide production in UCP1-ablated mice is lower than expected from wild type mitochondria with inhibited UCP1. The unexpectedly low superoxide production rates in cold-acclimated UCP1-ablated mitochondria can be explained by compensatory lower respiratory chain activity. There is evidence for lower complex I content in UCP1-ablated mitochondria (seen in Fig. 4), because the electron leak provoked by rotenone at the complex I flavin site is lower in UCP1-ablated than in wild type mitochondria. Other downstream components of the respiratory chain are also affected, because maximal succinate respiration in the presence of FCCP is also reduced in CA UCP1-ablated mitochondria (Fig. 2b). The failure to increase oxidative capacity in UCP1-ablated mice may protect the brown adipocytes from toxic superoxide levels. Indeed, we show that the protective function of UCP1 on potential superoxide production increases in the cold, and despite higher substrate turnover rates, superoxide production in wild type mitochondria is further reduced in the cold.
The direction of electron flow causing electron leak and superoxide formation is still controversially discussed. Superoxide production with NADH-linked substrates is generally associated with forward electron transfer. Measuring autofluorescence of NAD(P)H, the mitochondrial NAD(P)+ pool was ∼70% reduced by supplying a mixture of pyruvate/malate (100% in the presence of rotenone; see supplemental Fig. S3). Under these conditions, we found almost no generation of superoxide. Considering that superoxide production with the complex II-linked succinate was sensitive to GDP and diminished by rotenone, it may be suggested by current state-of-art definitions that superoxide in BAT mitochondria is likely caused by “reverse electron transport,” which can be potently diminished by UCP1 (Figs. 2d and 3d).
When respiring on palmitate with malate, we found a similar reduction state of the NAD(P)+ pool (∼70%) as with pyruvate/malate. Oxidizing palmitate in the presence of malate, 70% of the electrons are transferred to NAD+, whereas 30% of them are transferred to the electron transfer flavoprotein and complex II. Because complex I is unlikely to produce a significant amount of superoxide during forward transport in BAT mitochondria (as shown with pyruvate/malate) most of the superoxide during β-oxidation in UCP1-ablated mice can be addressed to reverse electron transport. Generation of superoxide by electron transfer flavoprotein per se may be of minor significance because UCP1-ablated mitochondria have less respiration/oxidative capacity in the cold but substantially produce more superoxide (Fig. 5).
Our data support physiological significance of UCP1 in blunting enhanced superoxide production during β-oxidation, the major pathway of substrate oxidation in brown adipose tissue during cold-induced thermogenesis. Pertaining to superoxide and the lack of cold-induced substrate oxidation in CA UCP1-ablated mice, superoxide may also act as a signaling molecule reporting the redox status of mitochondria, thereby adjusting mitochondrial biogenesis, as recently hypothesized (45). It has been emphasized that some reactive oxygen species are required to induce mitochondrial biogenesis via PGC-1α (46), but our results suggest that pathologically high reactive oxygen species concentrations inhibit respiration. Initial superoxide bursts during cold acclimation in UCP1-ablated mice may compromise the function of respiratory chain complexes, as seen for superoxide dismutase 2 knock-out mice (47). Further indirect evidence that superoxide negatively controls oxidative capacity was derived from superoxide dismutase 2 overexpression, which resulted in enhanced oxidative capacity (30).
The survival rates of UCP1-ablated mice are markedly reduced in the cold (11). Although local oxidative damage in BAT could be responsible for the shorter life span, the absence of UCP1 requires recruitment of other tissues for heat production with less mitochondrial uncoupling capacity. This might increase systemic radical damage, affecting life span.
Our results show the interrelationship of brown adipose tissue UCP1, energy dissipation, and superoxide production, which may become instrumental when considering human brown adipose tissue for the treatment of obesity. A model proposing activation of UCP1 directly by superoxide as a negative feedback, thereby preventing further superoxide production (27), is highly disputed (28–30). Independent of such a mechanism, we here demonstrate that UCP1 regulates mitochondrial superoxide production upon physiological challenge. We comprise our results in a model shown in Fig. 6. Mammalian UCP1 allows high oxidation rates in the absence of deleterious oxidative stress, and because of this dual role in heat production and superoxide reduction, evolution may have selected for the implementation of UCP1 in adaptive nonshivering thermogenesis of mammals. The role of recently discovered ancient UCP1 orthologues in ectotherms (48) may well be prevention of superoxide production prior to evolution of nonshivering thermogenesis.
FIGURE 6.
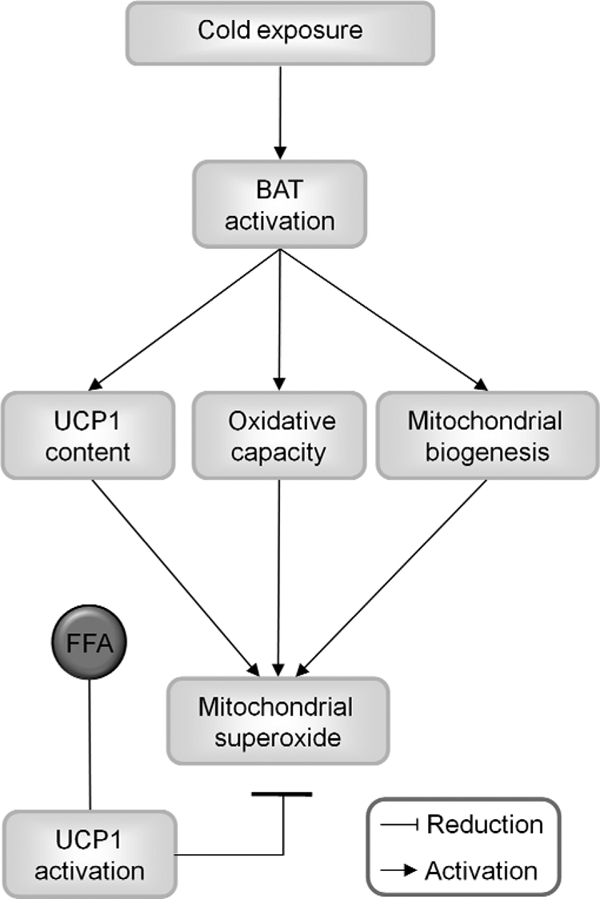
Physiological significance of mitochondrial uncoupling on mitochondrial superoxide production in brown adipose tissue mitochondria. Cold exposure activates brown adipose tissue for heat production, which is generally referred to as adaptive nonshivering thermogenesis. The heat output of the brown adipocytes is increased by stimulation of oxidative capacity and mitochondrial biogenesis. These factors would usually potently increase the probability of mitochondrial superoxide production. In BAT mitochondria, however, cold acclimation also stimulates UCP1 gene transcription and protein levels, and cold-induced free fatty acid (FFA) release activates UCP1, which dissipates proton motive force as heat. Based on our study, we put forward a model that proposes an additional physiological role of UCP1 by preventing cellular damage through the decrease of superoxide formation.
Supplementary Material
Acknowledgments
We thank Sigrid Stöhr and Sebastian Busse for excellent technical assistance and Prof. Martin Brand for helpful discussion.
This work was supported by Deutsche Forschungsgemeinschaft Grants HE 990/15-1 (to G. H. and C. W. M.) and JA 1884/2-1.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- BAT
- brown adipose tissue
- CA
- cold-acclimated
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- UCP
- uncoupling protein
- WA
- warm-acclimated
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- G3P
- glycerol-3-phosphate.
REFERENCES
- 1.Heldmaier G., Klaus S., Wiesinger H. (1990) Seasonal Adaptation of Thermoregulatory Heat Production in Small Mammals, Springer-Verlag, Berlin [Google Scholar]
- 2.Rothwell N. J., Stock M. J. (1983) Adv. Nutr. Res. 5, 201–220 [DOI] [PubMed] [Google Scholar]
- 3.Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., Kahn C. R. (2009) N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., Teule G. J. (2009) N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 5.Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerbäck S., Nuutila P. (2009) N. Engl. J. Med. 360, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 6.Cannon B., Vogel G. (1977) FEBS Lett. 76, 284–289 [DOI] [PubMed] [Google Scholar]
- 7.Rousset S., Alves-Guerra M., Mozo J., Miroux B., Cassard-Doulcier A., Bouillaud F., Ricquier D. (2004) Diabetes 53, (Suppl. 1) 130–135 [DOI] [PubMed] [Google Scholar]
- 8.Nicholls D. G., Locke R. M. (1984) Physiol. Rev. 64, 1–64 [DOI] [PubMed] [Google Scholar]
- 9.Klingenspor M. (2003) Exp. Physiol. 88, 141–148 [DOI] [PubMed] [Google Scholar]
- 10.Enerback S., Jacobsson A., Simpson E. M., Guerra C., Yamashita H., Harper M. E., Kozak L. P. (1997) Nature 387, 90–94 [DOI] [PubMed] [Google Scholar]
- 11.Golozoubova V., Hohtola E., Matthias A., Jacobsson A., Cannon B., Nedergaard J. (2001) FASEB J. 15, 2048–2050 [DOI] [PubMed] [Google Scholar]
- 12.Aydin J., Shabalina I. G., Place N., Reiken S., Zhang S. J., Bellinger A. M., Nedergaard J., Cannon B., Marks A. R., Bruton J. D., Westerblad H. (2008) FASEB J. 22, 3919–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granneman J. G., Burnazi M., Zhu Z., Schwamb L. A. (2003) Am. J. Physiol. Endocrinol. Metab. 285, E1230–E1236 [DOI] [PubMed] [Google Scholar]
- 14.Ukropec J., Anunciado R. P., Ravussin Y., Hulver M. W., Kozak L. P. (2006) J. Biol. Chem. 281, 31894–31908 [DOI] [PubMed] [Google Scholar]
- 15.Harman D. (1956) J. Gerontol. 11, 298–300 [DOI] [PubMed] [Google Scholar]
- 16.Harman D. (1972) Am. J. Clin. Nutr. 25, 839–843 [DOI] [PubMed] [Google Scholar]
- 17.Raha S., Robinson B. H. (2001) Am. J. Med. Genet. 106, 62–70 [DOI] [PubMed] [Google Scholar]
- 18.Wallace D. C. (1999) Science 283, 1482–1488 [DOI] [PubMed] [Google Scholar]
- 19.Cadenas E., Davies K. J. (2000) Free Radic. Biol. Med. 29, 222–230 [DOI] [PubMed] [Google Scholar]
- 20.Zhang L., Yu L., Yu C. A. (1998) J. Biol. Chem. 273, 33972–33976 [DOI] [PubMed] [Google Scholar]
- 21.Ratner P. L., Fisher M., Burkart D., Cook J. R., Kozak L. P. (1981) J. Biol. Chem. 256, 3576–3579 [PubMed] [Google Scholar]
- 22.Koza R. A., Kozak U. C., Brown L. J., Leiter E. H., MacDonald M. J., Kozak L. P. (1996) Arch. Biochem. Biophys. 336, 97–104 [DOI] [PubMed] [Google Scholar]
- 23.Drahota Z., Chowdhury S. K., Floryk D., Mrácek T., Wilhelm J., Rauchová H., Lenaz G., Houstek J. (2002) J. Bioenerg. Biomembr. 34, 105–113 [DOI] [PubMed] [Google Scholar]
- 24.Miwa S., Brand M. D. (2005) Biochim. Biophys. Acta. 1709, 214–219 [DOI] [PubMed] [Google Scholar]
- 25.Miwa S., St-Pierre J., Partridge L., Brand M. D. (2003) Free Radic. Biol. Med. 35, 938–948 [DOI] [PubMed] [Google Scholar]
- 26.Echtay K. S., Roussel D., St-Pierre J., Jekabsons M. B., Cadenas S., Stuart J. A., Harper J. A., Roebuck S. J., Morrison A., Pickering S., Clapham J. C., Brand M. D. (2002) Nature 415, 96–99 [DOI] [PubMed] [Google Scholar]
- 27.Echtay K. S., Esteves T. C., Pakay J. L., Jekabsons M. B., Lambert A. J., Portero-Otín M., Pamplona R., Vidal-Puig A. J., Wang S., Roebuck S. J., Brand M. D. (2003) EMBO J. 22, 4103–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Couplan E., del Mar Gonzalez-Barroso M., Alves-Guerra M. C., Ricquier D., Goubern M., Bouillaud F. (2002) J. Biol. Chem. 277, 26268–26275 [DOI] [PubMed] [Google Scholar]
- 29.Shabalina I. G., Petrovic N., Kramarova T. V., Hoeks J., Cannon B., Nedergaard J. (2006) J. Biol. Chem. 281, 13882–13893 [DOI] [PubMed] [Google Scholar]
- 30.Silva J. P., Shabalina I. G., Dufour E., Petrovic N., Backlund E. C., Hultenby K., Wibom R., Nedergaard J., Cannon B., Larsson N. G. (2005) EMBO J. 24, 4061–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klingenspor M., Ivemeyer M., Wiesinger H., Haas K., Heldmaier G., Wiesner R. J. (1996) Biochem. J. 316, 607–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mzilikazi N., Jastroch M., Meyer C. W., Klingenspor M. (2007) Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R2120–R2127 [DOI] [PubMed] [Google Scholar]
- 33.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 34.Reynafarje B., Costa L. E., Lehninger A. L. (1985) Anal. Biochem. 145, 406–418 [DOI] [PubMed] [Google Scholar]
- 35.Lambert A. J., Buckingham J. A., Brand M. D. (2008) FEBS Lett. 582, 1711–1714 [DOI] [PubMed] [Google Scholar]
- 36.Locke R. M., Rial E., Scott I. D., Nicholls D. G. (1982) Eur. J. Biochem. 129, 373–380 [DOI] [PubMed] [Google Scholar]
- 37.Lambert A. J., Brand M. D. (2004) J. Biol. Chem. 279, 39414–39420 [DOI] [PubMed] [Google Scholar]
- 38.Lambert A. J., Brand M. D. (2004) Biochem. J. 382, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brand M. D., Pakay J. L., Ocloo A., Kokoszka J., Wallace D. C., Brookes P. S., Cornwall E. J. (2005) Biochem. J. 392, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreyev AYu, Bondareva T. O., Dedukhova V. I., Mokhova E. N., Skulachev V. P., Volkov N. I. (1988) FEBS Lett. 226, 265–269 [DOI] [PubMed] [Google Scholar]
- 41.Andreyev AYu, Bondareva T. O., Dedukhova V. I., Mokhova E. N., Skulachev V. P., Tsofina L. M., Volkov N. I., Vygodina T. V. (1989) Eur. J. Biochem. 182, 585–592 [DOI] [PubMed] [Google Scholar]
- 42.Khailova L. S., Prikhodko E. A., Dedukhova V. I., Mokhova E. N., Popov V. N., Skulachev V. P. (2006) Biochim. Biophys. Acta. 1757, 1324–1329 [DOI] [PubMed] [Google Scholar]
- 43.Hittelman K. J., Lindberg O., Cannon B. (1969) Eur. J. Biochem. 11, 183–192 [DOI] [PubMed] [Google Scholar]
- 44.Richieri G. V., Anel A., Kleinfeld A. M. (1993) Biochemistry 32, 7574–7580 [DOI] [PubMed] [Google Scholar]
- 45.Murphy M. P. (2009) Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., Spiegelman B. M. (2006) Cell 127, 397–408 [DOI] [PubMed] [Google Scholar]
- 47.Melov S., Coskun P., Patel M., Tuinstra R., Cottrell B., Jun A. S., Zastawny T. H., Dizdaroglu M., Goodman S. I., Huang T. T., Miziorko H., Epstein C. J., Wallace D. C. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jastroch M., Wuertz S., Kloas W., Klingenspor M. (2005) Physiol. Genomics 22, 150–156 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.