

Abstract
More than 100 different mutations in Cu,Zn-superoxide dismutase (SOD1) are linked to a familial form of amyotrophic lateral sclerosis (fALS). Pathogenic mutations facilitate fibrillar aggregation of SOD1, upon which significant structural changes of SOD1 have been assumed; in general, however, a structure of protein aggregate remains obscure. Here, we have identified a protease-resistant core in wild-type as well as fALS-causing mutant SOD1 aggregates. Three different regions within an SOD1 sequence are found as building blocks for the formation of an aggregate core, and fALS-causing mutations modulate interactions among these three regions to form a distinct core, namely SOD1 aggregates exhibit mutation-dependent structural polymorphism, which further regulates biochemical properties of aggregates such as solubility. Based upon these results, we propose a new pathomechanism of fALS in which mutation-dependent structural polymorphism of SOD1 aggregates can affect disease phenotypes.
Keywords: Amyloid, Amyotropic Lateral Sclerosis (Lou Gehrig Disease), Protein Folding, Protein-Protein Interactions, Superoxide Dismutase (SOD), Protein Aggregation
Introduction
Misfolding of a protein molecule often causes its insoluble aggregation, and formation of inclusion bodies containing protein aggregates is a major pathological change in conformational diseases such as neurodegenerative disorders (1). Increasing evidence has suggested that structures/morphologies of protein aggregates affect disease phenotypes and that polymorphism of protein aggregates associates with phenotypic heterogeneity (2–5). Distinct molecular structures of protein aggregates will therefore play an important role in expression of different phenotypes observed in conformational diseases.
Among neurodegenerative disorders, more than a hundred dominant mutations in Cu,Zn-superoxide dismutase (SOD1) have been identified to cause a familial form of amyotrophic lateral sclerosis (fALS)4 (6). SOD1 binds a copper and a zinc ion and forms an intramolecular disulfide bond (7), all of which tightly regulate an antioxidant activity of SOD1 that converts superoxide into oxygen and hydrogen peroxide (8). SOD1 knock-out mice, however, show no fALS-like phenotypes, suggesting that an enzymatic role of SOD1 makes a minor contribution to neurodegeneration (9). Instead, SOD1 has been considered to gain toxic properties by fALS mutations, and one of those is the increased propensity of protein misfolding and aggregation (10). Among all SOD1-related fALS, a common pathological change is accumulation of detergent-insoluble SOD1 aggregates in spinal cords (10).
So far, much effort has been made to reveal a general mechanism of how >100 fALS mutations promote SOD1 aggregation. Most of fALS mutations destabilize a native structure of SOD1 (11, 12), which retards either metal binding or disulfide formation in SOD1 (13–15). A pathogenic consequence common to all fALS mutations in SOD1 has hence been proposed to increase an intracellular fraction of a metal-free SOD1 without a disulfide (apo-SOD1SH), which is the most aggregation-prone state (13). Overall structures of SOD1 appear to be preserved even after demetallation and disulfide reduction, but a significant structural disorder in the loop regions has been identified in soluble apo-SOD1SH (16). Such increased mobility of the loop regions in the apo-SOD1SH state has been considered to promote non-native interactions between SOD1s (17), possibly leading to insoluble aggregation.
During the aggregate formation, apo-SOD1SH has been predicted to undergo drastic structural changes, which include three-dimensional rearrangement of β-sheets called domain swapping (18). However, there is little experimental evidence to unveil the structure(s) that an SOD1 molecule adopts in the insoluble aggregates. In addition, less attention has been paid so far on any possible differences in structural properties among SOD1 aggregates with different fALS mutations. Given that aggregate polymorphism associates with different disease phenotypes in the other neurodegenerative disorders such as prion diseases (2, 3), an assumption on a “mutation-independent” structure of SOD1 aggregates should now be carefully examined; indeed, phenotypic heterogeneity has been reported in fALS patients with different SOD1 mutations (19).
In this study, we have experimentally identified a protease-resistant core structure in the SOD1 aggregates and found that three different regions (amino acids 1–30, 90–120, and 135–153) within an SOD1 primary sequence form a scaffold of a core in the aggregates. Interestingly, fALS mutations in SOD1 can modulate interactions of those three scaffold regions in a core structure, which produces mutation-dependent structural polymorphism of SOD1 aggregates. Furthermore, such mutation-dependent core structures lead to distinct morphological and biochemical properties of fALS-mutant SOD1 aggregates. Based upon this study, we propose a new pathomechanism of fALS in which mutation-dependent structures of SOD1 aggregates can affect the disease phenotypes.
EXPERIMENTAL PROCEDURES
Preparation of SOD1 Proteins
WT and fALS-mutant SOD1 cDNA were cloned in a multiple cloning site of a plasmid, pET15b (Novagen), using NdeI and SalI sites, where a SalI site is introduced between BamHI and Bpu1102I sites. SOD1 proteins were obtained as inclusion bodies in Escherichia coli, Rosetta, transformed with the above plasmid after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 6 h and purified using Proteus Midi IMAC (Pro-Chem Inc.) in the presence of 6 m guanidine hydrochloride. Purified SOD1 proteins were refolded by 100-fold dilution in a buffer containing 100 mm Na-Pi, 100 mm NaCl, 5 mm EDTA, pH 8.0 (NNE buffer), with 5 mm DTT and stirred at 4 °C overnight. Refolded protein samples were concentrated and ultracentrifuged at 110,000 × g for 30 min at 4 °C to remove insoluble materials. Protein concentration was spectroscopically determined from the absorbance at 280 nm (5,500 cm−1 m−1 of an extinction coefficient).
An N-terminal His tag in SOD1 was removed by treatment with thrombin immobilized with agarose (Calbiochem) in an NNE buffer at 37 °C at 100 rpm for an hour. After removal of immobilized thrombin by filtration and ultracentrifugation at 110,000 × g for 30 min, the protein concentration in a supernatant fraction was checked spectroscopically. Removal of a His tag was confirmed by SDS-PAGE as well as MALDI-TOF mass spectrometry (supplemental Fig. S1, A–C).
Characterization of SOD1 Aggregation
To examine aggregation of SOD1 proteins, 100 μm SOD1 in an NNE buffer containing 5 mm DTT was set in a 96-well plate and shaken at 37 °C at 1,200 rpm. Turbidity was monitored in a plate reader (ARVO MX, PerkinElmer Life Sciences) by measuring the absorbance around 405 nm. SOD1-insoluble aggregates were obtained as a pellet fraction after ultracentrifugation of the reaction mixtures at 110,000 × g for 30 min (supplemental Fig. S1H).
For the analysis of ThT fluorescence, SOD1 aggregates were resuspended in water with ultrasonication, and aliquots were loaded on an SDS-polyacrylamide gel and stained with Coomassie Brilliant Blue (supplemental Fig. S2A); thereby we can estimate SOD1 concentration (at a monomer basis) in aggregates. Using a 96-well plate, 3 μm SOD1 aggregates were then mixed with 25 μm ThT in an NNE buffer, and the fluorescence was measured in a plate reader (ARVO MX) with a CW lamp filter (440-nm cutoff) and an emission filter (486-nm cutoff).
To examine temperature dependence of solubility of SOD1 aggregates, 30 μm SOD1 aggregates were prepared in an NNE buffer containing 5 mm DTT and 1% Sarkosyl and incubated at either 37, 60, 80, or 100 °C for an hour. After ultracentrifugation at 110,000 × g for 30 min, a supernatant fraction was saved, and a pellet fraction was further redissolved in an NNE buffer with 2% SDS. Both fractions were then mixed with an SDS-PAGE sample buffer containing 5 mm TCEP, boiled, and loaded on a 12.5% SDS-polyacrylamide gel. Protein bands were visualized by staining with Coomassie Brilliant Blue, and intensity of SOD1 bands was analyzed by an ImageJ software. Band intensities of soluble and insoluble fractions are denoted as Is and Ii, respectively, and the fraction solubilized, F (%), is calculated as Is/(Is + Ii) × 100. F is then plotted against incubation temperature, T, and estimation of T½ was performed by fitting the data (Fig. 5C) with a sigmoidal function using Igor Pro version 4.0 (Wavemetrics) as shown in Equation 1,
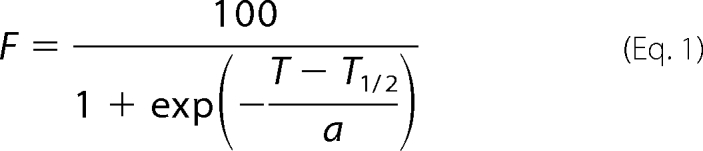 |
where a is a coefficient. Experiments were repeated three times to estimate errors.
FIGURE 5.

Distinct solubility of SOD1 aggregates with different fALS mutations in the presence of Sarkosyl. A, 2 μg of WT or L126X SOD1 aggregates were prepared in an NNE buffer with or without a detergent; either 1% Nonidet P-40 (NP-40), 1% Sarkosyl, or 1% SDS. After incubation at 37 °C for an hour, samples were ultracentrifuged at 110,000 × g for 30 min, and a supernatant (s) and a pellet (p) fraction were analyzed by a 12.5% SDS-polyacrylamide gel. B, temperature dependence of the WT or L126X aggregate solubility in the presence of 1% Sarkosyl. Experimental conditions were the same as described in A except the incubation temperature. C, effects of temperature on the aggregate solubility were further tested in all of the other fALS mutant SOD1 proteins, and fractions of solubilized SOD1 were estimated from its relative band intensities between supernatant and pellet on an SDS-polyacrylamide gel and plotted against incubation temperature. D, SOD1 aggregates were extracted from lumbar spinal cords of transgenic mice expressing G37R, G85R, and G93A SOD1 and incubated for an hour at indicated temperature in NNE buffer containing 1% Sarkosyl and 5 mm DTT. After ultracentrifugation at 110,000 × g for 30 min, supernatant (s) and pellet (p) fractions were probed by Western blotting analysis using sheep anti-SOD1 polyclonal antibody (Calbiochem). Percentages indicated below each gel image represent the fraction of solubilized SOD1 that is estimated from band intensities. E, T½ was obtained by fitting the data in C with a sigmoidal function (see “Experimental Procedures”) and plotted against fibril types defined in this study.
Electron Microscopy
To observe fibrillar morphologies of SOD1 aggregates with an electron microscope, we performed seeding reactions of SOD1 fibrillation, which can avoid shearing fibrils, namely 10 μm SOD1 aggregates were mixed with 100 μm soluble SOD1 and left at 37 °C for 3 days. Insoluble pellets were then collected by ultracentrifugation at 110,000 × g for 30 min and resuspended in water. SOD1 aggregates were adsorbed on 400-mesh grids coated by a glow-charged supporting membrane. After washed with pure water, negative staining with 1% uranyl acetate was performed. Images were obtained using an electron microscope (1200EX, JEOL). The width of a fibrillar structure was measured using a Photoshop 7.0 software (Adobe), and 10 different fibrils were examined to estimate errors.
Identification of a Core Region in SOD1 Aggregates
22.5 μg of SOD1 aggregates were resuspended in 100 μl of 50 mm Tris, 100 mm NaCl, 5 mm CaCl2, pH 8.0, mixed with 5 μg of Pronase (Calbiochem) and incubated at 37 °C for an hour. After ultracentrifugation at 110,000 × g for 30 min, supernatants were discarded, and the pellets were washed once with 150 μl of 50 mm Tris, 100 mm NaCl, pH 8.0. The final pellets were redissolved in 20 μl of 50 mm Tris, 500 mm NaCl, 6 m guanidine HCl, 5 mm EDTA, 5 mm DTT, pH 8.0, and desalted using NuTip C-18 (Glygen Co.). α-Cyano-4-hydroxycinnamic acid was chosen as a matrix, and MALDI-MS and MS/MS spectra were acquired using a 4800plus MALDI-TOF/TOF Analyzer (Applied Biosystems). Identification of peptides based upon the observed m/z values was performed using PAWS software (ProteoMetrics).
For modifications of SOD1 aggregates with Alexa555, 60 μm SOD1 aggregate was mixed with 300 μm Alexa555 in an NNE buffer containing 5 mm TCEP and incubated at room temperature for an hour in the dark. After ultracentrifugation at 110,000 × g for 30 min, the pellets were washed once with an NNE buffer and resuspended and sonicated in 10 mm Tris-HCl, pH 8.0, with 4 m urea. Modifications of soluble SOD1 with Alexa555 were also performed by incubating 60 μm SOD1 with 300 μm Alexa555 in an NNE buffer containing 5 mm TCEP. Followed by addition of 20% trichloroacetic acid, which quenches the modification reaction, the sample solution was centrifuged at 20,000 × g for 10 min. The pellets were washed once with acetone and redissolved in 10 mm Tris-HCl, pH 8.0, with 4 m urea.
To identify the modification sites in SOD1, 4.5 μg of soluble or aggregated SOD1 with or without Alexa555 modification was incubated with 0.1 μg of lysyl endopeptidase (Wako) in 10 mm Tris-HCl, pH 8.0, with 4 m urea. The samples were incubated at 30 °C for an hour, desalted/purified with NuTip C-18, and analyzed by a 4800plus MALDI-TOF/TOF analyzer. Although a chemical structure of Alexa555-C2-maleimide has not been available, we determined its mass (m/z 955.3, monoisotopic) by MALDI-TOF mass spectrometry.
Preparation of SOD1 Aggregates from Spinal Cords of Transgenic fALS Model Mice
Transgenic mice expressing SOD1 with G37R (established as LoxSOD1G37R mice) (20), G85R (SOD1G85R, a kind gift from Dr. Don Cleveland, University of California, San Diego) (21), and G93A (SOD1G93AG1H, obtained from The Jackson Laboratory) (22) mutations were housed in the animal facility of RIKEN Brain Science Institute and sacrificed at the end-stage of disease. End-stage was determined by hindlimb paralysis so severe that the mouse could not right itself within 20 s when placed on its side, an end point frequently used for the mutant SOD1-expressing mice (20) and one that was consistent with the requirements of the Animal Experiment Committee of the RIKEN Brain Science Institute. Detergent extraction of SOD1 aggregates from these murine lumbar spinal cords was performed as reported previously (23). In short, spinal cords were homogenized in TEN buffer (10 mm Tris, 1 mm EDTA, 100 mm NaCl, pH 8.0) and centrifuged at 800 × g for 10 min to remove initial debris. 1:10 volume of TEN buffer with 10% Nonidet P-40 was then added to the supernatant, which was further sonicated and ultracentrifuged at 110,000 × g for 30 min. The resultant pellets were resuspended and sonicated in TEN buffer containing 0.5% Nonidet P-40, 0.25% SDS, and 0.5% deoxycholate and then ultracentrifuged at 110,000 × g for 30 min. Final pellets from lumbar spinal cords were resuspended and sonicated in 100 μl of pure water per mouse. Out of a total of 100 μl, 20 μl was used for Pronase digestion experiments, and 6 μl was used for solubility assay in the presence of 1% Sarkosyl.
RESULTS
Core Structure of SOD1 Fibrillar Aggregates
As shown in our recent studies (13), SOD1 forms fibrillar aggregates only when copper and zinc ions as well as a disulfide bond are absent (i.e. apo-SOD1SH). To prepare SOD1 fibrillar aggregates, 100 μm soluble apo-SOD1SH was agitated at 1,200 rpm at 37 °C, and the aggregation reaction was monitored by the increase of solution turbidity (see WT in supplemental Fig. S1D). After the turbidity change reached a plateau, a sample solution was separated by ultracentrifugation into supernatant and pellets. SOD1 was detected only in the pellet fraction (see WT in supplemental Fig. S1H), showing that all SOD1 molecules were converted into insoluble aggregates. Furthermore, SOD1-insoluble aggregates increase the ThT fluorescence (see WT in supplemental Fig. S2C) and red-shift the electronic absorption spectrum of Congo Red (supplemental Fig. S2D), both of which are typical tinctorial changes to indicate amyloid-like fibrillar structures in protein aggregates. These data thus confirm that apo-SOD1SH forms amyloid-like fibrillar aggregates.
In general, protein fibrillar aggregates are composed of protease-resistant “core” and the associated “fuzzy coat” that is a susceptible region for proteolysis (24). We next attempted to identify which region(s) of an SOD1 amino acid sequence constitutes the protease-resistant core of SOD1 fibrillar aggregates. Apo-SOD1SH aggregates prepared above were treated with a nonspecific protease, Pronase, to digest and remove the fuzzy coat regions, and the remaining insoluble pellets obtained by ultracentrifugation were supposed to be a protease-resistant core of SOD1 aggregates. Insoluble pellets after Pronase treatment were then solubilized in 6 m guanidine hydrochloride, and mass peaks were obtained by MALDI-TOF mass spectrometry (supplemental Fig. S3A); in contrast, no mass peaks were observed (m/z 1500–30,000, data not shown) when experiments were done in soluble apo-SOD1SH. Based upon the observed mass with MS/MS analysis (supplemental Table S1), the peptides resistant to Pronase treatment are identified and mapped on an SOD1 primary sequence (Fig. 1A). As shown in Fig. 1A, multiple peptides are concentrated in one N-terminal (Ala1–Lys30, region A) and two C-terminal (Asp90–His120, region B; Thr135–Gln153, region C) regions. Soluble SOD1 has a three-dimensional structure with an immunoglobulin-fold comprising eight β-sheets, among which β1, β2, β6, β7, and β8 are involved in the formation of a core structure upon aggregation (Fig. 1B).
FIGURE 1.

MALDI-TOF mass spectrometric analysis of a core structure in SOD1 aggregate and experimental identification of SOD1 sequences with high aggregation propensities. A, peptides corresponding to the mass peaks detected are mapped on the primary sequence of SOD1. Numbers indicated above the primary sequence of SOD1 (shown as open bars) represent the amino residue numbers. Peptides were identified either by MS/MS analysis (thick bars) or mass (thin bars). Details of peptide identification are summarized in supplemental Table S1. B, three-dimensional structure of an SOD1 dimer with copper and zinc ions bound (Protein Data Bank code 1HL5). Regions A–C are colored red, blue, and green, respectively. C, after overnight agitation of 100 μm SOD1 fragments (ex1–2, 2–3, 3–4, and 4–5 as shown in A) at 37 °C, a sample solution was ultracentrifuged at 110,000 × g for 30 min to separate a supernatant (s) and a pellet (p) fraction, and these fractions were analyzed by a 15% SDS-polyacrylamide gel stained with Coomassie Brilliant Blue. D, fluorescence intensity was measured in the samples containing 25 μm ThT and 3 μm SOD1 fragments before (0 h, open bars) and after (13 h, filled bars) 1,200 rpm agitation at 37 °C. E and F, electron micrograms of the pellet fraction containing (E) ex1–2 and (F) ex4–5 aggregates.
To further test if these three regions are responsible for SOD1 aggregation, four overlapping SOD1 peptide fragments N-terminally fused with a His6 tag were prepared (Fig. 1A) as follows: Ala1–Ala55 (ex1–2), Glu24–Arg79 (ex2–3), Gly56–Val118 (ex3–4), and His80–Gln153 (ex4–5). Ex1–2 and ex4–5 peptides become insoluble after overnight agitation at 1,200 rpm, whereas ex2–3 and ex3–4 peptides remain in a supernatant fraction (Fig. 1C). Insoluble pellets of ex1–2 and ex4–5 but not a soluble form of these peptides increased the intensity of ThT fluorescence (Fig. 1D). By using an electron microscope, we have confirmed fibrillar morphology of ex1–2 and ex4–5 aggregates with ∼9 nm of its width (Fig. 1, E and F). Taken these results together, we have revealed that non-native interactions among regions A–C form a core structure of SOD1 fibrillar aggregates.
Effects of fALS Mutations on SOD1 Fibrillar Aggregation
We next asked whether pathogenic mutations affect a molecular structure of SOD1 aggregates. As performed in WT SOD1, aggregates of mutant SOD1 were prepared by constant agitation of 100 μm mutant apo-SOD1SH, and its aggregation reaction was monitored by the increase of solution turbidity (supplemental Fig. S1, D–G). Interestingly, not all of fALS mutations accelerate the reaction of aggregate formation. Given that SOD1 aggregation is significantly suppressed by the presence of copper and zinc ions as well as a disulfide bond (13), aggregation kinetics of apo-SOD1SH examined here (supplemental Fig. S1, D–G) will not correctly reflect that of SOD1 in the cellular environment where metal ions and disulfide bond could be introduced into mutant SOD1. Nonetheless, we would like to emphasize here that all mutant SOD1 proteins were eventually converted to insoluble aggregates when the turbidity change reached a plateau (supplemental Fig. S1H).
We have further confirmed the amyloid-like properties of mutant SOD1 aggregates by ThT fluorescence assay. After quantification of SOD1 aggregates by an SDS-PAGE analysis (supplemental Fig. S2, A and B), equal amounts of SOD1 aggregates were mixed with ThT (supplemental Fig. S2C). Although soluble SOD1 proteins showed no significant ThT fluorescence, all SOD1 aggregates tested here increased the intensity of ThT fluorescence, suggesting amyloid-like fibrillar structures in SOD1 aggregates. Furthermore, it is of note that each mutant SOD1 aggregate exhibits a distinct degree of the increase in the ThT fluorescence intensity (supplemental Fig. S2C). Given that the detailed mechanism of ThT binding to aggregates remains poorly understood, a molecular structure of the aggregate can hardly be inferred only from its ThT fluorescence intensity. These results, however, imply that each fALS-mutant SOD1 forms an insoluble aggregate with distinct structural/biochemical properties.
Core Structure of SOD1 Aggregate Is Dependent upon fALS Mutations
To reveal mutational effects on a molecular structure of aggregated SOD1, we have again identified Pronase-resistant core structures in fALS-mutant SOD1 aggregates by using MALDI-TOF mass spectrometry (supplemental Fig. S3, B–K and supplemental Table S1). As summarized in Fig. 2, all mutant SOD1 aggregates contain an N-terminal region (region A) as a core structure, suggesting its primary role in SOD1 aggregation. This is consistent with a previous study showing that an SOD1 fragment from Ala1 to Lys36 fused C-terminally with a yellow fluorescent protein forms aggregates in chick embryo spinal cord neural cells (25). In addition to such a common feature to all fALS-mutant SOD1s tested here, we noticed that a core structure in the aggregate is distinct among different fALS-mutant SOD1s and is classified into four types (Fig. 2 and Table 1). Type I includes WT and L144F, in which regions A–C include a core of aggregates. In A4V and G37R, which form type II aggregate, a core region is composed of regions A and C, but region B is not involved. G41D, G41S, and H46R can also be categorized as type II, in which the numbers of Pronase-resistant peptides covering region B were much less detected than those in the other types. Type III (G85R and G93R) shows a similar peptide map to type I, but a more extended C-terminal region participates in the formation of a core structure. In type IV (L126X and I149T), regions A and B but not C form a core in the aggregates. Although there may be more subtypes in the structural classification of SOD1 aggregates, our results have, for the first time, shown that fALS mutations can modulate non-native interactions among regions A–C to form distinct core structures in different mutant SOD1 aggregates.
FIGURE 2.

Core regions in aggregates of WT and fALS-mutant SOD1 proteins identified by MALDI-TOF mass analysis. SOD1G37R, SOD1G85R, and SOD1G93A indicate aggregates extracted from lumbar spinal cords of the corresponding transgenic mice, whereas the others are SOD1 aggregates prepared from the recombinant proteins with indicated fALS mutations. SOD1 aggregates were treated with Pronase, and the peptides resistant to digestion by Pronase were identified by MALDI-TOF mass spectrometry and mapped on the SOD1 primary sequence. Numbers indicated above the primary sequence of SOD1 (shown as an open bar) represent the amino residue numbers. Identification of mass peaks was performed based upon the observed mass values, and peptides shown as thick bars were further confirmed by MS/MS analysis. Details of peptide identification are summarized in supplemental Table S1 and supplemental Fig. S3, L–O.
TABLE 1.
Properties of SOD1 aggregates are dependent upon fALS mutations
| Type | Width of fibrils | T½ | Average onset of diseasea | Average duration of diseasea | |
|---|---|---|---|---|---|
| nm | °C | age | years | ||
| WT | I | 7.7 ± 1.1 | 57 ± 3.2 | ||
| A4V | II | 3.3 ± 0.4 | 48 ± 5.8 | 47.7 | 1.2 |
| G37R | II | 3.1 ± 0.3 | 53 ± 1.9 | 36.9 | 17.0 |
| G41D | II | NDb | 63 ± 1.8 | 45.2 | 14.1 |
| G41S | II | 3.0 ± 0.4 | 63 ± 1.8 | 47.9 | 1.0 |
| H46R | II | ND | 52 ± 2.7 | 45.6 | 17.6 |
| G85R | III | ND | 60 ± 4.4 | 55.5 | 6.0 |
| G93R | III | 4.3 ± 0.4 | 62 ± 3.0 | 35.0 | 5.3 |
| L126X | IV | 11 ± 1.1 | 43 ± 0.1 | 42.0c | 3.8c |
| L144F | I | 8.3 ± 1.4 | 68 ± 1.4 | 54.6 | 11.8 |
| I149T | IV | 10 ± 0.9 | 42 ± 0.9 | 37.6 | 2.7 |
We have also characterized Pronase-resistant peptides in the SOD1 aggregates extracted from lumbar spinal cords of transgenic mice expressing human SOD1 with G37R, G85R, and G93A mutations (supplemental Fig. S3, L–O). It is confirmed that regions A and C are involved in the core of all three in vivo SOD1 aggregates (SOD1G37R, SOD1G85R, and SOD1G93A in Fig. 2). In addition, a few peptides covering region B have been detected as aggregate core regions in in vivo SOD1G85R and SOD1G93A but not in in vivo SOD1G37R (Fig. 2). It is notable that no involvement of region B in the Pronase-resistant core is also characteristic of the in vitro G37R aggregates (see G37R in Fig. 2). Although maps of Pronase-resistant peptides are not precisely the same between in vitro and in vivo SOD1 aggregates, our results support some resemblance in the core regions between in vivo and in vitro SOD1 aggregates.
To get further insight into mutation-dependent structural polymorphism of SOD1 aggregates, we have tested reactivity of four Cys residues (Cys6, Cys57, Cys111, and Cys146) in soluble and aggregated SOD1 proteins toward chemical modifications. Compared with Cys residues in a fuzzy coat of aggregates, we suppose that Cys residues buried in an aggregate core are less reactive toward a thiol-specific modifier, Alexa Fluor 555-C2 maleimide (Alexa555), because of possible steric hindrance. Soluble or aggregated SOD1 with or without Alexa555 modifications was incubated in a buffer containing 4 m urea and 5 mm TCEP, where SOD1 aggregates are redissolved and any disulfide bonds are reduced. These SOD1 samples were then digested by lysyl endopeptidase and analyzed by MALDI-TOF mass spectrometry.
Among the mass peaks obtained, we have noted four proteolytic fragments, in particular Ala4–Lys23 (m/z 2115.1, monoisotopic, 2143.2 for A4V), Gly37–Lys70 (m/z 3592.9, average, 3692.1 for G37R), His71–Lys122 (m/z 5479.0, average), and Thr137–Gln153 (m/z 1587.8, monoisotopic, 1575.8 for I149T), for identification of Alexa555 modification at Cys6, Cys57, Cys111, and Cys146, respectively. Alexa555 modification adds 955.3 Da to the mass of the corresponding peptide. In soluble WT SOD1, mass peaks derived from the Alexa555-modified peptides (Gly37–Lys70, His71–Lys122, and Thr137–Gln153) were observed (left panels in Fig. 3, B–D), indicating that Alexa555 can access Cys57, Cys111, and Cys146. L126X, which is a truncated SOD1 at Leu126, does not produce a Thr137–Gln153 peptide (left panel in Fig. 3D). When WT SOD1 forms aggregates, the Alexa555-modified peptide spanning from Thr137 to Gln153 was no longer observed (right panel in Fig. 3D), whereas a mass peak from the corresponding unmodified peptide was detected (supplemental Fig. S4B). This result suggests that Cys146 becomes buried and not accessible by Alexa555 upon aggregation of WT SOD1. No modification of Alexa555 at Cys146 was also seen in A4V and G37R aggregates (right panel in Fig. 3D and supplemental Fig. S4B) and supports our findings that region C containing Cys146 constitutes a core structure of types I (WT) and II (A4V and G37R) aggregates (Fig. 2). In contrast, an Alexa555-modified peptide (Thr137–Gln153) was observed in I149T aggregates (right panel in Fig. 3D), which is consistent with no involvement of region C in a core structure of type IV fibrils (Fig. 2). A similar pattern of mass peaks was obtained between WT and A4V, but it is notable that A4V aggregates exhibit significantly larger peak intensity of the Alexa555-modified peptide (His71–Lys122) containing Cys111 than the other aggregates (right panel in Fig. 3C). This would be described by no involvement of region B in a core structure of A4V aggregates (Fig. 2). In all five SOD1 aggregates tested, Cys57 but not Cys6 is modified by Alexa555 (right panel in Fig. 3, A and B), but the corresponding unmodified peptide containing Cys6 was detected (supplemental Fig. S4A), which is again consistent with our findings that Cys6 but not Cys57 is involved in core structures of all four types of SOD1 aggregates (Fig. 2).
FIGURE 3.

Identification of SOD1 Cys residues that are modified with Alexa555. Alexa555-modified SOD1 (WT, A4V, G37R, L126X, or I149T) was digested with lysyl endopeptidase, and mass peaks from the following Cys-containing peptides modified with Alexa555 are shown: A, Ala4–Lys23 (Cys6) with an Alexa555; B, Gly37–Lys70 (Cys57) with an Alexa555; C, His71–Lys122 (Cys111) with an Alexa555; and D, Thr137–Gln153 (Cys146) with an Alexa555. In each panel, proteolytic digestion was performed using either soluble (left) or aggregated (right) SOD1 modified with Alexa555. Observed mass was shown in each spectrum, and calculated mass values of Alexa555-modified peptides are as follows: 3070.4 in Ala4–Lys23 (3098.5 for A4V), 4548.2 in Gly37–Lys70 (4647.3 for G37R), 6434.3 in His71–Lys122, and 2543.1 in Thr137–Gln153 (2531.1 for I149T). All of these mass peaks were not observed in unmodified SOD1 samples, confirming that the mass peaks shown in A–D are derived from Alexa555-modified peptides.
Mutation-dependent Polymorphism of SOD1 Fibrillar Aggregates
Given that a core structure is a building block of SOD1 aggregates, we expect that the aggregate morphology is also affected by mutation-dependent core structures. Although all mutant SOD1 aggregates tested here are fibrillar, the difference of fibril width is remarkable among mutant SOD1s (Fig. 4, A–K, and Table 1). For example, types I (WT and L144F, Fig. 4, A and J) and IV (L126X and I149T, Fig. 4, I and K) form relatively thicker fibrils (8–10 nm of its width) than the other mutant SOD1s, in which most of type II (A4V, G37R, and G41S, Fig. 4, B, C, and E) fibrils and type III (G93R, Fig. 4H) fibrils have ∼3 nm of the width. In G41D, H46R, and G85R (Fig. 4, D, F and G), it appears that thin fibers are stacked to form intertwined and thicker fibrils with complex shapes; therefore, the widths of these fibrils are difficult to examine. Although there will be other structural/environmental factors regulating morphologies of SOD1 aggregates, we have suggested that mutation-dependent core structures contribute to produce a variety of aggregate morphologies.
FIGURE 4.

Electron micrograms of (A) WT and (B–K) fALS-mutant SOD1 aggregates. A bar in each panel represents 50 nm.
New Biochemical Assay Method Reveals Mutation-dependent Solubility of SOD1 Aggregates
Our next question is whether any biochemical aspects of an SOD1 aggregate are affected by its structural polymorphism. In general, protein aggregates are insoluble in a physiological buffer solution, but addition of detergents to a buffer solution enhances solubility of aggregates. It has been reported that in vivo SOD1 aggregates purified from spinal cords of amyotrophic lateral sclerosis model mice are soluble in the presence of detergent SDS but not Nonidet P-40 (26). As shown in Fig. 5A, we have confirmed that our in vitro aggregates of WT apo-SOD1SH were not solubilized by 1% Nonidet P-40 but were completely soluble in the presence of 1% SDS. This is also the case in L126X apo-SOD1SH aggregates (Fig. 5A). Interestingly, when we tested another detergent called N-lauroylsarcosine (Sarkosyl), both WT and L126X aggregates were partially solubilized and exhibited different degrees of solubilization as follows: ∼20 and 40% in WT and L126X aggregates, respectively (Fig. 5A). We have thus noted solubility of aggregates in a Sarkosyl-containing buffer as a mutation-dependent biochemical feature of SOD1 aggregates.
We have further found that temperature is another factor to regulate the solubility of SOD1 aggregates in a Sarkosyl-containing buffer. As shown in Fig. 5B, increasing amounts of SOD1 aggregates become solubilized at higher temperatures, but the temperature dependence of solubilization is different between WT and L126X aggregates. For example, L126X aggregates are almost completely solubilized at 60 °C, where half-amounts of WT aggregates are still found in an insoluble pellet fraction. For quantitative analysis, we have defined T½ as temperature required for solubilization of half-amounts of aggregates. Table 1 summarizes T½ values of fALS-mutant SOD1 aggregates, which can be approximated by fitting the data (Fig. 5C) with a sigmoidal function (see “Experimental Procedures”). Among the fALS-mutant SOD1 proteins tested here, T½ was found to range from 42 °C (I149T) to 68 °C (L144F). We have also confirmed that in vivo SOD1 aggregates extracted from murine spinal cords also exhibit comparable temperature dependence of solubility to those of the in vitro ones; fractions of solubilized SOD1 in 1% Sarkosyl increases from around 30% (at 37 °C) to almost 100% (at 100 °C) (Fig. 5D).
According to our structural classification of SOD1 aggregates, we have found that type IV fibrils (L126X and I149T) exhibit significantly lower T½ than the other types (Fig. 5E). In type IV, region C is not involved in a core structure of aggregates (Fig. 2), suggesting a role of region C in increasing insolubility of SOD1 fibrils. Indeed, types I and III aggregates, in which regions A–C form a core structure (Fig. 2), have higher T½ than type IV (Fig. 5E). Accordingly, mutation-dependent interactions among regions A–C would regulate physical properties of SOD1 aggregates such as solubility.
DISCUSSION
Mutations in SOD1 have been identified as a cause of fALS, and insoluble aggregation of mutant SOD1 proteins is a common pathological change in this disease (10). We previously proposed a molecular mechanism of SOD1 aggregation, in which either metallation or disulfide formation in SOD1 completely suppresses its fibrillar aggregation (13). Furthermore, several groups including us have reported that a role of fALS mutations in facilitating SOD1 aggregation is to decrease the affinity for zinc ions and retard disulfide formation (13–15). Although significant structural changes of an SOD1 molecule have been assumed upon aggregate formation, it remains an open question how SOD1 changes its structure to form fibrillar aggregates. Here, we have identified a core structure in SOD1 aggregates and extended our SOD1 aggregation model to include a polymorphism of fALS-mutant SOD1 aggregates (Fig. 6).
FIGURE 6.

Our proposed model to describe mutation-dependent structural polymorphism of SOD1 aggregates. In secondary structural representation of SOD1 (left), regions A–C are colored red, blue and green, respectively. Rearrangement of these regions results in the formation of a core, and at least three different combinations of interactions among regions A–C are possible (middle). An exact alignment of β-sheets in the aggregates remains unknown; therefore, alignment of regions A–C in each schematic representation (middle) is still speculative. Interactions among aggregation core regions determine overall morphologies (right) and biochemical properties of SOD1 aggregates. Inset, average disease duration is plotted against average onset of disease. Data were taken from Refs. 31, 32. Type I/III, II, and IV aggregates are colored by black, red, and blue, respectively.
Molecular Mechanism to Produce Structurally Distinct SOD1 Aggregates
Based upon this study, it is likely that SOD1 aggregation occurs through unique and non-native interaction among three major parts of SOD1 (regions A–C, see Figs. 2 and 6). As mentioned in our previous study (13), a CD spectrum has suggested that SOD1 aggregates are composed mainly of β-sheet structures. Soluble apo-SOD1SH is also rich in β-sheets with an immunoglobulin fold (almost the same with Fig. 1B) but exhibits a CD spectrum different from that of SOD1 aggregates, suggesting significant structural changes of SOD1 upon its aggregation (13). Indeed, molecular dynamics simulations of WT SOD1 have proposed a domain swapping model in which drastic rearrangement of β-sheets leads to aggregation (18). In this model, non-native interactions possibly leading to aggregation occur among Ala1–Glu40, Glu100–His120, and the C termini, which correspond to regions A–C in this study, respectively (Fig. 1A). In silico and our in vitro/in vivo results have thus indicated that aggregation of SOD1 occurs not through a simple pile of apo-SOD1SH monomers but through rearrangement of β-sheets in an SOD1 molecule to realize non- native interactions among regions A–C (Fig. 6).
When a core in the aggregate contains fALS mutations (i.e. A4V, L126X, L144F, and I149T), non-native interactions among regions A–C will be affected. A computer algorithm, TANGO (27), predicts that A4V and L144F mutations increase aggregation propensities of regions A and C, respectively. These changes may describe formation of a distinct aggregation core in A4V (Fig. 2) or significantly high T½ of L144F aggregates (Fig. 5E). In I149T, an aggregation propensity of region C is predicted to significantly decrease (27), which is consistent with no involvement of region C in the core structure of I149T aggregates (Fig. 2). In addition, SOD1 forms type IV fibrils when truncated at Leu126 (L126X), confirming that region C is dispensable for formation of the aggregation core. However, it remains obscure how a distinct aggregation core is realized in the other SOD1s in which fALS mutations are located outside the core regions. Recent studies have suggested mutation-dependent structural dynamics of SOD1 (28, 29), which may be an important determinant for unique interactions among the core regions. We thus suppose that fALS mutations can directly or indirectly control non-native interactions among regions A–C and lead to formation of a distinct structure of SOD1 fibrils. Furthermore, in a native state of SOD1, regions A–C contain amino acid residues involved in dimerization, metal binding, and disulfide formation, respectively (Fig. 6). These post-translational processes therefore prohibit non-native interactions among regions A–C and suppress SOD1 aggregation, which is consistent with our previous studies (13).
Potential Roles of Structural Polymorphism of SOD1 Aggregates in Heterogeneous Phenotypes and Pathologies of fALS
Increasing evidence has suggested that a molecular structure of protein aggregates is tightly linked to a phenotype (5). For example, in transmissible spongiform encephalopathies, multiple strains with distinct disease phenotypes exist (2), and the emergence of such strains has been described by structural differences in fibrillar aggregates of a prion protein (3). Another example can be found in a neurodegenerative disease collectively called tauopathies, in which a microtubule-binding protein, Tau, forms insoluble fibrillar aggregates (30). Tauopathies include Alzheimer disease, Pick disease, and others; in each of these tauopathies, Tau forms fibrils with distinct morphologies such as paired helical filaments in Alzheimer disease and straight filaments in Pick disease (30). Importantly, albeit with less attention, different fALS mutations in SOD1 also associate with distinct disease phenotypes such as severity and age of disease onset (19, 31, 32). For example, duration of the disease depends upon fALS mutations and ranges from less than a year to more than 20 years (32). Because symptomatic changes follow appearance of SOD1 inclusions in fALS patients and rodent models (33), a possible mutation dependence of SOD1 aggregate structures may be relevant in understanding phenotypic heterogeneity in fALS.
Among SOD1 mutations examined here, there seems no correlation of disease onset with disease duration (Fig. 6, inset; Table 1) (31, 32). Based upon our structural classification of SOD1 aggregates, type IV aggregates seem to associate with relatively early onset and short duration of disease (Fig. 6, inset). Also, later disease-onset would be expected in type I/III mutant (L144F and G85R) and WT, but obviously the effects of the structural polymorphism of aggregates on disease phenotypes still need further investigations. For example, A4V and H46R mutations are well known to exhibit very severe and mild progression of the disease, respectively (32), but these two mutant SOD1 aggregates are categorized into the same type (type II) in our classification. Many biophysical factors such as affinity for metal ions (13) as well as protein structural stability (11) regulate the kinetics of SOD1 aggregation, which would further affect some of the disease phenotypes (34). Our in vitro and in vivo data therefore support roles of fALS mutations in structural and biochemical properties of SOD1 aggregates but also show that aggregate structure is not the only decisive factor regulating disease phenotypes.
Furthermore, it is notable that morphologies of SOD1 inclusions are variable among transgenic mice expressing human SOD1 with different fALS mutations (33). For example, formation of SOD1 inclusions is a major change in H46R and G85R SOD1 transgenic mice, although in G37R and G93A SOD1 severe mitochondrial damage is observed with lesser amounts of SOD1 inclusions (35, 36). In addition, thioflavin S, a fluorescent dye upon binding with amyloid-like fibrils, can stain SOD1 inclusions in G37R, G85R, and G93A (13, 37) but not L126X transgenic mice (38). Also in a Caenorhabditis elegans model, morphologically and biophysically distinct classes of mutant SOD1 aggregates have been found (39); FRAP analysis shows higher mobility of L126X aggregates than those of G85R and G93A aggregates in the body wall muscle cells. All these in vivo studies have thus suggested that fALS mutations affect structural properties of pathological SOD1 aggregates, and this study will provide a molecular basis to describe mutation-dependent structural and biochemical properties of SOD1 aggregates.
In summary, we have revealed mutation-dependent structural polymorphism of SOD1 aggregates. Multiple regions in a single SOD1 protein are responsible for formation of a core upon its fibrillar aggregation. We have further found that morphologies as well as biochemical properties of SOD1 aggregates are affected by a combination of interactions among the aggregate core regions (Fig. 6). In analogy with a protein folding process, we suppose that variable structures of protein aggregates are possible by alternatively or non-natively “folding” the peptide fragments involved in the core of aggregates. Disease-causing mutations would regulate such non-native folding pathways to form a distinct structure of protein aggregates and exert distinct cytotoxicity. Given that several regions within a single protein sequence are often predicted to have high aggregation propensities (27), our proposing model (Fig. 6) would be a general mechanism producing structural polymorphism in the aggregates of many other proteins.
Supplementary Material
Acknowledgments
We thank Dr. Shoji Watanabe for fruitful discussions. We also thank the Support Unit for Bio-material Analysis; RIKEN BSI Research Resources Center, especially Masaya Usui and Kaori Otsuki, for mass analysis; and Yuriko Sakamaki for electron micrographic observations.
This work was supported by Grant-in-aid for Scientific Research on Priority Areas (Research on Pathomechanisms of Brain Disorders) 17025044 (to N. N.), Grants-in-aid 20770130 (to Y. F.) and 21390274 (to K. Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a Health and Labour Science Research grant (to K. Y.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. S1–S4.
- fALS
- familial form of amyotrophic lateral sclerosis
- DTT
- dithiothreitol
- ThT
- thioflavin T
- WT
- wild type
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- MS/MS
- tandem mass spectrometry
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1.Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2.Bruce M. E., Fraser H. (1991) Curr. Top. Microbiol. Immunol. 172, 125–138 [DOI] [PubMed] [Google Scholar]
- 3.Jones E. M., Surewicz W. K. (2005) Cell 121, 63–72 [DOI] [PubMed] [Google Scholar]
- 4.Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., Tanaka M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka M., Chien P., Naber N., Cooke R., Weissman J. S. (2004) Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
- 6.Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X., et al. (1993) Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 7.McCord J. M., Fridovich I. (1969) J. Biol. Chem. 244, 6049–6055 [PubMed] [Google Scholar]
- 8.Furukawa Y., Torres A. S., O'Halloran T. V. (2004) EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reaume A. G., Elliott J. L., Hoffman E. K., Kowall N. W., Ferrante R. J., Siwek D. F., Wilcox H. M., Flood D. G., Beal M. F., Brown R. H., Jr., Scott R. W., Snider W. D. (1996) Nat. Genet. 13, 43–47 [DOI] [PubMed] [Google Scholar]
- 10.Bruijn L. I., Miller T. M., Cleveland D. W. (2004) Annu. Rev. Neurosci. 27, 723–749 [DOI] [PubMed] [Google Scholar]
- 11.Furukawa Y., O'Halloran T. V. (2005) J. Biol. Chem. 280, 17266–17274 [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furukawa Y., Kaneko K., Yamanaka K., O'Halloran T. V., Nukina N. (2008) J. Biol. Chem. 283, 24167–24176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayward L. J., Rodriguez J. A., Kim J. W., Tiwari A., Goto J. J., Cabelli D. E., Valentine J. S., Brown R. H., Jr. (2002) J. Biol. Chem. 277, 15923–15931 [DOI] [PubMed] [Google Scholar]
- 15.Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., Marklund S. L. (2006) Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 16.Hörnberg A., Logan D. T., Marklund S. L., Oliveberg M. (2007) J. Mol. Biol. 365, 333–342 [DOI] [PubMed] [Google Scholar]
- 17.Teilum K., Smith M. H., Schulz E., Christensen L. C., Solomentsev G., Oliveberg M., Akke M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 18273–18278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khare S. D., Wilcox K. C., Gong P., Dokholyan N. V. (2005) Proteins 61, 617–632 [DOI] [PubMed] [Google Scholar]
- 19.Andersen P. M., Nilsson P., Keränen M. L., Forsgren L., Hägglund J., Karlsborg M., Ronnevi L. O., Gredal O., Marklund S. L. (1997) Brain 120, 1723–1737 [DOI] [PubMed] [Google Scholar]
- 20.Boillée S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., Kollias G., Cleveland D. W. (2006) Science 312, 1389–1392 [DOI] [PubMed] [Google Scholar]
- 21.Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W. (1997) Neuron 18, 327–338 [DOI] [PubMed] [Google Scholar]
- 22.Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X. (1994) Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 23.Shaw B. F., Lelie H. L., Durazo A., Nersissian A. M., Xu G., Chan P. K., Gralla E. B., Tiwari A., Hayward L. J., Borchelt D. R., Valentine J. S., Whitelegge J. P. (2008) J. Biol. Chem. 283, 8340–8350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wischik C. M., Novak M., Thøgersen H. C., Edwards P. C., Runswick M. J., Jakes R., Walker J. E., Milstein C., Roth M., Klug A. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 4506–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghadge G. D., Wang L., Sharma K., Monti A. L., Bindokas V., Stevens F. J., Roos R. P. (2006) Neurobiol. Dis. 21, 194–205 [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N. G., Jenkins N. A., Borchelt D. R. (2003) Hum. Mol. Genet. 12, 2753–2764 [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Escamilla A. M., Rousseau F., Schymkowitz J., Serrano L. (2004) Nat. Biotechnol. 22, 1302–1306 [DOI] [PubMed] [Google Scholar]
- 28.Molnar K. S., Karabacak N. M., Johnson J. L., Wang Q., Tiwari A., Hayward L. J., Coales S. J., Hamuro Y., Agar J. N. (2009) J. Biol. Chem. 284, 30965–30973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw B. F., Durazo A., Nersissian A. M., Whitelegge J. P., Faull K. F., Valentine J. S. (2006) J. Biol. Chem. 281, 18167–18176 [DOI] [PubMed] [Google Scholar]
- 30.Lee V. M., Goedert M., Trojanowski J. Q. (2001) Annu. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 31.Prudencio M., Hart P. J., Borchelt D. R., Andersen P. M. (2009) Hum. Mol. Genet. 18, 3217–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q., Johnson J. L., Agar N. Y., Agar J. N. (2008) PLoS Biol. 6, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner B. J., Talbot K. (2008) Prog. Neurobiol. 85, 94–134 [DOI] [PubMed] [Google Scholar]
- 34.Lindberg M. J., Tibell L., Oliveberg M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16607–16612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagai M., Aoki M., Miyoshi I., Kato M., Pasinelli P., Kasai N., Brown R. H., Jr., Itoyama Y. (2001) J. Neurosci. 21, 9246–9254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe M., Dykes-Hoberg M., Culotta V. C., Price D. L., Wong P. C., Rothstein J. D. (2001) Neurobiol. Dis. 8, 933–941 [DOI] [PubMed] [Google Scholar]
- 37.Wang J., Xu G., Gonzales V., Coonfield M., Fromholt D., Copeland N. G., Jenkins N. A., Borchelt D. R. (2002) Neurobiol. Dis. 10, 128–138 [DOI] [PubMed] [Google Scholar]
- 38.Wang J., Xu G., Li H., Gonzales V., Fromholt D., Karch C., Copeland N. G., Jenkins N. A., Borchelt D. R. (2005) Hum. Mol. Genet. 14, 2335–2347 [DOI] [PubMed] [Google Scholar]
- 39.Gidalevitz T., Krupinski T., Garcia S., Morimoto R. I. (2009) PLoS Genet. 5, e1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.