

Abstract
Melzack and Wall’s Gate Control Theory of Pain laid the theoretical groundwork for a role of spinal inhibition in endogenous pain control. While the Gate Control Theory was based on the notion that spinal inhibition is dynamically regulated, mechanisms underlying the regulation of inhibition have turned out to be far more complex than Melzack and Wall could have ever imagined. Recent evidence indicates that an exquisitely sensitive form of regulation involves changes in anion equilibrium potential (Eanion), which subsequently impacts fast synaptic inhibition mediated GABAA, and to a lesser extent, glycine receptor activation, the prototypic ligand gated anion channels. The cation-chloride co-transporters (in particular NKCC1 and KCC2) have emerged as proteins that play a critical role in the dynamic regulation of Eanion which in turn appears to play a critical role in hyperalgesia and allodynia following peripheral inflammation or nerve injury. This review summarizes the current state of knowledge in this area with particular attention to how such findings relate to endogenous mechanisms of hyperalgesia and allodynia and potential applications for therapeutics based on modulation of intracellular Cl− gradients or pharmacological interventions targeting GABAA receptors
1 Introduction
Pain is a highly dynamic sensation. The enhanced sensitivity to pain that follows an injury or inflammation, generally known as hyperalgesia, is the archetypical expression of such plasticity. For over a century, hyperalgesic states have been interpreted as the consequence of the increased excitability of the peripheral and central nervous system induced by injury or inflammation. These enhancements of excitability are referred to as peripheral or central sensitization and can be produced by increased synaptic excitation, decreased synaptic inhibition (i.e. disinhibition), increased neuronal responsiveness, or any combination thereof.
This review focuses on disinhibition, and more specifically, on disinhibition caused by changes in Cl− regulation. Several studies have shown that hyperalgesia and allodynia are produced by pharmacologically blocking inhibition in the spinal cord (Loomis et al., 2001b; Malan et al., 2002a; Schoffnegger et al., 2008; Sherman and Loomis, 1994; Sherman and Loomis, 1995; Sherman and Loomis, 1996; Sivilotti and Woolf, 1994; Sorkin and Puig, 1996; Sorkin et al., 1998; Yaksh, 1989b) or through genetic changes that reduce inhibition (Ugarte et al., 2000). Conversely, increasing inhibition can reduce hyperalgesia and allodynia (Eaton et al., 1999; Hwang and Yaksh, 1997; Rode et al., 2005; Stubley et al., 2001). Indeed, such results are predicted by the Gate Control Theory of pain (Melzack and Wall, 1965). This influential theory proposed that in the superficial dorsal horn, afferent activity arriving along “large” (Aβ) fibers could reduce transmission of impulses in “small” (Aδ and C) afferents and thus reduce pain sensation. The original theory focused on presynaptic mechanisms of inhibition (see section 3), but subsequent work has revealed that postsynaptic mechanisms are also involved (see section 5).
Synaptic inhibition can be reduced through several mechanisms including reduction of transmitter release or number of receptors. However, the potency of synaptic inhibition can also be modulated through changes that are independent of the transmitter or the receptor. This is because GABAA and glycine receptors depend on the transmembrane Cl− gradient for their mechanism of action. The transmembrane Cl− gradient is maintained by co-transporters (see section 2.1). Changes in the expression and/or function of those co-transporters is an important pathophysiological mechanism responsible, at least in part, for disinhibition implicated in chronic pain and in other neurological disorders such as epilepsy. With respect to the processing of nociceptive information at the first synapse in the central nervous system, such changes affect both pre-and postsynaptic inhibition, although there are important distinctions based on differences in normal Cl− regulation between the two loci. Furthermore, derangement of Cl− regulation can lead to paradoxical excitation (rather than simply a reduction in inhibition) and requires special consideration for its therapeutic correction.
This article reviews the current state of knowledge on Cl− regulation and its impact on nociceptive processing in the spinal cord dorsal horn, together with the implications of recent observations for the development of new pain therapies. A particularly exciting aspect of these novel approaches is that they are not based on the classic interactions between transmitters and receptors but, rather, they consider changes in the ionic composition of cells that in turn lead to major alterations in synaptic function. This opens up new therapeutic possibilities that are as yet unexplored.
2 Importance of chloride regulation for synaptic inhibition
2.1 Chloride regulation by co-transporters
In order for ion channels to pass current, an electrochemical gradient must exist across the cell membrane. The direction and magnitude of current depends on the direction and steepness of that gradient. The transmembrane Cl− gradient is maintained primarily by cation-chloride co-transporters (for review see Payne et al. 2003). Among neurons, the two most important co-transporters for regulating intracellular Cl− concentration ([Cl−]in) are 1) NKCC1 (sodium-potassium-chloride co-transporter, that transports Cl− into the cell, and 2) KCC2 (potassium-chloride co-transporter, that transports Cl− out of the cell (Fig. 1). In both cases, transport is electroneutral (unlike channels, which produce a current) and only secondarily active (unlike pumps, which must hydrolyze ATP to function). For NKCC1, Cl− moves into the cell by following Na+, which flows down the Na+ gradient actively maintained by the Na+/K+-ATPase. For KCC2, Cl− moves out of the cell by following K+, which flows down the K+ gradient actively maintained by the Na+/K+-ATPase. Through this coupling, Cl− can be moved against its gradient without directly using energy.
Figure 1. Cation-chloride co-transporters and Cl− regulation.

NKCC1 transports Cl− into the cell whereas KCC2 transports Cl− out of the cell. In both cases, the process is electroneutral, i.e. there is no net charge transfer, and secondarily active, i.e. the co-transporters do not hydrolyze ATP themselves but instead rely on concentration gradients actively generated by the Na+/K+-ATPase (dotted arrows).
2.2 Mechanisms of inhibition: shunting vs. hyperpolarization
When a neuron’s membrane potential is near its resting value of around −70 mV, the electrochemical Cl− gradient across the membrane is normally quite small. This can be measured empirically as the anion reversal potential (Eanion) which is typically around −70 mV, although the exact value can vary depending on a variety of factors (see below). With Eanion near −70 mV, opening GABAA/glycine receptor-gated Cl− channels causes little if any hyperpolarization because of the negligible driving force, although opening those channels reduces the depolarization caused by concurrent excitatory input – this is known as shunting (Eccles, 1964). Shunting contrasts with hyperpolarization, such as that mediated by K+ channels, which pass significant outward current because the K+ reversal potential (near −90 mV) ensures a large driving force. With that said, one must understand that increased Cl− conductance can be inhibitory without causing gross hyperpolarization; indeed, primary afferent depolarization is normally inhibitory (see section 3.1).
In reality, the distinction between shunting- and hyperpolarizing-mechanisms of inhibition is artificial and potentially misleading. Eanion of −70 mV is still more negative than spike threshold (near −50 to −45 mV), which means increased Cl− conductance will produce outward or hyperpolarizing current at perithreshold potentials. In fact, this current, whose magnitude scales directly with depolarization away from Eanion (because of increased driving force) is the basis for shunting. Note that “Cl−” channels pass Cl− and, to a lesser extent, HCO3− anions (Kaila et al., 1989; Staley et al., 1995), but they do not pass the cations whose influx through AMPA and NMDA channels is responsible for glutamate-mediated excitation; in other words, shunting does not work by making the membrane leaky in a nonspecific way. It is more accurate to think of “shunting” as clamping or stabilizing the membrane potential near Eanion, since any deviation of membrane potential away from Eanion will increase the driving force and produce a current that pulls membrane potential back toward Eanion. In this sense, shunting constitutes a negative feedback mechanism whose strength reflects the amplitude of the Cl− conductance and whose set point reflects Eanion.
Computational studies indicate that Cl− flux may increase firing even if Eanion remains below spike threshold. Simulations in a lamina I neuron model with a spike threshold of −49 mV showed that shifting Eanion to −55 mV was sufficient to completely incapacitate inhibitory control of firing rate, while shifting Eanion to −50 mV caused increased firing (Fig. 2A) despite GABAA/glycine receptor-mediated input being incapable of independently causing suprathreshold depolarization (Prescott et al., 2006). In fact, at membrane potentials near or above threshold, GABAA/glycine receptor-mediated input reduced depolarization caused by AMPA/NMDA receptor-mediated input (Fig. 2B). This highlights a discrepancy between the modulation of firing rate and the modulation of membrane potential, thus revealing that membrane potential is not the only determinant of firing. As illustrated in Figure 2C, for a given membrane potential near or above threshold, the model with GABAA/glycine receptor-mediated input spiked faster than the model without that input. This is explained by shortening of the membrane time constant (τm) (Fig. 2D); recall that τm = RinCm, which means a reduction in input resistance (Rin) causes a reduction in τm assuming membrane capacitance (Cm) remains constant. The change in τm affects the filtering properties of the membrane (Fig. 2E) and thus allows a shunted neuron to charge its membrane faster, and therefore spike faster, than an unshunted neuron when both are equally depolarized (Fig. 2E inset).
Figure 2. Inhibition modulates firing rate through more than one mechanism.

Data are based on simulations in a model of a spinal lamina I neuron (see Prescott et al., 2006 for details). (A) Output firing rate (fout) is plotted against the rate of excitatory synaptic input (fexc). Gray line shows input-output curve for no inhibition. Black lines show input-output curve for inhibition (whose input frequency is proportional to fexc) for different values of Eanion. “Inhibitory” input reduces spiking when Eanion = −65 mV, but fails to reduce spiking when Eanion = −55 mV, and actually increases spiking when Eanion = −50 mV although spike threshold is −49 mV. (B) Despite its effect on firing rate, inhibition reduced average depolarization even for Eanion = −50 mV. Thus, there is a discrepancy between modulation of firing rate and modulation of membrane potential, which is explained in C. (C) For equivalent depolarization, the model with inhibition spiked faster than the model without inhibition, regardless of the value of Eanion. (D) The effect in C is explained by shortening of the membrane time constant (τm) that accompanies the increase in membrane conductance caused by inhibitory input. (E) The shunting-induced change in passive membrane properties influence how the neuron responds to inputs with different frequencies, as shown here by power spectral analysis of the voltage response to synaptic input. The important point is summarized in the inset, which shows that the inhibited neuron (with shorter τm, black line) charges its membrane faster, and is therefore able to spike faster, than the neuron without inhibition (gray line). Thus, inhibition can ironically increase spiking even while reducing depolarization. Modified from Prescott et al. (2006).
The precise biophysical mechanism through which “inhibitory” input modulates neuronal spiking is not as simple as depolarizing vs. hyperpolarizing, and we have not even begun to consider activity-dependent changes in Eanion (see section 2.3), variations in Eanion between neurons (Martina et al., 2001), variations in Eanion between different compartments within the same neuron (Khirug et al., 2008; Szabadics et al., 2006), unmasking of polysynaptic pathways (Keller et al., 2007; Schoffnegger et al., 2008; Torsney and MacDermott, 2006) or the fact that GABAB receptors gate K+ and Ca2+ rather than Cl− channels.
2.3 Activity-dependent changes in Eanion
Eanion is not a static value. In reality, transmembrane Cl− flux causes intracellular and, to a lesser extent, extracellular Cl− concentrations to change on a relatively fast time scale (10s to 100s of milliseconds), which in turn changes Eanion on the same time scale. The consequences of this are illustrated in Figure 3: A brief puff of GABA onto a lamina I neuron elicits a brief outward current (Fig. 3A) whereas a longer puff elicits an outward current that eventually inverts and becomes inward (Fig. 3B); a similar inversion is observed with repeated stimulation (Cordero-Erausquin et al., 2005). Significantly, these data came from a neuron whose measured value of Eanion (based on brief GABA puffs) was significantly more hyperpolarized than resting membrane potential. This illustrates that the value of Eanion depends on how it is measured, and a single measurement may fail to accurately reflect the strength or direction of inhibition in response to different stimulus protocols.
Figure 3. The relative contributions of Cl− and HCO3− flux to the direction of current.

(A) Trace shows response to 20 ms-long puff of GABA onto a lamina I neuron from a P10 rat. Cartoon above shows the channel and the relative magnitude of Cl− influx and HCO3− efflux. The net result is an outward current because the former is larger than the latter. (B) Trace shows response to 150 ms-long puff of GABA onto the same cell as in A. The current is initially outward but, over time, the Cl− gradient collapses, thus reducing the magnitude of Cl− influx until it eventually becomes smaller than HCO3− efflux. At that point, the current inverts and becomes inward. Modified from Coredero-Erausquin et al. (2005).
Sokal and Chapman (2003) observed that the GABAA receptor agonist muscimol significantly reduced initial responses to A- and C-fiber stimulation in control and nerve-injured rats. However, muscimol attenuated the response to repetitive C-fiber stimulation in control rats, but failed to do so in nerve-injured rate. One explanation is that, although nerve injury may not have altered the “resting” value of Eanion, the ability to maintain Cl− homeostasis was nonetheless compromised so that inhibition failed when the system was challenged more aggressively, i.e. with repeated stimulation (see Jin et al., 2005 for the significance of this issue in the context of epilepsy). This point is relevant for relating animal testing with the clinical reality of patients with mechanical allodynia: experimental determination of mechanical threshold by single, brief touches with a von Frey hair does not replicate the dynamic stimulation caused by clothes rubbing against the skin, a stimulus that is more spatially distributed (i.e. not punctate) and ongoing (i.e. not brief) and therefore more likely to cause spatial and temporal summation that could cause progressive, activity-dependent collapse of the Cl− gradient.
Another feature of the collapse in Cl− gradient is worth pointing out. As shown in Figure 3, “Cl−” channels pass both Cl− and HCO3− anions. This is why we refer to the reversal potential as Eanion rather than ECl, since both anion species must be taken into account when solving the Goldman-Hodgin-Katz equation. Under normal conditions, more Cl− flows into the cell than HCO3− flows out, which leads to a net outward current (Fig. 3A). If Cl− accumulates inside the cell, Eanion undergoes a depolarizing shift, eventually causing less Cl− to flow into the cell than HCO3− flows out, in which case the net current becomes inward (Fig. 3B) (Staley et al., 1995; Staley and Proctor, 1999). The transmembrane bicarbonate gradient does not collapse the same way the Cl− gradient does because intracellular HCO3− is constantly replenished by the actions of carbonic anhydrase, which catalyzes the reaction H2O + CO2 ⇋ HCO3− + H+ and carbon dioxide diffuses freely across the membrane. If inward current were caused by Cl− flux changing direction, Cl− efflux would mitigate the collapse in Cl− gradient; instead, the paradoxical excitation caused by HCO3− efflux does not stop the Cl− gradient from collapsing and, if anything, indirectly exacerbates it. Thus, although net current may change direction, this is because of a shift in the relative magnitudes of Cl− influx and HCO3− efflux and not because of inversion of Cl− flux. Such biophysical details may seem unimportant, but they have critical effects on the overall dynamics of the system.
Activity-dependent collapse of the Cl− gradient is liable to happen more rapidly in small intracellular volumes than in large intracellular volumes (Qian and Sejnowski, 1990). This is consistent with data from hippocampal pyramidal neurons, in which dendritic inhibition is compromised by accumulation of intracellular Cl− whereas somatic inhibition remains intact (Staley and Proctor, 1999). Similar data are not available in the spinal dorsal horn, but it is conceivable that different “types” of inhibition may be differentially compromised. Of course, this is particularly relevant in for presynaptic inhibition, since axons tend to have particularly small volumes. Moreover, KCC2 expression levels can vary between different cellular compartments, which explains why, in the axon initial segment of pyramidal neurons where NKCC1 levels are high but KCC2 levels are low, GABAergic input is excitatory (Khirug et al., 2008; Szabadics et al., 2006).
2.4 Developmental changes in Eanion and the robustness of Cl− homeostasis
It is now well established that upregulation of KCC2 expression is responsible for the hyperpolarizing shift in Eanion that occurs during development (Ben Ari, 2002; Rivera et al., 1999). In rat spinal lamina I neurons, Eanion appears to reach its “adult” level by the second postnatal week (Baccei and Fitzgerald, 2004) although full Cl− extrusion capacity is not reached until much later (Cordero-Erausquin et al., 2005); similar developmental changes are not observed in primary afferents (see section 3.1). Changes in nociceptive withdrawal thresholds parallel the changes in lamina I neuron Eanion values, being low in neonatal rats and only increasing to adult values by the second or third postnatal week (Falcon et al., 1996; Fitzgerald and Gibson, 1984; Jiang and Gebhart, 1998; Marsh et al., 1999; Teng and Abbott, 1998), although other developmental changes including changes in the number of neurons expressing GABA (Schaffner et al., 1993), changes in GABAA receptor subunit composition (Ma et al., 1993), and changes in the relative contribution of glycine and GABAA receptors (Baccei and Fitzgerald, 2004; Keller et al., 2001), may also contribute to changes in nociceptive threshold.
The developmental shift in Eanion is consistent with the results of Hathway et al. (2006) who showed that intrathecal application of the GABAA receptor antagonist gabazine decreased withdrawal threshold in postnatal day 21 (P21) rats but had the opposite effects in P3 rats; notably, however, the antinociceptive effect in P3 rats was abolished by spinalization. It would also be interesting to test prolonged or repeated stimulation given observations that GABA is not only depolarizing in neonates (because [Cl−]in is high), but also that [Cl−]in is less tightly regulated than in adults (Cordero-Erausquin et al., 2005). Thus, Eanion may shift more easily upon prolonged stimulation. Overall, inversion of the effects of GABAA receptor modulation seems to follow the same timeline as the developmental shift in Eanion.
3 GABA actions on primary afferent neurons and the role of NKCC1
The Gate Control Theory of pain, proposed in 1965 by Melzack and Wall, recognized that afferent inputs interact in the spinal dorsal horn and that this interaction can play a key role in the processing of pain-related information (see section 1). The most innovative aspects of the Gate theory were the idea of the gating of sensory inputs at the first synaptic relay and the proposal that the underlying mechanism was presynaptic interaction between primary sensory afferents in the superficial dorsal horn (see Cervero, 2005 for a recent review).
Primary afferent fibers interact in the spinal dorsal horn by generating PAD (primary afferent depolarization) on each other. Depolarization of the terminals of sensory afferents is mediated by GABAergic neurons that decrease excitatory transmitter release (see below). However, enhancement of PAD may also lead to generation of action potentials, thus transforming an inhibitory process into an excitatory one. In this way, Aβ-mediated PAD onto nociceptive afferents can, under normal circumstances, reduce pain by decreasing the effectiveness of their synaptic transmission but, following an injury, can evoke spiking in nociceptive terminals, resulting in touch-evoked pain or allodynia (Garcia-Nicas et al., 2006).
The mechanism that generates PAD in large myelinated afferent fibers in mammals is well established. Afferent input, or descending input from the brain, excites GABAergic spinal interneurons, which form axo-axonic synapses onto the central terminals of the primary afferent terminals. The primary afferent terminals express GABAA receptors, so that when GABA is released by the interneurons into the synaptic cleft, the Cl− channel associated with the receptor opens (Rudomin and Schmidt, 1999).
Primary afferent neurons maintain a high internal Cl− concentration because they express NKCC1 (Alvarez-Leefmans et al., 2001; Price et al., 2006) that transports Cl− into the cell using the energy of the Na+ gradient created by the Na-K-ATPase pump (Alvarez-Leefmans et al., 1988; Rocha-Gonzalez et al., 2008) (see section 2.1). Thus, when the Cl− channel associated with the GABAA receptor opens, there is a net efflux of anions that results in depolarization. A small depolarization of the membrane (which is typical of PAD) produces pre-synaptic inhibition because when an action potential arrives from the periphery along the axon of the primary afferent, the impact of the invading action potential is attenuated as a result of a depolarization-induced inactivation of voltage-gated Na+ channels and shunting caused by the increase in membrane conductance associated with open Cl− channels. Consequently, there is less transmitter release at the synapse between the primary afferent terminal and the second order neurons in the spinal dorsal horn.
3.1 Mechanisms of GABAergic excitation of primary sensory neurons
Primary sensory neurons maintain a high intracellular concentration of chloride ([Cl−]in) (Alvarez-Leefmans et al., 1988; Rocha-Gonzalez et al., 2008) and show depolarizing GABAA receptor currents throughout development and into maturity (Gilbert et al., 2007; Sung et al., 2000). As noted above, this feature contributes to what is commonly referred to as PAD, that reflects activation of pre-synaptic (i.e., on the central terminals of primary afferent neurons) GABAA receptors (Willis, 2006; Willis, 1999). While the polarity of this effect on GABAA channels is depolarizing, the net effect is shunting and reduction of neuronal excitability (Willis, 2006; Willis, 1999). In certain cases this depolarization can become large enough that antidromically conducting action potentials, commonly referred to as dorsal root reflexes (DRRs), can arise within the dorsal horn and produce neurogenic inflammation (Garcia-Nicas et al., 2001; Lin et al., 1999; Rees et al., 1994; Sluka et al., 1993). DRRs, like PAD, can be blocked by GABAA receptor antagonists, suggesting that they are mediated largely by GABAA receptor actions on the central terminals of nociceptive afferents (Garcia-Nicas et al., 2001; Lin et al., 1999; Lin et al., 2000; Rees et al., 1995). These DRRs can be blocked by spinal application of the NKCC1 inhibitor bumetanide, suggesting that NKCC1 contributes to their generation (Valencia-de Ita et al., 2006). While tissue injury is often associated with the emergence of DRRs, the exact mechanisms that underlie this transformation have yet to be delineated. Experiments with bumetanide suggest that NKCC1 is a key regulator of this effect; however, NKCC1 has not yet been localized to primary afferent terminals in the dorsal horn by immunohistochemistry or other methods. Moreover, direct measures of [Cl−]in have not been made at the central terminals of nociceptive afferents. Furthermore, the relative contribution of HCO3− anion flow through GABAA receptors on primary afferent neurons has not been established. Since these receptors are permeable to both Cl− and HCO3− (Kaila et al., 1989; Staley et al., 1995) it is possible that HCO3− contributes to GABAA-dependent depolarization in pathological states (see Fig. 3). It will be necessary to characterize the relative contribution of these two anions to injury-induced changes in GABAA signaling before it is possible to resolve to contribution of NKCC1 to the emergence of DRRs. The potential contribution of localized alterations in [Cl−]in are particularly important in light of recent findings demonstrating that within a single neuron, there can be considerably differences in dendritic, somatic and axonal GABAA reversal potentials that depend on the subcellular localization of NKCC1 and KCC2 (Khirug et al., 2008; Szabadics et al., 2006).
Similar to the central terminal of nociceptive afferents, GABA can also have an excitatory effect on peripheral nociceptive endings. The peripheral terminals of nociceptors contain GABAA receptor subunits and an intradermal injection of high-dose GABAA receptor agonist stimulates an increase in formalin-induced pain behaviors (Carlton et al., 1999). Keratinocytes are capable of synthesizing GABA (Ito et al., 2007), suggesting that peripheral mechanisms may be in place for GABAA-mediated excitation of sensory afferents in the periphery. A role for NKCC1 in GABAA receptor-mediated excitatory responses in the periphery has not been explored; however, NKCC1 localizes to sensory afferent axons in the sciatic nerve (Alvarez-Leefmans et al., 2001) and bumetanide inhibits formalin- and histamine-induced responses via a peripheral site of action (Granados-Soto et al., 2005; Willis et al., 2004).
GABAA receptor-mediated depolarization of nociceptive afferents appears to result from an elevated intracellular concentration of Cl−. Evidence in support of this mechanism and its causal link to NKCC1 come largely from studies on dissociated dorsal root ganglia (DRG) neurons in culture. The first indication that a loop diuretic (i.e., furosemide, piretanide or bumetanide)- sensitive mechanism was responsible for GABAA-mediated depolarization came from experiments in frog DRG neurons. While these authors observed a reduction in GABAA conductance with piretanide, they also observed a negative shift in Eanion, suggesting that piretanide was producing a decrease in [Cl−]in in these neurons (Wojtowicz and Nicoll, 1982). Subsequently, it was shown that furosemide, bumetanide and the Cl−/HCO3− exchanger inhibitor, disodium 4-acetamido-4′-isothiocyanato-stilben-2,2′-disulfonate (SITS), could also depress GABAA depolarization in cat DRG neurons although these studies did not find evidence for a shift in Eanion with these compounds (Gallagher et al., 1983). A later study in frog DRG neurons demonstrated that [Cl−]in was regulated by an electroneutral Na+-K+-2Cl− co-transporter that was sensitive to both furosemide and bumetanide (Alvarez-Leefmans et al., 1988). A study in DRG neurons isolated from NKCC1-KO mice implicated NKCC1 in GABAergic depolarization of DRG neurons because Eanion (these studies were conducted in the presence of HEPES, thus confounding contributions of HCO3−) was negatively shifted in neurons lacking NKCC1 (Sung et al., 2000). This study indicated that bumetanide mimicked the effects of knocking out NKCC1. However, as discussed below, the pharmacology of bumetanide is complex, with several potentially confounding off-target effects (Sung et al., 2000).
While the depolarizing effect of GABAA receptors in sensory neurons appears to be mediated, at least in part, by an NKCC1-sensitive mechanism, it is not clear how this mechanism can lead to excitation (i.e. action potential generation) rather than shunting inhibition (see also section 2.2). One hypothesis is that increases in NKCC1 activity are responsible for this effect because they lead to an increase in [Cl−]in which can increase GABAA-mediated depolarization above action potential threshold (section 3.2.1 – 3.2.2). Another possibility is that a small GABAergic depolarization can reach threshold for the activation of voltage-gated Ca2+ channels leading to an amplification of this depolarization. In a subset of nociceptive DRG neurons this appears to be the case. Neurons that express a prominent T-type Ca2+ current exhibit Ca2+ influx and spiking after GABAA receptor stimulation (Aptel et al., 2007). This effect is mediated by the CaV3.2 T-type Ca2+ channel subunit because it is absent in CaV3.2-KO mouse DRG neurons (Aptel et al., 2007). T-type Ca2+ channels, and CaV3.2 in particular, have come under consideration as targets for pain control because pain-related behaviors are reduced by CaV3.2 knockdown (Bourinet et al., 2005), because T-type Ca2+ channel inhibitors are effective in a variety of chronic pain models (Dogrul et al., 2003; Flatters and Bennett, 2004) and because T-type channels are strongly sensitized by redox agents (Nelson et al., 2005; Nelson et al., 2007; Todorovic et al., 2001; Todorovic et al., 2004). The missing link in this compelling set of data is that between T-type Ca2+ channels and NKCC1.
3.2 Modes of NKCC1 regulation as a mechanism of Cl− accumulation
A prominent hypothesis for GABAA receptor-mediated excitation of sensory neurons is that NKCC1 increases [Cl−]in to such an extent that GABAA-dependent depolarization reaches threshold for the activation of voltage-gated channels. For this to occur, mechanisms that can increase NKCC1 activity are required. NKCC1 activity can be controlled by phosphorylation, protein trafficking and by changes in intracellular concentrations of NKCC1 substrates.
3.2.1) Phosphorylation
Phosphorylation is a major regulatory mechanism of NKCC1-mediated Cl− transport (summarized in Fig. 4). NKCC1 is phosphorylated on the intracellular N-terminus (Darman and Forbush, 2002) leading to an increase in ion transport (Flemmer et al., 2002; Klein et al., 1999). A number of kinases including JNK (Klein et al., 1999), PKCδ (Liedtke and Cole, 2000; Liedtke and Cole, 2002), CamKIIα (Schomberg et al., 2001) and ERK (Liedtke and Cole, 2002) have been linked to NKCC1-dependent Cl− accumulation. NKCC1 activity can also be modified by mGluR and AMPA agonists and the mGluR-mediated upregulation of NKCC1 activity is attenuated by CamKIIα inhibition (Schomberg et al., 2001). However, JNK, Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) are the only kinases for which there is evidence in support of direct phosphorylation of NKCC1 (Klein et al., 1999).
Figure 4. Regulation of NKCC1.

Signaling events that may increase NKCC-mediated Cl− influx include phosphorylation (star 1), protein trafficking (star 2) or decreases in intracellular Na+ (star 3). Star 1: In contrast to KCC2, phosphorylation of NKCC1 results in an increase in activity, CaMKIIα (downstream of mGluR1/5), PKCδ and ERK have all been shown to increase NKCC1 activity, although it is not known if these kinases directly phosphorylate NKCC1. JNK also increases NKCC1 activity via direct phosphorylation of the co-transporter. A separate family of kinases, which are regulated by tonicity and cellular stress, positively modulate NKCC1. WNK1/4 act via SPAK and/or OSR1 to phosphorylate NKCC1. WNK3 also increases Cl− accumulation via NKCC1 and decreases KCC2 activity, which augments the net effect of NKCC1 stimulation in neurons that express both co-transporters. It is not known if WNK3 directly phosphorylates NKCC1 (and KCC2) or if it acts through SPAK/OSR1. SPAK and OSR1 interact with the N-terminus of NKCC1, where the co-transporter is phosphorylated, making these kinases ideal candidates as integrators of signaling that converges on NKCC1. Star 2: Dynamic regulation of the density of NKCC in the cell membrane may also influence rates of Cl− influx. An increase in intracellular cAMP concentration increases membrane delivery of NKCC2 in a Vamp2/3-dependent fashion. It is not known if this mechanism is also active at NKCC1. Star 3: A drop in the concentration of intracellular Na+ secondary to an increase in Na+/K+-ATPase may result in an increase in NKCC1 activity. This sequence of events may follow the increase in Na+ that occurs in association with an intense burst of neuronal activity. Star A, B and C: Persistent nociceptor activation caused by an intracolonic irritant injection causes an increase in dorsal horn NKCC1 phosphorylation (Star A) and an increase in NKCC1 in the cell membrane (Star B). It is not known if nociceptor spiking associated with this stimulus also contributes to an increase in NKCC1 activity in the dorsal horn (Star C).
Evidence that SPAK and OSR1 may associate with NKCC1 comes from yeast-two-hybrid studies. Both SPAK and OSR1 phosphorylate NKCC1 in the N-terminal domain of the protein (Dowd and Forbush, 2003; Gagnon et al., 2006a; Piechotta et al., 2002). Interestingly, in the nervous system, SPAK appears to act as a scaffold for NKCC1 and p38-MAPkinase (Piechotta et al., 2003). Moreover, an alternative translational start-site for SPAK (which is expressed in the CNS) yields a protein lacking the kinase domain suggesting that SPAK might act solely as a scaffold in certain brain regions (Piechotta et al., 2003).
Another family of kinases called with no lysine (K), or WNKs, are regulators of neuronal Cl− balance via the dual regulation of NKCC1 and KCC2. Given that phosphorylation of these two transporters has opposite effects on activity (Kahle et al., 2005), WNKs can produce rapid increases in [Cl−]in as has been demonstrated for WNK3 which stimulates NKCC1 activity and decreases KCC2 activity. These properties make WNK3 an attractive target for the control of the polarity of GABAergic responses in the nervous system (Kahle et al., 2005); however, it is not known if WNK3 directly phosphorylates NKCC1. WNK1 and WNK4 have similar effects on NKCC1 and, in the case of these kinases, it is known that they act via SPAK and/or OSR1 (Gagnon et al., 2006b; Moriguchi et al., 2005). The activity of SPAK, OSR1 and WNK kinases are controlled by changes in tonicity and other forms of cellular stress (Gagnon et al., 2006b; Moriguchi et al., 2005). It remains to be seen how these kinases contribute to NKCC1 activity in the nervous system.
3.2.2) Protein Trafficking
While the primary mechanism for control of NKCC1 activity appears to be phosphorylation there is evidence that trafficking of NKCCs to the plasma membrane can also influence their activity (summarized in Fig. 4). These modes of activation appear to be at least partially non-overlapping as phosphorylation of NKCC1 via WNK activation has no effect on membrane targeting of the co-transporter (Gagnon et al., 2006b). Evidence of NKCC translocation comes primarily from studies of NKCC2, a kidney-specific isoform. In the thick ascending limb of the loop of Henle, cAMP increases NKCC2 activity and also increases NKCC2 localization to the plasma membrane (Ortiz, 2006). This effect is completely blocked by tetanus toxin suggesting that this translocation is vesicle associated membrane protein (VAMP)-2 and -3 mediated (Ortiz, 2006). While similar studies have not been conducted with cAMP-dependent NKCC1 trafficking, a persistent painful stimulus can cause a long-lasting increase in plasma membrane-associated NKCC1 in the dorsal horn of the spinal cord (Galan and Cervero, 2005). Taken together, these studies suggest that trafficking of NKCC1 may represent another mode of increasing NKCC1 activity in neuronal tissues.
3.2.3) Intracellular Na+ and Cl− concentrations
A recent study in rat DRG neurons suggests that NKCC1 activity in DRG neurons is regulated via a negative feedback loop that is dependent on [Cl−]in (Rocha-Gonzalez et al., 2008). Importantly, in DRG neurons at rest, the [Cl−]in is high enough for GABAA-mediated depolarization, but below the concentration that could be attained if NKCC1 reached thermodynamic equilibrium (Rocha-Gonzalez et al., 2008). Thus, as suggested by the previous discussion, an increase in NKCC1 activity would drive an additional increase in [Cl−]in. Interestingly, Cl− accumulation in DRG neurons is only determined, in part, by the concentration of extracellular Na+, as DRG neurons previously depleted of [Cl−]in are able to re-establish a resting concentration of this anion in the absence of extracellular Na+ (Rocha-Gonzalez et al., 2008). This finding is supported by earlier studies of cat DRG neurons where a long-lasting absence of extracellular Na+ did not negatively influence GABAA-dependent depolarization of these neurons (Gallagher et al., 1978). Collectively, these findings indicate that an as yet to be discovered mechanisms may be involved in Cl− accumulation in DRG neurons.
NKCC1 activity can also be controlled by changes in [Na+]in especially under conditions where spike activity is present (summarized in Fig. 4). In neonatal CA1 pyramidal neurons, trains of action potentials elicit an increase in [Cl−]in that is dependent on Na+/K+-ATPase activity that effectively lowers [Na+]in (Brumback and Staley, 2008). Hence, in the setting of intense nociceptor spiking, it is feasible that a Na+/K+-ATPase-dependent decrease in [Na+]in can cause a persistent NKCC1-dependent increase in [Cl−]in leading to enhanced GABAA-mediated depolarization of these neurons.
3.3 NKCC1 Expression in Sensory Ganglia
Early studies on the pharmacology of Cl− accumulation in mammalian and amphibian DRG neurons suggested that loop diuretic-sensitive Na+-K+-2Cl− co-transporters were expressed by DRG neurons (Alvarez-Leefmans et al., 1988; Gallagher et al., 1983; Wojtowicz and Nicoll, 1982). A subsequent study in mice demonstrated that NKCC1 showed a membrane-associated immunohistochemical staining pattern in native DRG neurons that was completely absent in NKCC1-KO mice (Sung et al., 2000). Alvarez-Leefmans and colleagues used an antibody that recognizes both NKCC1 and NKCC2 to show that the majority of DRG neurons in frogs, cats and rats show a membrane-associated NKCC immunohistochemical staining pattern consistent with studies in mice (Alvarez-Leefmans et al., 2001). These authors also showed that NKCC-immunoreactivity localized to the peripheral axons of DRG neurons in the sciatic nerve (Alvarez-Leefmans et al., 2001). Quantitative analysis of NKCC1 mRNA expression in the DRG and trigeminal ganglia (TG) indicates that NKCC1 mRNA expression is largely confined to small diameter, peripherin- and TRPV1-positive DRG and TG neurons (Price et al., 2006).
These authors did not detect NKCC2 mRNA expression in the DRG or TG. Interestingly, these authors also observed NKCC1 mRNA and protein in NG2-positive satellite glial cells in the DRG with an immunohistochemical staining pattern striking similar to the “membrane-associated” NKCC-immunoreactivity pattern that had been described by other authors (Price et al., 2006). Another, more recent study measured [Cl−]in and NKCC1 immunoreactivity in mouse DRG neurons showing that while [Cl−]in decreased over development, NKCC1 mRNA expression levels did not change and the majority of DRG neurons contained NKCC1-immunoreactivity (Gilbert et al., 2007). Moreover, these authors found that there was considerable heterogeneity in [Cl−]in among adult mouse DRG neurons that was not related to neuron size (Gilbert et al., 2007). Taken together, while there appears to be consensus that NKCC1 is present in DRG neurons and contributes to the regulation of [Cl−]in (at least at the cell soma) there are still a number of inconsistencies that have yet to be resolved.
3.4 Experimental evidence for a role of NKCC1 in nociceptive processing
3.4.1) Behavioral/Biochemical Findings
The first studies to demonstrate a role for NKCC1 in nociceptive processing indicated that NKCC1 plays a role in thermal nociception. NKCC1-KO mice show increased latencies to withdrawal from noxious heat in the hot-plate test (Sung et al., 2000) and also in the tail-flick test (Laird et al., 2004). NKCC1 is expressed by nociceptive DRG neurons (Price et al., 2006; Sung et al., 2000), suggesting that this phenotype may be related to alterations in heat responses by heat-sensing nociceptors; however, NKCC1-KO mice have major deficits in motor coordination (Sung et al., 2000), hence, further studies are needed to gain a fuller understanding of the role of NKCC1 in thermal nociception.
Several other studies have suggested that NKCC1 contributes to other nociceptive modalities as well as itch. In human skin, bumetanide and furosemide, both of which block NKCC1, inhibit itch and flare responses to histamine (Willis et al., 2004). In the formalin model of tissue injury induced pain, peripheral bumetanide attenuated phase I and II behavioral responses and intrathecal delivery attenuated phase II responses (Granados-Soto et al., 2005). These spinal effects were mimicked by piretanide and furosemide (inhibitors of both NKCCs and KCCs) and the effects of bumetanide were not blocked by naloxone. Spinally applied bumetanide also inhibits neurogenic inflammation and DRRs evoked by mechanical and electrical stimulation following capsaicin injection into the hind paw (Valencia-de Ita et al., 2006).
NKCC1 is hypothesized to play a role in touch-evoked pain, or allodynia, and subsequent studies have focused largely on examining this possibility. NKCC1-KO mice show normal mechanical hyperalgesia in response to an intradermal capsaicin injection. On the other hand, they demonstrate a reduction in allodynia evoked by brushing the affected hindpaw (Laird et al., 2004). In contrast to effects seen in NKCC1-KO mice, in rats, intradermal capsaicin injection-evoked mechanical allodynia and hyperalgesia were both blocked by spinal application of the NKCC1 blocker bumetanide (Valencia-de Ita et al., 2006). Further studies from the Cervero group have focused largely on the role of NKCC1 in a model of referred allodynia that is stimulated by injecting irritants into the colon. This paradigm stimulates a robust allodynia in the abdominal region that is dependent on central processing (Galan et al., 2003; Laird et al., 2001). Intracolonic capsaicin injection stimulates NKCC1 phosphorylation in the spinal dorsal horn that peaks within 10 min of injection and is transient in nature (Galan and Cervero, 2005).
Longer lasting changes were observed in membrane delivery of NKCC1 in the dorsal spinal cord (lasting for at least 180 min) and these were not dependent on transcriptional alterations (Galan and Cervero, 2005). These results suggest that phosphorylation of NKCC1 might play a role in the initiation of allodynia whereas trafficking is involved in maintenance of the allodynic state. The role of NKCC1 in referred allodynia in this model was further examined in pharmacological studies. The NKCC1 inhibitor bumetanide was injected intrathecally prior to, or after, intracolonic capsaicin injection. In both cases, intrathecal bumetanide attenuated abdominal allodynia over a time period consistent with the pharmacokinetics of the compound (Pitcher et al., 2007).
The role of spinal TRPV1 receptors, which localize to the central terminals of nociceptors in the dorsal horn, has been explored in relation to NKCC1. Intrathecal injection of a TRPV1 antagonist blocked referred allodynia evoked by intracolonic capsaicin and an intrathecal injection of an endogenous TRPV1 agonist stimulated allodynia. The allodynia evoked by the intrathecal injection of the TRPV1 agonist was blocked by the NKCC1 inhibitor bumetanide (Pitcher et al., 2007). Further experiments will be required to elucidate mechanisms of how TRPV1 stimulation signals to NKCC1; however, these experiments suggest that visceral irritation causes the stimulation of spinal TRPV1 receptors that then cause an NKCC1-dependent referred allodynia (Pitcher et al., 2007). It should also be noted that the interpretation of these data must be considered in terms of a lack of selectivity in the effects of bumetanide, as discussed in section 4.1.
3.4.2) Changes in NKCC1 expression
Another mode through which NKCC1 could affect nociception is via changes in expression in acute or chronic pain states. In a rodent model of arthritis, NKCC1 mRNA expression was not changed in the ipsilateral DRG while increases were observed in the ipsilateral spinal dorsal horn (Morales-Aza et al., 2004). Another study utilized the intradermal complete Freund’s adjuvant (CFA) model to assess changes in NKCC1 protein expression up to 7 days following CFA injection. These authors did not observe any change in NKCC1 protein expression at any time point post-CFA injection (Zhang et al., 2008). The number of dorsal horn neurons expressing NKCC1 also does not change up to 60 min after formalin injection into the hindpaw (Nomura et al., 2006). While neither of these studies identified evidence of changes in NKCC1 expression, both observed a decrease in KCC2 expression in the dorsal horn of the spinal cord (Nomura et al., 2006; Zhang et al., 2008) similar to what has been observed following peripheral nerve injury (Coull et al., 2003) (see section 5). Possible changes in NKCC1 expression in the DRG or dorsal spinal cord have not been assessed in neuropathic pain models; however, axotomy of vagal afferents or facial motoneurons does not alter NKCC1 mRNA expression in regions associated with afferent input from these nerve injuries (Nabekura et al., 2002; Toyoda et al., 2003).
3.5 Other roles for NKCC1 in sensory neurons
In addition to the role of NKCC1 in GABAA-mediated depolarization of DRG neurons, NKCC1 also appears to play a role in neurite outgrowth in PC12 cells and DRG neurons. In PC12 cells, nerve growth factor (NGF) increases NKCC1 expression and NKCC1 knockdown drastically reduces NGF-mediated neurite outgrowth (Nakajima et al., 2007). In DRG neurons, axotomy increases [Cl−]in in an NKCC1-dependent fashion and the velocity of neurite outgrowth of axotomized neurons is blunted in DRG neurons that lack NKCC1 (Pieraut et al., 2007). This effect is mimicked by bumetanide application in neurons obtained from wild-type mice suggesting that NKCC1 plays a crucial role in DRG neuron recovery following axotomy (Pieraut et al., 2007).
4 Current limitations in experimental approaches to study NKCC1
4.1 Pharmacological considerations
Investigations that seek to establish the causality of NKCC1 in sensory function face a number of challenges and limitations. One of the most significant limitations concerns the selectivity of the pharmacological antagonists or inhibitors that are currently available. Benzmetanide, bumetanide, piretanide and furosemide are all inhibitors of NKCC1. Of these, bumetanide is the most specific for NKCC1 and is considered the prototypic agent for pharmacologic investigations of the role of NKCC1 (Payne et al., 2003; Russell, 2000). However, none of these drugs, bumetanide included, can be considered specific for NKCC1. They are also able to inhibit NKCC2, and other anionic exchange mechanisms such as Cl−/HCO3− exchange, Cl− channels, and KCC family members (Russell, 2000).
Concerns about the ability of these drugs to discriminate between NKCC1 and NKCC2 are minimal because NKCC2 is localized exclusively to the kidney, and is therefore absent from the central nervous system and DRG (Gilbert et al., 2007; Haas and Forbush, 2000; Price et al., 2006). Rather, the significant concern is the ability of these drugs to discriminate between NKCC1 and KCC2, particularly following intrathecal administration, since both co-transporters play crucial roles in neuronal Cl− regulation (see section 2.1). Recent reports that DRG neurons are labeled after intrathecal administration of some fluorescent probes suggest that the actions of intrathecally administered drugs are not necessarily confined to the spinal cord, but may also extend to the DRG (Luo et al., 2005). However, inhibition of KCC2 in the DRG or central terminals of primary afferent neurons is unlikely to confound interpretation of the actions of intrathecally administered NKCC1 inhibitors. This conclusion stems from several reports in which KCC2 mRNA or protein could not be localized in rat DRG (Coull et al., 2003; Price et al., 2006). However, there is one report of weak expression of KCC2 mRNA in the DRG of the mouse (Gilbert et al., 2007).
Of far greater concern to the interpretation of drug effects is the high concentration of KCC2 in the dorsal horn. Here, loss of selectivity and inadvertent inhibition of KCC2, with a concomitant production of allodynia or hyperalgesia (Coull et al., 2003), could confound interpretation of the effects of intrathecally administered NKCC1 inhibitors. Literature reports of the Ki for bumetanide-induced inhibition of NKCC1 or the Kd for binding of [3H]bumetanide to NKCC1 range from 0.07 μM to as high as 8.7 μM. The Ki of bumetanide for inhibition of KCC2 is estimated to be 25–50 μM (Payne et al., 2003). Thus, bumetanide may have at best ~500-fold and at worst three- to five-fold selectivity for NKCC1 over KCC2. Unfortunately, no estimates of the Ki or Kd of bumetanide for NKCC1 or KCC2 in tissue from mammalian DRG or dorsal horn are available. Nonetheless, both Russell (2000) and others (Payne et al., 2003) suggest that selectivity for NKCC1 is likely retained at bumetanide concentrations of 10 μM or less.
Difficulties in interpretation of pharmacological manipulation of NKCCs or KCCs also come from the possibility that blockers of these co-transporters also interact, as antagonists, at GABAA receptors. The first indication that cation-Cl− co-transporter inhibitors have an effect on the GABAA complex came from studies in the frog spinal cord. Furosemide, at high concentrations (1 mM), inhibited dorsal root potentials (DRP) evoked by GABA with an accompanying movement of Eanion toward the resting potential but with a large depression of the DRP suggesting a blockade of GABAA receptors (Nicoll, 1978). Later studies, also in the frog spinal cord, showed that piretanide (1 mM) similarly depressed DRPs but, in this case, with only a small change in Eanion (Wojtowicz and Nicoll, 1982). In both of these studies, co-transporter inhibitors were applied for long periods of time (up to 1 hr). Subsequent experiments in preparations isolated from cat showed that short application (5 min) of bumetanide (250 μM –500 μM) or furosemide (250 μM – 1 mM) depressed GABAA-dependent responses of cat DRG neurons without any change in Eanion (Gallagher et al., 1983), further substantiating the hypothesis that cation-Cl− co-transporter inhibitors interfere with GABAA receptor function in a manner independent from their primary pharmacological mechanism. Finally, work in NKCC1-KO mice demonstrated that bumetanide (10 μM), the prototypical NKCC1 inhibitor, depressed GABAA conductance at concentrations relevant for NKCC1 inhibition (Sung et al., 2000). This finding further clouds interpretation of results obtained with bumetanide although the mechanism of action of this (and other) compound at GABAA receptors is currently unknown. A better understanding of how cation-Cl− co-transporter inhibitors influence GABAA receptor conductance (and at which concentrations) will be paramount to advancing this area of research.
4.2 Limitations of immunohistochemical techniques
Another limitation has to do with the paucity of suitable, well-defined antibodies for the detection and localization of NKCC1 protein in the spinal cord and DRG. The monoclonal T4 and R5 antibodies developed by Lytle, Forbush and colleagues (Flemmer et al., 2002; Lytle et al., 1995; Lytle and Forbush, 1996) have proven highly useful for detection of NKCC1 (T4) and phosphorylated NKCC1 (T4 and R5) by Western blotting methods. Rabbit polyclonal antibodies directed against the N terminal or the C terminal of NKCC1 have proven useful for labeling in other tissue, such as stomach, cerebellum and choroid plexus (McDaniel et al., 2005; Price et al., 2006). However, these antibodies have not proven to be highly reliable for immunohistochemical localization or visualization of NKCC1 protein in neurons of the spinal cord or DRG (Price et al., 2006, but see Alvarez-Leefmans et al., 2001) despite the presence of mRNA in neurons (Price et al., 2006). The absence of antibodies for immunohistochemistry has forced an over reliance on measurements of mRNA, with attendant concerns as to how well changes in levels of message reflect changes in levels of protein and the difficulties of conducting in situ hybridization studies in concert with immunohistochemical labeling. Thus, a number of key questions and controversies remain unresolved including (1) whether NKCC1 detected in the dorsal horn (Galan and Cervero, 2005; Morales-Aza et al., 2004) is restricted to neurons, to glia, or solely to the central terminals of primary afferent neurons, and (2) whether NKCC1 is differentially distributed among different populations of primary afferent neurons in the DRG (Price et al., 2006) or is present in the majority of neurons (Alvarez-Leefmans et al., 2001). Finally, excellent antibodies will also be required for pre or postembedding immunohistochemistry and electron microscopy in order to better understand whether and how persistent pain states alter the trafficking of NKCC1 to the plasma membranes of dorsal horn neurons or central terminals of primary afferent neurons. This approach has been effectively used for studies of the delta opioid receptor and persistent pain states (Cahill et al., 2007). Such knowledge will be critical for proper interpretation of the results of studies in which the co-transporter has been inhibited or genetically deleted, and for future studies of how its function is modulated after injury.
5 Postsynaptic inhibition of spinal dorsal horn neurons
The original Gate Control Theory emphasized the role of presynaptic inhibition (Melzack and Wall, 1965) (see section 3), but inhibition acting postsynaptically on dorsal horn neurons is also critical for controlling the flow of sensory information from the periphery through the spinal cord to the brain (Brown et al., 1987; De Koninck and Henry, 1994; Kato et al., 2006; Lin et al., 1996a; Todd and Spike, 1993; Yoshimura and Nishi, 1995). Unlike primary afferent cells, which lack KCC2 (Coull et al., 2003; Price et al., 2006), spinal dorsal horn neurons maintain low [Cl−]in because of their relatively high level of KCC2 expression – at least this is true in the adult (see section 2.4) under normal conditions (see below). With low [Cl−]in, Eanion is maintained at around −70 mV.
5.1 Pathological changes in Eanion
A depolarizing shift in Eanion can contribute to hyperexcitability in a variety of disease states (De Koninck, 2007; Payne et al., 2003). Cohen et al. (2002) demonstrated the importance of Eanion for seizures, and subsequent work has shown that downregulation of KCC2 or upregulation of NKCC1 encourages seizures (Dzhala et al., 2005; Woo et al., 2002) and that expression of KCC2 and NKCC1 is down- and upregulated, respectively, in the hippocampus of epileptic patients (Huberfeld et al., 2007; Munoz et al., 2007; Palma et al., 2006). It is, therefore, not surprising that benzodiazepines and barbiturates are often ineffective for controlling seizures in patients with temporal lobe epilepsy (Kahle and Staley, 2008) or in neonates (Rennie and Boylan, 2003). This has the interesting and clinically relevant consequence that blocking NKCC1 with bumetanide improves the efficacy of phenobarbital in a neonatal seizure model (Dzhala et al., 2008).
In the spinal cord, downregulation of KCC2 has been shown to contribute to neuropathic pain following peripheral nerve injury (Coull et al., 2003), and is caused by BDNF released from activated microglia (Coull et al., 2005; Tsuda et al., 2005). Other studies have reported reduction in KCC2 expression in the dorsal horn in various pain models (Miletic and Miletic, 2007; Nomura et al., 2006; Zhang et al., 2008) although there is also evidence for increased NKCC1 expression (Morales-Aza et al., 2004). This is consistent with several studies showing that many features of neuropathic pain can be reproduced by pharmacologically blocking inhibition in the spinal cord (see section 1). The converse, that increasing inhibition can reduce neuropathic pain (Eaton et al., 1999; Hwang and Yaksh, 1997; Rode et al., 2005; Stubley et al., 2001) seems intuitive but requires additional consideration.
Although augmenting inhibition may seem like a good way to correct disinhibition, the success of such an intervention depends on the mechanism of disinhibition as well as the degree of disinhibition. Indeed, prolonging activation of GABAA receptors with a benzodiazepine should mitigate the disinhibition caused by reduction of GABA release and/or by reduced GABAA receptor expression or function, but the same benzodiazepine may be less effective, completely ineffective, or even paradoxically excitatory if disinhibition results from accumulation of intracellular Cl− (see section 6). Computer simulations indicate that increasing the strength of GABAergic transmission mitigates the effects of shifting Eanion to −60 mV, but becomes ineffective when Eanion rises to −55 mV or higher (Fig. 5A) (Prescott et al., 2006). Furthermore, as GABAergic transmission is increased in order to offset the shift in Eanion, the system becomes more and more prone to abrupt decompensation insofar as even a small change in Eanion can cause a large increase in spiking (Fig. 5B); this is critical if you consider that even modest synaptic activity can lead to activity-dependent shifts in Eanion (see section 2.3), especially if Cl− homeostatic mechanisms have been weakened by KCC2 downregulation (see section 2.4).
Figure 5. Ability to correct disinhibition by increasing inhibition depends on the degree of change in Eanion.

Data are based on same model described in Figure 2. (A) For Eanion = −70 mV, even modest inhibition reduced the slope of the input-output curve. Rate of inhibitory synaptic input (finh) is expressed relative to excitation, where α = finh/fexc. For Eanion = −60 mV, reduction in potency of inhibition could be compensated by increased GABAergic transmission. For Eanion = −55 mV, this compensation failed (i.e. all dotted/dashed curves have roughly the same slope as the solid curve representing no inhibition). For Eanion = −50 mV, increasing GABAergic transmission paradoxically increased spiking. (B) Even if increasing GABAergic transmission managed to prevent frank disinhibition, the highly compensated system was prone to abrupt decompensation (i.e. small additional shifts in Eanion could cause substantial increases in firing). Graph here shows firing rate with inhibition (fout) relative to firing rate without inhibition (fout0) for different ratios of excitation to inhibition, as in A. Modified from Prescott et al. (2006).
6 Role of GABAA receptors in allodynia and hyperalgesia
As detailed in the preceding sections of this review, there is compelling evidence to suggest that tissue injury results in a depolarizing shift in the anion equilibrium potential (Eanion) which, depending on the magnitude of the shift, may be associated with either a simple loss of inhibition or the emergence of excitation associated with the activation of Cl− channels such as the GABAA receptor. Indeed, direct electrophysiological evidence indicates that, following nerve injury, there is a depolarizing shift in Eanion caused by reduced KCC2 expression in superficial dorsal horn neurons; this shift in Eanion enables GABA to elicit action potentials in a subset of neurons (Coull et al., 2003; Coull et al., 2005) (see section 5.1). In contrast, both acute noxious stimuli (e.g. peripheral administration of capsaicin) and more persistent inflammatory stimuli (e.g. kaolin-carrageenan or CFA) produce changes in GABAA receptor-mediated signaling at the central terminals of sensory neurons that appear to reflect an increase in NKCC1 activity. While direct evidence of a neuronal activity and/or inflammation-induced increase in [Cl−]in within the central terminal of sensory neurons is still lacking, the combination of data from NKCC1 null mutant mice (Laird et al., 2004), anatomical studies on differential distribution of NKCC1 (Price et al., 2006) and activity dependent mobilization of this transporter (Galan and Cervero, 2005), and pharmacological studies with the NKCC1 antagonist bumetanide (Valencia-de Ita et al., 2006), all provide compelling support for such a mechanism (see section 3). Thus, despite differences in the underlying mechanisms (KCC2 vs NKCC1) and site (dorsal horn neuron vs primary afferent) of a depolarizing shift in Eanion, this mechanism should contribute to the pain and/or hypersensitivity associated with nerve injury, acute noxious stimuli, and persistent inflammation. However, as detailed below, both clinical and pre-clinical behavioral pharmacology studies in which this prediction has been tested, suggest that a simple shift in Eanion is insufficient to account for results obtained.
Because much of both clinical and preclinical data relevant to the impact of a shift in Eanion on nociceptive behavior involves the use of GABAA receptor agonists and antagonists, a brief discussion of GABAA receptor structure-function properties is in order. These channels are composed of 5 distinct subunits that are assembled to form a Cl− and HCO3− permeable channel. Nineteen GABAA receptor subunits have been cloned, including α (1–6), β (1–3), γ(1–3), δ, ε, θ, π and ρ (1–3) (Farrant and Nusser, 2005). However, GABAA receptors are generally composed of 2α, 2β and 1 other subunit. The specific combination of subunits determines the biophysical and pharmacological properties of the channel, its location in the plasma membrane, and phosphorylation-dependent modulation. For example, the presence of the ε subunit appears to confer spontaneous activity to the receptor (Wagner et al., 2005). α4/6 and δ containing receptors appear to be high affinity (i.e. μM) slowly desensitizing and targeted extrasynaptically (Kullmann et al., 2005; Semyanov et al., 2004); these receptors appear to underlie tonic currents in the brain (Semyanov et al., 2004) and spinal cord (Takahashi et al., 2006). A site formed between α and γ2 subunits confers classical benzodiazepine sensitivity and is most commonly found in low affinity (i.e., mM) synaptic receptors (Johnston, 2005; Sieghart and Sperk, 2002). The β2 subunit is a target for Akt-dependent phosphorylation which results in a dramatic increase in functional receptors in the membrane (Mody and Pearce, 2004). Finally, while ρ can function as a 5th subunit in a heteromeric channel, this subunit can form functional homomeric channels, historically been referred to as GABAC receptors with unique pharmacological properties.
6.1 Changes in GABAA-mediated effects in the presence of injury or inflammation
Despite what appears to be a significant degree of heterogeneity in sensory neuron [Cl−]in (Gilbert et al., 2007) and the presence of a GABAA receptor-dependent PAD observed following action potential invasion of the superficial dorsal horn (Eccles et al., 1963), the net effect of both endogenous and exogenous GABAA receptor activation in the spinal cord dorsal horn in the absence of tissue injury is antinociception. That is, spinal administration of GABAA receptor antagonists result in thermal and mechanical hypersensitivity (i.e., hyperalgesia and allodynia) (Anseloni and Gold, 2008; Loomis et al., 2001a; Sivilotti and Woolf, 1994; Yaksh, 1989a) suggesting that endogenous GABAA receptor activation is not only inhibitory, but contributes to the establishment of nociceptive threshold. Conversely, spinal administration of GABAA receptor agonists are analgesic (Anseloni and Gold, 2008; Caba et al., 1994; Dirig and Yaksh, 1995; Roberts et al., 1986). Of note, while GABAA receptor mediated modulation of sensitivity to mechanical stimuli is clearly manifest (Anseloni and Gold, 2008), modulation of thermal stimuli is less pronounced, if detectable at all (Hammond and Drower, 1984; Roberts et al., 1986). This modality selectivity may reflect the differential involvement of low threshold mechano-receptors to mechanical nociceptive threshold, afferents that, according to the anatomical evidence, receive more extensive pre-synaptic GABAergic input (Todd, 2002). Finally, while Eanion appears to be normally well above action potential threshold in a small subpopulation of afferents, the functional consequences of GABA-mediated excitation of these afferents, remains to be determined. DRR activity may, however, be more common in the absence of tissue injury (i.e., see Hains et al this volume) than originally reported (Rees et al., 1995).
As discussed above, there are injury-induced changes in the regulation of [Cl−]in that can influence GABA mediated effects. However, the behavioral consequences of a change in GABAA mediated signaling appear to depend on a number of factors including the type of injury and the mode of receptor activation. For example, intense nociceptor activation following a peripheral injection of capsaicin is sufficient for the induction of DRR activity that can be blocked by GABAA receptor antagonists (Lin et al., 1999; Weng and Dougherty, 2005; Willis, 1999). Furthermore, there is at least one report supporting the suggestion that inflammatory allodynia reflects a GABAA receptor dependent activation of nociceptive afferents by low-threshold Aβ-fibers (Garcia-Nicas et al., 2001); although other investigators have failed to detect evidence in support of such a GABAA receptor dependent coupling between low-threshold and nociceptive afferents (Schoffnegger et al., 2008; Weng and Dougherty, 2005). These short term effects do not appear to be specific for capsaicin, as similar changes in DRR activity and neurogenic inflammation were described within hours of intra-articular kaolin-carrageenan injection (Rees et al., 1995; Sluka et al., 1993; Sluka et al., 1995). In contrast, spontaneous nociceptive behavior is facilitated by GABAA receptor antagonists (Kaneko and Hammond, 1997) and inhibited by GABAA receptor agonists (Dirig and Yaksh, 1995; Kaneko and Hammond, 1997) in the formalin model. This behavioral profile is mirrored by changes in activity in dorsal horn neurons (Green and Dickenson, 1997). Given evidence that formalin evoked nociceptive behavior depends, at least in part, on capsaicin sensitive afferents (Peterson et al., 1997), and that afferent activity evoked during phase I of the formalin test (McCall et al., 1996) is comparable to that evoked by capsaicin (Schmelz et al., 2000), differences in GABAA receptor dependent signaling in these acute models is unlikely to depend on the initial afferent activity or which fibers are activated. An alternative possibility is that there are differences in the GABAergic circuitry underlying the regulation of spontaneous and evoked afferent input to the spinal cord. If true, one would predict that GABAA receptor antagonists should inhibit mechanical hypersensitivity observed following formalin injection.
Data from nerve injury models are no less perplexing. As detailed in section 5.1, the cellular processes underlying peripheral nerve injury-induced shift in GABAA (and glycine) receptor signaling has been worked out in exquisite detail. Importantly, behavioral data supports the involvement of activated microglia, P2X4 expression, BDNF release and TrkB activation in the nerve-injury mechanical hypersensitivity (Coull et al., 2005). This chain of events leads to a depolarizing shift in Eanion that results in reduced inhibition (or even paradoxical excitation) that should increase the transmission of nociceptive signals through lamina I (see Fig. 5). However, behavioral data indicate that spinal GABAA receptor activation continues to be antinociceptive. That is, nerve injury-induced mechanical hypersensitivity is decreased by GABAA (Hwang and Yaksh, 1997; Malan et al., 2002b) and benzodiazepine receptor agonists (Knabl et al., 2008) (the latter of which should facilitate endogenous GABAA receptor mediated signaling).
One explanation for these apparently discrepant observations is that only a subpopulation of superficial dorsal horn neurons is critical for the transmission of nociceptive information. For example, neurons expressing the neurokinin-1 (NK-1) receptor appear serve such a role in the rodent (Khasabov et al., 2002), yet they only constitute ~10% of the total population, and therefore could have been overlooked in studies documenting the nerve injury induced shift in Eanion (Coull et al., 2003; Coull et al., 2005). Consistent with this suggestion, GABAA receptor activation continues to inhibit the peripheral nerve injury-induced induction of an excitatory link between low threshold afferent input and nociceptive neurons in the superficial dorsal horn (Schoffnegger et al., 2008). Along the same line of reasoning, it is also possible that deep dorsal horn neurons play a dominant role in mediating the nociceptive behavior assessed in rodent models, and normal inhibitory GABAergic signaling may be preserved in these neurons in the presence of nerve injury. Consistent with these suggestions, there was no evidence of a muscimol (GABAA receptor agonist)-induced activation of deep (lamina V–VI) dorsal horn neurons in either the presence or absence of nerve injury, despite a significant GABAA receptor mediated inhibition of both C- and A-delta-evoked activity (Sokal and Chapman, 2003).
Pre-synaptic inhibition that persists in the presence of nerve injury suggests a third potential explanation for the antinociceptive efficacy of GABAA receptor agonists in the presence of nerve injury. Such a mechanism would account for the analgesic efficacy of GABAA receptor agonists in the spinal cord, at least as manifest in rodent models in the form of changes in reflexive behaviors (i.e., withdrawal from mechanical or thermal stimuli). An implication of this possibility is that if a post-synaptic change in Eanion and the associated responses to GABAA receptor activation contribute to changes in nociceptive processing, they should be detectable with the appropriate assay. Given recent evidence suggesting that there may be distinct ascending pathways underlying sensory/discriminative and affective components of pain (Braz et al., 2005), it may be possible to detect an influence of a post-synaptic change in Eanion with nociceptive assays based on operant paradigms, that may be more sensitive in the assessment of the affective dimension of nociception (Vierck et al., 2008; Vierck et al., 2004).
While these operant experiments have yet to be performed in rodent nerve injury models, there are clinical data that speak to the potential role of nerve injury-induced changes in GABA signaling. The benzodiazepine receptor agonist midazolam is the only compound in its class that is approved for spinal administration in patients. It is still widely used in surgical settings as an adjunct to local anesthetics and opioids for peri- and post-operative pain management (Sandby-Thomas et al., 2008), although both pre-clinical (Edwards et al., 1990; Goodchild and Serrao, 1987; Kyles et al., 1995) and clinical (Salonia et al., 2006) data indicate the compound has full analgesic efficacy when administered alone. Well-controlled clinical trials are clearly needed to confirm the analgesic efficacy of spinal midazolam for the treatment of neuropathic pain. Nevertheless, in the data available, there is no evidence that midazolam has pro-nociceptive activity in neuropathic pain patients (Borg and Krijnen, 1996). Of note, as illustrated in Figure 5, a small depolarizing shift in Eanion will result in a decrease in the potency of inhibition, but this does not necessarily lead to frank excitation. Consequently, the antinociceptive actions of midazolam may still reflect facilitation of inhibitory (although less so) synaptic transmission.
The changes in GABAA receptor signaling observed in the presence of persistent inflammation are similar to those described following peripheral administration of capsaicin and intra-articular injection of kaolin-carrageenan. That is, GABAA receptor antagonists become analgesic and, at least at low doses, the GABAA receptor agonist exacerbates ongoing hyperalgesia (Anseloni and Gold, 2008). The persistent inflammation-induced shift in the behavioral impact of GABAA receptor antagonists suggests that persistent inflammatory hyperalgesia does not simply reflect a loss of endogenous GABAA receptor mediated inhibition (Lin et al., 1996b), but the emergence of a pro-nociceptive role for this transmitter/receptor system. Importantly, there appears to be no shift in the actions of midazolam, which, as suggested above, has full analgesic efficacy in both pre-clinical (Anseloni and Gold, 2008) and clinical (Borg and Krijnen, 1996; Kim and Lee, 2001; Serrao et al., 1992; Wu et al., 2005) models of persistent inflammation. It is important to note that while off target effects of midazolam have been described (Zhao et al., 1996), the analgesic effects of spinal midazolam administration appear to be mediated by the central benzodiazepine receptor (i.e., the GABAA receptor) as these effects are antagonized by specific loss-of-function mutations (Knabl et al., 2008) and antagonists (Anseloni and Gold, 2008) of the central benzodiazepine receptor.
6.2 “Paradoxical” actions of benzodiazepines following inflammation or injury
A depolarizing shift in Eanion could account for the shift in the behavioral consequences of both GABAA receptor antagonists and low doses of the GABAA receptor agonist muscimol. Furthermore, as discussed above, a host of complex mechanisms ranging from activity dependent changes in Eanion, heterogeneity in the degree of change in Eanion (among patients or in an animal model), to changes in the ability to maintain [Cl−]in, may influence the behavioral response to exogenous manipulation of GABAergic transmission in the presence of tissue injury. This complexity in the face of species differences, differences in testing paradigms (that would elicit different degrees of activity-dependent changes), and/or different stimulus modalities assessed (e.g., mechanical stimulation activates spinal inhibitory neurons more strongly than thermal stimulation (Furue et al., 1999)) may contribute to differences in behavioral outcomes associated with pharmacological manipulations in the presence of tissue injury. However, results obtained with midazolam do pose a paradox worthy of further consideration.
If midazolam is acting through a central benzodiazepine receptor, then it should facilitate GABAA-mediated neurotransmission and consequently exacerbate inflammatory hyperalgesia. Consideration of a simple circuit diagram representing neuronal connectivity within the spinal cord dorsal horn suggests two explanations for the midazolam paradox (Fig. 6A). One explanation is that GABA is acting on at least two distinct sets of neurons. If benzodiazepine receptors are differentially distributed such that they are present in a higher density on an output neuron and there is a depolarizing shift in Eanion in a different set of neurons (e.g., an excitatory interneuron presynaptic to the output neuron), then midazolam should retain analgesic efficacy despite a depolarizing shift in Eanion that confers a pro-nociceptive influence to at least a component of the GABA circuit. In support of this model, there is evidence that at Eanion does not change in at least some dorsal horn neurons (at least following nerve injury) (Schoffnegger et al., 2008; Sokal and Chapman, 2003). There is also evidence that GABAA receptors with a benzodiazepine receptor are differentially distributed (Knabl et al., 2008; Rudolph and Mohler, 2006).
Figure 6. The midazolam paradox.
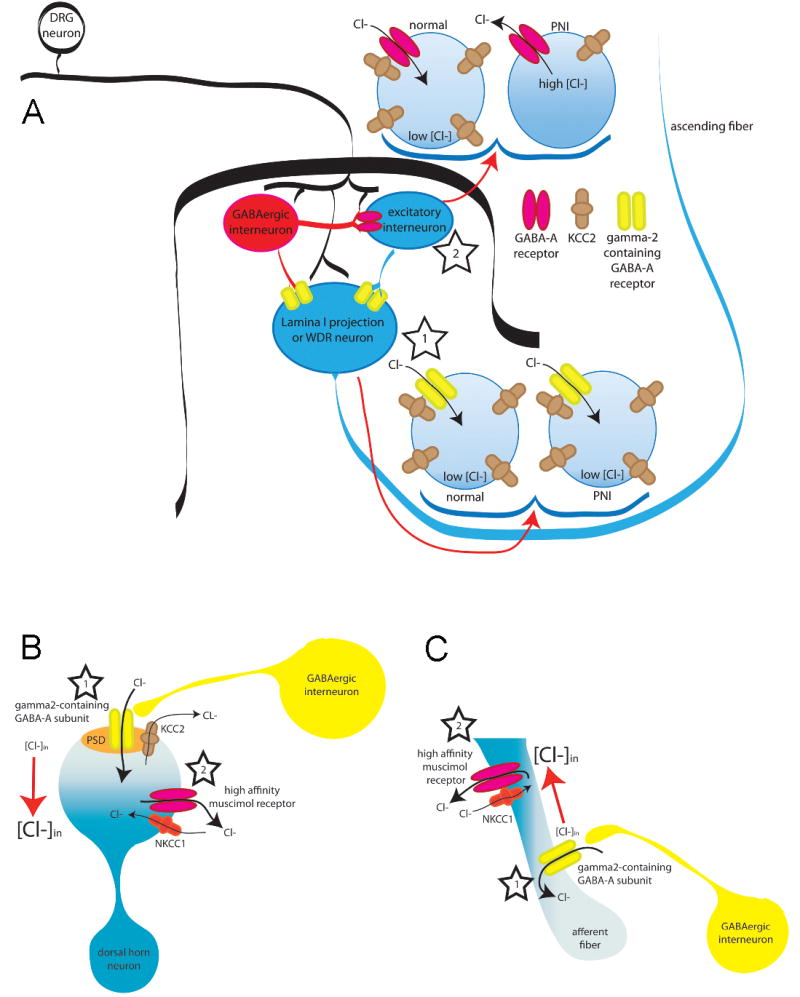
There are at least 3 models that could explain the apparent paradox associated with the continued analgesic efficacy of benzodiazepine receptor agonist in the face of a shift in the valence of GABAA receptor antagonist effects in the presence of persistent inflammation. All 3 require a functional separation between depolarizing shifts in Cl− equilibrium potential and γ2 containing GABAA receptors (i.e., those receptors responsive to benzodiazepines). Panel A: Separation in two subpopulations of dorsal horn neurons: In this model, γ2 containing GABAA receptors are enriched in a subpopulation of dorsal horn neurons critical for the establishment of nociceptive threshold and/or the perception of pain (i.e., dorsal horn projection neuron, star 1). There is no shift in Eanion in these neurons, so that GABAA receptor activation is inhibitory, and facilitation of this receptor activation by benzodiazepines is analgesic (star 1). The γ2-containing neuron is post-synaptic to both GABAergic input and excitatory interneurons. A depolarizing shift in Eanion in these excitatory interneurons results in a shift in the valence of GABAA signaling and a GABA-mediated facilitation of nociceptive behavior (star 2). The density of γ2-containing GABAA receptors in these interneurons would have to be relatively low such that benzodiazepines do not facilitate excitatory GABAergic transmission. The majority of lamina I–II neurons are interneurons (~90%) and these neurons may account for reduced KCC2 expression and excitatory synaptic GABAA responses (star 2) observed following peripheral nerve injury (PNI). Such a model could also account for changes observed in the presence of inflammation if the GABAA receptors on these excitatory interneurons have a higher affinity for muscimol. Panel B: Separation within a dorsal horn neuron: A differential distribution of γ2 containing GABAA receptors and cation-Cl− co-transporters within a dorsal horn neuron critical for nociceptive behavior and the perception of pain could underlie relatively localized changes in Eanion. In this model, there would be little or no change in Eanion around γ2 containing receptors following tissue injury (star 1), whereas a depolarizing shift in Eanion near receptors with a high affinity for muscimol would confer the facilitation of nociceptive transmission observed in the presence of persistent inflammation (star 2). Importantly, such a model is unlikely to account for changes observed following PNI given that a depolarizing shift in Eanion appears to impact the response to both exogenous and endogenous GABAA receptor activation, suggesting that the shift in Eanion is relatively uniform in these neurons. Panel C: Separation with the central terminals of primary afferent neurons. This model is analogous to that described for Panel B, except that the separation of γ2-containing receptors and depolarizing shifts in Eanion occur in the primary afferent. A subset of dorsal horn interneurons make axo-axonic synapses onto incoming afferent fibers (star 1). While the subcellular localization of GABAA subunits and/or NKCC1 is not known in these afferent axons, mechanisms involving the localization of midazolam-sensitive (γ2-containing) GABAA receptors coupled with differences in the localization of inflammation-induced plasticity in Cl− gradients (via changes in NKCC1 expression, localization or activity) could yield pharmacological profiles that account for the preservation of antinociceptive effects of midazolam observed in the presence of both inflammation and nerve injury (star 2).
Another explanation for the midazolam paradox is that different GABAA receptor subtypes are differentially coupled to local changes in Eanion in the same neuron. In this model, benzodiazepine receptors would remain coupled to a hyperpolarized Eanion, while GABAA receptors without a benzodiazepine binding site would be coupled to a depolarizing shift in Eanion. Given evidence for the differential subcellular distribution of γ2 subunit containing receptors (i.e., those with a benzodiazepine receptor) with γ2 containing receptors preferentially targeted to synapses (Rudolph and Mohler, 2006), a depolarizing shift in Eanion around extrasynaptic receptors would have to underlie the pro-nociceptive actions of GABAA receptor activation observed in the presence of inflammation (Fig. 6B and C). In support of this model, there is recent evidence for the differential subcellular distribution of NKCC1 and KCC2 (Khirug et al., 2008; Szabadics et al., 2006) which could underlie local, subcellular variations in Eanion. Furthermore, given recent evidence that the potency of muscimol may be higher at extrasynaptic GABAA receptors (You and Dunn, 2007), this model would account for the “U- shaped” dose-response curve obtained with muscimol in the presence of inflammation: low doses of the agonist preferentially activated the pro-nociceptive extrasynaptic receptors, while higher concentrations ultimately activated the antinociceptive synaptic receptors (Anseloni and Gold, 2008). Additional data is clearly needed, however, to distinguish between these and other possibilities.
There are clearly a number of issues yet to be resolved concerning injury-induced changes in the regulation of Eanion and the impact of these changes in what is normally inhibitory circuitry in the spinal cord. However, data in hand also clearly illustrate a number of important issues for our understanding of pain and its treatment. First, injury-induced changes in Eanion and inhibitory signaling play a critical role in the manifestation of persistent pain. Second, specific mechanisms underlying these changes depend on the type of injury. And third, the spinal benzodiazepine receptor should be pursued as a target for the treatment of both inflammatory and neuropathic pain.
7 Closing remarks
While Melzack and Walls Gate Control Theory first laid the foundations for a major role for GABAergic neurotransmission in pain control, recent advances have made it clear that pain control via manipulation of GABAergic and glycinergic transmission in the dorsal horn is subject to nuances in receptor localization, subunit composition and Cl− gradients that are only now beginning to be fully appreciated. A better understanding of Cl− (and HCO3−) regulation and its impact on synaptic transmission under various pathological conditions is likely to reveal new therapeutic opportunities for pain control.
Acknowledgments
TJP is supported by startup funds from the University of Arizona and by the University of Arizona Foundation. FC is the holder of a CIHR Research Chair. Work in his lab is supported by CIHR, FRSQ and CFI. MSG is supported by 7R01NS044992-05 from NIH. DLH is supported by R01DA016430 from NIH. SAP is supported by startup funds from the University of Pittsburgh.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Alvarez-Leefmans FJ, Gamino SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurones studied with ion-selective microelectrodes. J Physiol. 1988;406:225–46. doi: 10.1113/jphysiol.1988.sp017378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Leon-Olea M, Mendoza-Sotelo J, Alvarez FJ, Anton B, Garduno R. Immunolocalization of the Na(+)-K(+)-2Cl(−) cotransporter in peripheral nervous tissue of vertebrates. Neuroscience. 2001;104:569–82. doi: 10.1016/s0306-4522(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Anseloni VC, Gold MS. Inflammation-Induced Shift in the Valence of Spinal GABA-A Receptor-Mediated Modulation of Nociception in the Adult Rat. J Pain. 2008 doi: 10.1016/j.jpain.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aptel H, Hilaire C, Pieraut S, Boukhaddaoui H, Mallie S, Valmier J, Scamps F. The Cav3.2/alpha1H T-type Ca2+ current is a molecular determinant of excitatory effects of GABA in adult sensory neurons. Mol Cell Neurosci. 2007;36:293–303. doi: 10.1016/j.mcn.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Baccei ML, Fitzgerald M. Development of GABAergic and glycinergic transmission in the neonatal rat dorsal horn. J Neurosci. 2004;24:4749–57. doi: 10.1523/JNEUROSCI.5211-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Borg PA, Krijnen HJ. Long-term intrathecal administration of midazolam and clonidine. Clin J Pain. 1996;12:63–68. doi: 10.1097/00002508-199603000-00012. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–24. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JM, Nassar MA, Wood JN, Basbaum AI. Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron. 2005;47:787–93. doi: 10.1016/j.neuron.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Brown AG, Koerber HR, Noble R. Actions of trains and pairs of impulses from single primary afferent fibres on single spinocervical tract cells in cat. J Physiol (Lond) 1987;382:313–329. doi: 10.1113/jphysiol.1987.sp016369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl- cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J Neurosci. 2008;28:1301–12. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caba M, Gonzalez-Mariscal G, Beyer C. Perispinal progestins enhance the antinociceptive effects of muscimol in the rat. Pharmacol Biochem Behav. 1994;47:177–182. doi: 10.1016/0091-3057(94)90128-7. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Coggeshall RE. Peripheral GABA(A) receptors: evidence for peripheral primary afferent depolarization. Neuroscience. 1999;93:713–22. doi: 10.1016/s0306-4522(99)00101-3. [DOI] [PubMed] [Google Scholar]
- Cervero F. The Gate Theory Then and Now. In: Merskey H, editor. The Paths of Pain. IASP Press; Seattle: 2005. pp. 33–48. [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–21. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cordero-Erausquin M, Coull JA, Boudreau D, Rolland M, De Koninck Y. Differential maturation of GABA action and anion reversal potential in spinal lamina I neurons: impact of chloride extrusion capacity. J Neurosci. 2005;25:9613–9623. doi: 10.1523/JNEUROSCI.1488-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–42. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–50. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- De Koninck Y, Henry JL. Prolonged GABAA-mediated inhibition following single hair afferent input to single spinal dorsal horn neurones in cats. J Physiol. 1994;476:89–100. [PMC free article] [PubMed] [Google Scholar]
- De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol. 2007;7:93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Yaksh TL. Intrathecal baclofen and muscimol, but not midazolam, are antinociceptive using the rat-formalin model. J Pharmacol Exp Ther. 1995;275:219–227. [PubMed] [Google Scholar]
- Dogrul A, Gardell LR, Ossipov MH, Tulunay FC, Lai J, Porreca F. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain. 2003;105:159–68. doi: 10.1016/s0304-3959(03)00177-5. [DOI] [PubMed] [Google Scholar]
- Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278:27347–53. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Plunkett JA, Martinez MA, Lopez T, Karmally S, Cejas P, Whittemore SR. Transplants of neuronal cells bioengineered to synthesize GABA alleviate chronic neuropathic pain. Cell Transplant. 1999;8:87–101. doi: 10.1177/096368979900800102. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Schmidt R, Willis WD. Pharmacological Studies On Presynaptic Inhibition. J Physiol. 1963;168:500–30. doi: 10.1113/jphysiol.1963.sp007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC. The Physiology of Synapses. Springer-Verlag; Berlin: 1964. [Google Scholar]
- Edwards M, Serrao JM, Gent JP, Goodchild CS. On the mechanism by which midazolam causes spinally mediated analgesia. Anesthesiology. 1990;73:273–277. doi: 10.1097/00000542-199008000-00015. [DOI] [PubMed] [Google Scholar]
- Falcon M, Guendellman D, Stolberg A, Frenk H, Urca G. Development of thermal nociception in rats. Pain. 1996;67:203–8. doi: 10.1016/0304-3959(96)03070-9. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M, Gibson S. The postnatal physiological and neurochemical development of peripheral sensory C fibres. Neuroscience. 1984;13:933–44. doi: 10.1016/0306-4522(84)90107-6. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain. 2004;109:150–61. doi: 10.1016/j.pain.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl otransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–8. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Furue H, Narikawa K, Kumamoto E, Yoshimura M. Responsiveness of rat substantia gelatinosa neurones to mechanical but not thermal stimuli revealed by in vivo patch-clamp recording. J Physiol. 1999;521(Pt 2):529–535. doi: 10.1111/j.1469-7793.1999.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol. 2006a;26:689–98. doi: 10.1128/MCB.26.2.689-698.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. Volume sensitivity of cation-Cl- cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am J Physiol Cell Physiol. 2006b;290:C134–42. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- Galan A, Cervero F, Laird JM. Extracellular signaling-regulated kinase-1 and -2 (ERK 1/2) mediate referred hyperalgesia in a murine model of visceral pain. Brain Res Mol Brain Res. 2003;116:126–34. doi: 10.1016/s0169-328x(03)00284-5. [DOI] [PubMed] [Google Scholar]
- Galan A, Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience. 2005;133:245–52. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Gallagher JP, Higashi H, Nishi S. Characterization and ionic basis of GABA-induced depolarizations recorded in vitro from cat primary afferent neurones. J Physiol. 1978;275:263–82. doi: 10.1113/jphysiol.1978.sp012189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JP, Nakamura J, Shinnick-Gallagher P. The effects of temperature, pH and Cl-pump inhibitors on GABA responses recorded from cat dorsal root ganglia. Brain Res. 1983;267:249–59. doi: 10.1016/0006-8993(83)90877-6. [DOI] [PubMed] [Google Scholar]
- Garcia-Nicas E, Laird JM, Cervero F. Vasodilatation in hyperalgesic rat skin evoked by stimulation of afferent A beta-fibers: further evidence for a role of dorsal root reflexes in allodynia. Pain. 2001;94:283–91. doi: 10.1016/S0304-3959(01)00365-7. [DOI] [PubMed] [Google Scholar]
- Garcia-Nicas E, Laird JM, Cervero F. GABA-A receptor blockade reverses the injury-induced sensitization of Nociceptor Specific (NS) neurons in the spinal dorsal horn of the rat. Journal of Neurophysiology. 2006 doi: 10.1152/jn.00377.2006. in press. [DOI] [PubMed] [Google Scholar]
- Gilbert D, Franjic-Wurtz C, Funk K, Gensch T, Frings S, Mohrlen F. Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int J Dev Neurosci. 2007;25:479–89. doi: 10.1016/j.ijdevneu.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Goodchild CS, Serrao JM. Intrathecal midazolam in the rat: evidence for spinally-mediated analgesia. Br J Anaesth. 1987;59:1563–1570. doi: 10.1093/bja/59.12.1563. [DOI] [PubMed] [Google Scholar]
- Granados-Soto V, Arguelles CF, Alvarez-Leefmans FJ. Peripheral and central antinociceptive action of Na+-K+-2Cl- cotransporter blockers on formalin-induced nociception in rats. Pain. 2005;114:231–238. doi: 10.1016/j.pain.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Green GM, Dickenson A. GABA-receptor control of the amplitude and duration of the neuronal responses to formalin in the rat spinal cord. Eur J Pain. 1997;1:95–104. doi: 10.1016/s1090-3801(97)90067-7. [DOI] [PubMed] [Google Scholar]
- Haas M, Forbush B., 3rd The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515–34. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- Hammond DL, Drower EJ. Effects of intrathecally administered THIP, baclofen and muscimol on nociceptive threshold. Eur J Pharmacol. 1984;103:121–125. doi: 10.1016/0014-2999(84)90197-3. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JH, Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain. 1997;70:15–22. doi: 10.1016/s0304-3959(96)03249-6. [DOI] [PubMed] [Google Scholar]
- Ito K, Tanaka K, Nishibe Y, Hasegawa J, Ueno H. GABA-synthesizing enzyme, GAD67, from dermal fibroblasts: evidence for a new skin function. Biochim Biophys Acta. 2007;1770:291–6. doi: 10.1016/j.bbagen.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Jiang MC, Gebhart GF. Development of mustard oil-induced hyperalgesia in rats. Pain. 1998;77:305–13. doi: 10.1016/S0304-3959(98)00110-9. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl- extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Johnston GA. GABA(A) receptor channel pharmacology. Curr Pharm Des. 2005;11:1867–1885. doi: 10.2174/1381612054021024. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci U S A. 2005;102:16783–8. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Staley K. Altered neuronal chloride homeostasis and excitatory GABAergic signaling in human temporal lobe epilepsy. Epilepsy Curr. 2008;8:51–53. doi: 10.1111/j.1535-7511.2008.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Pasternack M, Saarikoski J, Voipio J. Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J Physiol. 1989;416:161–81. doi: 10.1113/jphysiol.1989.sp017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Hammond DL. Role of spinal gamma-aminobutyric acidA receptors in formalin-induced nociception in the rat. J Pharmacol Exp Ther. 1997;282:928–938. [PubMed] [Google Scholar]
- Kato G, Yasaka T, Katafuchi T, Furue H, Mizuno M, Iwamoto Y, Yoshimura M. Direct GABAergic and glycinergic inhibition of the substantia gelatinosa from the rostral ventromedial medulla revealed by in vivo patch-clamp analysis in rats. J Neurosci. 2006;26:1787–1794. doi: 10.1523/JNEUROSCI.4856-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AF, Coull JA, Chery N, Poisbeau P, De Koninck Y. Region-specific developmental specialization of GABA-glycine cosynapses in laminas I–II of the rat spinal dorsal horn. J Neurosci. 2001;21:7871–7880. doi: 10.1523/JNEUROSCI.21-20-07871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22:9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–9. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Lee YM. Intrathecal midazolam increases the analgesic effects of spinal blockade with bupivacaine in patients undergoing haemorrhoidectomy. Br J Anaesth. 2001;86:77–79. doi: 10.1093/bja/86.1.77. [DOI] [PubMed] [Google Scholar]
- Klein JD, Lamitina ST, O’Neill WC. JNK is a volume-sensitive kinase that phosphorylates the Na-K-2Cl cotransporter in vitro. Am J Physiol. 1999;277:C425–31. doi: 10.1152/ajpcell.1999.277.3.C425. [DOI] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Ruiz A, Rusakov DM, Scott R, Semyanov A, Walker MC. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog Biophys Mol Biol. 2005;87:33–46. doi: 10.1016/j.pbiomolbio.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyles AE, Waterman AE, Livingston A. Antinociceptive activity of midazolam in sheep. J Vet Pharmacol Ther. 1995;18:54–60. doi: 10.1111/j.1365-2885.1995.tb00551.x. [DOI] [PubMed] [Google Scholar]
- Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain. 2001;92:335–42. doi: 10.1016/S0304-3959(01)00275-5. [DOI] [PubMed] [Google Scholar]
- Laird JM, Garcia-Nicas E, Delpire EJ, Cervero F. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett. 2004;361:200–3. doi: 10.1016/j.neulet.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Liedtke CM, Cole TS. PKC signaling in CF/T43 cell line: regulation of NKCC1 by PKC-delta isotype. Biochim Biophys Acta. 2000;1495:24–33. doi: 10.1016/s0167-4889(99)00146-9. [DOI] [PubMed] [Google Scholar]
- Liedtke CM, Cole TS. Activation of NKCC1 by hyperosmotic stress in human tracheal epithelial cells involves PKC-delta and ERK. Biochim Biophys Acta. 2002;1589:77–88. doi: 10.1016/s0167-4889(01)00189-6. [DOI] [PubMed] [Google Scholar]
- Lin Q, Peng YB, Willis WD. Role of GABA receptor subtypes in inhibition of primate spinothalamic tract neurons: difference between spinal and periaqueductal gray inhibition. J Neurophysiol. 1996a;75:109–123. doi: 10.1152/jn.1996.75.1.109. [DOI] [PubMed] [Google Scholar]
- Lin Q, Peng YB, Willis WD. Inhibition of primate spinothalamic tract neurons by spinal glycine and GABA is reduced during central sensitization. J Neurophysiol. 1996b;76:1005–1014. doi: 10.1152/jn.1996.76.2.1005. [DOI] [PubMed] [Google Scholar]
- Lin Q, Wu J, Willis WD. Dorsal root reflexes and cutaneous neurogenic inflammation after intradermal injection of capsaicin in rats. J Neurophysiol. 1999;82:2602–11. doi: 10.1152/jn.1999.82.5.2602. [DOI] [PubMed] [Google Scholar]
- Lin Q, Zou X, Willis WD. Adelta and C primary afferents convey dorsal root reflexes after intradermal injection of capsaicin in rats. J Neurophysiol. 2000;84:2695–8. doi: 10.1152/jn.2000.84.5.2695. [DOI] [PubMed] [Google Scholar]
- Loomis CW, Khandwala H, Osmond G, Hefferan MP. Coadministration of intrathecal strychnine and bicuculline effects synergistic allodynia in the rat: an isobolographic analysis. J Pharmacol Exp Ther. 2001a;296:756–761. [PubMed] [Google Scholar]
- Loomis CW, Khandwala H, Osmond G, Hefferan MP. Coadministration of intrathecal strychnine and bicuculline effects synergistic allodynia in the rat: an isobolographic analysis. J Pharmacol Exp Ther. 2001b;296:756–61. [PubMed] [Google Scholar]
- Luo M, Zhang D, Ma S, Huang Y, Shuster SJ, Porreca F, Lai J. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Molecular Pain. 2005;1:29. doi: 10.1186/1744-8069-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle C, Xu JC, Biemesderfer D, Forbush B., 3rd Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol. 1995;269:C1496–505. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- Lytle C, Forbush B., 3rd Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol. 1996;270:C437–48. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- Ma W, Saunders PA, Somogyi R, Poulter MO, Barker JL. Ontogeny of GABAA receptor subunit mRNAs in rat spinal cord and dorsal root ganglia. J Comp Neurol. 1993;338:337–359. doi: 10.1002/cne.903380303. [DOI] [PubMed] [Google Scholar]
- Malan TP, Mata HP, Porreca F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology. 2002a;96:1161–7. doi: 10.1097/00000542-200205000-00020. [DOI] [PubMed] [Google Scholar]
- Malan TP, Mata HP, Porreca F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology. 2002b;96:1161–1167. doi: 10.1097/00000542-200205000-00020. [DOI] [PubMed] [Google Scholar]
- Marsh D, Dickenson A, Hatch D, Fitzgerald M. Epidural opioid analgesia in infant rats I: mechanical and heat responses. Pain. 1999;82:23–32. doi: 10.1016/S0304-3959(99)00028-7. [DOI] [PubMed] [Google Scholar]
- Martina M, Royer S, Pare D. Cell-type-specific GABA responses and chloride homeostasis in the cortex and amygdala. J Neurophysiol. 2001;86:2887–2895. doi: 10.1152/jn.2001.86.6.2887. [DOI] [PubMed] [Google Scholar]
- McCall WD, Tanner KD, Levine JD. Formalin induces biphasic activity in C-fibers in the rat. Neurosci Lett. 1996;208:45–8. doi: 10.1016/0304-3940(96)12552-0. [DOI] [PubMed] [Google Scholar]
- McDaniel N, Pace AJ, Spiegel S, Engelhardt R, Koller BH, Seidler U, Lytle C. Role of Na-K-2Cl cotransporter-1 in gastric secretion of nonacidic fluid and pepsinogen. Am J Physiol Gastrointest Liver Physiol. 2005;289:G550–60. doi: 10.1152/ajpgi.00095.2005. [DOI] [PubMed] [Google Scholar]
- Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–9. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- Miletic G, Miletic V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain. 2007 doi: 10.1016/j.pain.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Morales-Aza BM, Chillingworth NL, Payne JA, Donaldson LF. Inflammation alters cation chloride cotransporter expression in sensory neurons. Neurobiol Dis. 2004;17:62–9. doi: 10.1016/j.nbd.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005;280:42685–93. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- Munoz A, Mendez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48:663–673. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–7. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Miyazaki H, Niisato N, Marunaka Y. Essential role of NKCC1 in NGF-induced neurite outgrowth. Biochem Biophys Res Commun. 2007;359:604–10. doi: 10.1016/j.bbrc.2007.05.133. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci. 2005;25:8766–75. doi: 10.1523/JNEUROSCI.2527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez-Reyes E, Lee JH, Shin HS, Todorovic SM. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci. 2007;27:8250–60. doi: 10.1523/JNEUROSCI.1800-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. The blockade of GABA mediated responses in the frog spinal cord by ammonium ions and furosemide. J Physiol (Lond) 1978;283:121–32. doi: 10.1113/jphysiol.1978.sp012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura H, Sakai A, Nagano M, Umino M, Suzuki H. Expression changes of cation chloride cotransporters in the rat spinal cord following intraplantar formalin. Neurosci Res. 2006;56:435–40. doi: 10.1016/j.neures.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Ortiz PA. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: role of VAMP. Am J Physiol Renal Physiol. 2006;290:F608–16. doi: 10.1152/ajprenal.00248.2005. [DOI] [PubMed] [Google Scholar]
- Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci USA. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Peterson MA, Basbaum AI, Abbadie C, Rohde DS, McKay WR, Taylor BK. The differential contribution of capsaicin-sensitive afferents to behavioral and cardiovascular measures of brief and persistent nociception and to Fos expression in the formalin test. Brain Res. 1997;755:9–16. doi: 10.1016/s0006-8993(97)00068-1. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–9. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Garbarini N, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl- cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem. 2003;278:52848–56. doi: 10.1074/jbc.M309436200. [DOI] [PubMed] [Google Scholar]
- Pieraut S, Laurent-Matha V, Sar C, Hubert T, Mechaly I, Hilaire C, Mersel M, Delpire E, Valmier J, Scamps F. NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J Neurosci. 2007;27:6751–9. doi: 10.1523/JNEUROSCI.1337-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MH, Price TJ, Entrena JM, Cervero F. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain. 2007;3:17. doi: 10.1186/1744-8069-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SA, Sejnowski TJ, De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain. 2006 doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Hargreaves KM, Cervero F. Protein expression and mRNA cellular distribution of the NKCC1 cotransporter in the dorsal root and trigeminal ganglia of the rat. Brain Res. 2006;1112:146–58. doi: 10.1016/j.brainres.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian N, Sejnowski TJ. When is an inhibitory synapse effective? Proc Natl Acad Sci USA. 1990;87:8145–8149. doi: 10.1073/pnas.87.20.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. Do dorsal root reflexes augment peripheral inflammation? Neuroreport. 1994;5:821–4. doi: 10.1097/00001756-199403000-00021. [DOI] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. The role of glutamate and GABA receptors in the generation of dorsal root reflexes by acute arthritis in the anaesthetized rat. J Physiol. 1995;484(Pt 2):437–45. doi: 10.1113/jphysiol.1995.sp020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie JM, Boylan GB. Neonatal seizures and their treatment. Curr Opin Neurol. 2003;16:177–181. doi: 10.1097/01.wco.0000063768.15877.23. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–5. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Roberts LA, Beyer C, Komisaruk BR. Nociceptive responses to altered GABAergic activity at the spinal cord. Life Sci. 1986;39:1667–74. doi: 10.1016/0024-3205(86)90164-5. [DOI] [PubMed] [Google Scholar]
- Rocha-Gonzalez HI, Mao S, Alvarez-Leefmans FJ. Na+,K+,2Cl-cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J Neurophysiol. 2008 doi: 10.1152/jn.01007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode F, Jensen DG, Blackburn-Munro G, Bjerrum OJ. Centrally-mediated antinociceptive actions of GABA(A) receptor agonists in the rat spared nerve injury model of neuropathic pain. Eur J Pharmacol. 2005;516:131–138. doi: 10.1016/j.ejphar.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr Opin Pharmacol. 2006;6:18–23. doi: 10.1016/j.coph.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Rudomin P, Schmidt RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res. 1999;129:1–37. doi: 10.1007/s002210050933. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–76. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Salonia A, Suardi N, Crescenti A, Colombo R, Rigatti P, Montorsi F. General versus spinal anesthesia with different forms of sedation in patients undergoing radical retropubic prostatectomy: results of a prospective, randomized study. Int J Urol. 2006;13:1185–90. doi: 10.1111/j.1442-2042.2006.01524.x. [DOI] [PubMed] [Google Scholar]
- Sandby-Thomas M, Sullivan G, Hall JE. A national survey into the peri-operative anaesthetic management of patients presenting for surgical correction of a fractured neck of femur. Anaesthesia. 2008;63:250–8. doi: 10.1111/j.1365-2044.2007.05328.x. [DOI] [PubMed] [Google Scholar]
- Schaffner AE, Behar T, Nadi S, Smallwood V, Barker JL. Quantitative analysis of transient GABA expression in embryonic and early postnatal rat spinal cord neurons. Brain Res Dev Brain Res. 1993;72:265–276. doi: 10.1016/0165-3806(93)90192-d. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Schmid R, Handwerker HO, Torebjork HE. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain. 2000;123(Pt 3):560–571. doi: 10.1093/brain/123.3.560. [DOI] [PubMed] [Google Scholar]
- Schoffnegger D, Ruscheweyh R, Sandkuhler J. Spread of excitation across modality borders in spinal dorsal horn of neuropathic rats. Pain. 2008;135:300–10. doi: 10.1016/j.pain.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Schomberg SL, Su G, Haworth RA, Sun D. Stimulation of Na-K-2Cl cotransporter in neurons by activation of Non-NMDA ionotropic receptor and group-I mGluRs. J Neurophysiol. 2001;85:2563–75. doi: 10.1152/jn.2001.85.6.2563. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Serrao JM, Marks RL, Morley SJ, Goodchild CS. Intrathecal midazolam for the treatment of chronic mechanical low back pain: a controlled comparison with epidural steroid in a pilot study. Pain. 1992;48:5–12. doi: 10.1016/0304-3959(92)90125-U. [DOI] [PubMed] [Google Scholar]
- Sherman SE, Loomis CW. Morphine insensitive allodynia is produced by intrathecal strychnine in the lightly anesthetized rat. Pain. 1994;56:17–29. doi: 10.1016/0304-3959(94)90146-5. [DOI] [PubMed] [Google Scholar]
- Sherman SE, Loomis CW. Strychnine-dependent allodynia in the urethane-anesthetized rat is segmentally distributed and prevented by intrathecal glycine and betaine. Can J Physiol Pharmacol. 1995;73:1698–1705. doi: 10.1139/y95-733. [DOI] [PubMed] [Google Scholar]
- Sherman SE, Loomis CW. Strychnine-sensitive modulation is selective for non-noxious somatosensory input in the spinal cord of the rat. Pain. 1996;66:321–330. doi: 10.1016/0304-3959(96)03063-1. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Sperk G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol. 1994;72:169–79. doi: 10.1152/jn.1994.72.1.169. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Willis WD, Westlund KN. Joint inflammation and hyperalgesia are reduced by spinal bicuculline. Neuroreport. 1993;5:109–12. doi: 10.1097/00001756-199311180-00003. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Rees H, Westlund KN, Willis WD. Fiber types contributing to dorsal root reflexes induced by joint inflammation in cats and monkeys. J Neurophysiol. 1995;74:981–9. doi: 10.1152/jn.1995.74.3.981. [DOI] [PubMed] [Google Scholar]
- Sokal DM, Chapman V. Effects of spinal administration of muscimol on C- and A-fibre evoked neuronal responses of spinal dorsal horn neurones in control and nerve injured rats. Brain Res. 2003;962:213–20. doi: 10.1016/s0006-8993(02)04057-x. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Puig S. Neuronal model of tactile allodynia produced by spinal strychnine: effects of excitatory amino acid receptor antagonists and a mu-opiate receptor agonist. Pain. 1996;68:283–292. doi: 10.1016/s0304-3959(96)03130-2. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Puig S, Jones DL. Spinal bicuculline produces hypersensitivity of dorsal horn neurons: effects of excitatory amino acid antagonists. Pain. 1998;77:181–190. doi: 10.1016/S0304-3959(98)00094-3. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–81. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABA(A) receptor function by the kinetics of Cl- and HCO3- transport. J Physiol. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubley LA, Martinez MA, Karmally S, Lopez T, Cejas P, Eaton MJ. Only early intervention with gamma-aminobutyric acid cell therapy is able to reverse neuropathic pain after partial nerve injury. J Neurotrauma. 2001;18:471–477. doi: 10.1089/089771501750171092. [DOI] [PubMed] [Google Scholar]
- Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci. 2000;20:7531–8. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory Effect of GABAergic Axo-Axonic Cells in Cortical Microcircuits. Science %R 10.1126/science.1121325. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Tokunaga A, Yamanaka H, Mashimo T, Noguchi K, Uchida I. Two types of GABAergic miniature inhibitory postsynaptic currents in mouse substantia gelatinosa neurons. Eur J Pharmacol. 2006;553:120–128. doi: 10.1016/j.ejphar.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Teng CJ, Abbott FV. The formalin test: a dose-response analysis at three developmental stages. Pain. 1998;76:337–47. doi: 10.1016/S0304-3959(98)00065-7. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC. The localization of classical transmitters and neuropeptides within neurons in laminae I–III of the mammalian spinal dorsal horn. Prog Neurobiol. 1993;41:609–45. doi: 10.1016/0301-0082(93)90045-t. [DOI] [PubMed] [Google Scholar]
- Todd AJ. Anatomy of primary afferents and projection neurones in the rat spinal dorsal horn with particular emphasis on substance P and the neurokinin 1 receptor. Exp Physiol. 2002;87:245–9. doi: 10.1113/eph8702351. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, Zorumski CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Redox modulation of peripheral T-type Ca2+ channels in vivo: alteration of nerve injury-induced thermal hyperalgesia. Pain. 2004;109:328–39. doi: 10.1016/j.pain.2004.01.026. [DOI] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Ohno K, Yamada J, Ikeda M, Okabe A, Sato K, Hashimoto K, Fukuda A. Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J Neurophysiol. 2003;89:1353–62. doi: 10.1152/jn.00721.2002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Ugarte SD, Homanics GE, Firestone LL, Hammond DL. Sensory thresholds and the antinociceptive effects of GABA receptor agonists in mice lacking the beta3 subunit of the GABA(A) receptor. Neuroscience. 2000;95:795–806. doi: 10.1016/s0306-4522(99)00481-9. [DOI] [PubMed] [Google Scholar]
- Valencia-de Ita S, Lawand NB, Lin Q, Castaneda-Hernandez G, Willis WD. Role of the Na+-K+-2Cl- cotransporter in the development of capsaicin-induced neurogenic inflammation. J Neurophysiol. 2006;95:3553–61. doi: 10.1152/jn.01091.2005. [DOI] [PubMed] [Google Scholar]
- Vierck CJ, Hansson PT, Yezierski RP. Clinical and pre-clinical pain assessment: are we measuring the same thing? Pain. 2008;135:7–10. doi: 10.1016/j.pain.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Vierck CJ, Jr, Kline Rt, Wiley RG. Comparison of operant escape and innate reflex responses to nociceptive skin temperatures produced by heat and cold stimulation of rats. Behav Neurosci. 2004;118:627–35. doi: 10.1037/0735-7044.118.3.627. [DOI] [PubMed] [Google Scholar]
- Wagner DA, Goldschen-Ohm MP, Hales TG, Jones MV. Kinetics and spontaneous open probability conferred by the epsilon subunit of the GABAA receptor. J Neurosci. 2005;25:10462–1048. doi: 10.1523/JNEUROSCI.1658-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HR, Dougherty PM. Response properties of dorsal root reflexes in cutaneous C fibers before and after intradermal capsaicin injection in rats. Neuroscience. 2005;132:823–31. doi: 10.1016/j.neuroscience.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Willis EF, Clough GF, Church MK. Investigation into the mechanisms by which nedocromil sodium, frusemide and bumetanide inhibit the histamine-induced itch and flare response in human skin in vivo. Clin Exp Allergy. 2004;34:450–5. doi: 10.1111/j.1365-2222.2004.01898.x. [DOI] [PubMed] [Google Scholar]
- Willis WD. John Eccles’ studies of spinal cord presynaptic inhibition. Prog Neurobiol. 2006;78:189–214. doi: 10.1016/j.pneurobio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Willis WD., Jr Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res. 1999;124:395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- Wojtowicz JM, Nicoll RA. Selective action of piretanide on primary afferent GABA responses in the frog spinal cord. Brain Res. 1982;236:173–81. doi: 10.1016/0006-8993(82)90043-9. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–68. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Wu YW, Shiau JM, Hong CC, Hung CP, Lu HF, Tseng CC. Intrathecal midazolam combined with low-dose bupivacaine improves postoperative recovery in diabetic mellitus patients undergoing foot debridement. Acta Anaesthesiol Taiwan. 2005;43:129–34. [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989a;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989b;37:111–23. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Nishi S. Primary afferent-evoked glycine- and GABA-mediated IPSPs in substantia gelatinosa neurones in the rat spinal cord in vitro. J Physiol. 1995;482:29–38. doi: 10.1113/jphysiol.1995.sp020497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Dunn SM. Identification of a domain in the delta subunit (S238-V264) of the alpha4beta3delta GABAA receptor that confers high agonist sensitivity. J Neurochem. 2007;103:1092–1101. doi: 10.1111/j.1471-4159.2007.04817.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu LY, Xu TL. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience. 2008;152:502–10. doi: 10.1016/j.neuroscience.2007.12.037. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Hertz L, Code WE. Effects of benzodiazepines on potassium-induced increase in free cytosolic calcium concentration in astrocytes: interactions with nifedipine and the peripheral-type benzodiazepine antagonist PK 11195. Can J Physiol Pharmacol. 1996;74:273–277. [PubMed] [Google Scholar]