

Abstract
Fragile X syndrome (FXS) causes mental impairment and autism through transcriptional silencing of the Fmr1 gene, resulting in the loss of the RNA-binding protein fragile X mental retardation protein (FMRP). Cortical pyramidal neurons in affected individuals and Fmr1 knock-out (KO) mice have an increased density of dendritic spines. The mutant mice also show defects in synaptic and experience-dependent circuit plasticity, which are known to be mediated in part by dendritic spine dynamics. We used in vivo time-lapse imaging with two-photon microscopy through cranial windows in male and female neonatal mice to test the hypothesis that dynamics of dendritic protrusions are altered in KO mice during early postnatal development. We find that layer 2/3 neurons from wild-type mice exhibit a rapid decrease in dendritic spine dynamics during the first 2 postnatal weeks, as immature filopodia are replaced by mushroom spines. In contrast, KO mice show a developmental delay in the downregulation of spine turnover and in the transition from immature to mature spine subtypes. Blockade of metabotropic glutamate receptor (mGluR) signaling, which reverses some adult phenotypes of KO mice, accentuated this immature protrusion phenotype in KO mice. Thus, absence of FMRP delays spine stabilization and dysregulated mGluR signaling in FXS may partially normalize this early synaptic defect.
Introduction
Various forms of autism and mental impairment share in common an abnormality in dendritic spines (Marin-Padilla, 1972; Kaufmann and Moser, 2000). Spine dysgenesis has been characterized most extensively in fragile X syndrome (FXS), the most common form of inherited mental impairment (Garber et al., 2008). FXS is attributable to transcriptional silencing of the Fmr1 gene, which results in the absence of the fragile X mental retardation protein (FMRP). FMRP is an RNA-binding protein at spine synapses that regulates the translation of several mRNAs important for neuronal development and plasticity (Bassell and Warren, 2008; De Rubeis and Bagni, 2010). Dendritic spines in the brains of individuals with FXS are abnormally long, thin, and tortuous (Rudelli et al., 1985). The same synaptic defect occurs in the Fmr1 knock-out (KO) mouse model of FXS (Comery et al., 1997).
Because filopodia, the earliest dendritic protrusions, are also thin and sometimes long (Yuste and Bonhoeffer, 2004), it has been suggested that FXS might be caused by a failure in the transition from filopodia to spines (Comery et al., 1997; Portera Cailliau and Yuste, 2001). Early protrusions also differ from mature spines on the basis of their shorter lifetime and greater motility (Dailey and Smith, 1996; Lendvai et al., 2000; Portera-Cailliau et al., 2003; Holtmaat et al., 2009). Therefore, the immature-looking dendritic spines in FXS might be unusually dynamic, but this has not been carefully examined. Because sensory deprivation leads to changes in protrusion dynamics in neonatal mice (Lendvai et al., 2000), alterations in spine turnover in Fmr1 KO mice might explain their deficits in experience-dependent plasticity (Dölen et al., 2007; Bureau et al., 2008). In addition, protrusion dynamics are important for synaptogenesis (Ziv and Smith, 1996; Luikart et al., 2008), so the observed reduction of spine synapses in Fmr1 KO mice (Antar et al., 2006) could reflect defects in spine motility or turnover.
Fmr1 KO mice also exhibit excessive group I metabotropic glutamate receptor (mGluR) signaling (Huber et al., 2002). A mechanistic link between this unchecked activation of mGluRs and the spine defect in FXS has been postulated (Bear et al., 2004). Pharmacologic stimulation of mGluRs in neurons in vitro leads to immature, filopodia-like protrusions that resemble those in FXS (Vanderklish and Edelman, 2002; Abu-Elneel et al., 2008). Furthermore, dampening mGluR signaling can rescue the abnormal spine phenotype in Fmr1 KO mice (Dölen et al., 2007; de Vrij et al., 2008). However, whether mGluRs also play a role in spine dynamics or in regulating the density of immature protrusions in vivo has not yet been established.
We used in vivo two-photon time-lapse imaging of green fluorescent protein (GFP)-expressing cortical neurons in neonatal mice to address two questions: First, are spine density and length affected in the intact neocortex of neonatal Fmr1 KO mice? Second, are dendritic protrusion size and turnover abnormally regulated in mutant mice during early postnatal development, and if so, can such defects be reversed by blocking mGluR signaling? We find that early dendritic protrusions in wild-type (WT) mice stabilize into mature spines during the first 2 postnatal weeks, whereas those in KO mice remain highly unstable during that period, consistent with a developmental delay of spine maturation in FXS. Pharmacological inhibition of mGluR5 did not correct the abnormal protrusion turnover, but uncovered new immature phenotypes in KO mice. We conclude that FMRP plays a role in spine maturation and that the upregulated mGluR signaling in mutant mice masks some immature protrusion phenotypes but not others at early developmental stages.
Materials and Methods
All experimental protocols were conducted according to the National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at University California, Los Angeles.
Mice, constructs, and reagents.
Fmr1 KO mice in a C57BL/6 background were obtained from Dr. William Greenough (University of Illinois at Urbana-Champaign, Urbana, IL). The experimenters (A. Cruz-Martín and M. Crespo) were blind to genotyping until after the analysis was completed. Genotypes were determined by PCR analysis of DNA extracted from tail samples using previously described primers (Dutch-Belgian Fragile X Consortium, 1994). As controls, we used either wild-type littermates or wild-type mice from different litters. A plasmid containing the EGFP coding sequence (pCAG-GFP; Addgene plasmid 11150) (Matsuda and Cepko, 2004) was used for electroporation. All DNA was purified and concentrated using QIAGEN plasmid preparation kits and dissolved in 10 mm Tris-HCl, pH 8.0.
In utero electroporation.
Progenitor cells of future layer 2/3 (L2/3) neurons in barrel cortex were transfected via in utero electroporation as previously described (Saito and Nakatsuji, 2001). Timed-pregnant mice at embryonic day 16 (E16) were deeply anesthetized using an isoflurane–oxygen mixture [4.5% (v/v) for induction and 1.5% (v/v) for maintenance] delivered via nose cone using an anesthesia regulator (SurgiVet). After a midline low abdominal incision, the uterine horns were exposed and individual embryos were handled gently with forceps. Approximately 1 μl of DNA solution (containing 0.65–1.0 μg/μl plasmid and 0.1% Fast Green) was pressure injected with a Picospritzer (General Valve) through the uterine wall into the left lateral ventricle of all embryos using pulled-glass capillaries (Sutter Instrument). Next, the head of each embryo was placed between custom-made tweezer-type copper electrodes and two to three square electric pulses (40 V, 50 ms long) were delivered at 500 ms intervals using a custom-built electroporator. The embryos were placed back in the abdominal cavity, and the wall and skin of the dam's abdominal cavity were sutured. Finally, the dams were allowed to recover in a warm chamber for 1 h and then returned to their cage.
Surgery.
Glass-covered cranial windows for chronic imaging were implanted over the barrel field in primary somatosensory cortex at postnatal day 7 (P7) to P12 and P21–P24 as previously described (Portera-Cailliau et al., 2005; Mostany and Portera-Cailliau, 2008; Holtmaat et al., 2009). Briefly, mice were deeply anesthetized with an isoflurane–oxygen mixture and given the analgesic carprofen (5 mg/kg) subcutaneously. After a midline scalp incision, a thin layer of cyanoacrylate-based glue was applied to the exposed skull surface with the exception of the craniotomy region. A small craniotomy (2–2.5 mm diameter) was made with a pneumatic dental drill (Henry Schein) over the left somatosensory cortex (0.5 mm posterior from bregma and 2–3 mm lateral from the midline). The dura was kept moist with sterile saline. Next, a sterile 3 mm glass coverslip was gently laid over the dura (without using agarose) and glued to the skull with cyanoacrylate glue. Dental acrylic was then applied throughout the skull surface up to the edges of the coverslip and the skin. A titanium bar was also embedded in the dental acrylic to help secure the mouse onto the stage of the microscope for imaging, to eliminate movement artifact from breathing. The animals were allowed to recover in a heated chamber and woke up within 20 min after stopping the flow of anesthetic.
Pharmacology.
2-Methyl-6-phenylethynyl pyridine hydrochloride (MPEP) (technically, Inc.) was a gift from the FRAXA Research Foundation. MPEP aliquots were prepared fresh by dissolving it in sterile saline (final concentration of 1 mg/ml). Mice were injected intraperitoneally with MPEP (30 mg/kg) daily from P7 up to the day of the imaging experiment at P10–P12 (i.e., 3–5 d of treatment). The last dose was administered 1 h before imaging. For both WT and KO mice, chronic administration of MPEP did not have a significant effect on their weight (p > 0.05, Tukey's test) (data not shown).
Imaging.
All imaging was performed with a custom-built two-photon microscope, using a Ti:sapphire laser (Chameleon Ultra II; Coherent) tuned to 910 nm. The objective (40×, 0.8 numerical aperture, water-immersion), tube lens, and trinoc were from Olympus, and the photomultiplier tube was from Hamamatsu. For imaging, we used ScanImage software (Pologruto et al., 2003) written in MATLAB (The MathWorks). Excitation power measured at the back aperture of the objective was typically between 20 and 40 mW and was adjusted to achieve near-identical levels of fluorescence within each imaging session. Acute imaging sessions began after a recovery period of at least 1 h and were accomplished under isoflurane anesthesia [1% (v/v) maintenance]. During an imaging session, two to five regions of interest (ROIs) per animal were selected along the dendritic tufts of GFP-expressing L2/3 pyramidal neurons. All imaged dendrites were in L1 (within first 100 μm below the dura mater). Each ROI was collected at high ScanImage zoom and consisted of a stack of images (∼20–40 optical sections, separated axially by 1 μm), where the resolution for each optical section (512 × 512 pixels) was 0.11 μm/pixel. For time-lapse imaging, we collected stacks every 10 min (Lendvai et al., 2000) over 60 min (P7–P12) or 120 min (P21–P24) sessions. Care was taken to achieve close to identical fluorescence levels across imaged regions within an experiment and across different imaging sessions in different animals. In some of the figure panels, distracting processes (e.g., axons) were digitally removed in Adobe Photoshop for display purposes only.
Data analysis.
We analyzed imaging data from N = 4, 5, 4, and 4 WT mice at P7–P8, P10–P12, P10–P12 treated with MPEP, and P21–P24, as well as N = 4, 5, 6, and 3 KO mice at P7–P8, P10–P12, P10–P12 treated with MPEP, and P21–P24, respectively. Data on dendritic protrusion morphology, density, and dynamics were obtained using protrusion analysis software written in MATLAB [kindly provided by Tim O'Connor and Karel Svoboda (Janelia Farm, Howard Hughes Medical Institute, Ashburn, VA)]. This analysis is done on the raw image stacks (except for a median filter; radius, 1–2), and the presence or absence of a protrusion was determined by inspecting individual slices from the entire stack of images. However, because of the lower resolution of two-photon microscopy in the axial plane, only dendritic protrusions that were clearly projecting laterally were included in the analysis (Holtmaat et al., 2009). For a dendritic protrusion to be considered new or lost, it had to clearly protrude out of the shaft by at least four pixels (>0.44 μm), which corresponded to the noise on either side of the dendritic shaft. Measurements of protrusion length (from protrusion tip to shaft) were accomplished by manually drawing a line through the center of the protrusion to the shaft of the dendrite for each frame the protrusion was present. Motility was calculated as the absolute difference in length of protrusions from frame-to-frame, divided by the total number of frames. Lifetime (in minutes) was calculated as the number of frames in which a particular protrusion was visible over a 60 min imaging session. The rates of protrusion gained and lost (F gain and F lost) were defined, respectively, as the fraction of protrusions that appeared and disappeared between two successive frames, relative to the total protrusion number. Protrusion turnover was defined as the sum of the protrusions lost and gained divided by twice the total protrusion number. To quantify the size of dendritic protrusions, the integrated fluorescence intensity [total integrated brightness (TIB)] of each entire protrusion at the best focal plane was divided by the mean fluorescence intensity of the adjacent dendritic shaft to correct for varying imaging conditions. Repeating this analysis with a maximum-intensity projection of all sections containing the protrusion (instead of the best focal section) yielded very similar results (data not shown).
For the unbiased cluster analysis, we used the k-means test in MATLAB, where every single protrusion from WT mice from P7 to P24 was automatically sorted into three separate categories based on three parameters: TIB, lifetime, and motility. To compare the relative abundance of filopodia (F), protospine (PS), and mature spine (MS) categories in WT and KO mice, we resorted protrusions in WT based on arbitrary cutoffs for TIB and lifetime inspired from the k-means results (see Fig. 5 D). Thus, protrusions with TIB ≥90 arbitrary units (a.u.) were labeled as PS, those with TIB <90 a.u. and lifetime ≤40 min were labeled F, and those with TIB <90 a.u and lifetime >40 min were called MS.
Figure 5.
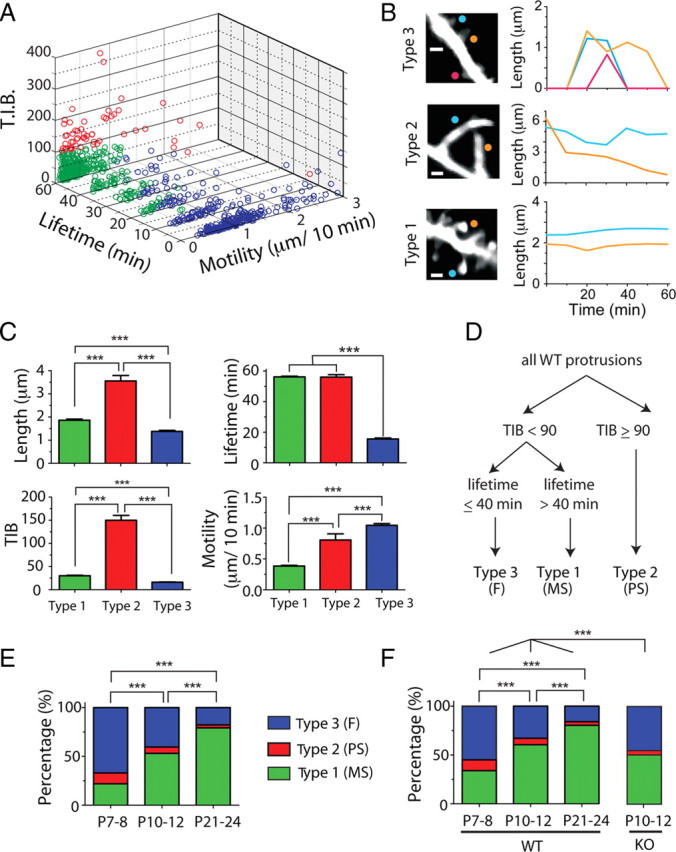
Overabundance of immature protrusion subtypes in Fmr1 KO mice. A, Cluster analysis of all WT protrusions from P7 to P24, based on lifetime, motility, and TIB (total integrated brightness of GFP). Protrusions were segregated into three groups using a k-means test in MATLAB clustering algorithm (see Materials and Methods). B, Representative examples of the three types of protrusions and their length over time in time-lapse imaging sessions. Note that type 2 protrusions are longer and brighter than the others. C, Average length, TIB, lifetime, and motility for each of the three types of protrusions. Types 1, 2, and 3 institutively fall into three well established types of protrusion: mature spines (MS), protospines (PS), and filopodia (F). ***p < 0.001, one-way ANOVA with Tukey's test in C, E, and F. D, Manual sorting of protrusions based on parameters inspired from cluster analysis. These cutoffs were then applied to the protrusions from KO mice. E, Relative abundance of individual protrusion subclasses at different postnatal ages in WT mice as determined by k-means test (includes all protrusions). F, Relative abundance of individual protrusion subclasses in WT and Fmr1 KO mice using cutoffs inspired by k-means test (A) for TIB and lifetime.
To determine whether or not protrusions had heads at their distal tip, we used custom routines in NIH ImageJ and Igor Pro (Wavemetrics). First, we quantified the raw pixel intensity values (supplemental Fig. 5, blue circles, available at www.jneurosci.org as supplemental material) along a manually drawn line through the long axis of each protrusion, at the best focal plane in the stack of images. To reduce noise, these values were then binomially smoothed (dashed red line), and the derivative of the raw intensity (green solid line) was calculated with respect to distance. A headless protrusion was identified if the derivative function did not cross zero at the y-axis (pixel intensity). In contrast, if the derivative function crossed zero twice at the y-axis (because of higher pixel values at the dendrite shaft and at center of the head compared with the neck), the protrusion was considered to have a head.
All statistical analyses were performed with GraphPad Prism (GraphPad Software), and error bars in graphs represent the SEM, where n = number of dendrites, except in Figures 5, C, E, and F, and 6, C and D, where n = number of spines. In Figures 3, 4, and 6 B, the box-and-whisker plots show the average (+) and the median (horizontal line), and the whiskers represent the 10 and 90 percentile boundaries. To determine statistical significance, we used a one-way ANOVA followed by Tukey's multiple-comparison tests (see Figs. 1 C,D, 2 B–D, 3 B,C, 4 B–D, 5 C,E,F, 6 B,C), a Kolmogorov–Smirnov test (see Fig. 1 E), a two-way ANOVA followed by Bonferroni's test (see Figs. 2 E, 4 E), or a t test (see Fig. 6 D).
Figure 6.

Dysregulated mGluR signaling in neonatal Fmr1 KO mice—mGluR antagonist uncovers other immature dendritic protrusion phenotypes but fails to rescue abnormal TOR. A, Representative images showing WT and KO dendrites (top and bottom, respectively) that were treated chronically with MPEP (30 mg/kg, i.p.). B, Density, length, motility, and turnover (TOR) of protrusions after treatment with MPEP. The mGluR antagonist caused a specific decrease in the density, increase in the average length, and a higher motility of protrusions in KO mice. However, MPEP failed to rescue the abnormal TOR phenotype in KO animals and did not affect baseline TOR levels in WT mice. *p < 0.05, one-way ANOVA with Tukey's test in B and C. C, Relative abundance of individual protrusion subclasses in Fmr1 KO mice with and without MPEP treatment using cutoffs inspired by k-means test for TIB and lifetime (Fig. 5 D). The distribution of different subtypes of protrusions in KO mice treated with MPEP resembled most that of WT protrusions at P7–P8. ***p < 0.001. D, TIB for Fmr1 KO mice at P10–P12 with and without MPEP treatment. The average (dashed line) and SEM (shaded area) values for WT mice at P7–P8 and P10–P12 are shown for comparison. ***p < 0.001, Student's t test.
Figure 3.

Length and density of protrusions develop normally in Fmr1 KO mice. A, High-resolution images of dendritic protrusions at different postnatal ages (best projections; ∼8–12 optical sections, 1 μm apart). Compared with WT mice (gray bars), dendrites in mutant mice (blue circles) appeared to have an abundance of thin protrusions (arrows) and fewer mushroom spines with heads (arrowheads) (for detailed quantification of this phenotype, see also Fig. 5). The asterisks point to large knobby protrusions that are usually seen in WT mice at P7–P8. B, C, Density and length of dendritic protrusions at different postnatal ages. Each blue circle indicates a different dendrite from a KO mouse. Control values from WT mice are indicated by gray box-and-whisker plots that show the average (+) and the median (horizontal line). The whiskers represent the 10 and 90 percentile boundaries. *p < 0.05, one-way ANOVA followed by Tukey's test. There were no significant differences between WT and Fmr1 KO mice.
Figure 4.
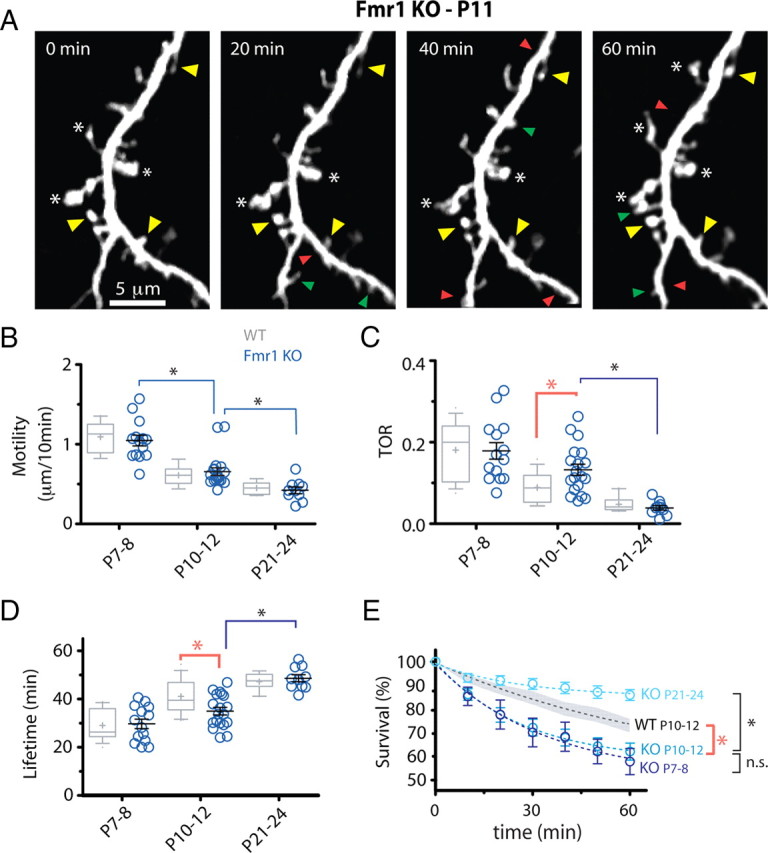
Protrusions in Fmr1 KO mice are abnormally unstable. A, Representative in vivo time-lapse imaging of dendritic protrusions in Fmr1 KO mice at P11. Imaged are best projections (9–11 slices, 1 μm apart). Note the high prevalence of immature knobby protrusions (asterisks) and high turnover. Images were collected every 10 min but only one-half of the time points are shown for simplicity. Added, lost, and stable spines are labeled with green, red, and yellow arrowheads, respectively. B, Motility of dendritic protrusions develops normally in KO mice (blue circles). *p < 0.05, one-way ANOVA followed by Tukey's test for B–D. C, D, TOR and lifetime of dendritic protrusions at different postnatal ages. The developmental change in protrusion turnover and lifetime was delayed in mutant mice compared with WT mice. This transient defect in turnover resulted in an abnormal decrease in protrusion lifetime only in P10–P12 KO mice. E, Survival graphs for dendritic protrusions over a 60 min imaging session at different postnatal ages. The graph for WT protrusions at P10–P12 (average, dashed black line; SEM, gray shadow) is also shown for comparison. Note the developmental delay in protrusion turnover maturation in KO mice, as shown by nearly identical survival curves at P7–P8 and P10–P12 (t 1/2 = 67.9 and 72.6 min, respectively; *p < 0.05, two-way ANOVA with Bonferroni's posttest).
Figure 1.

Developmental regulation of early dendritic protrusion density and length in wild-type mice. A, Low-magnification view of two L2/3 pyramidal cells in the somatosensory cortex of a P10 WT mouse that was sparsely labeled with GFP via in utero electroporation and imaged with two-photon microscopy in vivo. The image is a maximum intensity projection of ∼150 slices (3 μm apart). The inset shows the side view (yz projection) of the same cells. Scale bars, 25 μm. The boxed region in red (shown at higher magnification in B, middle panel) is an example of a dendritic region of interest from the apical tuft that was chosen for time-lapse imaging. B, High-magnification view of representative dendritic branches at the three postnatal ages examined. Images are best projections (∼5–7 optical sections, 1 μm apart). Throughout development, thin protrusions (arrows) are gradually replaced with larger spines (arrowheads), typical of mature dendrites. Note also the presence of long and very bright protrusions (asterisks) at P7–P8. C, Density of dendritic protrusions at different postnatal ages. Each square indicates a different dendrite. The largest increase in protrusion density occurs after P10–P12. *p < 0.05, one-way ANOVA followed by Tukey's multiple-comparison test in C and D. D, Length of dendritic protrusions changes only slightly during postnatal development and remains constant after P10–P12. E, Frequency distribution histogram of average protrusion length at P7–P8 and P21–P24 (*p < 0.05, Kolmogorov–Smirnov test). Very long protrusions (>4 μm) represent 8% of all protrusions at P7–P8 but are absent at P21–P24. The distribution of protrusion lengths at P10–P12 (data not shown) was also significantly different from that of P7–P8 and P21–P24 mice.
Figure 2.
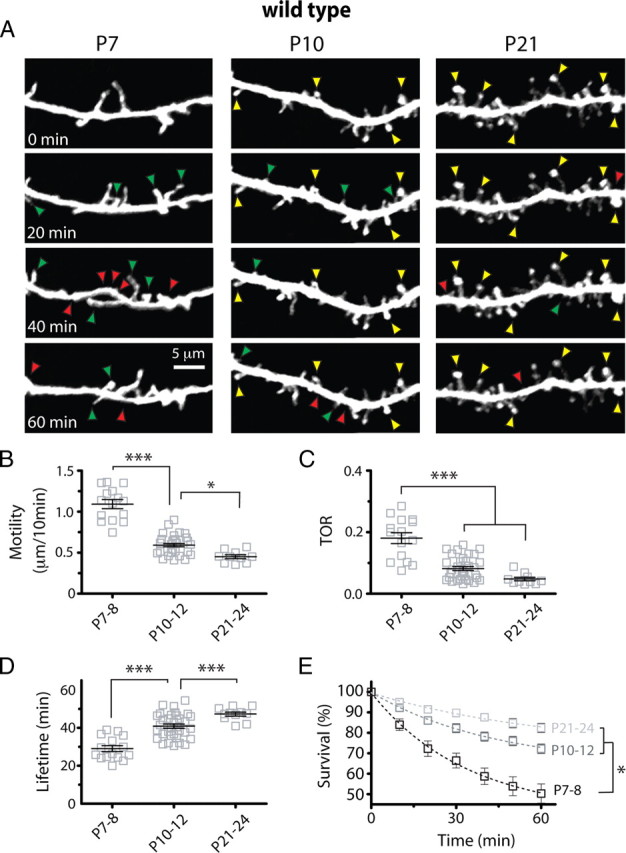
Downregulation of early protrusion dynamics between P7–P8 and P10–P12. A, High-resolution in vivo time-lapse two-photon imaging of dendritic protrusions. Images are best projections (∼6–14 optical sections, 1 μm apart) collected every 10 min (only one-half of the time points are shown for simplicity). Examples of gained (green arrowheads), lost (red arrowheads), and stable (yellow arrowheads) protrusions are indicated. B–D, Motility, TOR, and lifetime of dendritic protrusions at different postnatal ages (***p < 0.001, one-way ANOVA with Tukey's test). Protrusions stabilize quickly between P7–P8 and P10–P12. E, Survival graphs for dendritic protrusions over a 60 min time-lapse imaging session at different postnatal ages (t 1/2 = 53.6, 125.2, and 204.2 min at P7–P8, P10–P12, and P21–P24, respectively; *p < 0.05, two-way ANOVA with Bonferroni's posttest).
Results
Developmental regulation of early dendritic protrusions in vivo
We examined various aspects of protrusion dynamics in pyramidal neurons from WT and KO mice during early cortical development with in vivo two-photon microscopy, in an effort to maintain intact circuits with preserved sensory inputs. We focused on the first postnatal weeks because this time period is characterized by a sharp increase in synapse formation (Micheva and Beaulieu, 1996), a rapid phase of experience-dependent fine-tuning of intracortical circuitry (Simons and Land, 1987; Fox, 1992; Lendvai et al., 2000; Stern et al., 2001; Maravall et al., 2004), and a major shift in network dynamics (Golshani et al., 2009; Rochefort et al., 2009).
L2/3 pyramidal neurons were labeled with GFP using in utero electroporation in mice at E16. This resulted in a sparse labeling of dendrites, which was ideal for high-resolution in vivo two-photon imaging (Fig. 1 A). In a series of control experiments, we characterized the action potential firing and passive membrane properties of GFP-transfected neurons and found them to be indistinguishable from those of untransfected neighboring neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Imaging experiments were started at various postnatal ages to encompass the developmental changes in the length, density, and dynamics of early dendritic protrusions in the apical tufts of L2/3 neurons in mouse somatosensory cortex. The density of protrusions increased nearly threefold throughout postnatal development from 0.27 ± 0.03 protrusions/μm (mean ± SEM) at P7–P8 to 0.99 ± 0.05 protrusions/μm at P21–P24 (p < 0.001, Tukey's multiple-comparison test; n = 15, 22, and 10 dendrites for WT mice at P7–P8, P10–P12, and P21–P24, respectively) (Fig. 1 B,C).
The shape and size of protrusions also changed throughout postnatal development. At the earliest ages imaged (P7–P8), there was an abundance of long and headless protrusions (Fig. 1 B, arrows), as previously described for immature neurons at these ages in vivo (Fiala et al., 1998; Lendvai et al., 2000) and in vitro (Dailey and Smith, 1996; Ziv and Smith, 1996; Portera-Cailliau et al., 2003). After the first week, most protrusions resembled mushroom-like spines (Fig. 1 B, arrowheads) typical of mature dendrites (Bourne and Harris, 2008). However, in comparison with the dramatic changes in protrusion density, the average length of protrusions changed only slightly during postnatal development, decreasing from 2.17 ± 0.13 μm at P7–P8 to 1.65 ± 0.06 μm at P21–P24 (p < 0.001, Tukey's test) (Fig. 1 D). This difference was mostly attributable to the disappearance of extremely long protrusions between the first and fourth postnatal weeks. Indeed, although as many as 7.9% of the earliest protrusions at P7–P8 reached lengths of >4 μm, the length of protrusions never exceeded 4 μm after P21 (Fig. 1 E) (p < 0.01, all age comparisons, Kolmogorov–Smirnov test).
We next examined the developmental regulation of protrusion dynamics in WT mice by imaging dendritic branches of L2/3 neurons in barrel cortex at 10 min intervals. These time-lapse imaging experiments allowed us to quantify protrusion motility, turnover and lifetime (Fig. 2). Early dendritic protrusions were the most motile at P7–P8 (1.09 ± 0.05 μm/10 min) but subsequently stabilized quickly, such that by the end of the third postnatal week motility was reduced by 58% (P21–P24, 0.45 ± 0.02 μm/10 min; p < 0.001 vs P7–P8, Tukey's test) (Fig. 2 A,B).
In addition to being highly motile, protrusions in the youngest mice quickly emerged and disappeared from dendrites (Fig. 2 A,C). Turnover ratio (TOR) (defined as the total number of protrusions gained and/or lost, divided by twice the total number of protrusions) was highest at P7–P8 when, on average, 18% of protrusions were either added or gained every 10 min. TOR then quickly decreased more than threefold to a value of ∼4% per 10 min by P21–P24 (p < 0.001, Tukey's test) (Fig. 2 C). Most of this downregulation in protrusion turnover occurred rapidly between P7–P8 and P10–P12, in contrast to the change in protrusion density, which increased most dramatically between P10–P12 and P21–P24. Similarly, we observed a 62% increase in protrusion lifetime (Fig. 2 D) and a 64% increase in the survival fraction of protrusions from P7–P8 to P21–P24 (Fig. 2 E). This developmental decrease in TOR was associated with proportional decreases in the rates of addition (F gained) and elimination (F lost) of protrusions (p < 0.001 for both, Tukey's test) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). When we extended time-lapse imaging at P21–P24 to 2 h, we found that 94% of protrusions that reached 1 h were still present at the 2 h time point (data not shown). Of note, protrusion turnover for pyramidal neurons in vivo is one order of magnitude lower than in acute brain slices (Portera-Cailliau et al., 2003), which underscores the effects of slicing on protrusion dynamics and synaptogenesis.
Normal length and density of protrusions in FMR1 KO mice in vivo
It has been shown that the abnormalities in length and density of dendritic spines observed in barrel cortex of Fmr1 KO mice are most noticeable in the first postnatal week (Nimchinsky et al., 2001). Unexpectedly, we did not see any significant differences in either the density or length of dendritic protrusions between WT and KO mice (p > 0.05, WT vs KO for all age comparisons, Bonferroni's posttest; N/n = 4/14, 5/19, and 3/10 mice/dendritic branches from KO mice at P7–P8, P10–P12, and P21–P24, respectively) (Fig. 3). Just as WT mice, KO mice also exhibited a gradual increase in protrusion density from 0.24 ± 0.03 protrusions/μm at P7–P8 to 0.89 ± 0.04 protrusions/μm at P21–P24, as well as a steady decrease in protrusion length from 2.31 ± 0.24 μm at P7–P8 to 1.63 ± 0.05 μm at P21–P24 (p < 0.05, Tukey's test) (Fig. 3 B,C).
Protrusions in Fmr1 KO mice are abnormally unstable
It has been suggested that the presence of unusually long and thin dendritic spines (reminiscent of filopodia) in adults with FXS or other forms of autism and mental impairment may be a sign of arrested dendritic development (Comery et al., 1997; Kaufmann and Moser, 2000; Portera Cailliau and Yuste, 2001; Fiala et al., 2002). However, we and others have argued that motility and turnover are better than morphological criteria (e.g., shape, length) at distinguishing immature filopodial protrusions from mature spines (Ziv and Smith, 1996; Portera-Cailliau et al., 2003; Holtmaat and Svoboda, 2009). Therefore, to test the hypothesis that dendritic spines are abnormally immature in FXS, we examined the dynamics of early dendritic protrusions with in vivo imaging in Fmr1 KO (Fig. 4 A; supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
The motility of early dendritic protrusions in KO mice gradually decreased throughout postnatal development (P7–P8, 1.05 ± 0.07 μm/10 min, n = 14; P10–P12, 0.66 ± 0.05 μm/10 min, n = 19; P21–P24, 0.42 ± 0.04 μm/10 min, n = 10), but there were no differences compared with WT mice (p > 0.05, Bonferroni's posttest) (Fig. 4 B).
We also observed a developmental downregulation of protrusion turnover in KO mice, but this was delayed compared with WT mice, as it was only apparent between P10–P12 and P21–P24 (0.13 ± 0.01 over 10 min of imaging at P10–P12 vs 0.04 ± 0.01 at P21–P24; p < 0.001) (Fig. 4 C). In fact, TOR at P10–P12 was 44% higher in KO mice than in WT (0.13 ± 0.01 in KO vs 0.09 ± 0.01 in WT; p < 0.05, Bonferroni's posttest). This higher TOR in KO mice at P10–P12 was attributable to a balanced increase in the addition and elimination of dendritic protrusions compared with WT mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), which may explain why protrusion density was unaffected in KO mice.
In keeping with the TOR result, both the lifetime of early dendritic protrusions (35.0 ± 1.57 min in KO vs 41.1 ± 1.49 min in WT; p < 0.01) (Fig. 4 D) and the survival fraction over 60 min (62.4 ± 2.45% in KO vs 72.4 ± 2.94% in WT; p < 0.05) (Fig. 4 E) were abnormally regulated at P10–P12 in KO mice. The lifetime and survival graphs for KO mice at P7–P8 and P10–P12 were indistinguishable from each other (p > 0.05, Bonferroni's posttest) (Fig. 4 D,E), consistent with the idea that neurons in KO mice failed to downregulate their protrusion dynamics between the first and second postnatal weeks.
Early dendritic protrusions in Fmr1 KO mice do not transition into mature spines
As part of their maturation process, in addition to becoming more stable, early dendritic protrusions undergo significant morphological transformations. Thin filopodia are eventually replaced by mushroom and stubby spines. Several intermediate protrusion types may exist (Irwin et al., 2000) along a so-called filopodium–spine continuum where dynamic parameters (e.g., motility and lifetime) can be used to distinguish between different protrusion intermediates (Portera-Cailliau et al., 2003). In particular, the concept of protospines has been used to describe protrusions that contain characteristics of both immature and mature protrusions: they are long and motile, but they also have longer lifetimes than filopodia because they have established one or more synaptic contacts (Dailey and Smith, 1996; Marrs et al., 2001). Such protospines may contain several varicosities along their length, but lack a single large head that is typical of mushroom spines. In addition, it has been remarked that, in adult mice, thin filopodia-like spines with high turnover tend to be smaller than the more stable mushroom spines (Holtmaat et al., 2005). Thus, size and dynamics may be important independent parameters to distinguish between different types of protrusions throughout development.
First, we compared the size of early dendritic protrusions in WT mice at different postnatal ages to determine whether protrusion size correlates with maturation stage in early postnatal mice. Since GFP brightness is monotonically related to the volume in single spines (Holtmaat et al., 2005), we quantified the fluorescence intensity (TIB) (see Materials and Methods) at different postnatal ages in WT mice (n = 242, 428, and 163 protrusions at P7–P8, P10–P12, and P21–P24, respectively). This analysis revealed a large heterogeneity in protrusion size at P10–P12 (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material), but, unexpectedly, not a developmental increase in protrusion size (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Instead, protrusion brightness was highest at P7–P8 (P7–P8, 44.9 ± 5.1 a.u.; P10–P12, 29.9 ± 1.5 a.u.; P21–P24, 30.2 ± 2.2 a.u.; P7–P8 vs P10–P12, p < 0.001, Tukey's test). We conclude that, at least during early development, size alone is not good predictor of protrusion maturity.
Next, we used an unbiased cluster analysis approach (see Materials and Methods) to systematically segregate all protrusions in WT mice from P7 to P24 (n = 833) into three distinct categories (Fig. 5 A). A priori, we predicted that these three groups would represent the intuitively defined groups of F, PS, and MS. Careful inspection of the cluster analysis indicated that protrusions in one of the clusters (type 2) (Fig. 5 A, red circles) were clearly segregated from the others based on their large size (high TIB values). Type 2 protrusions were very long and had relatively high motility (immature characteristics), but were relatively stable and had unique shapes with multiple swellings (Fig. 5 B,C), and occasionally several branches. Thus, we reasoned that type 2 protrusions fit the characteristics of the PS category. The other two types had similar overall size (TIB, <90 a.u.) but differed in their motility and especially in their lifetime. Type 1 protrusions (Fig. 5 A, green circles) were more stable (in terms of lifetime) and exhibited less motility, whereas the opposite was true for type 3 protrusions (blue circles) (Fig. 5 A–C). Moreover, type 1 protrusions often had large heads typical of mushroom spines, whereas type 3 protrusions looked more like filopodia (Fig. 5 B). We concluded that type 1 and type 3 protrusions corresponded to MS and F categories, respectively.
The relative prevalence of each of these categories was also developmentally regulated in the expected pattern: gradual disappearance of F and PS protrusions and increasing prevalence of the MS type (Fig. 5 E). In addition, the relative abundance of PS (the largest protrusions) decreased from 11.2 to 6.5% between P7–P8 and P10–P12 (p < 0.001, χ2), which explains the drastic developmental decrease in TIB (Fig. 5 C; supplemental Fig. 4B, available at www.jneurosci.org as supplemental material).
To compare the relative frequency of F, PS, and MS in WT and Fmr1 KO mice, we segregated all of the imaged protrusions into the three separate categories using cutoffs determined from the differences in TIB and lifetime that were apparent in the cluster analysis (Fig. 5 D) (see Materials and Methods). We focused the analysis on the P10–P12 time point because this was the age when KO and WT mice differed in the stability of spines. This approach was validated when it revealed a nearly identical pattern of the relative abundance of F, PS, and MS in WT mice at different postnatal ages as the cluster analysis had (Fig. 5 F). Furthermore, in WT mice, MS protrusions had 30% more spine heads than F spines (p < 0.001, Fisher's test) (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material) (see Materials and Methods), suggesting that this classification scheme was correctly identifying widely recognized protrusion subtypes. Importantly, the distribution of protrusion subtypes at P10–P12 in Fmr1 KO mice was different from that of WT mice, because mutants had more F and fewer MS (Fig. 5 F).
Chronic inhibition of mGluR signaling exaggerates spine immaturity in KO mice
Abnormal signaling through group I mGluRs has been proposed as one of the mechanisms underlying symptoms in FXS (Bear et al., 2004). In support of this theory, pharmacological (Chuang et al., 2005; Yan et al., 2005) or genetic (Dölen et al., 2007) manipulation of mGluRs in KO mice reverses many of the morphological, physiological, and behavioral deficits of the mutant mice. We therefore investigated the effects on dendritic spines of the mGluR5-specific inverse agonist MPEP, which acts as a potent noncompetitive mGluR5 antagonist by inhibiting both agonist-induced activation of mGluR5 and its constitutive activity (Carroll et al., 2001). Specifically, we asked whether chronic administration of MPEP (30 mg/kg i.p.) for at least 3 d before in vivo imaging at P10–P12 could correct the abnormal protrusion TOR down to WT levels. A similar strategy proved beneficial in reducing seizures and correcting a behavioral phenotype in Fmr1 KO mice (Yan et al., 2005).
We first asked whether mGluR5 plays any role in regulating dendritic protrusions in WT mice at P10–P12, which is the age when we observed the largest differences in protrusion dynamics between WT and mutant mice. Chronic administration of MPEP in WT mice (n = 12 dendrites) changed neither the density nor the length of early dendritic protrusions (density, WTcontrol, 0.42 ± 0.02 protrusions/μm; WTMPEP, 0.47 ± 0.04 protrusions/μm; p > 0.05; length, WTcontrol, 1.80 ± 0.07 μm; WTMPEP, 1.73 ± 0.09 μm; p > 0.05, Tukey's test) (Fig. 6 A,B). This was not surprising because WT mice with a 50% reduction in mGluR5 have normal spine density (Dölen et al., 2007). Furthermore, neither the motility nor the turnover of protrusions was affected by chronic MPEP administration (motility, WTcontrol, 0.61 ± 0.03 μm/10 min; WTMPEP, 0.56 ± 0.03 μm/10 min; p > 0.05; TOR, WTcontrol, 0.09 ± 0.09; WTMPEP, 0.07 ± 0.01; p > 0.05, Tukey's test) (Fig. 6 C,D). In agreement with these results, the lifetime of WT protrusions was also unaffected by MPEP treatment (WTcontrol, 41.1 ± 1.5 min; WTMPEP, 40.6 ± 1.7 min; p > 0.05, Tukey's test) (data not shown).
We also examined the effects of chronic MPEP administration on protrusion dynamics in KO mice (n = 21 dendrites). First, we found that the significant differences in TOR and lifetime between KO and WT mice were also apparent in these additional experiments in which both sets of animals were treated with MPEP (Fig. 6 B), confirming our previous findings (Fig. 4). Importantly, this also means that MPEP failed to rescue the abnormal TOR phenotype in KO animals (KOcontrol, 0.13 ± 0.01; KOMPEP, 0.13 ± 0.01; p > 0.05, Tukey's test) (Fig. 6 D). Similarly, the abnormally short protrusion lifetime in KO mice was not corrected by MPEP (KOcontrol, 35.0 ± 1.6 min; n = 19; KOMPEP, 35.1 ± 1.7 min; p > 0.05, Tukey's test) (data not shown).
In contrast, we observed a KO-specific 33% increase in protrusion length by MPEP (KOcontrol, 1.60 ± 0.08; n = 19 dendrites; KOMPEP, 2.12 ± 0.15; p < 0.01, Tukey's test) (Fig. 6 B). Interestingly, the length of protrusions in MPEP-treated KO mice at P10–P12 was similar to the length of protrusions in KO or WT mice at P7–P8 (p > 0.05, Tukey's test), suggesting that mGluR signaling in mutant mice may mask the abnormally immature length of protrusions.
Two additional differences between WT and KO mice appeared at P10–P12 after treatment with MPEP. Namely, we observed a 36% lower protrusion density in treated KO mice (WTMPEP, 0.47 ± 0.04 protrusions/μm; KOMPEP, 0.30 ± 0.02 protrusions/μm; p < 0.05, Tukey's test) (Fig. 6 B) and a 32% increase in motility (WTMPEP, 0.56 ± 0.03 μm/10 min; KOMPEP, 0.74 ± 0.03 μm/10 min; p < 0.05, Tukey's test) (Fig. 6 B). These differences were brought out, not by changes triggered by MPEP in WT mice, but rather by MPEP-specific effects on KO mice. This was most obvious in the case of protrusion density, which was 23% lower in MPEP-treated KO mice than in control KO mice (KOcontrol, 0.39 ± 0.04/μm; KOMPEP, 0.30 ± 0.02/μm; p = 0.06, Tukey's test) (Fig. 6 A,B); the trend was not significant for motility (p = 0.11). The values for density and motility in MPEP-treated KO mice again corresponded approximately to the density and motility values for WT and KO mice at P7–P8, suggesting that blockade of mGluR signaling reverts protrusions in KO mice to an even more immature state.
When we segregated protrusions in MPEP-treated KO mice according to the cutoffs for TIB and lifetime derived from the cluster analysis (Fig. 5 D), we found an increase in the fraction of filopodia and protospines, as well as a concomitant decrease in the fraction of mature spines compared with their P10–P12 counterparts (Fig. 6 C). Indeed, the relative abundance of each subtype matched very closely that of protrusions in WT mice at P7–P8. Furthermore, the size of protrusions (TIB) at P10–P12 was much higher in Fmr1 KO mice after chronic MPEP treatment and resembled that of WT protrusions at P7–P8 (P10–P12, KOcontrol, 27.0 ± 1.91 a.u.; n = 534; vs KOMPEP, 45.7 ± 3.70 a.u.; n = 191; p < 0.001, t test) (Fig. 6 D). Thus, MPEP treatment accentuated the immature appearance of dendritic protrusions in Fmr1 KO mice but did not change their abnormally high turnover.
Discussion
Developmental delay in the maturation of dendritic protrusions in Fmr1 KO: an early synaptic phenotype in FXS
FXS is a developmental disorder in which symptoms of autism and intellectual dysfunction manifest in early childhood. Pathologically, the salient defect in adults with this disorder is an abnormally high density of dendritic spines that are also unusually long and therefore appear somewhat immature. Surprisingly, little is known about the development of dendritic protrusions in early postnatal KO mice, during the time when dendritic protrusions transition from immature filopodia to mature spines.
We characterized the maturation of dendritic protrusions in the intact somatosensory cortex of WT and Fmr1 KO mice during a critical period in early postnatal development of experience-dependent plasticity and rapid synaptogenesis. Because spine turnover is intimately linked to changes in sensory experience (Holtmaat and Svoboda, 2009), we examined the maturation of dendritic protrusions in the intact neocortex of living Fmr1 KO mice with in vivo imaging.
We find that protrusions of L2/3 neurons in KO mice exhibit an abnormally high turnover compared with those in WT mice at P10–P12 and that the mutant mice have an overabundance of immature protrusion subtypes (filopodia). This is the most compelling evidence of a developmental delay in the maturation of dendritic protrusions in FXS. It is possible that similar defects in spine turnover occur even earlier in L5 pyramidal neurons, because they mature before L2/3 neurons. We conclude that FMRP plays a crucial role in cortical circuit assembly and function by regulating protrusion turnover to stabilize dendritic spines during early postnatal development. The bulk of the developmental decrease in protrusion turnover in WT mice occurs rapidly between P7–P8 and P10–P12. In contrast, the increase in protrusion density occurs later, between P10–P12 and P21–P24. Thus, the mechanisms regulating spine turnover versus density are probably distinct, and FMRP seems to play a more important role in regulating protrusion turnover than in promoting spinogenesis.
Our study failed to detect any differences in the length or density of dendritic protrusions between WT and KO mice during early postnatal development. This is in contrast with a previous study in fixed tissue that reported an increase in the length and density of dendritic spines in L5 pyramidal neurons from barrel cortex of KO mice during the first postnatal weeks (Nimchinsky et al., 2001). This apparent discrepancy could be attributable to differences in the neuronal subtype examined (L2/3 vs L5 pyramidal neurons) or to the type of preparation (in vivo vs fixed tissue). Indeed, the previous developmental study did not detect differences in protrusion density or length in living pyramidal neurons in cultured slices (Nimchinsky et al., 2001). It should also be noted that spine alterations, although real in FXS and the mouse model, are subject to variability based on several parameters, including the brain region and cell type being examined, or the genetic background and age of the mouse.
Implications for synaptogenesis, synaptic strength, and circuit plasticity
What are the implications of abnormal protrusion maturation in FXS? The appearance and disappearance of dendritic protrusions is thought to enhance the ability of the dendrite to sample the extracellular environment in search for suitable presynaptic terminals to establish early synapses (Ziv and Smith, 1996; Bonhoeffer and Yuste, 2002). Indeed, dendritic filopodia can make transient contacts with axons (Ziv and Smith, 1996; Lohmann and Bonhoeffer, 2008) and form immature synapses (Fiala et al., 1998). Furthermore, the developmental downregulation of filopodial dynamics correlates with the end of synapse formation (Ziv and Smith, 1996), and perturbations in protrusion dynamics result in altered synaptogenesis (Kayser et al., 2008; Luikart et al., 2008). Therefore, the failure of spines to stabilize during the critical period strongly suggests that protrusions in Fmr1 KO mice might have problems in maintaining the proper balance between stable and dynamic connections that is necessary to establish mature synapses. This is certainly the case in vitro, where the absence of FMRP induces synapse loss (Antar et al., 2006; Pfeiffer and Huber, 2007).
Spine dynamics are also important for synaptic and experience-dependent plasticity (Lendvai et al., 2000; Yuste and Bonhoeffer, 2001; Holtmaat et al., 2006). Interestingly, the transient defect of reduced L4→L3 experience-dependent synaptic plasticity in the mutant mice (Bureau et al., 2008) fits well with the timing of the protrusion defects we describe (both take place in the second postnatal week) and the two could be linked.
We also observed a relative paucity of mature spines in KO mice (Fig. 5 F). Previous work has established a strong correlation between protrusion size and synaptic strength (Matsuzaki et al., 2001; Zito et al., 2009), and modulating synaptic strength results in changes in spine volume (Lang et al., 2004; Matsuzaki et al., 2004; Zhou et al., 2004; Kopec et al., 2006). Thus, the overabundance of small filopodia protrusions in KO mice implies that synapses, on average, could be weaker in KO mice, at least during the first 2 postnatal weeks. Since thin spines with high turnover often lack synapses when they first appear (Knott and Holtmaat, 2008), this also could explain the reduced numbers of synapses on spines seen in KO mice (Antar et al., 2006).
Overall, our findings of abnormal protrusion maturation could explain a variety of circuit defects in the mutant mice, including deficits in neocortical long-term potentiation (LTP) (Li et al., 2002; Zhao et al., 2005; Wilson and Cox, 2007) and spike timing-dependent plasticity (STDP) (Desai et al., 2006; Meredith et al., 2007). For example, the immature dendritic protrusions in KO mice may not express the L-type calcium channels or proper synaptic machinery necessary for STDP (Meredith et al., 2007). Interestingly, increasing the number of action potentials paired with EPSPs during LTP induction can increase postsynaptic calcium entry into spines and restore plasticity in KO mice, suggesting that the mechanisms mediating LTP are not absent in mutant mice, but rather that a higher threshold is required for induction (Meredith et al., 2007).
Regulation of early dendritic protrusions in Fmr1 KO mice by mGluRs
Pharmacological dampening of mGluR signaling did not reverse the abnormal spine turnover in Fmr1 KO mice. In contrast, three new abnormal protrusion phenotypes appeared in mutant mice with MPEP treatment: an increase in length, a lower density, and a higher motility of protrusions. In addition, MPEP triggered an increase in the fraction of filopodia and protospines (protrusions of MPEP-treated KO mice at P10–P12 resembled those in untreated WT and KO mice at P7–P8). Thus, blocking mGluRs in mutant mice results in a greater degree of spine immaturity.
This result was unexpected, because reducing mGluR5 expression or signaling in KO mice can rescue many other abnormalities, including their high spine density (Dölen et al., 2007). There are several possible explanations for this discrepancy. First, the regulation of protrusion density and dynamics may involve different signaling pathways. Second, it is possible that mGluRs have different effects on spines in mature versus developing neurons. Overall, we interpret our results to mean that excessive signaling through mGluRs in FXS may mask some aspects of the relative immaturity of protrusions triggered by the absence of FMRP. If so, this mechanism might correct or compensate for inherent problems in motility, length, and density, but perhaps not turnover (which appears to involve mGluR-independent mechanisms). Still, our data are consistent with the idea that mGluRs are dysregulated in FXS, as proposed previously (Bear et al., 2004). Indeed, many other defects in Fmr1 KO mice, such as their propensity to seizures (Yan et al., 2005), or their high level of AMPA receptor (AMPA-R) internalization (Nakamoto et al., 2007), are partially or completely rescued by MPEP. Additionally, although we did not observe a difference in spine density between WT and KO mice, spine density was reduced by MPEP in Fmr1 KO preferentially. A similar KO-specific effect of MPEP has previously been reported in dissociated hippocampal neurons, but only on filopodia (de Vrij et al., 2008), which is also consistent with the mGluR theory.
Interestingly, brain extracts from Fmr1 KO show a selective reduction in mGluR5 content in synaptic plasma membranes, compared with WT mice (Giuffrida et al., 2005). Thus, excessive mGluR-dependent signaling during development appears to trigger a decrease in the density of mGluRs, perhaps through a feedback mechanism (e.g., receptor desensitization and internalization). This reduction in the density of mGluRs in mutant animals might explain the MPEP-specific effects we observed in Fmr1 KO mice.
Our finding of longer protrusions in KO mice after MPEP administration may be surprising because acute stimulation of mGluRs in vitro has been shown to elongate dendritic spines (Vanderklish and Edelman, 2002; Abu-Elneel et al., 2008). Differences in the experimental paradigm (in vitro vs in vivo) or in how mGluR signaling was modulated (acutely vs chronically) could explain the apparent discrepancy.
Role of FMRP in promoting spine stabilization
Our data are consistent with the notion that FMRP promotes spine maturation and stabilization. One way that FMRP could regulate synaptic stabilization is through delivery and/or degradation of specific mRNAs for proteins that are known to regulate dendritic spines in response to synaptic activity (Bagni and Greenough, 2005; Bassell and Warren, 2008). For example, FMRP binds to and regulates the stability of PSD-95 mRNA, which is also synaptically localized (Zalfa et al., 2007). Furthermore, in response to mGluR activation, cortex-derived synaptoneurosomes from Fmr1 KO mice show deficient synthesis of PSD-95 and αCaMKII proteins (Muddashetty et al., 2007), which are normally enriched in dendritic spines and are implicated in learning and memory through their effects on synaptic plasticity (Silva et al., 1992; Migaud et al., 1998). Because PSD-95 can regulate the cell surface expression of AMPA-R (Colledge et al., 2003), FMRP can directly or indirectly regulate AMPA-R trafficking during mGluR-dependent plasticity, which will affect synaptic strength (Ronesi and Huber, 2008). During development, the absence of FMRP may critically impair synaptogenesis, because the resulting low levels of PSD-95 and AMPA-R (or other critical proteins) in spines might impair their ability to select synaptic partners and subsequently stabilize. Indeed, our observations of delayed spine stabilization in Fmr1 KO mice presumably reflect this postsynaptic defect in protein translation and trafficking.
However, because FMRP has also been localized to axons and their growth cones (Antar et al., 2006), loss of FMRP could also conceivably affect the presynaptic compartment, especially during development. For example, FMRP might be involved in the sculpting of axonal arbors, as it facilitates axon pruning in Drosophila (Tessier and Broadie, 2008). This might explain why the L4→L3 cortical projection in barrel cortex is more diffuse in Fmr1 KO mice (Bureau et al., 2008). In turn, this could be explained in part by the observation that growth cones from Fmr1 KO mice are much less dynamic than in WT controls (Antar et al., 2006). More relevant to our study, one also wonders whether defects in axon development in Fmr1 KO mice might explain the immature dendritic protrusion phenotype that we observed. For instance, it is possible that during normal development axons contacting dynamic dendritic protrusions trigger FMRP-mediated signaling cascades necessary for spine stabilization (including synthesis of PSD-95, CaMKII, or catenins), and this critical balance in bidirectional signaling between presynaptic and postsynaptic elements might be missing in FXS. All of these are interesting speculations that should be tested in the coming years.
Concluding remarks: stabilizing spines as a therapeutic tool in FXS
The delay in spine maturation that we report is the earliest cortical deficit in Fmr1 KO mice and suggests that FMRP is essential in regulating spine dynamics and synapse formation. It is conceivable that intellectual deficits or autistic traits in individuals with FXS could be attributable to defects in experience-dependent wiring of the cortex triggered by abnormal spine dynamics. If so, it would be interesting to investigate whether strategies to restore normal spine turnover might correct these symptoms in FXS if implemented early in development. We find that blocking mGluR signaling in young animals does not correct the instability of spines and in fact appears to worsen the immature protrusion phenotype. But several other signaling pathways have been identified as regulators of spine size and/or turnover, including cadherin/catenins (Togashi et al., 2002; Abe et al., 2004; Abu-Elneel et al., 2008). Pharmacologic or genetic approaches aimed at these pathways in the mutant mice at early postnatal ages, before more serious and/or irreversible circuit deficits emerge, might be of therapeutic interest in FXS if they could reverse the maturational defect in dendritic protrusions.
Footnotes
This work was supported by National Institute of Child Health and Human Development–National Institutes of Health (NIH) Grant 5R01 HD054453 (C.P.-C.) and a Diversity Supplement (A.C.-M.), the University of California President's Postdoctoral Fellowship Program (A.C.-M.), Minority Access to Research Careers Undergraduate Student Training in Academic Research NIH–National Institute of General Medical Sciences (M.C.), and the FRAXA and Dana Foundations, as well as by the generous support of The Fu-Hsing and Jyu-Yuan Chen Family Foundation. We thank Tara Chowdhury and James Anstey for maintaining the Fmr1 KO colony and genotyping the mice, Adrian Cheng for help with MATLAB, Mike Tranfaglia (from FRAXA) for providing MPEP, Karel Svoboda and Tim O'Connor for the spine analysis software, and William Greenough for providing the Fmr1 KO mice. We are grateful to Felix Schweizer, Peyman Golshani, and members of the Portera-Cailliau Laboratory, especially Ricardo Mostany, for helpful discussions and feedback on this manuscript.
References
- Abe K, Chisaka O, Van Roy F, Takeichi M. Stability of dendritic spines and synaptic contacts is controlled by alpha N-catenin. Nat Neurosci. 2004;7:357–363. doi: 10.1038/nn1212. [DOI] [PubMed] [Google Scholar]
- Abu-Elneel K, Ochiishi T, Medina M, Remedi M, Gastaldi L, Caceres A, Kosik KS. A delta-catenin signaling pathway leading to dendritic protrusions. J Biol Chem. 2008;283:32781–32791. doi: 10.1074/jbc.M804688200. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP traficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Müller T, Pin JP, Prézeau L. BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol. 2001;59:965–973. [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Bagni C. Fragile X mental retardation protein control of neuronal mRNA metabolism: insights into mRNA stability. Mol Cell Neurosci. 2010;43:43–50. doi: 10.1016/j.mcn.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Desai NS, Casimiro TM, Gruber SM, Vanderklish PW. Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol. 2006;96:1734–1745. doi: 10.1152/jn.00221.2006. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fox K. A critical period for experience-dependent synaptic plasticity in rat barrel cortex. J Neurosci. 1992;12:1826–1838. doi: 10.1523/JNEUROSCI.12-05-01826.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16:666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida R, Musumeci S, D'Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci. 2005;25:8908–8916. doi: 10.1523/JNEUROSCI.0932-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golshani P, Gonçalves JT, Khoshkhoo S, Mostany R, Smirnakis S, Portera-Cailliau C. Internally mediated developmental desynchronization of neocortical network activity. J Neurosci. 2009;29:10890–10899. doi: 10.1523/JNEUROSCI.2012-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69. doi: 10.1016/j.neuron.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G, Holtmaat A. Dendritic spine plasticity—current understanding from in vivo studies. Brain Res Rev. 2008;58:282–289. doi: 10.1016/j.brainresrev.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Bonhoeffer T. A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron. 2008;59:253–260. doi: 10.1016/j.neuron.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Zhang W, Wayman GA, Kwon CH, Westbrook GL, Parada LF. Neurotrophin-dependent dendritic filopodial motility: a convergence on PI3K signaling. J Neurosci. 2008;28:7006–7012. doi: 10.1523/JNEUROSCI.0195-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Stern EA, Svoboda K. Development of intrinsic properties and excitability of layer 2/3 pyramidal neurons during a critical period for sensory maps in rat barrel cortex. J Neurophysiol. 2004;92:144–156. doi: 10.1152/jn.00598.2003. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44:625–629. doi: 10.1016/0006-8993(72)90324-1. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci. 2001;4:1006–1013. doi: 10.1038/nn717. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD. Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron. 2007;54:627–638. doi: 10.1016/j.neuron.2007.04.028. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. A craniotomy surgery procedure for chronic brain imaging. J Vis Exp. 2008;12:pii, 680. doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci. 2007;27:3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera Cailliau C, Yuste R. On the function of dendritic filopodia (in Spanish) Rev Neurol. 2001;33:1158–1166. [PubMed] [Google Scholar]
- Portera-Cailliau C, Pan DT, Yuste R. Activity-regulated dynamic behavior of early dendritic protrusions: evidence for different types of dendritic filopodia. J Neurosci. 2003;23:7129–7142. doi: 10.1523/JNEUROSCI.23-18-07129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Weimer RM, De Paola V, Caroni P, Svoboda K. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 2005;3:e272. doi: 10.1371/journal.pbio.0030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochefort NL, Garaschuk O, Milos RI, Narushima M, Marandi N, Pichler B, Kovalchuk Y, Konnerth A. Sparsification of neuronal activity in the visual cortex at eye-opening. Proc Natl Acad Sci U S A. 2009;106:15049–15054. doi: 10.1073/pnas.0907660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Metabotropic glutamate receptors and fragile X mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 2008;1:pe6. doi: 10.1126/stke.15pe6. [DOI] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67:289–295. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Land PW. Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature. 1987;326:694–697. doi: 10.1038/326694a0. [DOI] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K. Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron. 2001;31:305–315. doi: 10.1016/s0896-6273(01)00360-9. [DOI] [PubMed] [Google Scholar]
- Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008;135:1547–1557. doi: 10.1242/dev.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1639–1644. doi: 10.1073/pnas.032681099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A. 2007;104:2454–2459. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10:578–587. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]