

Abstract
The modular nature of nicotinamide adenine dinucleotide (NAD)-mimicking inosine monophsophate dehydrogenase (IMPDH) inhibitors has prompted us to investigate novel mycophenolic adenine dinucleotides (MAD) in which 1,2,3-triazole linkers were incorporated as isosteric replacements of the pyrophosphate linker. Synthesis and evaluation of these inhibitors led to identification of low nanomolar inhibitors of human IMPDH and more importantly the first potent inhibitor of IMPDH from Mycobacterium tuberculosis (mtIMPDH). Computational studies of these IMPDH enzymes helped rationalize the observed structure–activity relationships. Additionally, the first cloning, expression, purification and characterization of mtIMPDH is reported.
Keywords: Nicotinamide adenine dinucleotide; Mycophenolic adenine dinucleotide; Inosine monophosphate dehydrogenase; Copper-mediated azide–alkyne 1,3-dipolar cyclization (CuAAC, “click reaction”); Mycobacterium tuberculosis
Introduction
Nicotinamide adenine dinucleotide (NAD)-dependent inosine monophosphate dehydrogense (IMPDH), which converts inosine monophosphate (IMP) into xanthosine monophosphate (XMP), is a key enzyme in the de novo synthesis of guanine nucleotides. Inhibition of IMPDH depletes the supply of guanine nucleotides that are required for the growth and proliferation of cells and constitutes a powerful strategy for the treatment of cancers and autoimmune diseases, as well as viral-, protozoal-, and bacterial infections.1 There are two human IMPDH isoforms, hIMPDH1 and hIMPDH2, with high (84%) sequence similarity. The hIMPDH1, long considered as a house-keeping enzyme, is involved in diverse biological processes, such as angiogenesis2, translation regulation3 and DNA-binding.4 By contrast, the hIMPDH2 is up-regulated in cancer cells representing an attractive anti-cancer target.5, 6 IMPDH from non-human species, such as parasites and microbes, has received growing attention as potential targets for antimicrobial drug discovery. For instance, potent inhibitors of Cryptosporidium parvum IMPDH have been recently discovered through high-throughput screening and selected hits have been subjected to structure-activity analysis.7, 8 IMPDH from Mycobacterium tuberculosis (Mtb) has been proposed as a potential target to combat tuberculosis (TB).9-11 The World Health Organization (WHO) has estimated that one-third of the world’s population, nearly 2 billion people, is infected with TB. The emergence of multidrug and extensively drug resistant TB strains represents a serious and unsolved public health problem that requires the identification of drugs ideally with new mechanisms of action.
The enzymatic mechanism of hIMPDH has been extensively studied and is illustrated in Scheme 1.1, 12 After binding of the substrates IMP and NAD, a covalent thioimidate enzyme-substrate adduct with IMP (E–IMP) is formed with concomitant production of NADH. After dissociation of NADH, the thioimidate complex (E–IMP) is hydrolyzed to afford the product XMP. Both the substrate (IMP) binding site and the cofactor (NAD) binding domain have been targeted for the rational development of IMPDH inhibitors. Inhibitors that target the NAD binding site can interact with the three subsites of the cofactor binding domain; the nicotinamide binding subsite (N-subsite), the adenosine binding subsite (A-subsite), and the pyrophosphate binding subsite (P-subsite). Mycophenolic acid (1, MPA, Figure 1) is a clinically used IMPDH inhibitor that binds to the N-subsite as well as a portion of the P-subsite. Tiazofurin (2, TR) is another well-known IMPDH inhibitor that is bioactivated into the corresponding tiazofurin adenine dinucleotide (TAD) and interacts with all three subsites of the NAD cofactor binding domain.
Scheme 1.

RP - ribofuranosyl-5′-monophosphate, IMP - inosine monophosphate, Enz - enzyme, E·IMP-IMP/IMPDH adduct, E-XMP* - IMPDH/XMP thioimidate intermediate, XMP - xanthosine monophosphate
Figure 1.

IMPDH inhibitors.
We have previously described a series of mycophenolic adenine dinucleotide (MAD) as potent IMPDH inhibitors such as C2-MAD and its analogues (3a–c, Figure 2), and C4-MAD (4, Figure 2) that contain methylenebis(phosphonates) as isosteres of the metabolically labile pyrophosphate moiety of NAD.13-15 These modular inhibitors interact with all three of the N-, A-, and P-subsites. In efforts to replace the negatively charged methylenebis(phosphonate) linkers in these compounds, we recently reported the synthesis of analogues containing a non-ionic methylenebis(sulfonamide) linker (5a–d, Figure 3).16, 17 Significantly, the methylenebis(sulfonamide) MAD analogues 5a–d showed potency against hIMPDH comparable to that of compound 3a demonstrating the remarkable promiscuity of the P-subsite. This finding prompted us to explore 1,2,3-triazole as a non-ionic isosteric replacement of the methylenebis(phosphonate) linker in compound 3a, and we predicted that these triazole linkers would maintain the overall geometric positioning of the mycophenolic and adenosine moieties in their respective N- and A-subsites within IMPDH.
Figure 2.

Compounds 3a, 4 and methylenebis(phosphonate) analogues.
Figure 3.

Mycophenolic adenine methylenebis(sulfonamide)s.
Herein we report the design and synthesis of a novel series of triazole-linked mycophenolic adenine inhibitors as NAD mimics. These inhibitors were biochemically evaluated against both human IMPDH isoforms as well as the Mycobacterium tuberculosis IMPDH. Additionally, we describe the first cloning, expression, purification, and characterization of M. tuberculosis IMPDH (mtIMPDH). Molecular docking of these compounds into the reported crystal structures of hIMPDHs and homology model of mtIMPDH was used to rationalize the observed structure–activity relationships (SAR).
Results and discussion
Design and synthesis
We designed a series of novel triazole-linked IMPDH inhibitors (Figure 4) containing aminomethyl-1,2,3-triazole or oxymethyl-1,2,3-triazole moieties, which serve as a conformationally constrained isosteric replacement of the pyrophosphate linker (Figure 4). As shown in Figure 4, compound 6 could be considered a truncated MPA derivative wherein a two-carbon side chain was attached to adenosine via an aminomethyl-1,2,3-triazole linker. In compound 7 a four-carbon side chain was incorporated, a design based on our previous observation that compound 4 showed inhibitory activity similar to that of compound 3a. In compounds 8 and 9, an oxymethyl-1,2,3-triazole linker was used, with an ether linkage at the 5′ position of adenosine.
Figure 4.

Triazole-linked MAD analogues.
Our modeling studies (vide infra) based on the crystal structure of compound 3a/hIMPDH2 complex indicated that compound 3a analogues containing a rigid 1,2,3-triazole ring were accommodated in the P-subsite of IMPDH. In addition, the 1,4-substitution pattern of the triazole ring projects the truncated MPA subunit and adenosine motifs into their respective N- and A-subsites. Furthermore, a 1,2,3-triazole linker can be prepared through a copper-(I)-catalyzed azide–alkyne cycloaddition (CuAAC) coupling reaction, which involves a highly efficient and chemoselective coupling between an azide and an alkyne allowing preparation of the requisite triazoles in a highly convergent fashion.8, 18-20 This approach would enable us to expeditiously probe the factors that influence the potency and isoform selectivity of IMPDH inhibitors that target human enzymes as well as those from other species.
Preparation of compound 6 began with aldehyde 1017 wherein the phenol was protected as a 2-(trimethylsilyl)ethoxymethyl (SEM) ether. Reductive amination of propargylamine and aldehyde 10 afforded alkyne 11 (Scheme 2). CuAAC coupling reaction between alkyne 11 and 5′-azido-5′-deoxy-2′,3′-O-isopropylidene adenosine 1221 yielded protected triazole 13. Removal of the SEM and isopropylidene protecting groups was accomplished with 80% aqueous TFA to provide target compound 6. Compound 7 was prepared from aldehyde 1414 that was first protected under phase-transfer catalysis conditions22 to give protected aldehyde 15 with a side chain longer that of aldehyde 10. In analogy to the preparation of compound 6, homologated aldehyde 15 was further elaborated to give compound 7 (Scheme 3).
Scheme 2.

a
a Reagents and conditions: (a) propargylamine, NaBH(OAc)3, HOAc, dichloroethane, 86%; (b) CuSO4·5H2O, sodium ascorbate, tBuOH/H2O (1:1), 77%; (c) 80% (v/v) TFA, H2O, 86%.
Scheme 3.

a
a Reagents and conditions: (a) SEMCl, Adogen 464, NaOH, CH2Cl2/H2O (1:1), 88%; (b) propargylamine, NaBH(OAc)3, HOAc, dichloroethane, 65%; (c) CuSO4·5H2O, sodium ascorbate, tBuOH/H2O (1:1), 64%; (d) 80% (v/v) TFA, H2O, 80%.
For the synthesis of compound 8, aldehyde 10 was converted to azide 19 through a three-step process involving reduction to alcohol 18 followed by mesylation and subsequent nucleophilic displacement with sodium azide. The alkyne coupling partner was prepared by debenzoylation of N-benzoyl-2′,3′-O-isopropylidene-5′-O-propargyl adenosine (20), which was prepared as previously described.23 CuAAC reaction between azide 19 and 2′,3′-O-isopropylidene-5′-O-propargyl adenosine (21) catalyzed by Cu (I) in a mixture of tert-butyl alcohol and water afforded protected triazole 22 (Scheme 4). Removal of the SEM and isopropylidene protecting groups with aqueous TFA furnished compound 8.
Scheme 4.

a
a Reagents and conditions: (a) i. MsCl, Et3N, THF, 0 °C; ii. NaN3, DMF, 80 °C, 37% for 2 steps; (b) NH, MeOH, 92%; (c) CuSO4·5H2O, sodium ascorbate, tBuOH/H2O (1:1), 86%; (d) 80% (v/v) TFA, H2O, 48%.
As depicted in Scheme 5, compound 9 was prepared in an analogous fashion from azide 24 and alkyne 21. The azide coupling partner was prepared from aldehyde 15 by sequential reduction to alcohol 23, mesylation, and azidation to afford 24. The resulting azide 24 comprised an approximately 6:3:1 mixture of allylic azides in chloroform as judged by 1H NMR. Careful examination of the spectrum revealed the existence of two terminal azides which accounted for 60% and 10% of the mixture, respectively, presumably favoring azide 24 with the Z geometry of the double bond. This observation indicated that facile [3,3]-sigmatropic rearrangement24-26 of allylic azide 24 not only shifted the position of azido group but also compromised the geometry of the double bond, resulting in a mixture of allylic azides in rapid equilibrium. Azide 24 and its allylic isomers were subjected to a CuAAC reaction with alkyne 21 to provide a mixture of isomeric triazoles 25, which could not be separated. After deprotection with aqueous TFA, the desired compound 9 was successfully purified as a single isomer by trituration with hot methanol. Since the geometry of the trisubstituted Z-olefin in the precursor azide 24 had been compromised by the allylic rearrangement, we performed nuclear overhauser effect (NOE) experiments on the final compound 9. Selective NOE interactions are shown in Figure 6. The allylic methyl protons showed a correlation with protons attached to methylene carbon a while a correlation was observed between the vinylic proton and protons on methylene carbon b, confirming the Z geometry of the double bond as shown in compound 9.
Scheme 5.

a
a Reagents and conditions: (a) NaBH4, CeCl3·7H2O, MeOH, H2O, 57%; (b) i. MsCl, Et3N, THF, 0 °C; ii. NaN3, DMF, 60 °C, 85% for 2 steps; (c) CuSO4·5H2O, sodium ascorbate, tBuOH/H2O (1:1); (d) 80% (v/v) TFA, H2O, 5% for 2 steps.
Figure 6.
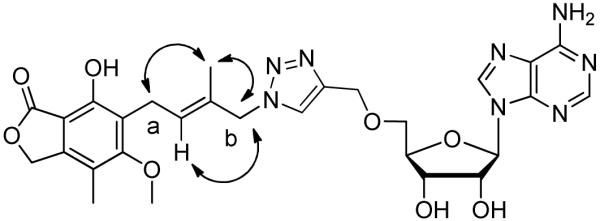
NOE correlations observed for compound 9.
Cloning, expression and purification of mtIMPDH, hIMPDH1, and hIMPDH2
The three IMPDH paralogs guaB1, guaB2, and guaB3 were annotated in the genome of Mycobacterium tuberculosis by Cole and co-workers.9 The guaB2 was identified as an essential gene by Himar1-based transposon mutagenesis in M. tuberculosis H37Rv whereas guaB1 was identified as non-essential and guaB3 was not studied.10 The guaB2 gene was amplified by PCR from M. tuberculosis H37Rv genomic DNA then cloned into pET28b to generate a N-terminal (His)6-tagged fusion protein and overexpressed, and purified as described in the Experimental Section.
Apparent steady-state kinetic parameters of mtIMPDH were determined for both NAD and IMP (Table 1). Because of the reduced rates observed at high NAD concentrations the kinetic data were fit to a substrate-inhibited model to provide a KM and kcat of 1005 μM and 0.53 s−1 for NAD at saturating concentrations of IMP. The substrate inhibition constant, Ki, was 5.0 mM for NAD, which is 5-fold higher than the KM value. By comparison, the Ki of NAD with respect to hIMPDH2 was reported as 590 μM, which is approximately 100-fold higher than the respective KM value for NAD.27 The KM of 78 μM for IMP was determined at subsaturating concentrations of NAD, due to substrate inhibition by NAD and the kinetic data were fit to the Michaelis-Menten model. The kcat for mtIMPDH is similar to the values for hIMPDH1 and hIMPDH2. However, the KM (IMP) for mtIMPDH is 4 to 20-fold higher than the human enzymes while the KM(NAD) for mtIMPDH is 14 and 168-fold higher than hIMPDH1 and hIMPDH2 respectively.
Table 1.
Michaelis–Menten Parameters for M. tuberculosis and Human Type I and II IMPDH
In order to simplify purification of hIMPDH1 and hIMPDH2, the corresponding plasmids pH1 and pHIA5 (generously provided by Prof. Liz Hedstrom) were subcloned into pRK793 to generate expression constructs containing TEV-cleavable N-terminal (His)6-tagged MBP fusion proteins, which were overexpressed as described in the Experimental Section. Both hIMPDH1 and hIMPDH2 were purified by standard immobilized metal affinity chromatography and the N-terminal (His)6-tagged maltose binding protein (MBP) fusions were cleaved with TEV protease to afford wild-type hIMPDH1 and hIMPDH2 that possessed catalytic activity commensurate with the reported values.27, 28
Biochemical evaluation
Triazole-linked MAD analogues 6–9 were evaluated as inhibitors of hIMPDH1 and hIMPDH2 (Table 2). Compound 6 was a modest inhibitor of both isoforms with low micromolar Kiapp values. However compound 7, which has a longer side chain, was found to be a potent nanomolar inhibitor with Kiapp values against the type 1 and the type 2 isoform approximately 180- and 60-fold lower, respectively, than those for compound 6. This finding differs from our previous observation that a shorter compound 3a (Figure 2) was nearly equipotent as the corresponding longer compound 4. Compound 8, which contains an oxymethyltriazole linker, was 10-fold more potent than compound 6 in spite of the fact that both compounds are almost identical in the overall length. Furthermore, compound 9 showed increased potency over the corresponding shorter analogue 8, albeit to an extent much lower than the enhancement observed for compound 7 versus 6. Interestingly, compounds 7 and 9 shared almost identical activity against either hIMPDH1 or hIMPDH2.
Table 2.
Biological evaluations of IMPDH inhibitors
| Compound |
KiapphIMPDH1 (μM) |
KiapphIMPDH2 (μM) |
KiappmtIMPDH (μM) |
|---|---|---|---|
| 1 | 0.033a | 0.007a | 62 |
| 3a | 0.33a | 0.25a | >100 |
| 3b | 0.066a | 0.11a | >100 |
| 3c | 0.016a | 0.038a | >100 |
| 4 | 0.52b | 0.38b | >100 |
| 5a | 0.35c | 0.17c | >100 |
| 5b | 0.66c | 0.31c | >100 |
| 5c | 0.52c | 0.18c | >100 |
| 5d | 0.82c | 0.44c | >100 |
| 6 | 14 | 2.2 | >100 |
| 7 | 0.077 | 0.034 | >100 |
| 8 | 1.5 | 0.20 | >100 |
| 9 | 0.070 | 0.044 | 1.5d (2.2)e |
These findings indicate that the linker length is a key factor that influences the activity against hIMPDHs. This trend was not unexpected as previous studies have demonstrated that NAD binds to hIMPDHs in an extended conformation. This preferred binding mode could account for the activity observed for compounds 6–9, which were designed to mimic NAD, the natural substrate. It was expected that a rigid 1,2,3-triazole ring substituted at the 1 and 4 positions would allow for a desired fitting of the mycophenolic derivative and adenosine moieties in their respective N- and A-subsites of the human enzymes. Conceivably the adenine ring of our triazole-linked inhibitors is sandwiched between aromatic residues H253 and F282 of hIMPDH2 in the A-subsite as it was found in the crystal structures of the complexes of compound 3a/hIMPDH2 and NAD/hIMPDH2. Not surprisingly, compounds 7 and 9 showed increased activity in comparison with compounds 6 and 8, respectively, since 7 and 9 resemble very well the length of NAD.
In addition to the importance of linker length, the charge state in the linker region can also affect the activity. The aminomethyl 1,2,3-triazole linker present in compounds 6 and 7 was expected to bind to the P-groove that is occupied by the negatively charged pyrophosphate moiety of NAD. Therefore it is reasonable to speculate that a positive charge that is generated through a protonation of the secondary amine present in compound 6 might cause unfavorable interactions with surrounding amino acid residues. However, in compound 7 an elongation of the linker might position the proposed positive charge in such a way that it now could not disturb the ligand-enzyme interactions, leading to a highly potent inhibitor. By contrast, in compounds 8 and 9 the linker region is neutral under physiological conditions. Under these circumstances, the linker length determines the inhibitory activity, with compound 9 better mimicking NAD and consequently a more active inhibitor.
Next, we evaluated compounds 6–9 and a panel of IMPDH inhibitors previously developed in our laboratory including MPA (1), methylenebis(phosphonate) MAD analogues 3a–c, and methylenebis(sulfonamide) MAD analogues 5a–c against mtIMPDH (Table 2). MPA was a weak inhibitor with a Kiapp value of 62 μM. Nevertheless, compound 3a and its analogues 3b–3c were completely inactive against mtIMPDH even though they were all low nanomolar inhibitors of hIMPDHs. Compound 3a analogues in which the truncated MPA and adenosine are connected through a methylenebis(sulfonamide) bridge (5a–d) also displayed no activity against mtIMPDH. Among the triazole derivatives 6–9, only 9 displayed significant inhibition. Further characterization of compound 9 was performed to determine its mode of inhibition by varying each substrate with the other substrate maintained at a fixed concentration (Figure 7). Compound 9 displayed uncompetitive inhibition with both NAD and IMP providing Kiu values of 1.5 ± 0.1 and 2.2 ± 0.1 μM, respectively. The observed inhibition modality suggests that 9 has a strong preference for the E–XMP* intermediate consistent with previous studies with mycophenolic acid.1
Figure 7.

Inhibition studies with 9. Initial rate data of variable amounts of inhibitor and either IMP (A) or NAD (B). The fixed substrate was held at 100 μM for IMP and 500 μM for NAD. 9 was used at concentrations of 0 μM ([●]), 3.33 μM ([○]), and 10 μM ([▼]). The inhibitor is uncompetitive with respect to both substrates with Ki(IMP) 2.2 ± 0.1 μM and Ki(NAD) 1.5 ± 0.1 μM.
Computational modeling
To understand the binding selectivity hIMPDH1 and hIMPDH2 versus mtIMPDH, compound 3a was modeled into the NAD binding sites of all three IMPDHs with bound IMP (Figure 8). The per residue interaction energy between compound 3a to individual IMPDH residues within the NAD binding sites are shown in Figure 9. Examination of the interactions revealed variations in the NAD binding domain that might account for compound 3a’s lack of activity against mtIMPDH. Firstly in the solved X-ray crystal structure of compound 3a/hIMPDH2 complex, the adenine ring was involved in an aromatic ring stacking between F282 and H253, which were replaced by energetically similar Y282 and R253 in the modeled compound 3a/hMPDH1 complex. In contrast, in the A-subsite of mtIMPDH, the adenine recognition site consists of L291 and V261, both of which are incapable of aromatic ring stacking with the adenine moiety. In addition, the hydrophobic A-subsite appears to be shallower than those of hIMPDHs (Figure 10), reducing its capacity to accommodate the adenine ring. Taken together, these variations suggest that the adenine moiety fails to contribute significantly to the binding affinity as has been observed in hIMPDHs.
Figure 8.

Comparison between the NAD binding sites of hIMPDHs and mtIMPDH.
Figure 9.

Per residue interaction energy in kcal/mol between compound 3a and IMPDH.
Figure 10.

A) Modeled mode of binding of compound 9 in mtIMPDH. B) Per residue interaction energy in kcal/mol between compound 9 and mtIMPDH.
Furthermore, in the N-subsite of mtIMPDH E458 exhibits an unfavorable 1.5 kcal/mol toward compound 3a binding primarily through long range electrostatic repulsions with the negatively charged methylenebis(phosphonate) group in the P-subsite. In hIMPDH1 and hIMPDH2, the neutral Q334 and Q441 lack such long-range repulsions, but instead engage in favorable hydrogen bonding interactions with the phenolic hydroxyl group of the mycophenolic moiety. In short, lack of aromatic stacking of the adenine moiety together with unfavorable electrostatic interactions might explain the inability of compound 3a and its analogues to inhibit mtIMPDH.
Next we examined compound 9 in an attempt to rationalize its activity against mtIMPDH. While the variations in the A- and N-subsites might render compound 3a inactive against mtIMPDH, the residues in the P-subsite provide key clues to the activity of compound 9. Since the 1,2,3-triazole linker is neutral, the unfavorable electrostatic interactions with compound 3a, which are elicited by E458 and D283, are abrogated in the compound 9/mtIMPDH complex. More importantly, the interactions in the P-subsites contribute positively to the binding energy. Analysis of the P-subsite of IMPDHs revealed that hIMPDHs have a large and shallow region with a floor formed by S275 and S276, which are important in the selective binding to the methylenebis(phosphonate) linker in compound 3a. But in mtIMPDH these two serines are replaced by T284 and A285, respectively. The methyl groups of T284 and A285 create an extension of the hydrophobic binding pocket of the N-subsite, and allow favorable binding of the four-carbon side of the mycophenolic moiety present in compound 9. In short a neutral linker such as 1,2,3-triazole induces favorable interactions and concomitantly avoids undesired charge-charge interactions with mtIMPDH. The gain of energy could potentially offset the loss of energy due to the absence of π – π stacking in the A-subsite and the hydrogen bonding in the P-subsite, leading to compound 9 as a potent inhibitor of mtIMPDH. Therefore it is reasonable to speculate that the electronic nature and proper positioning of the linker, especially the mycophenolic side chain portion, represent promising elements for further chemical modifications in order to enhance the selectivity against mtIMPDH.
Conclusions
Our earlier studies on IMPDH inhibitors that bind at the cofactor binding domain have indicated that these NAD mimics can be divided into three modules, targeting the N-, A-, and P-subsites, respectively. We have also demonstrated these three modules can be modified and optimized individually. In this study we have shown that a 1,4-disubstituted 1,2,3-triazole can mimic the natural pyrophosphate moiety in NAD, resulting in compounds 6–9 that possessed potent activity against human IMPDHs. This finding further validates the premise that the P-subsite is relatively promiscuous and can accommodate various modified linkers. Our preliminary SAR suggests that the linker length plays a key role on activity but other factors such as the charge state and the position of the charge state within the linker region should also be taken into account. The cloning, expression, purification, and characterization of M. tuberculosis IMPDH (mtIMPDH) have allowed us to evaluate these triazole-linked inhibitors and other known NAD mimics against mtIMPDH, leading to the identification of compound 9 as the first potent inhibitor of this enzyme. Our modeling study has revealed the structural variations between hIMPDH and mtIMPDH in the A- and N-subsites, which could account for the lack of activity against mtIMPDH for compound 3a and its analogues. Significantly, we have identified the charge state and the position of hydrophobic portion in the linker region as potential key factors that contribute to the activity of compound 9.
In summary, our study has produced potent inhibitors of hIMPDHs and has provided clues for the design of selective inhibitors of mtIMPDH. This work has also validated our general design of inhibitors based the modular nature of IMPDH enzymes, an approach which is expected to find broad applications in exploring NAD mimics as inhibitors of NAD-utilizing enzymes.
Experimental Section
General methods
All commercial reagents (Sigma-Aldrich, Acros) were used as provided unless otherwise indicated. An anhydrous solvent dispensing system (J. C. Meyer) using 2 packed columns of neutral alumina was used for drying THF, Et2O, and CH2Cl2, while 2 packed columns of molecular sieves were used to dry DMF. Solvents were dispensed under argon. Flash chromatography was performed with Ultra Pure silica gel (Silicycle) with the indicated solvent system. Analytical HPLC was performed on a Varian Microsorb column (C18, 5 μ, 4.6×250 mm) with a flow rate of 0.5 mL/min. All target compounds possessed a purity of greater than 95% as determined by HPLC. Nuclear magnetic resonance spectra were recorded on a Varian 600 MHz with Me4Si or signals from residual solvent as the internal standard for 1H. Chemical shifts are reported in ppm, and signals are described as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broad singlet), and dd (double doublet). Values given for coupling constants are first order. High resolution mass spectra were recorded on an Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface.
5-Methoxy-4-methyl-6-(2-(prop-2-yn-1-ylamino)ethyl)-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (11)
A solution of aldehyde 1017 (450 mg, 1.23 mmol), propargylamine (0.12 mL. 1.9 mmol), NaB(OAc)3H (785 mg, 3.70 mmol) and HOAc (70 μL, 1.2 mmol) in dry dichloroethane (10 mL) was allowed to stir at rt for 4 h. After addition of NaHCO3 (840 mg), the mixture was stirred for 30 min and then diluted with CH2Cl2 (30 mL) and H2O (10 mL). The organic layer was separated and concentrated. The residue was purified by silica gel column chromatography (2%-6% MeOH/CH2Cl2) to give alkyne 11 as a yellow syrup (432 mg, 86%). 1H NMR (CDCl3, 600 MHz) δ 5.41 (s, 2H), 5.12 (s, 2H), 3.85 (t, J = 8.4 Hz, 2H), 3.81 (s, 3H), 3.44 (d, J = 1.8 Hz, 2H), 2.96-2.92 (m, 4H), 2.21-2.16 (m, 4H), 0.98 (t, J = 8.4 Hz, 2H), 0.02 (s, 9H). HRMS calcd for C21H32NO5Si 406.2044 (M+H)+, found 406.2074.
6-(2-(((1-(5′-Deoxy-2′,3′-O,O-isopropylidene-adenosin-5′-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)ethyl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (13)
To a solution of alkyne 11 (210 mg, 0.52 mmol) in tert-BuOH (4 mL) and H2O (4 mL) were added CuSO4·5H2O (6.0 mg, 0.024 mmol), sodium ascorbate (21 mg, 0.10 mmol) and then azide 1221 (345 mg, 1.04 mmol). The mixture was allowed to stir at rt for 3 h and concentrated. The residue was purified by preparative thin-layer chromatography (1000 micron, 5%-10% MeOH/CH2Cl2) to give protected triazole 13 as a pale solid (293 mg, 77%). 1H NMR (CD3OD, 600 MHz) δ 8.22 (s, 1H), 8.15 (s, 1H), 7.64 (s, 1H), 6.21 (s, 1H), 5.45 (d, J = 6.0 Hz, 1H), 5.34 (s, 2H), 5.24-5.18 (m 3H), 4.82-4.78 (m, 2H), 4.60-4.54 (m, 1H), 3.88-3.75 (m, 7H), 2.95 (t, J = 7.5 Hz, 2H), 2.76 (t, J = 7.5 Hz, 2H), 2.18 (s, 3H), 1.58 (s, 3H), 1.36 (s, 3H), 0.89 (t, J = 8.4 Hz, 2H), 0.32 (s, 9H). HRMS calcd for C34H48N9O8Si 738.3389 (M+H)+, found 738.3393.
6-(2-(((1-(5′-Deoxy-adenosin-5′-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)ethyl)-7-hydroxy-5-methoxy-4-methylisobenzofuran-1(3H)-one (6)
A solution of protected triazole 13 (230 mg, 0.31 mmol) in TFA (4 mL) and H2O (1 mL) was allowed to stir at rt overnight. The mixture was concentrated and co-evaporated with MeOH. The solid obtained was triturated with hot hexanes and then dissolved in MeOH (ca. 2 mL). Diethyl ether (30 mL) was added and the precipitate was filtered and washed with diethyl ether to give triazole 6 as a pale solid (184 mg, 86%). 1H NMR (CD3OD, 600 MHz) δ 8.37 (s, 1H), 8.33 (s, 1H), 8.02 (s, 1H), 6.04 (d, J = 4.2 Hz, 1H), 5.26 (s, 2H), 4.91-4.87 (m, 1H), 4.72 (t, J = 4.8 Hz, 2H), 4.45 (t, J = 5.1 Hz, 1H), 4.44-4.39 (m, 1H), 4.34 (d, J = 14.4 Hz, 1H), 4.31 (d, J = 14.4 Hz, 1H), 3.81 (s, 3H), 3.22 (t, J = 7.2 Hz, 2H), 3.09 (t, J = 7.8 Hz, 2H), 2.16 (s, 3H). HRMS calcd for C25H30N9O7 568.2262 (M+H)+, found 568.2290.
(E)-4-(6-Methoxy-7-methyl-3-oxo-4-((2-(trimethylsilyl)ethoxy)methoxy)-1,3-dihydroisobenzofuran-5-yl)-2-methylbut-2-enal (15)
To a mixture of NaOH (100 mg, 2.5 mmol) in CH2Cl2 (4 mL) and H2O (4 mL) were added Adogen 464 (0.41 mL, 0.28 mmol) and aldehyde 1414 (380 mg, 1.38 mmol). After stirring for 20 min, 2-(trimethylsilyl)ethoxymethyl chloride (SEMCl, 0.44 mL, 2.5 mmol) was added dropwise. The resulting mixture was allowed to stir at rt for 3 h, and the organic layer was separated and then concentrated. The residue was purified by silica gel column chromatography (10%-30% EtOAc/hexanes) to give aldehyde 15 as a clear syrup (492 mg, 88%). 1H NMR (CDCl3, 600 MHz) δ 9.38 (s, 1H), 6.56 (t, J = 6.9 Hz, 1H), 5.45 (s, 2H), 5.16 (s, 2H), 3.85-3.80 (m, 4H), 3.78 (s, 3H), 2.12 (s, 3H), 1.93 (s, 3H), 0.95 (t, J = 8.4 Hz, 2H), 0.00 (s, 9H). HRMS calcd for C21H30O6NaSi 429.1703 (M+Na)+, found 429.1704.
(E)-5-Methoxy-4-methyl-6-(3-methyl-4-(prop-2-yn-1-ylamino)but-2-en-1-yl)-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (16)
In a manner similar to that described for the preparation of alkyne 11, aldehyde 15 (353 mg, 0.87 mmol) and propargylamine (50 μL, 0.87 mmol) underwent reductive amination to give alkyne 16 as a yellowish syrup (251 mg, 65%). 1H NMR (CDCl3, 600 MHz) δ 5.44 (t, J = 6.9 Hz, 1H), 5.41 (s, 2H), 5.13 (s, 2H), 3.86 (t, J = 8.4 Hz, 2H), 3.78 (s, 3H), 3.51 (d, J = 6.6 Hz, 2H), 3.35 (d, J = 1.8 Hz, 2H), 3.21 (s, 2H), 2.22-2.16 (m 4H), 1.85 (s, 3H), 1.44 (brs, 1H), 0.97 (t, J = 8.7 Hz, 2H), 0.02 (s, 9H). HRMS calcd for C24H36NO5Si 446.2357 (M+H)+, found 446.2396.
6-((E)-4-(((1-(5′-Deoxy-2′,3′-O,O-isopropylidene-adenosin-5′-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)-3-methylbut-2-en-1-yl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (17)
In a manner similar to that described for the preparation of protected triazole 13, alkyne 16 (238 mg, 0.53 mmol) and azide 12 (354 mg, 1.06 mmol) underwent a click reaction to give protected triazole 17 as a pale solid (265 mg, 64%). 1H NMR (CD3OD, 600 MHz) δ 8.21 (s, 1H), 8.15 (s, 1H), 7.58 (s, 1H), 6.19 (s, 1H), 5.45 (d, J = 6.0 Hz, 1H), 5.37 (t, J = 6.6 Hz, 1H), 5.34 (s, 2H), 5.22 (s, 2H), 5.18 (dd, J = 5.7, 3.9 Hz, 1H), 4.80-4.72 (m, 2H), 4.56-4.51 (m, 1H), 3.81 (t, J = 8.1 Hz, 2H), 3.78 (s, 3H), 3.70 (d, J = 14.4 Hz, 1H), 3.65 (d, J = 13.8 Hz, 1H), 3.51 (d, J = 6.6 Hz, 2H), 3.08 (s, 2H), 2.18 (s, 3H), 1.82 (s, 3H), 1.56 (s, 3H), 1.35 (s, 3H), 0.91 (t, J = 8.4 Hz, 2H), 0.02 (s, 9H). HRMS calcd for C37H52N9O8Si 778.3708 (M+H)+, found 778.3706.
6-((E)-4-(((1-(5′-Deoxy-adenosin-5′-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)-3-methylbut-2-en-1-yl)-7-hydroxy-5-methoxy-4-methylisobenzofuran-1(3H)-one (7)
In a manner similar to that described for the preparation of triazole 6, protected triazole 17 (80 mg, 0.10 mmol) was deprotected under acidic conditions. Triazole 7 was obtained as a pale solid after a preparative thin-layer chromatography (59 mg, 80%). 1H NMR (CD3OD, 600 MHz) δ 8.14 (s, 1H), 8.08 (s, 1H), 7.78 (s, 1H), 5.94 (d, J = 3.6 Hz, 1H), 5.60 (brs, 1H), 5.23 (s, 2H), 4.94-4.86 (m, 1H), 4.82-4.78 (m, 1H), 4.47 (t, J = 4.8 Hz, 1H), 4.35 (brs, 1H), 4.07 (d, J = 13.8 Hz, 1H), 4.01 (d, J = 15.0 Hz, 1H), 3.77 (s, 3H), 3.63-3.58 (m, 1H), 3.50-3.44 (m, 4H), 2.14 (s, 3H), 1.88 (s, 3H). HRMS calcd for C28H34N9O7 608.2575 (M+H)+, found 608.2626. HRMS calcd for C28H34N9O7 608.2581 (M+H)+, found 608.2581.
6-(2-Azidoethyl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (19)
To a solution of alcohol 1817 (1.05 g, 2.84 mmol) and Et3N (0.34 mL, 4.4 mmol) in anhydrous THF (20 mL) at 0 °C was added dropwise MsCl (0.80 mL, 5.7 mmol). The mixture was allowed to stir at 0 °C for 40 min and warm to rt. After the reaction mixture was diluted with EtOAc (100 mL), the resulting solution was washed with water (2×30 mL) and brine (2×30 mL). The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and re-dissolved in anhydrous DMF (10 mL). After NaN3 (380 mg, 5.84 mmol) was added at rt, the resulting mixture was heated at 80 °C for 2 h and cooled to rt. The mixture was then diluted with EtOAc (100 mL) and washed with water (2×30 mL) and brine (2×30 mL). The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and the residue was purified by silica gel column chromatography (10%-30% EtOAc/hexanes) to give azide 19 as a wax-like solid (418 mg, 37%). 1H NMR (CDCl3, 600 MHz) δ 5.42 (s, 2H), 5.14 (s, 2H), 3.88-3.82 (m, 5H), 3.48 (t, J = 7.5 Hz, 2H), 3.04 (t, J = 7.5 Hz, 2H), 2.20 (s, 3H), 0.98 (t, J = 8.4 Hz, 2H), 0.30 (s, 9H). HRMS calcd for C18H27N3O5NaSi 416.1612 (M+Na)+, found 416.1620.
2′,3′-O,O-Isopropylidene-5′-O-propargyl adenosine (21)
A solution of alkyne 2023 (1.28 g, 2.85 mmol) in methanolic ammonia (ca. 7N, 30 mL) was allowed to stir at rt overnight and then concentrated. The residue was purified by silica gel column chromatography to give alkyne 21 as a white solid (903 mg, 92%). 1H NMR (CDCl3, 600 MHz) δ 8.38 (s, 1H), 8.04 (s, 1H), 6.20 (d, J = 1.8 Hz, 1H), 5.79 (s, 2H), 5.33 (d, J = 5.4 Hz, 1H), 5.02 (dd, J = 6.6, 2.4 Hz, 1H), 4.52 (dd, J = 7.2, 3.6 Hz, 1H), 4.13 (d, J = 1.8 Hz, 2H), 3.78 (dd, J = 10.2, 3.6 Hz, 1H), 3.73 (dd, J = 10.2, 4.8 Hz, 1H), 2.44 (s, 1H), 1.64 (s, 3H), 1.40 (s, 3H). HRMS calcd for C16H20N5O4 346.1515 (M+H)+, found 346.1512.
6-(2-(4-(2′,3′-O,O-Isopropylidene-adenosin-5′-yl)methyl-1H-1,2,3-triazol-1-yl)ethyl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (22)
In a manner similar to that described for the preparation of protected triazole 13, alkyne 21 (361 mg, 0.92 mmol) and azide 19 (260 mg, 0.75 mmol) underwent a click reaction to give protected triazole 22 as a light orange solid (476 mg, 86%). 1H NMR (CD3OD, 600 MHz) δ 8.23 (s, 1H), 8.18 (s, 1H), 7.65 (s, 1H), 6.19 (d, J = 1.8 Hz, 1H), 5.35-5.30 (m, 3H), 5.20 (s, 2H), 5.00 (d, J = 6.0 Hz, 1H), 4.68-4.57 (m, 2H), 4.53 (s, 2H), 4.47-4.44 (m, 1H), 3.81 (t, J = 8.4 Hz, 2H), 3.76 (s, 3H), 3.65 (dd, J = 10.8, 3.6 Hz, 1H), 3.60 (dd, J = 10.8, 4.0 Hz, 1H), 3.36-3.34 (m, 2H), 2.14 (s, 3H), 1.60 (s, 3H), 1.38 (s, 3H), 0.90 (t, J = 8.1 Hz, 2H), 0.03 (s, 9H). HRMS calcd for C34H47N8O9Si 739.3235 (M+H)+, found 739.3234.
6-(2-(4-(Adenosin-5′-yl)methyl-1H-1,2,3-triazol-1-yl)ethyl)-7-hydroxy-5-methoxy-4-methylisobenzofuran-1(3H)-one (8)
In a manner similar to that described for the preparation of triazole 6, protected triazole 22 (348 mg, 0.47 mmol) was deprotected under acidic conditions to give triazole 8 as a light orange solid (128 mg, 48%). 1H NMR (DMSO-d6, 600 MHz) δ 9.70 (s, 1H), 8.32 (s, 1H), 8.16 (brs, 1H), 8.04 (s, 1H), 7.35 (brs, 2H), 5.89 (d, J = 5.4 Hz, 1H), 5.48 (brs, 1H), 5.24 (brs, 3H), 4.58-2.52 (m,3H), 4.87 (t, J = 7.5 Hz, 2H), 4.13 (t, J = 4.2 Hz, 1H), 4.02 (dd, J = 7.8, 4.2 Hz, 1H), 3.68 (dd, J = 10.8, 3.6 Hz, 1H), 3.64-3.58 (m, 4H), 3.14 (t, J = 7.5 Hz, 2H), 2.04 (s, 3H). HRMS calcd for C25H29N8O8 569.2102 (M+H)+, found 569.2134.
(E)-6-(4-Hydroxy-3-methylbut-2-en-1-yl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (23)
To a solution of protected aldehyde 15 (1.08 g, 2.67 mmol) in MeOH (40 mL) and H2O (0.5 mL) at 0 °C was added CeCl3·7H2O (1.02 g, 2.74 mmol) and then NaBH4 (280 mg, 7.40 mmol) in portions. After the addition of NaBH4 was complete, the mixture was allowed to warm to rt and stir for 1 h. The mixture was concentrated and the residue was dissolved in EtOAc (100 mL) and H2O (30 mL). The mixture was acidified with 1N HCl solution to pH ≈ 4. The organic layer was separated and washed with H2O (2×30 mL) and brine (60 mL), and dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was purified by silica gel column chromatography to give protected alcohol 23 as a clear syrup (621 mg, 57%). 1H NMR (CDCl3, 600 MHz) δ 5.50 (t, J = 6.6 Hz, 1H), 5.41 (s, 2H), 5.13 (s, 2H), 3.99 (s, 2H), 3.85 (t, J = 8.4 Hz, 2H), 3.79 (s, 3H), 3.52 (d, J = 6.6 Hz, 2H), 2.19 (s, 3H), 1.85 (s, 3H), 0.97 (t, J = 8.4 Hz, 2H), 0.02 (s, 9H). HRMS calcd for C21H33O6Si 409.2046 (M+H)+, found 409.2040.
6-((E)-4-(4-(2′,3′-O,O-Isopropylidene-adenosin-5′-yl)methyl-1H-1,2,3-triazol-1-yl)-3-methylbut-2-en-1-yl)-5-methoxy-4-methyl-7-((2-(trimethylsilyl)ethoxy)methoxy)isobenzofuran-1(3H)-one (25)
To a solution of protected alcohol 23 (729 mg, 1.78 mmol) and Et3N (0.50 mL, 3.6 mmol) in THF (20 mL) at 0 °C was added MsCl (0.21 mL, 2.7 mmol). The resulting mixture was allowed to stir at 0 °C for 1 h and warm to rt. After diluted with EtOAc (100 mL), the mixture was washed with H2O (2×30 mL) and brine (2×30 mL), and dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was dissolved in dry DMF (10 mL). After addition of NaN3 (240 mg, 3.69 mmol), the reaction mixture was heated at 60 °C for 2 h and cooled to rt. The mixture was diluted with EtOAc (100 mL), washed with H2O (2×30 mL) and brine (2×30 mL), and dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was purified by column chromatography (10-30% EtOAc/hexanes) to give protected azide 24 as a clear syrup (656 mg, 85%, a mixture of isomers as indicated by 1H NMR) which was used directly for the subsequent CuAAC reaction.
6-((E)-4-(4-(Adenosin-5′-yl)methyl-1H-1,2,3-triazol-1-yl)-3-methylbut-2-en-1-yl)-7-hydroxy-5-methoxy-4-methylisobenzofuran-1(3H)-one (9)
To a mixture of protected azide 24 (603 mg, 1.39 mmol) in tert-BuOH (5 mL) and H2O (5 mL) were added CuSO4·5H2O (12 mg, 0.048 mmol), sodium ascorbate (45 mg, 0.23 mmol) and then alkyne 21 (320 mg, 0.93 mmol). The mixture was allowed to stir for 2 h at rt and concentrated. The residue was then dissolved in TFA (4 mL) and H2O (1 mL) and the mixture was allowed to stir at rt overnight. After concentration, the residue was co-evaporated with MeOH and purified by preparative thin-layer chromatography (1000 micron, 10% MeOH/CH2Cl2) to give triazole 9 as a pale solid (84 mg). A portion of this solid (20 mg) in MeOH (ca. 5 mL) was heated at 60 °C overnight and allowed to stir at rt for 7 h. The solid was filtered and washed with MeOH to give pure triazole 9 as an off-white solid (7.5 mg, estimated yield 5% based on alkyne 21). 1H NMR (DMSO-d6, 600 MHz) δ 9.49 (s, 1H), 8.29 (s, 1H), 8.14 (s, 1H), 7.95 (s, 1H), 7.27 (s, 2H), 5.89 (d, J = 5.4 Hz, 1H), 5.48 (d, J = 5.4 Hz, 1H), 5.42 (t, J = 6.6 Hz, 1H), 5.18-5.30 (m, 3H), 4.86 (s, 2H), 4.50-4.60 (m, 3H), 4.13 (brd, J = 3.0 Hz, 1H), 4.02 (brd, J = 4.2 Hz, 1H), 3.69 (dd, J = 10.5, 3.9 Hz, 1H), 3.65 (s, 3H), 3.61 (dd, J =10.8, 4.8 Hz, 1H), 2.06 (s, 3H), 1.64 (s, 3H). HRMS calcd for C28H33N8O8 609.2412 (M+H)+, found 609.2412.
Molecular modeling
All modeling were carried out using the Schrodinger modeling package.29 The solved X-ray crystallographic structure of hIMPDH in complex with 6-Cl-IMP (PDB: 1JCN) and hIMPDH2 in complex compound 3a (PDB: 1NF7) were taken from the Protein Data Bank. The missing hydrogen atoms were added to both X-ray structures followed by energy minimization using OPLS 2005 forcefield to optimize all hydrogen bonding networks. Since the structure of the mtIMPDH has not yet been determined, the structure of mtIMPDH (GuaB2, Rv3411c) from Mycobacterium. tuberculosis was homology modeled based the solved X-ray crystallographic structure of Streptococcus pyogenes (Sp.) IMPDH in complex with inosine at 1.9 Å resolution (PDB: 1ZFJ).30 An initial sequence alignment of Mycobacterium tuberculosis, Thermotoga maritima, Streptococcus pyogenes, and Homo sapiens Type 1 and 2 IMPDH was performed using clustalW to identify the structural conserved regions (see supplemental Figure S1). The homology model of mtIMPDH was then carried out based on this sequence alignment and the conserved structural regions of Streptococcus pyogenes, with particular emphasis placed on the NAD binding site. The mtIMPDH homology model was validated by comparison of the structure and the recently solved X-ray crystal structure of Cryptosporidium parvum IMPDH at 2.8Å (PDB: 3KHJ),31 which showed consistent overlap of key residues within the NAD binding site. Compound 3a was modeled into the binding site via superpositioning of the compound 3a/hIMPDH2 complex onto the binding site of hIMPDH1 and mtIMPDH. Compound 9 was modeled by the replacement of the methylene(bisphosphonate) linker in compound 3a. To account for solvent effect and protein flexibility, the ligand binding site of each model was further refined by restraint energy minimization using OPLS 2005 forcefield32 within 20 Å TIP3P33 surface constrained water sphere. To identify key amino residues involved in ligand binding, the non-bond per residue interaction energies between each modeled ligand to individual IMPDH residues within the NAD binding sites were evaluated with a constant dielectric constant of 4.
Cloning, Protein Overexpression, Purification, and Enzyme Assays
General
Chemically competent E. coli Mach1 and BL21 STAR (DE3), plasmids pCR2.1-TOPO and pENTR/D-TOPO, and Gateway LR Clonase were purchased from Invitrogen (Carlsbad, CA, USA). The TEV protease expression vector pRK793 was obtained from Addgene (plasmid number 8827) and expressed as previously described.34 Restriction enzymes were purchased from New England Biolabs (Ipswich, MA, USA). PrimeSTAR HS DNA polymerase was purchased from TAKARA Bio Inc (Otsu, Shiga, Japan). Primers for PCR were obtained from Integrated DNA Technologies (Coralville, IA, USA). The expression vector pDEST-HisMBP and pET28b were purchased from Addgene (Cambridge, MA, USA) and EMD biosciences (San Diego, CA, USA) respectively. Ni-NTA, and DNA purification/isolation kits was obtained from Qiagen Sciences (Germantown, MD, USA). NAD and IMP as well as all biological buffers and components were purchased from Sigma Aldrich (St. Louis, MO, USA). Enzymatic activity, kinetic parameters, and inhibition assays were performed on a Molecular Devices M5e multi-mode plate reader. (Sunnyvale, CA, USA).
Cloning, expression, and purification of mtIMPDH
The Mtb IMPDH gene guaB2 was amplified by PCR from H37Rv genomic DNA using the primers CACCCATATGTCCCGTGGCATGTCC and CCAAGCTTAGCGCGCGTAGTAGTTG. The product was initially cloned into the PCR capture vector pCR2.1 TOPO (Invitrogen) and then subcloned into pET28b (Novagen) using the NdeI and HindIII sites contained within the primers. The resulting plasmid pCDD100 expresses GuaB2 with an N-terminal His tag for ease of purification. pCDD100 was transformed into BL21 STAR (DE3). Overnight cultures grown in LB supplemented with kanamycin (50 μg/mL) were uses to inoculate 1 L of LB with the same concentration of kanamycin. Cultures were grown to an OD600 of 0.7 at 37 °C and induced with 0.4 mM IPTG. The temperature was lowered to 30 °C and the cultures were grown an additional 4 h. The culture was centrifuged and the cell pellets were resuspended in 20 mL lysis buffer (50 mM HEPES, 300 mM NaCl, 10 mM imidazole, pH 8.0) containing 2 mg/mL lysozyme. After 30 min incubation on ice, the solution was sonicated at 0 °C with four bursts (2 min, 30% duty cycle, power 6) with a 1 min break between each burst (Branson Sonifier 250). The lysate was centrifuged at 50,000 × g for 10 min then 2 mL of 50% Ni-NTA was added to the cleared supernatant. After incubating at 4 °C for 1 h, the lysate was loaded onto a column and the flow through was collected. The column was washed with 16 mL of wash buffer (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, pH 8.0) and eluted with 3 mL of elution buffer (50 mM HEPES, 300 mM NaCl, 250 mM imidazole, pH 8.0) of which the first 0.5 mL was discarded. The protein solution was desalted on a PD-10 column into storage buffer (50 mM Tris pH 7.5, 1 mM DTT, 100 mM KCl, 10% glycerol). Protein concentrations were determined by the Bradford method using BSA as a standard and stored at −80 °C.35
Cloning, expression, and purification of hIMPDH type I and II
To simplify expression and purification of hIMPDH type I and II, new expression vectors were constructed to allow metal affinity column purification and affinity tag cleavage to yield the native enzymes. Expression vectors pH1 and pHIA5 containing IMPDH I and II without affinity tags were kindly provided by Prof. Liz Hedstrom (Brandeis University). For expression the genes were amplified using the forward primers CACCGAAAACCTGTATTTTCAGATGGCGGACTACCTGATC and CACCGAAAACCTGTATTTTCAGATGGCCGACTACCTGATTAG for IMPDH I and II respectively, which attaches an N-terminal TEV protease site that cleaves before the first methionine residue yielding fully native protein and the reverse primers CTCAGTACAGCCGCTTTTC and CTCAGAAAAGCCGCTTCTC. The resulting PCR products were cloned into pENTR/D-TOPO (Invitrogen) to create the gateway entrance vectors pCDD026 and pCDD027. In order to enhance solubility of the proteins the destination vector pDEST-HisMBP (gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli were attained from Addgene, plasmid number 11085) was recombined with pCDD026 and pCDD027 using the gateway system (Invitrogen) to give pCDD032 and pCDD033, which express a fusion protein of TEV cleavable IMPDH I or IMPDH II with an N-terminal His tagged MBP solubility enhancer.36
For overexpression and purification of IMPDH I and II, the plasmids pCDD032 and pCDD033 were transformed into BL21 STAR (DE3) (Invitrogen). Starter cultures (5 mL) grown in ZYM-505237 for 6 hours were used to inoculate 100 mL ZYM-5052 containing ampicillin (100 μg/mL) and grown at 30 °C to an OD600 of 15. The culture was centrifuged and the cell pellet was resuspended in 20 mL lysis buffer (50 mM HEPES, 300 mM NaCl, 10 mM imidazole, pH 8.0) containing 2 mg/ml lysozyme. After 30 min incubation on ice, the solution was sonicated at 0 °C with four bursts (2 min, 30% duty cycle, power 6) with a 1 min break between each burst (Branson Sonifier 250). The lysate was centrifuged at 50,000 × g for 10 min then 2 mL of 50% Ni-NTA was added to the cleared supernatant. After incubating at 4 °C for 1 h, the lysate was loaded onto a column and the flow through was collected. The column was washed with 16 mL of wash buffer (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, pH 8.0) and eluted with 3 mL of elution buffer (50 mM HEPES, 300 mM NaCl, 250 mM imidazole, pH 8.0) of which the first 0.5 mL was discarded. The protein solution was desalted on a PD-10 column into TEV cleavage buffer (50 mM Tris·HCl, pH 8.0, 0.5 mM EDTA, 300 mM NaCl, 1 mM DTT). TEV protease was added to a final concentration of 0.15 mg/ml and the proteins were allowed to cleave overnight at 4 °C. The cleaved proteins were purified by adding 500 μL of 50% Ni-NTA and incubating at 4 °C for 1 h to bind the His-tagged MBP and His-tagged TEV protease. After the incubation, the Ni-NTA resin was filtered out of solution and the proteins were dialyzed twice against buffer A (50 mM Tris pH 7.5, 1 mM DTT, 100 mM KCl, 10% glycerol) and stored at −80 °C. Protein concentrations were determined by measuring the specific activity of the enzymes as previously reported.38
Enzyme Assays
IMPDH inhibition assays were performed under initial velocity conditions in a total volume of 100 μL at 25 °C for 5 minutes and the production of NADH was monitored by following changes in absorbance at 340 nm on a Molecular Devices M5e multimode plate reader. Assays were set up in duplicate and contained either hIMPDH type 1 (100 nM), hIMPDH type 2 (30 nM), or mtIMPDH (400 nM) in reaction buffer (50 mM Tris, pH 8.0, 100 mM KCl, 1 mM DTT, 100 μM IMP, 100 μM NAD). Since compound 1 exhibited a large Kiapp of 62 μM against mtIMPDH with a corresponding Hill-slope of 1.5, indicative of aggregation and non-specific inhibition,39 we additionally evaluated 1 against the reaction buffer containing 0.1% Trition X-100; however, the Kiapp value was not affected. A two-fold dilution series of inhibitors in DMSO was added to UV clear 96 well half-area plates (Greiner) to give a final DMSO concentration of 1%. The Kiapp values were calculated using the Hill equation (eq 1). For inhibitors that displayed tight-binding inhibition (Kiapp < 10·[E]), the Kiapp values were calculated using the Morrison equation (eq 2). In these equations, the fractional activity (vi/v0) versus inhibitor concentration were fit by non-linear regression analysis using GraphPad Prism version 4.0 to obtain Kiapp values, where vi is the reaction velocity at a given [I] and v0 is the reaction velocity of the DMSO control. The steady-state kinetic parameters KM and kcat of NAD for mtIMPDH were determined by measuring the initial velocity as a function of NAD concentration from 10 mM down to 78 μM at saturating IMP concentrations (1 mM) to provide a saturation curve, which was fit by nonlinear regression analysis to the substrate inhibition equation. The steady-state kinetic parameters of IMP for mtIMPDH were determined by measuring the initial velocity as a function of IMP concentration from 1.5 mM down to 2 μM at sub-inhibitory NAD concentrations (3 mM) and the data were fit to the Michaelis-Menten equation. Reactions to determine the Kiapp and mode of inhibition of compound 9 with mtIMPDH were set up essentially as described above. Initial velocities were measured with increasing concentrations of IMP from 1.4 μM to 1 mM (0.5 mM NAD) or NAD from 94 μM to 3 mM (100 μM IMP) with or without inhibitor (0, 3.33, 10 μM). mtIMPDH was held constant at 0.15 μM for all reactions and the data were fit to competitive, uncompetitive, noncompetitive and mixed models of inhibition using the enzyme kinetics module of SigmaPlot and the model with the highest r2 value was selected.
| (1) |
| (2) |
Supplementary Material
Figure 5.

Steady-state kinetics of mtIMPDH. (A) Initial velocity vs. [IMP]. Data were fit to the Michaelis–Menten equation. (B) Initial velocity vs. [NAD]. Data were fit to the Michaelis–Menten substrate inhibition equation. Each reaction contained mtIMPDH at 75 nM, 50 mM Tris, pH 8.0, 100 mM KCl, 1 mM DTT and either 3 mM NAD (for curve A) or 1 mM IMP (for curve B).
Acknowledgment
This research was supported by USDOD ARMY grant W81XWH-05-01-0216 and by the Center for Drug Design in the Academic Health Center of the University of Minnesota to K.P. and the NIH (AI070219) to C.C.A. We thank Prof. Liz Hedstrom for generously providing plasmids pH1 and pHIA5. The University of Minnesota Supercomputing Institute provided all the necessary computational resources. We thank Drs. Takashi Tsuji and Kunisuke Izawa of Ajinomoto Co., Inc., Japan for a gift of MPA.
Abbreviations
- NAD
nicotinamide adenine dinucleotide
- IMPDH
inosine monophsophate dehydrogenase
- IMP
inosine monophosphate
- XMP
xanthosine monophosphate
- hIMPDH
human inosine monophsophate dehydrogenase
- mtIMPDH
Mycobacterium tuberculosis inosine monophsophate dehydrogenase
- TB
tuberculosis
- MPA
mycophenolic acid
- TAD
tiazofurin adenine dinucleotide
- MAD
mycophenolic adenine dinucleotide
- SAR
structure–activity relationship
- CuAAC
copper-(I)-catalyzed azide–alkyne cycloaddition
- SEM
2-(trimethylsilyl)ethoxymethyl
- TFA
trifluoroacetic acid
- NMR
nuclear magnetic resonance
- NOE
nuclear overhauser effect
Footnotes
Supporting Information Available: Table of HPLC purities of compounds 6–9; alignment of Mycobacterium tuberculosis, Thermogota maritima, Streptococcus pyogenes, and Homo sapiens Type I & II IMPDH sequences; and structure comparison of the mtIMPDH homology model and the recently solved X-ray crystal structure of Cryptosporidium parvum IMPDH (PDB: 3KHJ). This material is available free of charge via internet at http://pubs.acs.org.
References
- (1).Hedstrom L. IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chong CR, Qian DZ, Pan F, Wei Y, Pili R, Sullivan DJ, Jr., Liu JO. Identification of type 1 inosine monophosphate dehydrogenase as an antiangiogenic drug target. J. Med. Chem. 2006;49:2677–2680. doi: 10.1021/jm051225t. [DOI] [PubMed] [Google Scholar]
- (3).Mortimer SE, Xu D, McGrew D, Hamaguchi N, Lim HC, Bowne SJ, Daiger SP, Hedstrom L. IMP dehydrogenase type 1 associates with polyribosomes translating rhodopsin mRNA. J. Biol. Chem. 2008;283:36354–36360. doi: 10.1074/jbc.M806143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).McLean JE, Hamaguchi N, Belenky P, Mortimer SE, Stanton M, Hedstrom L. Inosine 5′-monophosphate dehydrogenase binds nucleic acids in vitro and in vivo. Biochem. J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen L, Pankiewicz KW. Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr. Opin. Drug Discov. Devel. 2007;10:403–412. [PubMed] [Google Scholar]
- (6).Chen L, Petrelli R, Felczak K, Gao G, Bonnac L, Yu JS, Bennett EM, Pankiewicz KW. Nicotinamide adenine dinucleotide based therapeutics. Curr. Med. Chem. 2008;15:650–670. doi: 10.2174/092986708783885282. [DOI] [PubMed] [Google Scholar]
- (7).Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem. Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- (10).Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- (11).Shu Q, Nair V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev. 2008;28:219–232. doi: 10.1002/med.20104. [DOI] [PubMed] [Google Scholar]
- (12).Hedstrom L, Gan L. IMP dehydrogenase: structural schizophrenia and an unusual base. Curr. Opin. Chem. Biol. 2006;10:520–525. doi: 10.1016/j.cbpa.2006.08.005. [DOI] [PubMed] [Google Scholar]
- (13).Lesiak K, Watanabe KA, Majumdar A, Powell J, Seidman M, Vanderveen K, Goldstein BM, Pankiewicz KW. Synthesis of a methylenebis(phosphonate) analogue of mycophenolic adenine dinucleotide: a glucuronidation-resistant MAD analogue of NAD. J. Med. Chem. 1998;41:618–622. doi: 10.1021/jm970705k. [DOI] [PubMed] [Google Scholar]
- (14).Pankiewicz KW, Lesiak-Watanabe KB, Watanabe KA, Patterson SE, Jayaram HN, Yalowitz JA, Miller MD, Seidman M, Majumdar A, Prehna G, Goldstein BM. Novel mycophenolic adenine bis(phosphonate) analogues as potential differentiation agents against human leukemia. J. Med. Chem. 2002;45:703–712. doi: 10.1021/jm0104116. [DOI] [PubMed] [Google Scholar]
- (15).Chen L, Gao G, Felczak K, Bonnac L, Patterson SE, Wilson D, Bennett EM, Jayaram HN, Hedstrom L, Pankiewicz KW. Probing binding requirements of type I and type II isoforms of inosine monophosphate dehydrogenase with adenine-modified nicotinamide adenine dinucleotide analogues. J. Med. Chem. 2007;50:5743–5751. doi: 10.1021/jm070568j. [DOI] [PubMed] [Google Scholar]
- (16).Chen L, Gao G, Bonnac L, Wilson DJ, Bennett EM, Jayaram HN, Pankiewicz KW. Methylenebis(sulfonamide) linked nicotinamide adenine dinucleotide analogue as an inosine monophosphate dehydrogenase inhibitor. Bioorg. Med. Chem. Lett. 2007;17:3152–3155. doi: 10.1016/j.bmcl.2007.03.035. [DOI] [PubMed] [Google Scholar]
- (17).Chen L, Petrelli R, Olesiak M, Wilson DJ, Labello NP, Pankiewicz KW. Bis(sulfonamide) isosters of mycophenolic adenine dinucleotide analogues: inhibition of inosine monophosphate dehydrogenase. Bioorg. Med. Chem. 2008;16:7462–7469. doi: 10.1016/j.bmc.2008.06.003. [DOI] [PubMed] [Google Scholar]
- (18).Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- (19).Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- (20).Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, Aldrich CC. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: antibacterial nucleosides effective against Mycobacterium tuberculosis. J. Med. Chem. 2008;51:7495–7507. doi: 10.1021/jm8008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu F, Austin DJ. Synthesis of 5′-functionalized adenosine: suppression of cyclonucleoside formation. Tetrahedron Lett. 2001;42:3153–3154. [Google Scholar]
- (22).van Heerden FR, van Zyl JJ, Rall GJH, Brandt EV, Roux DG. Phase-transfer catalysis: A general method of methoxymethylation of the hydroxyl function. Tetrahedron Lett. 1978;19:661–662. [Google Scholar]
- (23).Jawalekar AM, Meeuwenoord N, Cremers JS, Overkleeft HS, van der Marel GA, Rutjes FP, van Delft FL. Conjugation of nucleosides and oligonucleotides by [3+2] cycloaddition. J. Org. Chem. 2008;73:287–290. doi: 10.1021/jo702023s. [DOI] [PubMed] [Google Scholar]
- (24).Gagneux A, Winstein S, Young WG. Rearrangement of allylic azides. J. Am. Chem. Soc. 1960;82:5956–5957. [Google Scholar]
- (25).Feldman AK, Colasson B, Sharpless KB, Fokin VV. The allylic azide rearrangement: achieving selectivity. J. Am. Chem. Soc. 2005;127:13444–13445. doi: 10.1021/ja050622q. [DOI] [PubMed] [Google Scholar]
- (26).Kale TA, Distefano MD. Diazotrifluoropropionamido-containing prenylcysteines: syntheses and applications for studying isoprenoid-protein interactions. Org. Lett. 2003;5:609–612. doi: 10.1021/ol026752a. [DOI] [PubMed] [Google Scholar]
- (27).Wang W, Hedstrom L. Kinetic mechanism of human inosine 5′-monophosphate dehydrogenase type II: random addition of substrates and ordered release of products. Biochemistry. 1997;36:8479–8483. doi: 10.1021/bi970226n. [DOI] [PubMed] [Google Scholar]
- (28).Mortimer SE, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem. J. 2005;390:41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Maestro 8.5, P., version 2.0, Macromodel 9.6. Schrodinger, LLC; New York, NY: 2008. [Google Scholar]
- (30).Zhang R, Evans G, Rotella FJ, Westbrook EM, Beno D, Huberman E, Joachimiak A, Collart FR. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:4691–4700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- (31).Macpherson IS, Kirubakaran S, Gorla SK, Riera TV, D’Aquino JA, Zhang M, Cuny GD, Hedstrom L. The structural basis of Cryptosporidium -specific IMP dehydrogenase inhibitor selectivity. J. Am. Chem. Soc. 2010;132:1230–1231. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- (33).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- (34).Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- (35).Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- (36).Nallamsetty S, Austin BP, Penrose KJ, Waugh DS. Gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Sci. 2005;14:2964–2971. doi: 10.1110/ps.051718605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- (38).Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid-resistant mutants of inosine-5′-monophosphate dehydrogenase. J. Biol. Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]
- (39).Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.