

Abstract
Purpose
To investigate the role of HtrA2/Omi, a nuclear-encoded mitochondrial serine protease with a proapoptosis function, under H2O2-induced oxidative stress in human RPE, in the Ccl2−/− Cx3cr1−/− double-knockout (DKO) mouse retina, and the HtrA2/Omi-deficient mice.
Methods
Oxidative stress was induced in ARPE-19 cells by 1 mM H2O2 for 2 hours. HtrA2/Omi and caspase-3 expression was evaluated using RQ-PCR, immunohistochemistry, or Western blot. Cell viability was detected by MTT assay. HtrA2/Omi expression in the subcellular components and activated caspase-3 were measured. These processes were also evaluated in cells treated with UCF-101, an HtrA2/Omi inhibitor or in cells subjected to RNAi against HtrA2/Omi. Oxidative stress was assayed and compared in retinas of DKO and wild-type (WT) mice by determining serum NADPH oxidase subunits and nitrite levels. Transmission electron microscopy was used to view the retinal ultrastructure of the HtrA2/Omi-deficient mice.
Results
H2O2-induced oxidative damage resulted in HtrA2/Omi translocation from mitochondria to cytosol, leading to RPE cell apoptosis via a caspase-mediated pathway. Treatment of RPE cells with UCF-101 reduced the cytosolic translocation of HtrA2/Omi, attenuated caspase-3 activation, and decreased apoptosis. After specific HtrA2 downregulation, increased cell viability was measured in H2O2-treated ARPE-19 cells. Retina of DKO mice exhibit increased oxidative stress and upregulation of HtrA2/Omi. Fewer and abnormal mitochondria were found in HtrA2/Omi−/− photoreceptors and RPE.
Conclusions
These findings suggest that HtrA2/Omi is related to RPE apoptosis due to oxidative stress, which may play an important role in the integrity of mitochondria and the pathogenesis of AMD.
Age-related macular degeneration (AMD), a neurodegenerative disease of the central retina, is the leading cause of blindness in industrialized countries1 and among the elderly throughout the world.2 Although the exact pathogenesis of AMD remains unclear, the current pathophysiologic concept points to the critical role of cumulative oxidative damage to the retinal pigment epithelium (RPE).3,4 The RPE not only participates in photoreceptor metabolism by transporting nutrients from the choroid into the retina, but also removes waste products from the retina, thereby acting as a metabolic gatekeeper between the photoreceptors and choriocapillaris.5 These cells are involved in maintaining retinal homeostasis by phagocytizing the discs shed from the photoreceptor outer segments.6 Deliberate disruption of these processes in experimental animal models has been shown to result in retinal degeneration,7 and defects in the RPE contribute to the initiation and progression of AMD.8,9
Oxidative damage due to an imbalance between the generation and elimination of reactive oxygen species (ROS) is a major cause of RPE damage in AMD.3,4 High oxygen tension in the macula, exposure to light, and the RPE-mediated processes of phagocytosis and lipid peroxidation increase ROS generation in the RPE.10 Accordingly, oxidative stress may promote AMD pathogenesis by interfering with RPE function, decreasing RPE junctional integrity, enhancing RPE expression of proinflammatory and proangiogenic cytokines, and/or promoting RPE apoptosis.5,11,12 Mitochondria is a major producer of ROS and is particularly prone to ROS injury.13
HtrA2/Omi belongs to the family of high temperature requirement protein A (HtrA) serine proteases conserved from bacteria to humans.14,15 Another member of the family, HtrA1, has been reported to possess several single-nucleotide polymorphisms that are associated with increased risk of AMD.16–21 HtrA1 is a stress-inducible serine protease that has dual functions in both extracellular protein degradation and intracellular growth or survival. It may therefore play a role in angiogenesis or RPE and photoreceptor atrophy in AMD.22–24
Human HtrA2/Omi has been described both as a chaperone protein and as a serine protease responsible for cleavage of denatured proteins at elevated temperatures. HtrA2 is expressed as a 45-kDa precursor protein that is cleaved on post-translational translocation to the mitochondria to generate the mature 36-kDa protein. The mature protein is then localized to the mitochondrial intermembrane space, where it remains until subsequent translocation to the cytosol in response to apoptotic stimuli.25 Once in the cytosol, HtrA2/Omi promotes cell death by two different mechanisms. It can either bind to an inhibitor of apoptosis proteins (IAPs) via its amino terminal, reaper-related motif, and thus, induce caspase activity, or it can mediate caspase-independent death through its own protease activity.26–29
In this study, we sought to determine the role of HrtA2/Omi in oxidatively damaged RPE cells and to assess HtrA2/Omi expressions in the retina of the Ccl2−/−/Cx3cr1−/− murine model showing oxidative stress-related retinal degeneration and HtrA2/Omi−/− mice.
Materials and Methods
The study was conducted in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All animal experiments were performed under the protocols approved by the National Eye Institute Institutional Animal Care and Use Committee.
Cell Cultures
Adult human RPE cells (ARPE-19) were obtained from the American Type Culture Collection (Manassas, VA). This cell line is not transformed and has structural and functional properties characteristic of RPE cells in vivo. The RPE cells were cultured in DMEM/F12 medium (1:1; Sigma-Aldrich, St. Louis, MO) containing 10% fetal bovine serum (Invitrogen, Inc., Grand Island, NY), 100 U/mL penicillin G potassium, and 0.1 mg/mL streptomycin sulfate (Sigma-Aldrich). The cells were cultured at 37°C in humidified 5% CO2 conditions and were split when approximately 90% confluent. The cells propagated rapidly and were used for experiments at passages 3 to 5.
Animals
The development of Ccl2−/−/Cx3cr1−/− (DKO) mice has been de scribed previously.30,31 These DKO mice were characterized by a broad spectrum of AMD-like disease with early onset and high penetrance. Fundoscopy revealed deep retinal focal drusenlike lesions, histology demonstrated RPE and photoreceptor degeneration, and biochemistry revealed increased A2E accumulation and altered mRNA and protein levels of endoplasmic reticulum proteins.30,31 Wild-type (WT) mice were of C57BL/6 background.
HtrA2/Omi−/− mice, which developed Parkinson disease, were generated by using standard embryonic stem cell culture and gene-targeting techniques as previously.32
Drugs and Chemicals
To induce oxidative stress in this study, we made fresh 1 mM hydrogen peroxide (H2O2)–conditioned medium by dissolving 30% H2O2 (Fisher Scientific, Fair Lawn, NJ) in growth medium just before use. The HtrA2/Omi inhibitor, 5-[5-(2-nitrophenyl) furfuryliodine]-1,3-diphenyl-2-thiobarbituric acid (UCF-101) (Calbiochem, Darmstadt, Germany), was dissolved in DMSO (Mediatech, Inc., Herndon, VA) as a stock solution (20 mM) and used at a final concentration of 1 and 2 μM in growth medium. The vehicle group was treated with the DMSO solution dissolved in growth medium at the same ultimate concentrations. Before stimulation with H2O2/UCF-101, the cells were serum starved for 24 hours. All analyses were performed 2 hours after 1 mM H2O2 exposure.
Small RNAi System and Transfection
Small interfering (si)RNAs were synthesized by Dharmacon Research, Inc. (Lafayette, CO). The siRNAHtrA2/Omi consisted of a mixture of four siRNA duplexes targeting four different regions of the HtrA2/Omi mRNA (ON-TARGETplus siRNA SMARTpool human HtrA2/Omi, L-006014-00). A pool of four nontargeting siRNA duplexes was used as a negative control (siRNAcontrol). Transfection of cells with siRNA duplexes was then performed (DharmaFECT1; Dharmacon Research, Inc.). ARPE19 cells were transfected with 20 nM siRNAcontrol and siRNAHtrA2/Omi for 48 hours. The cells were incubated in the siRNA-conditioned medium for the first 24 hours with additional 24-hour incubation in regular medium. The downregulation of HtrA2/Omi was confirmed by Western blot analysis. Subsequently, cells in both groups were stressed for four more hours with 1 mM H2O2. The siRNA study was repeated six times and averaged.
Flow Cytometry
ARPE19 cells were treated with UCF-101 or HtrA/Omi RNAi as previously described. After treatment, the cells were harvested, washed twice with ice-cold PBS, and resuspended in PBS containing 0.25% BSA at a density of 1 × 106/mL. Mitochondrial depolarization was assayed with a JC-1 kit (no. M34152; Invitrogen, Eugene OR). The data were collected with a flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA).
Cell Viability by MTT Assay
Cell viability was assessed by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay to determine the proportion of living cells in control, H2O2-treated, and H2O2 + UCF-101-treated ARPE-19 cells. After exposure to the control or one of the test solutions (H2O2, and/or UCF-101, and/or siRNA reagent) for the indicated time periods, 96-well cultures were incubated with 50 μg/mL MTT at a dilution of 1:10 for 4 hours at 37°C. At the end of the incubation, the MTT solution was removed, and the cells were dissolved in 200 μL DMSO. The proportion of viable cells was determined by measuring the optical density (OD) of each sample at 570 nm with an ELISA plate reader (GE Health Care, Uppsala, Sweden). For repeat studies, six wells were exposed to each solution. The mean optical densities for each group of cultures were compared.
Quantitative RT-PCR
Total RNA extracted (RNeasy Mini Kit; Qiagen, Hilden, Germany) from ARPE-19 cells, retina-RPE tissue of DKO or wild-type mice, was treated with DNase I (Qiagen). Five-microgram RNA from control and H2O2-treated ARPE-19 cells or retina-RPE was used for cDNA synthesis (Superscript II RNase H Reverse Transcriptase; Invitrogen) and RQ-PCR (Mx3000 Real-Time PCR System and Brilliant SYBR Green QPCR Master Mix; Stratagene, La Jolla, CA). Primers of HtrA2 transcripts were synthesized by and supplied as a gene expression assay kit (RT2 Real-Time; Super-Array Bioscience Corp., Frederick, MD). Real-time PCR amplification of β-actin, with primers 5′-TCCCCCAACTTGAGATGTAT-GAAG-3′ and 5′-AACTGGTCTCAAGTCAGTGTACAGG-3′, served as an internal control. The average change (x-fold) resulting from HtrA2 gene manipulation was again normalized to the transcript level of the control groups in both the cell and mice experiments. Three samples were analyzed from each group, and each sample was analyzed three times.
Immunohistochemistry
Expression of HtrA2, XIAP, and activated caspase-3 was measured in the control, H2O2-treated, and H2O2 + UCF-101-treated ARPE-19 cells. The cells were fixed with 4% paraformaldehyde and blocked in 10% goat serum solution. Rabbit anti-HtrA2/Omi antibody (R&D Systems, Minneapolis, MN) and rabbit anti-human activated caspase-3 antibody (Abcam Inc., Cambridge, MA) were used as the primary antibodies. Secondary antibodies were Alexa Fluor 555 goat anti-rabbit or goat anti-mouse IgG. Nuclei were stained with DAPI (diamidino-phenyl-indole; Invitrogen). Staining assays for each primary antibody were repeated at least three times. Confocal microscopy (Leica, Wetzlar, Germany) was used to evaluate immunoreactivity. Expression of HtrA2 in the ocular frozen sections of wide-type and DKO mice was analyzed with a similar protocol.
Measurement of Serum Nitrite and NADP/NADPH Concentration
Serum nitrite concentration was measured by modified Griess colorimetric reaction.33 Briefly, 20 μL of serum was diluted with distilled water to 100 μL and mixed with equal volume of Griess reagent, containing 1.0% sulfanilamide, 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride and 2.5% phosphoric acid, at room temperature. The absorbency was read after 10 minutes in a spectrophotometer at 550 nm and analyzed by reference to a standard nitrite quantitative curve.
Serum NADP/NADPH was measured using the NADP+/NADPH assay kit (ECNP-1000; BioAssay System, Hayward, CA), based on a glucose dehydrogenase cycling reaction.34 The intensity of the reduced product color, measured at 565 nm, is proportionate to the NADP+/NADPH concentration in the sample.
Isolation of Cell Mitochondria and Cytosol
Twenty million resuspended ARPE-19 cells treated with H2O2, H2O2 + vehicle, or H2O2 + UCF-101 were washed with cold PBS at 850g for 5 minutes, and the cytosolic and mitochondrial fractions were isolated according to a modification of the manufacturer’s procedure (Mitochondria Isolation Kit for Cultured Cells; Pierce, Rockford, IL).
Western Blot Analysis
ARPE-19 cells (1 × 106/150-mm dish or 5 × 105/100-mm dish) were seeded for 4 days. After stimulation with H2O2/UCF-101/siRNA, the cells were washed twice with PBS and lysed in 1× RIPA lysis buffer (Upstate, Lake Placid, NY), containing 50 mM Tris-HCl (pH 7.4), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 mM Na3VO4, and 1 mM NaF. Lysates were ultrasonically homogenized and clarified by centrifugation at 16,000g for 30 minutes at 4°C with collection of the resultant supernatant. Protein concentrations were determined by the Bradford method with bovine serum albumin use as the standard. In the animal experiments, neuroretina-RPE cells of DKO mice were dissected and washed twice with PBS and lysed in 1× RIPA lysis buffer.
Total cell lysates were resolved by SDS-polyacrylamide gels, transferred to polyvinylidene difluoride membranes (Invitrogen), and detected with rabbit anti-human HtrA2 antibody (1:2000) and mouse anti-human XIAP (1:2000; Abcam Inc.). The blots were subsequently incubated with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase. Images were developed by using the enhanced chemiluminescence system (Pierce). GAPDH (Invitrogen), COX IV, and tubulin (Invitrogen) were used as the loading control for human cytosol, mitochondria, and mice tissues, respectively.
Transmission Electron Microscopy
Three eyes of HtrA2/Omi−/− and three eyes of WT mice at 30 days of age were harvested for transmission electron microscopy (TEM). After fixation in 4% glutaraldehyde-10% formalin, the retina, RPE, choroid, and sclera were dissected and embedded in epoxy resin (LX-112; LADD Research Industries, Burlington, VT). Ultrathin sections were stained with uranyl acetate and lead citrate. The tissues were examined with a transmission electron microscope (JEM-100B; JEOL, Tokyo, Japan).
Statistical Analysis
The results of the MTT, serum NADP/NADPH, and nitrite assays were entered into a worksheet program (Excel Office 2000; Microsoft Corp., WA). Multiple comparisons among experimental groups were made by one-way analysis of variance (ANOVA) with the level of significance set at P < 0.05. The values are presented as the mean ± SD or SE.
Results
Effect of H2O2 Treatment on Expression of HtrA2/Omi and Promotion of HtrA2/Omi Translocation from Mitochondria to Cytosol in ARPE-19 Cells
Compared with the control cells, ARPE-19 cells treated with H2O2 exhibited a mild increase in HtrA2/Omi transcript levels (2.49-fold change relative to control and protein expression (Fig. 1). Moreover, H2O2 treatment led to a significant decrease in processed HtrA2/Omi in the mitochondria and a significant increase in cytosolic HtrA2/Omi (Fig. 2), suggesting that H2O2-induced oxidative stress promotes translocation of processed HtrA2/Omi from the mitochondria to the cytosol. GAPDH and Cox IV were used as internal controls to verify equivalent cytosolic and mitochondrial protein loading, respectively.
Figure 1.
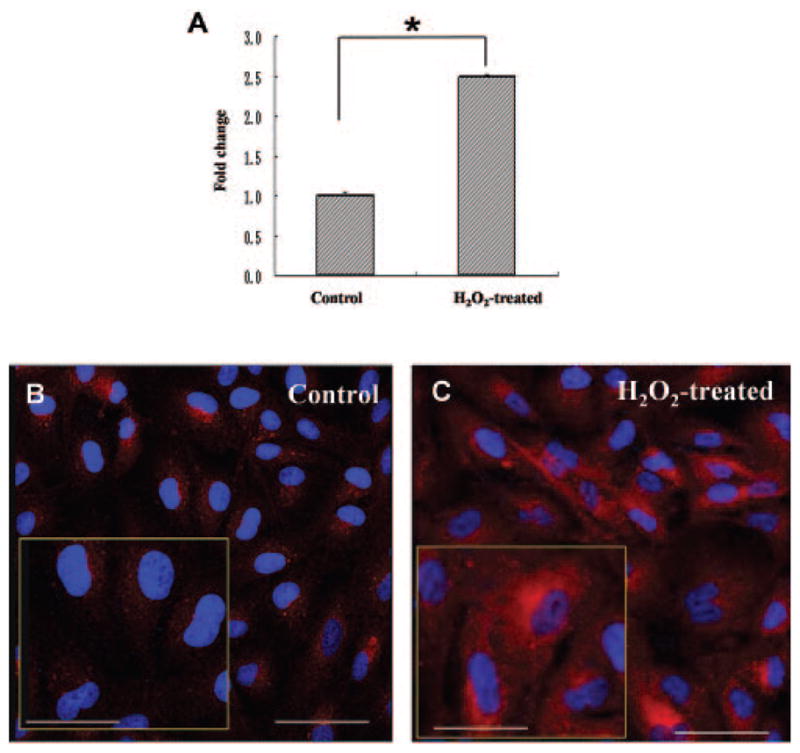
Increased HtrA2/Omi transcript and protein expression in H2O2-treated RPE. ARPE-19 cells were treated with 1 mM H2O2 for 2 hours. The cells were then harvested, and total RNA was isolated and converted to cDNA. (A) RQ-PCR analysis was then performed to determine the levels of HtrA2/Omi transcript. Bars represent the mean ± SD of three independent experiments indicating change (x-fold) compared with untreated cells. The cells were fixed after incubation with 1 mM H2O2 for 2 hours and then stained with rabbit anti-human HrtA2/Omi (red). The nuclei were stained with DAPI (blue). Immunoreactivity shows a mild HtrA2/Omi expression in ARPE-19 cells of (B) the control group and (C) the H2O2-treated group showing a strong increase in HtrA2/Omi expression. Insets: high-power images. Scale bar: (B, C) 100 μm; insets: 50 μm.
Figure 2.

Translocation of HtrA2/Omi from mitochondria to cytosol in H2O2-treated RPE. ARPE-19 cells were left untreated (control) or were treated with 1 mM H2O2 for 2 hours. The cells were then harvested, and the cytosolic and mitochondrial fractions were isolated. Lysates of cytosolic and mitochondrial fractions were separated on 4% to 12% SDS-polyacrylamide gels, and protein expression was detected by immunoblot with rabbit anti-HtrA2/Omi and anti-GAPDH (loading control for total protein and cytosol) or anti-COX IV (loading control for mitochondrial fraction) antibodies. Quantification of protein band density was also performed for each experiment with change relative to protein levels in untreated (control) ARPE-19 cells. The level of processed HtrA2/Omi significantly decreased in the mitochondria (A) and increased in the cytosol (B) of H2O2-treated cells.
Association of H2O2 Treatment with Increased Degradation of Apoptosis Inhibitor XIAP in ARPE-19 Cells
A prior study indicated that HtrA2/Omi activation is associated with decreased levels of X-linked inhibitor of apoptosis (XIAP), suggesting that HrA2/Omi itself cleaves and degrades XIAP.25 We examined the cytosol lysates from control and H2O2-treated cells for XIAP protein levels. XIAP immunoreactivity was evident in Western blots as a single band at a molecular mass of 47 kDa. A significant reduction of XIAP protein was observed in cell lysates treated with H2O2, suggesting that degradation of XIAP occurred after oxidative stress induced by H2O2 (Figs. 3A, 3B). Immunoblot analysis with anti-GADPH antibody confirmed equivalent loading of cell lysates in each lane. These results suggest that oxidative stress-induced cytosolic translocation of HtrA2/Omi is associated with decreased XIAP levels, and thus suggests that HtrA2/Omi may correlate with a proapoptotic cellular environment.
Figure 3.

XIAP degradation and caspase-3 activation after H2O2-induced oxidative stress. (A) Total protein samples were prepared as described in Figure 2 and were detected by immunoblot with rabbit anti-XIAP antibody or GAPDH (loading control). XIAP was decreased in H2O2-induced cells compared with that of untreated cells, indicating that release of mature HtrA2 into the cytosol leads to cleavage and inactivation of XIAP, an inhibitor of apoptosis, thus triggering the cascade of apoptosis. (B) Quantification of XIAP protein levels are represented graphically. (C, D) Cells were fixed after incubation with 1 mM H2O2 for 2 hours and then stained with rabbit anti-human caspase-3 (red). The nuclei were stained with DAPI (blue). The intensities of immunostaining for the activated subunits of caspase-3 were higher in H2O2-treated cells (D) than in untreated control cells (C). Scale bar, 50 μm.
Effect of H2O2 Treatment on Caspase-3 in Cytosol of ARPE-19 Cells
We have thus far demonstrated that H2O2-induced oxidative injury leads to HtrA2/Omi translocation and XIAP degradation. To determine whether these seemingly proapoptotic events are associated with caspase activation, we performed immunohistochemistry for caspase-3-activated subunits. The intensities of immunoreactivity against the activated subunits of caspase-3 were higher in H2O2-treated cells than in untreated control cells (Figs. 3C, 3D). These findings suggest that an increase in HtrA2/Omi after H2O2-induced oxidative stress results in activation of caspase-dependent apoptotic pathways.
Protection of ARPE-19 Cells from H2O2-Induced Oxidative Injury by UCF-101
Given that our findings implied an association between HtrA2/Omi activation and translocation and the proapoptotic state of RPE cells after H2O2-induced oxidative stress, we attempted to strengthen the link between HtrA2/Omi and apoptosis by evaluating whether HtrA2/Omi inhibition by UCF-101 could protect survival of RPE cells after oxidative stress. ARPE-19 cells were treated with 1 mM H2O2 + vehicle or 1 mM H2O2 + UCF-101 (1 or 2 μM) in medium and incubated at 37°C for 2 hours. Western blot analysis showed that UCF-101 treatment led to decreased translocation of HtrA2/Omi from the mitochondria to the cytosol (Fig. 4A). Treatment with 1 μM UCF-101 also increased ARPE-19 cell viability after H2O2-induced oxidative injury, as determined by the MTT assay. The cytoprotective effect increased proportionally as the concentration of UCF-101 increased from 1 to 2 μM, although the change was not statistically significant (Fig. 4B). Notably, a further increase in UCF-101 concentration (>5 μM) no longer offered protection but instead caused a toxic cellular effect (data not shown). Altogether, these results suggest that blocking HtrA2/Omi provides consistent molecular rescue and a modest increase in RPE cell viability under H2O2-induced oxidative stress.
Figure 4.

Decreased HtrA2/Omi expression and increased cell viability after UCF-101 treatment. Mitochondrial and cell lysates were also collected from ARPE-19 cells treated with 1 mM H2O2 + vehicle, 1 mM H2O2 + 1μM UCF-101, or 1 mM H2O2 + 2 μM UCF-101 for 2 hours. The lysates were separated on SDS-polyacrylamide gels and protein expression was detected by immunoblot with rabbit anti-HtrA2/Omi antibody. (A) UCF-101 (1 μM) significantly increased mitochondrial processed HtrA2/Omi levels (37 kDa) but decreased cytosolic HtrA2/Omi levels, suggesting decreased mitochondria-to-cytosol translocation after UCF-101 treatment. This effect was even more prominent with 2 μM UCF-101. (B) Cells from all three groups were also subjected to the MTT cell-viability assay. Data shown are the mean ± SD of results in the three independent experiments, indicating percentage of cell viability compared with that in cells treated with vehicle only. Treatment with UCF-101 increased cell viability in a dose-dependent manner. *Significant difference (P < 0.05). Mit, Mitochondria.
Next we investigated the expression level of activated caspase-3 in UCF-101 and vehicle-treated groups. Immunostaining and immunoblot analysis showed that after H2O2-induced oxidative stress, activated caspase-3 expression levels were lower in UCF-101-treated RPE cells than in those treated with vehicle (Fig. 5). Flow cytometry revealed that after UCF-101 treatment, the membrane potential of mitochondria was less compromised after oxidative stress injury (Fig. 6). The data demonstrated that HtrA2/Omi inhibition by UCF-101 ameliorated cellular injury via inhibition of the caspase-3-mediated apoptosis pathway.
Figure 5.

Decreased activation of caspase-3 after UCF-101 treatment. Cells were fixed after incubation with 1 mM H2O2 + vehicle, 1 mM H2O2 +1 μM UCF-101, or 1 mM H2O2 + 2 μM UCF-101 and then stained with rabbit antibodies against the activated subunits of caspase-3. The nuclei were stained with DAPI (blue). Immunostaining shows (A) positive immunoreactivity against caspase-3 in ARPE19 cells. (B) A significant decrease in the activation of caspase-3 with 1 μM UCF-101 treatment. (C) This effect was even more pronounced after treatment with 2 μM UCF-101. Scale bar: 37.5 μm. (D) Total protein samples were detected by immunoblot with rabbit anti-activated caspase-3 or GAPDH (loading control). Activated caspase-3 was increased in H2O2-induced cells compared with that of untreated cells and decreased after UCF-101 treatment. (E) Quantification of activated caspase-3 protein levels.
Figure 6.

Protective effects against apoptosis after UCF-101 treatment in H2O2-induced oxidative stress. (A) x-Axis: JC-1 green, representative of depolarized mitochondria; y-axis: JC-1 red, representative of healthy mitochondria. The results showed that 93.8% of cells stained with JC-1 red but not with JC-1 green, showing that the mitochondria were healthy; 2.81% cells stained both green and red, indicating compromised mitochondria; 2.72% stained green but showed relatively negative red staining, showing total loss of the integrity of the mitochondrial membrane. (B) Only 16.7% cells were with healthy mitochondria after H2O2 treatment. As suggested in (C), UCF101 protected ARPE19 cells from H2O2-induced apoptosis, 68.7% H2O2 + UCF101-treated cells had healthy mitochondria. Moreover, it was preventive but did not rescue the cells.
Protection of ARPE-19 Cells from Oxidative Injury by Specific HtrA2/Omi Inhibition with RNA Interference
Compared with cells subjected to nonspecific control siRNA, ARPE-19 cells subjected to siRNA directed against HtrA2/Omi exhibited decreased protein levels of both precursor and processed HtrA2, as determined by Western blot (Fig. 7A). Down-regulation of HtrA2, in turn, resulted in increased ARPE-19 cell viability after H2O2-induced oxidative injury, as determined by the MTT assay (Fig. 7B).
Figure 7.

Increased cell viability in H2O2-treated ARPE cells after specific HtrA2 downregulation by RNA interference. (A) Total protein samples in HtrA2-specific interference cells (lanes 1 and 2 from the left), negative control transfected cells (lane 3), and mock-transfected cells (lane 4) were prepared as described in Figure 6 and were detected by immunoblot with rabbit anti-HtrA2 antibody or GAPDH (loading control). The expression of HtrA2 is decreased significantly after transfection, compared with the negative control and mock group. Lanes 1 and 2: results of independent experiments by MTT cell viability assay. Representative images are shown. (B) H2O2 treatment for 2 or 4 hours in HtrA2-downregulated cells and mock transfected cells were also subjected to the MTT cell viability assay. Data shown are mean ± SD of results in three independent experiments indicating the percentage of cell viability compared with that in cells without H2O2 treatment.
Increased Oxidative Stress in DKO Mice
The level of serum NO in wild-type mice was 7.47 ± 0.42 nM (mean ± SE), compared with 8.99 ± 1.14 nM (mean ± SE) in age-matched DKO mice. NADP/NADPH expression (the ratio) was 6.86 ± 3.92 (mean ± SE) in DKO and 0.40 ± 0.04 (mean ± SE) in age-matched WT mice. NAD(P)H oxidase-derived oxidative stress is a critical biomarker for oxidative stress. These results show that DKO mice are subjected to increased oxidative stress. Moreover, as reported previously, retinal lipofuscin, which is a byproduct of the oxidative processes, is increased in DKO mice.30,31 These data suggest an major role for increased oxidative stress in the DKO murine model of AMD.
HtrA2/Omi Transcript and Protein in DKO Mice
Given that DKO mice exhibit increased oxidative stress and in vitro studies demonstrated increased HtrA2/Omi expression in RPE cells subjected to oxidative stress, we examined the expression of HtrA2/Omi in WT and DKO mice. HtrA2/Omi was expressed at a low level in the neuroretina of wild-type mice (Figs. 8A, 8C, 8E). The immunoreactivity was strongest in the photoreceptor inner segments, where mitochondria are abundant. In contrast, a marked increase in HtrA2/Omi expression was observed in both the neuroretina and RPE of DKO mice compared with the control animals (Figs. 8B, 8D, 8F). Western blot analysis also demonstrated an increase in HtrA2/Omi protein in DKO mice, compared with that in the control wild-type (Figs. 9A, 9B). RQ-PCR revealed that HtrA2/Omi transcript levels were 3.5-fold higher in the retinal-RPE cells of DKO mice, compared with those of the wild-type (Fig. 9C). Thus, HtrA2/Omi may mediate some of the pathologic changes seen in these mice.
Figure 8.
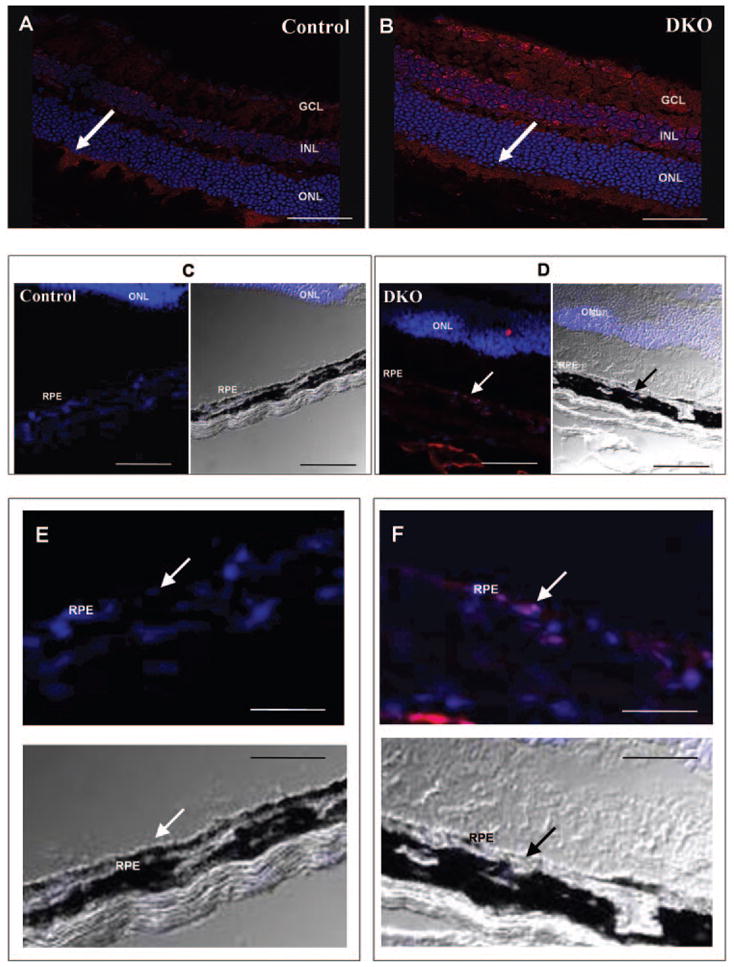
Increased HtrA2/Omi protein expression in DKO mice. Photomicrographs of HtrA2/Omi (red, arrows) in the retina, RPE, and choroid. (A, C, E) HtrA2/Omi was weakly positive in the nerve fiber layer, inner nuclear layer, and inner segment of the normal WT retina but negative in WT RPE. (B, D, F) Enhanced expression of HtrA2/Omi was apparent in DKO retina and RPE. Nuclei were stained with DAPI. Scale bar: (A, B) 50 μm; (C, D) 125 μm; (E, F) 250 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Figure 9.
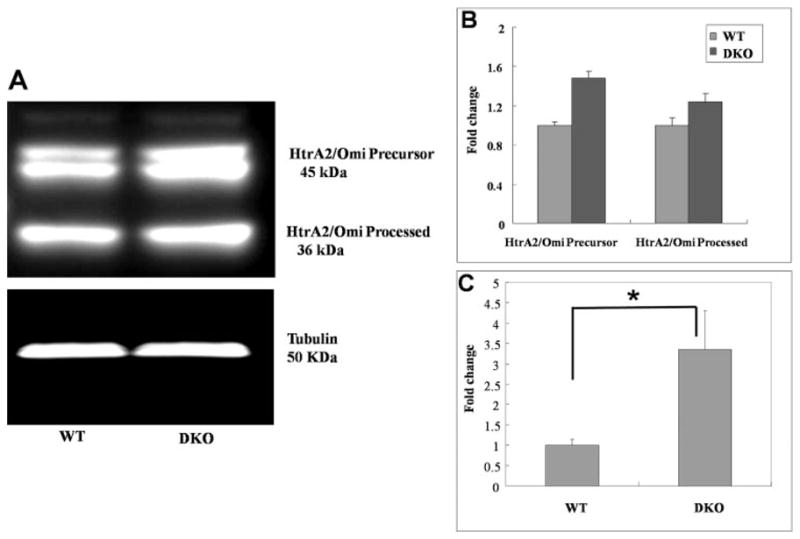
Increased HtrA2/Omi protein and transcripts in DKO mice. (A, B) SDS-polyacrylamide gels demonstrated a slight increase in HtrA2/Omi protein expression in DKO mice eye, compared with the WT. (C) RQ-PCR for HtrA2/Omi mRNA showed significantly higher HtrA2/Omi transcripts in DKO, compared with wild-type control mice. *P < 0.05.
Abnormal Mitochondria in HtrA2−/− Mice
The amount of mitochondria in the photoreceptors and RPE was decreased in the HtrA2/Omi−/− mice compared with WT. The most notable difference was seen in the photoreceptor inner segments, where mitochondria are abundant. A significant decrease in mitochondria was seen in the HtrA2/Omi−/− mice: 22.8 ± 4.15 (mean ± SD)/image in WT mice versus 15.2 ± 3.11/image in knockout mice (P = 0.01; Fig. 10A). Furthermore, the photoreceptors and RPE of the HtrA2/Omi−/− mice contained ballooning (intracristal swelling), branching, and/or fusion of mitochondrial cristae (Figs. 10B, 10C). Cytoplasmic organelle degeneration, vacuolation, and secondary lysosomes were also found in the neuroretina and RPE of the HtrA2−/− mice.
Figure 10.

Disorganized mitochondria in the retina of HtrA2−/− mice. TEM of the photoreceptors of wild-type and HtrA2-deficient mice. (A) Wild-type mouse at 30 days of age: mitochondria (gray arrows) appeared normal with regular cristae (black arrows). (B) HtrA2 deficient mouse at 30 days of age: degenerative mitochondria (gray arrows) contained dilated cristae (black arrows). (C) HtrA2-deficient mouse at 30 days of age showed severely damaged mitochondria (gray arrow) with loss of most cristae. (D) Compared with wild-type mice, HtrA2−/− mice showed a statistically significant decrease in the amount of mitochondria detected by TEM in the photoreceptors. *P < 0.05.
Discussion
Compelling evidence indicates that apoptosis plays a critical role in the pathogenesis of RPE-related diseases including AMD.4,5,35 AMD pathology is characterized by drusen, thickened Bruch’s membrane, apoptosis of RPE and photoreceptors, and, in the wet subtype of AMD, choroidal neovascularization.8 The process of drusen formation in AMD seems to parallel apoptotic processes.36 Moreover, apoptosis is involved in the early outgrowth of choroidal neovascular membranes in wet AMD, and in the development of geographic atrophy in dry AMD.
Severe oxidative stress, which has long been associated with AMD, has been reported to predispose RPE cells to apoptosis and could therefore be involved in the pathogenesis of AMD.37 Because of the high oxygen tension, intense light exposure, and high concentrations of polyunsaturated fatty acids in photoreceptor outer segments as well as the oxidative process of photoreceptor phagocytosis, RPE cells are exposed to a high level of oxidative stress. The role of oxidative damage in AMD pathogenesis is further supported by studies demonstrating an increase in levels of antioxidant proteins in human eyes at advanced stages of AMD,38 and a report from the Age-Related Eye Disease Study (AREDS) has shown that antioxidant vitamins and/or zinc significantly reduces the risk of development of choroidal neovascularization.39
Two interconnected pathways, one extrinsic and another intrinsic, trigger apoptosis.40,41 Extrinsic signaling through death receptors on the cell surface leads to the formation of the death-inducing signaling complex (DISC), whereas intrinsic signaling occurs mainly through mitochondria leading to the formation of the apoptosome. Mitochondria are the key organelles in apoptotic cell death.42 Mutations within the mitochondrial genome have been shown to accumulate with age and are common in neurodegenerative diseases. Various pro-apoptotic stimuli increase outer mitochondrial membrane permeability, allowing the release of multiple mitochondrial pro-apoptotic proteins from the intermembrane space into the cytosol. These proteins include cytochrome c; caspase-2, -3, -7, and -9; and apoptosis-inducing factor (AIF).43 HtrA2/Omi is identified as a proapoptosis mitochondrial protein that is released from mitochondria and directly binds to and inhibits the caspase-inhibitory activity of XIAP.25
In this study, we demonstrated the participation of HtrA2/Omi in RPE cell death from H2O2-induced oxidative stress. In normal conditions, HtrA2/Omi primarily remains in the mitochondrial fraction. H2O2 induced apoptosis is mediated, at least in part, by HtrA2/Omi, which translocates from the mitochondria to the cytosol and subsequently induces apoptosis. We also demonstrated that inhibition of HtrA2/Omi by the HtrA2/Omi serine protease inhibitor UCF-101 or by RNAi reversed apoptosis.
We translated our in vitro findings to an animal model of AMD. The DKO murine model of AMD exhibits increased oxidative stress, as determined by NADPH oxidase subunit expression, serum NO, and retinal lipofuscin.30,31 This increased oxidative stress is associated with increased levels of cytosolic, activated HtrA2/Omi, suggesting that HtrA2/Omi may play a role in the pathogenesis of AMD, similar to that in DKO mice. These findings are consistent with prior in vivo studies demonstrating that HtrA2/Omi translocates from mitochondria to the cytosol in mouse cardiac cells after ischemia and reperfusion.44
Translocation of HtrA2 from mitochondria to the cytosol leads to inactivation of XIAP. Recent findings demonstrate that HtrA2/Omi inactivates XIAP by both direct binding to XIAP through its reaper motif and by serine–protease degradation.25 In the present study, we showed that the increase in cytosolic HtrA2/Omi after H2O2 treatment correlates with a significant decrease in XIAP levels, presumably by HtrA2/Omi-mediated XIAP inhibition and degradation. Decreased inhibition of apoptosis by XIAP frees caspase-3 to trigger the caspase-dependant apoptotic pathway, thus increasing apoptotic damage to the RPE cells. Our finding confirms that HtrA2/Omi plays a critical role in oxidative stress-induced RPE cell apoptosis, an important factor in the pathogenesis of RPE-related diseases including AMD. In fact, the findings of increased oxidative stress and elevated HtrA2/Omi mRNA and protein levels in the retina-RPE tissues of the DKO mice further suggest a role for HtrA2/Omi in AMD pathogenesis.
We also evaluated the potential of the HtrA2/Omi inhibitor UCF-101 to protect RPE cells from H2O2-induced oxidative stress. Our result shows that UCF-101 treatment decreases the translocation of HtrA2/Omi from the mitochondria to the cytosol after H2O2-induced oxidative stress, which correlates with an amelioration in caspase-3 activation and accordingly, an increase in RPE cell viability. UCF-101 found by screening a combinatorial library with bacterially made Omi-(134 – 458) protease and fluorescein-casein as a generic substrate,45 has a specific activity against HtrA2/Omi with very little cross reactivity with various other serine proteases. Our RNAi data further confirm the cytoprotective effect of HtrA2/Omi, as RNAi-induced downregulation of this molecule resulted in a significant improvement in cell viability in the face of oxidative stress. Therefore, we have provided strong evidence that anti-HtrA2 treatment exerts cytoprotective effects by inhibiting HtrA2/Omi translocation to the cytosol and preventing Htra2/Omi-mediated activation of the apoptosis pathway, at least partly by attenuation of XIAP degradation.
Of interest, HtrA2/Omi can play dual roles in mammalian cells, acting not only to induce apoptosis under cellular stress, but also to maintain mitochondrial homeostasis under normal condition.46 Indeed, several caspase-independent death effectors including apoptosis-inducing factor, endonuclease G, and the HtrA2/Omi serine protease are released from the mitochondrial intermembrane space on permeabilization of the outer membrane.40 Such proteins also have important roles in cellular redox metabolism and/or mitochondrial biogenesis. As a general rule, it thus appears that cell-death-relevant proteins, especially those involved in the core of the apoptosis-executing machinery, have a dual function in life and death. On induction of apoptosis, HtrA2/Omi is released into the cytosol where it promotes cell death.28,47 On the other hand, HtrA2/Omi has been shown to play a protective role against stress in neurons,48,49 and a loss of function mutation in the gene encoding HtrA2/Omi is reported in human Parkinson disease.50 Given the role of mitochondrial integrity in apoptosis, paradoxically, under nonstress conditions, the inhibition of HtrA2/Omi may actually enhance susceptibility to apoptosis and may explain the increased degeneration of striatal neurons in HtrA2/Omi−/− mice.32 These mice allow us to examine the direct effect of HtrA2/Omi gene on ocular tissues. We found abnormal mitochondria in HtrA2/Omi−/− retina, a similar observation in the substantia nigra of these mice.32 It is possible that HtrA2/Omi protease is essential for the maintenance of mitochondrial function in normal cells not committed to apoptosis. Indeed, we documented loss and abnormal mitochondria in human AMD eyes.51 Taken together, these findings suggest that HtrA2/Omi has a complex role in cell death; under normal conditions HtrA2/Omi expression may be beneficial in mitochondrial integrity; however, under oxidative stress, enhanced HtrA2/Omi expression may become harmful to mitochondrial activity. The precise characterization of HtrA2/Omi-induced cell death requires further evaluation in more physiological and pathologic conditions.
In summary, we have demonstrated, for the first time, that in vitro oxidative stress promotes mitochondria-to-cytosol translocation of HtrA2/Omi in APRE-19 cells, leading to decreased XIAP, increased activity of caspase-3, and ultimately, decreased cell viability. In addition, inhibition of HtrA2/Omi either pharmacologically or by RNAi leads to cell protection by inhibiting cytosolic translocation of HtrA2/Omi and antagonizing the caspase-dependent apoptosis pathway. Therapeutic interventions that inhibit HtrA2/Omi expression, translocation, or protease activity may therefore represent novel therapeutic strategies for RPE and RPE-related diseases, including AMD, in which apoptotic cell death is one of the key pathologic components.
Acknowledgments
Supported by the National Institutes of Health Intramural Research Foundation.
Footnotes
Disclosure: X. Ding, None; M. Patel, None; D. Shen, None; A.A. Herzlich, None; X. Cao None; R. Villasmil, None; K. Klupsch, None; J. Tuo, None; J. Downward, None; C.-C. Chan, None
References
- 1.Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 2.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration: emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King A, Gottlieb E, Brooks DG, Murphy MP, Dunaief JL. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem Photobiol. 2004;79:470–475. doi: 10.1562/le-03-17.1. [DOI] [PubMed] [Google Scholar]
- 4.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 5.Binder S, Stanzel BV, Krebs I, Glittenberg C. Transplantation of the RPE in AMD. Prog Retin Eye Res. 2007;26:516–554. doi: 10.1016/j.preteyeres.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Bird AC. The Bowman lecture: towards an understanding of age-related macular disease. Eye. 2003;17:457–466. doi: 10.1038/sj.eye.6700562. [DOI] [PubMed] [Google Scholar]
- 7.Gal A, Li Y, Thompson DA, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 8.Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 9.Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina: relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–1699. [PubMed] [Google Scholar]
- 10.Cai J, Nelson KC, Wu M, Sternberg P, Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 11.Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- 12.Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:675–684. doi: 10.1167/iovs.03-0351. [DOI] [PubMed] [Google Scholar]
- 13.Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- 14.Gray CW, Ward RV, Karran E, et al. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur J Biochem. 2000;267:5699–5710. doi: 10.1046/j.1432-1327.2000.01589.x. [DOI] [PubMed] [Google Scholar]
- 15.Savopoulos JW, Carter PS, Turconi S, et al. Expression, purification, and functional analysis of the human serine protease HtrA2. Protein Expr Purif. 2000;19:227–234. doi: 10.1006/prep.2000.1240. [DOI] [PubMed] [Google Scholar]
- 16.Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, Camp NJ, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–993. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- 18.Cameron DJ, Yang Z, Gibbs D, et al. HTRA1 variant confers similar risks to geographic atrophy and neovascular age-related macular degeneration. Cell Cycle. 2007;6:1122–1125. doi: 10.4161/cc.6.9.4157. [DOI] [PubMed] [Google Scholar]
- 19.Mori K, Horie-Inoue K, Kohda M, et al. Association of the HTRA1 gene variant with age-related macular degeneration in the Japanese population. J Hum Genet. 2007;52:636–641. doi: 10.1007/s10038-007-0162-1. [DOI] [PubMed] [Google Scholar]
- 20.Tam PO, Ng TK, Liu DT, et al. HTRA1 variants in exudative age-related macular degeneration and interactions with smoking and CFH. Invest Ophthalmol Vis Sci. 2008;49:2357–2365. doi: 10.1167/iovs.07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuo J, Ross R, Reed G, et al. The HtrA1 promoter polymorphism, smoking, and age-related macular degeneration in multiple case-control samples. Ophthalmology. 2008;115:1891–1898. doi: 10.1016/j.ophtha.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oka C, Tsujimoto R, Kajikawa M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development. 2004;131:1041–1053. doi: 10.1242/dev.00999. [DOI] [PubMed] [Google Scholar]
- 23.Grau S, Richards PJ, Kerr B, et al. The role of human HtrA1 in arthritic disease. J Biol Chem. 2006;281:6124–6129. doi: 10.1074/jbc.M500361200. [DOI] [PubMed] [Google Scholar]
- 24.Lehner T. Special regulatory T cell review: the resurgence of the concept of contrasuppression in immunoregulation. Immunology. 2008;123:40–44. doi: 10.1111/j.1365-2567.2007.02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins LM. The serine protease Omi/HtrA2: a second mammalian protein with a Reaper-like function. Cell Death Differ. 2002;9:699–701. doi: 10.1038/sj.cdd.4401044. [DOI] [PubMed] [Google Scholar]
- 26.Deveraux QL, Reed JC. IAP family proteins: suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 28.Hegde R, Srinivasula SM, Zhang Z, et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 29.Martins LM, Iaccarino I, Tenev T, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 30.Tuo J, Bojanowski CM, Zhou M, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan CC, Ross RJ, Shen D, et al. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martins LM, Morrison A, Klupsch K, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding A, Nathan C, Stuehr D. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages: comparison of activating cytokines and evidence for independent production. J Immunol. 1988;131:2407–2412. [PubMed] [Google Scholar]
- 34.Zhao Z, Hu X, Ross C. Comparison of tissue preparation methods for assay of nicotinamide coenzymes. Plant Physiol. 1987;84:987–988. doi: 10.1104/pp.84.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haddad WM, Coscas G, Soubrane G. Age-related macular degeneration and apoptosis (in French) J Fr Ophtalmol. 2003;26:307–311. [PubMed] [Google Scholar]
- 36.Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasahara E, Lin LR, Ho YS, Reddy VN. SOD2 protects against oxidation-induced apoptosis in mouse retinal pigment epithelium: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:3426–3434. doi: 10.1167/iovs.05-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The AREDS Study Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrido C, Kroemer G. Life’s smile, death’s grin: vital functions of apoptosis-executing proteins. Curr Opin Cell Biol. 2004;16:639–646. doi: 10.1016/j.ceb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]
- 42.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 43.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 44.Liu HR, Gao E, Hu A, et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111:90–96. doi: 10.1161/01.CIR.0000151613.90994.17. [DOI] [PubMed] [Google Scholar]
- 45.Cilenti L, Lee Y, Hess S, et al. Characterization of a novel and specific inhibitor for the pro-apoptotic protease Omi/HtrA2. J Biol Chem. 2003;278:11489–11494. doi: 10.1074/jbc.M212819200. [DOI] [PubMed] [Google Scholar]
- 46.Liu ML, Liu MJ, Kim JM, Kim HJ, Kim JH, Hong ST. HtrA2 interacts with A beta peptide but does not directly alter its production or degradation. Mol Cells. 2005;20:83–89. [PubMed] [Google Scholar]
- 47.Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 48.Plun-Favreau H, Klupsch K, Moisoi N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 49.Liu MJ, Liu ML, Shen YF, et al. Transgenic mice with neuron-specific overexpression of HtrA2/Omi suggest a neuroprotective role for HtrA2/Omi. Biochem Biophys Res Commun. 2007;362:295–300. doi: 10.1016/j.bbrc.2007.07.118. [DOI] [PubMed] [Google Scholar]
- 50.Bogaerts V, Nuytemans K, Reumers J, et al. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat. 2008;29:832–840. doi: 10.1002/humu.20713. [DOI] [PubMed] [Google Scholar]
- 51.Ding X, Patel M, Chan C. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28:1–18. doi: 10.1016/j.preteyeres.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]