

Abstract
A series of new resveratrol analogues were designed and synthesized and their inhibitory activities against aromatase were evaluated. The crystal structure of human aromatase (PDB 3eqm) was used to rationalize the mechanism of action of the aromatase inhibitor 32 (IC50 0.59 µM) through docking, molecular mechanics energy minimization, and computer graphics molecular modeling, and the information was utilized to design several very potent inhibitors, including compounds 82 (IC50 70 nM) and 84 (IC50 36 nM). The aromatase inhibitory activities of these compounds are much more potent than that for the lead compound resveratrol, which has an IC50 of 80 µM. In addition to aromatase inhibitory activity, compounds 32 and 44 also displayed potent QR2 inhibitory activity (IC50 1.7 µM and 0.27 µM, respectively) and the high-resolution X-ray structures of QR2 in complex with these two compounds provide insight into their mechanism of QR2 inhibition. The aromatase and quinone reductase inhibitors resulting from these studies have potential value in the treatment and prevention of cancer.
Keywords: Resveratrol analogues, Aromatase, Quinone reductase 2
1. Introduction
Resveratrol (3,5,4’-trihydroxystilbene) (Figure 1) was first isolated from the roots of the white hellebore lily Veratrum grandiflorum O. Loes in 1940.1 This compound is a naturally occurring phytoalexin produced by a wide range of plants (at least 72 plant species) in response to environmental stress or pathogenic attack. Since the discovery of its cardioprotective activity in 1992, resveratrol research has steadily accelerated. This increased attention eventually resulted in the discovery of its cancer chemopreventive properties.2 Resveratrol was found to be capable of interfering with all three steps of carcinogenesis (initiation, promotion, and progression).2
Figure 1.

Chemical Structures of Resveratrol and Compound JH-2–29
Resveratrol has been reported to exert a variety of biological activities. Some of these include antioxidant, anti-inflammatory, anti-infective, anti-ischemic, cardioprotective, neuroprotective, anti-aging (prolongs lifespan), anti-obesity, anti-viral, and cancer chemopreventive effects.3 These effects are mediated through several biological receptors, including cyclooxygenase (COX), lipooxygenase (LOX), nuclear factor kappa-beta (NF-kB), quinone reductase 1 (QR1), quinone reductase 2 (QR2), ornithine decarboxylase (ODC), and aromatase.
In the United States, breast cancer is the most commonly diagnosed type of cancer in women and is the second leading cause of death from cancer in women. Approximately 180,000 new cases of breast cancer were detected among women in the United States in 2008.4 About two-thirds premenopausal and three-fourths postmenopausal breast cancer patients have estrogen-dependent cancer.5
In hormone-dependent breast cancer, estrogens play a critical role in stimulating breast cancer cell proliferation.6 High levels of estrogens promote the progression of breast cancer. Two main strategies have been devised by medicinal chemists to control or block the pathological activity of estrogens.7 The first strategy involves the design and synthesis of estrogen receptor antagonists, which has yielded useful anticancer and chemopreventive drugs such as tamoxifen and raloxifene.8,9 Aromatase, a pivotal enzyme responsible for the conversion of androgens to estrogens, is "the other" attractive biological target for the development of new agents for the treatment of breast cancer.10–14 As the enzyme responsible for the final step of the estrogen biosynthetic pathway, selective inhibition of aromatase will not interfere with the production of other steroids in the pathway (e.g. adrenal corticoids).15,16 Therefore, aromatase is a useful therapeutic target in the treatment or prevention of estrogen-dependent breast cancers.17
Quinone reductase 2 (QR2) is a cytosolic FAD-dependent flavoenzyme that catalyzes the reduction of quinones by reduced N-alkyl- and N-ribosylnicotinamides.18 Recent studies have indicated that QR2 may transform certain quinone substrates into more highly reactive species that are capable of causing increased cellular damage.19–21 Therefore, it is hypothesized that inhibition of QR2 in certain cases may lead to protection of cells against these reactive species. Several QR2 inhibitors have been reported in the literature22 and the X-ray crystal structure of QR2 in complex with resveratrol has been determined.23,24 These observations motivated the present study.
Both aromatase and QR2 have been targeted for discovery and identification of chemopreventive agents.24 An array of resveratrol analogues have been synthesized in our laboratory in an attempt to discover new inhibitors of these two enzymes. The trans stilbene JH-2–29 (Figure 1, Scheme 7), with a para-amino group, was found to exhibit versatile biological activities including nitric oxide synthase inhibition, aromatase inhibition (IC50 22 µM), and inhibition of TNF-α-induced NF-κB activity, and was therefore selected as a lead compound for further optimization. In order to improve the potency and selectivity of JH-2–29, a limited number of methoxylated compounds were synthesized and observed empirically to enhance aromatase inhibitory activity. In particular, compound 32 (Figure 2) was found to possess significant aromatase inhibitory activity, with an IC50 value of 0.59 µM, which is comparable to the clinically useful aromatase inhibitor 2-aminoglutethimide (IC50 0.27 µM) (Figure 2).18 Molecular modeling was used to investigate the mechanism of action, and a variety of structurally related stilbenes were designed and synthesized based on the results. Several of these synthetic derivatives were found to inhibit aromatase activity with IC50 values in the sub-micromolar range. In addition, two compounds, 32 and 44, were discovered to exhibit potent QR2 inhibitory activity, and X-ray crystallographic analysis revealed these two compounds bind to QR2 in an orientation similar to that of resveratrol, but both compounds demonstrate greater inhibitory potency than resveratrol.
Figure 7.

High-resolution X-ray Structures of human QR2 in complex with resveratrol (A), compound 32 (B) and compound 44 (C). All panels are presented in walleyed stereoviews showing the FAD cofactor and the active site residues Asn161 and Thr71. Active site water molecules are shown as red spheres and hydrogen bonds are shown as dashed lines. 2Fo-Fc electron density maps are contoured to 1 σ. Carbon atoms and bonds are colored blue for resveratrol, green for compound 32 and cyan for compound 44. The X-ray structure of the QR2-resveratrol complex is from PDB code 1SG0.22
Figure 2.

Chemical Structures of Compound 32 and Aminoglutethimide
2. Results and Discussion
2.1. Chemistry
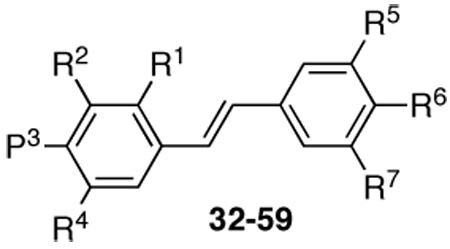
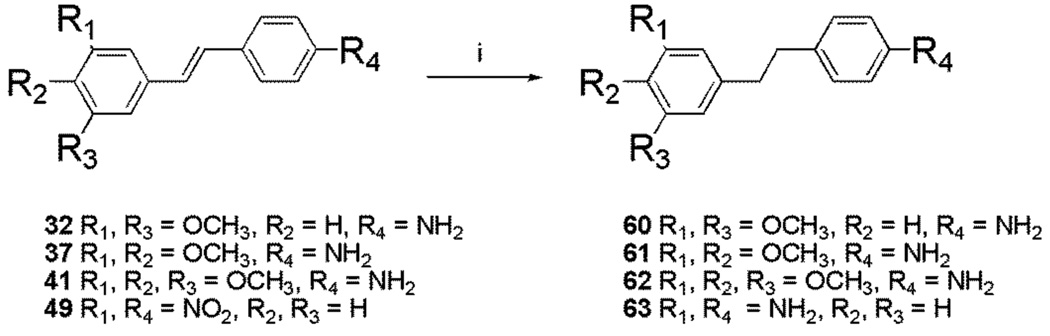
Preparation of the trans stilbene derivatives 32–59 was accomplished mainly by means of the Wittig reaction between the appropriately substituted aromatic aldehydes 1–12 and methyl triphenylphosphonium bromide, and the following Heck reaction between appropriately substituted vinylbenzenes 11–22 and iodobenzenes 23–31 as shown in Scheme 1 and Table 1. The coupling constants of the vinylic protons of the trans stilbenes were about 16 Hz. The double bonds of the analogues 32, 37, 41 and 49 were reduced by catalytic hydrogenation to provide the corresponding derivatives 60, 61, 62 and 63 (Scheme 2). Dinitro analogues 50 and 52 were reduced with stannous chloride to provide the respective polyamines 64 and 65 in satisfactory yields (Scheme 3). The acetate groups of compounds 39 and 55 were hydrolyzed by sodium methoxide in methanol to yield phenol derivatives 66 and 67 (Scheme 4). The tert-butyldimethylsilyl (TBDMS)-protected vinylbenzene 69 (Scheme 5) reacted with 4-iodoaniline in a standard Heck reaction, but the yield was very low. The method of Angela et al. was used and the TBDMS group of intermediate compound 69 was cleaved by tetrabutylammonium fluoride (TBAF) to afford the diphenol 70, which was acetylated to produce 71. Heck reaction between compound 71 and 4-iodoaniline or 4-iodonitrobenzene led to the respective trans-stilbenes 72 and 73. Acetate hydrolysis yielded the desired compounds 74 and 75.25 The synthetic route to imidazole analogue 82 began with the preparation of phosphonium salt 78 from aldehyde 5, and ketone 80 from commercially available compound 79 (Scheme 6). The building blocks 78 and 80 were then combined by the Wittig reaction in the presence of potassium carbonate and 18-crown-6,26,27 and the resulting E and Z isomers were separated and purified by chromatography to provide the nitro analogue 82. Reduction by stannous chloride resulted in the amine derivative 84 (Scheme 6). The double bond conformation of compound 82 was confirmed by 2D 1H-1H NOESY spectroscopy (Figure 3).
Table 1.
Structures of Compounds 32–59
 | |||||||
| Compd | R1 | R2 | R3 | R4 | R5 | R6 | R7 |
| 32 | H | OCH3 | H | OCH3 | H | NH2 | H |
| 33 | OCH3 | H | OCH3 | H | H | NH2 | H |
| 34 | OCH3 | H | OCH3 | H | NH2 | H | H |
| 35 | OCH3 | H | H | OCH3 | H | NH2 | H |
| 36 | OCH3 | OCH3 | H | H | H | NH2 | H |
| 37 | H | OCH3 | OCH3 | H | H | NH2 | H |
| 38 | H | OCH3 | OCH3 | H | H | CN | H |
| 39 | H | OCH3 | OCH3 | H | H | OAc | H |
| 40 | H | OCH3 | OCH3 | H | H | F | H |
| 41 | H | OCH3 | OCH3 | OCH3 | H | NO2 | H |
| 42 | H | OCH3 | OCH3 | OCH3 | NH2 | H | H |
| 43 | H | OCH3 | OCH3 | OCH3 | H | CH2NH2 | H |
| 44 | H | OCH3 | OCH3 | OCH3 | H | NH2 | H |
| 45 | OCH3 | OCH3 | OCH3 | H | H | NH2 | H |
| 46 | OCH3 | OCH3 | H | OCH3 | H | NH2 | H |
| 47 | H | -OCH2O- | H | NH2 | H | H | |
| 48 | H | -OCH2O- | H | H | NH2 | H | |
| 49 | H | H | NO2 | H | NO2 | H | H |
| 50 | H | NO2 | H | H | H | NH2 | H |
| 51 | H | OCH3 | H | H | H | NH2 | H |
| 52 | H | H | NO2 | H | NO2 | H | NO2 |
| 53 | H | OCH3 | H | OCH3 | H | NO2 | H |
| 54 | H | OCH3 | H | OCH3 | NH2 | H | H |
| 55 | H | OCH3 | H | OCH3 | H | OAc | H |
| 56 | H | OCH3 | H | OCH3 | H | CH2NH2 | H |
| 57 | H | OCH3 | H | OCH3 | H | F | H |
| 58 | H | OCH3 | H | OCH3 | H | Cl | H |
| 59 | H | OCH3 | H | OCH3 | H | CN | H |
Figure 3.

Region of a 2D 1H-1H NOESY spectrum of 82. The red circle indicates the NOE signal of H8 and H17/21.
Figure 4.

(A) Ribbon diagram illustrating the hypothetical complex structure of human aromatase with compound 32 bound to the active site, and the hydrogen bonds are labeled as yellow broken lines. (B) Potential interactions between compound 32 and Ile305, Asp309, Thr310, Trp224, Val370, Met374 in human aromatase active site. The hydrogen bonds are labeled as yellow broken lines.
Figure 5.

Design of the novel resveratrol analogues based on the docking result of compound 32.
Figure 6.

Potential interactions between compound 84 and Ile305, Asp309, Thr310, Trp224, Val370, Met374 and heme in human aromatase active site. The hydrogen bonds are labeled as yellow broken lines.
2.2. Mechanism
The aromatase inhibitory activity of the resveratrol analogue 32 (IC50 0.59 µM) in comparison with resveratrol itself (IC50 80 µM) led to an investigation of the mechanism of action of compound 32. Recently, Ghosh et al. solved the crystal structure of human aromatase in complex with androgen.28 Compound 32 was docked into the binding site of Ghosh’s structure (PDB code 3eqm) using the GOLD (Genetic Optimization for Ligand Docking) program,29–31 and the energy minimized. In the resulting hypothetical structure (Figure 4), the amine group on ring A hydrogen bonds with the carbonyl group of Ile305 and carboxyl group of Asp309, which plays a critical role in catalysis.28 Ring A is involved in a π-π interaction with the indole ring of Trp224, and a CH-π interaction with methyl group of Thr310. Ring B forms a CH-π interaction with methyl group of Val370. The methoxyl group on ring B is involved in a potential hydrogen bond with the backbone NH of Met374. All of the residues mentioned above are included in the catalytic cleft of aromatase.28
The docking and energy minimization results provide a good foundation for design of new analogues. In order to minimize the possible cytotoxicity contributed by the aniline part of compound 32, the amino group on ring A was replaced by hydrogen bond-donating groups, such as hydroxy and aminomethyl groups, which may form hydrogen bonds with Asp309 and Ile305 (Figure 5). As shown in Figure 4, the backbone carbonyl group of residue Leu477, a potential hydrogen bond acceptor located near ring B, suggested the addition of hydrogen bond donating groups on ring B. Met374, working as a hydrogen bond donor, also suggests incorporation of hydrogen bond acceptors on ring B. In order to probe the function of the stilbene double bond, compounds with single bonds were also synthesized. Many aromatase inhibitors have imidazole or triazole groups that can coordinate to the iron atom,32–36 and these were therefore added to the stilbene framework and the resulting structures were modeled in complex with aromatase.
2.3. Aromatase Inhibitory Activity
An aromatase inhibition assay was performed on synthetic analogues of compound JH-2–29, as described in the Experimental Section. The inhibition percentage at a concentration of 20 µg/mL and the IC50 values of the more active compounds are reported in Table 2. Most of the resveratrol analogues with an amino group on the para position of ring A (32, 33, 35, 36, 37, 44, 45, 46, 48, 51, 64, 65, 72 and 74) consistently exhibited significant activity, with IC50 values in the 0.59 to 14.51 µM range. Substitution of the para amino group with nitro, halogen, hydroxy, nitrile, acetyl and aminomethyl groups made the resulting compounds inactive (compounds 38, 39, 40, 41, 43, 52, 53, 55, 56, 57, 58, 59, 66, 67 and 75). Changing the position of amino group on ring A also led to reduced activity (compounds 34, 42 and 54). Presumably, altering the position of the amino group is expected to influence its ability to simultaneously hydrogen bond with the Asp309 carboxylate and the backbone carbonyl oxygen of Ile305 (Figure 4). Double bond reduction impacted the activity positively (37 vs. 61, 49 vs. 63, and 41 vs. 62) or negatively (32 vs. 60) to some extent. Within the series, the position of amino group on ring A was of greater importance than that of methoxy groups (ring B), and a para amino group was better than an ortho or meta amino group.
Table 2.
Inhibition of the Human Aromatase by Resveratrol Analogues
| Cpd | % inhib (20 µg/mL)a |
IC50 (µM)b |
Cpd | % inhib (20 µg/mL)a |
IC50 (µM)b |
Cpd | % inhib (20 µg/mL)a |
IC50 (µM)b |
|---|---|---|---|---|---|---|---|---|
| 32 | 92.91 | 0.59 ± 0.07 | 47 | 77.6 | 21.86 ± 0.44 | 62 | 89.67 | 2.79 ± 0.11 |
| 33 | 91.14 | 0.76 ± 0.04 | 48 | 81.80 | 8.49 ± 0.08 | 63 | 87.01 | 3.21 ± 0.09 |
| 34 | 76.27 | 11.14 ± 3.09 | 49 | 63.71 | 8.33 ± 0.81 | 64 | 88.68 | 7.47 ± 0.79 |
| 35 | 80.94 | 1.82 ± 0.03 | 50 | 38.08 | N/Tc | 65 | 88.04 | 8.51 ± 0.64 |
| 36 | 91.22 | 0.98 ± 0.12 | 51 | 89.90 | 3.08 ± 0.44 | 66 | 13.18 | N/Tc |
| 37 | 78.93 | 14.51 ± 0.20 | 52 | 4.21 | N/Tc | 67 | 23.52 | N/Tc |
| 38 | 31.38 | N/Tc | 53 | 0 | N/Tc | 72 | 86.39 | 2.94 ± 0.43 |
| 39 | 5.33 | N/Tc | 54 | 44.25 | N/Tc | 73 | 35.82 | N/Tc |
| 40 | 7.56 | N/Tc | 55 | 22.19 | N/Tc | 74 | 94.01 | 5.0 ± 1.17 |
| 41 | 6.74 | N/Tc | 56 | 19.65 | N/Tc | 75 | 45.23 | N/Tc |
| 42 | 83.50 | 16.42 ± 0.21 | 57 | 15.38 | N/Tc | 80 | 94.92 | 0.19 ± 0.08 |
| 43 | 12.01 | N/Tc |
58 | 14.65 | N/Tc | 81 | 94.20 | 1.84 ± 0.02 |
| 44 | 90.86 | 0.90 ± 0.08 | 59 | 18.33 | N/Tc | 82 | 91.4 | 0.07 ± 0.02 |
| 45 | 88.94 | 3.57 ± 0.11 | 60 | 90.66 | 2.76 ± 0.33 | 84 | 93.6 | 0.036 ± 0.003 |
| 46 | 81.26 | 2.28 ± 0.28 | 61 | 92.49 | 0.67 ± 0.07 |
The inhibition percentage was tested at a concentration of 20 µg/mL of each compound.
The IC50 values (µM) are the concentrations corresponding to 50% inhibition of aromatase.
N/T means not tested (in general, compounds with low percent inhibitions in the initial screening at 20 µg/mL were not evaluated more extensively to determine their IC50 values).
Compound 84 was the most potent aromatase inhibitor, with an IC50 value of 36 nM. As expected, molecular modeling indicated that one of the imidazole nitrogens coordinates with the iron atom of heme, which plays a critical role in catalysis (Figure 6).28
KM and Vmax values for those analogues demonstrated to inhibit aromatase can be found in Table 3. Vmax values for each inhibitor are quite similar to each other and to the Vmax of the control (34.8 pmol/min/mg protein), indicating that these resveratrol analogues are competitive inhibitors of CYP19.
Table 3.
KM and Vmax Values for Resveratrol Analogues Active as Inhibitors of Aromatasea
| Compound Number | KM (µM) | Vmax |
|---|---|---|
| 32 | 7.44 ± 0.06 | 35.2 ± 0.33 |
| 33 | 10.68 ± 0.19 | 35.6 ± 0.18 |
| 34 | 2.87 ± 0.08 | 35.1 ± 0.31 |
| 35 | 8.21 ± 0.24 | 34.2 ± 0.21 |
| 36 | 5.92 ± 0.14 | 34.8 ± 0.41 |
| 37 | 2.57 ± 0.13 | 34.8 ± 0.15 |
| 42 | 2.35 ± 0.10 | 34.4 ± 0.26 |
| 44 | 6.25 ± 0.11 | 35.4 ± 0.27 |
| 45 | 7.48 ± 0.17 | 34.9 ± 0.34 |
| 46 | 9.31 ± 0.21 | 34.6 ± 0.29 |
| 47 | 2.19 ± 0.05 | 34.9 ± 0.24 |
| 48 | 7.59 ± 0.09 | 35.1 ± 0.29 |
| 49 | 6.94 ± 0.07 | 34.8 ± 0.29 |
| 51 | 8.75 ± 0.16 | 35 ± 0.08 |
| 60 | 8.35 ± 0.13 | 34.8 ± 0.25 |
| 61 | 8.72 ± 0.18 | 34.7 ± 0.16 |
| 62 | 8.53 ± 0.16 | 34.7 ± 0.26 |
| 63 | 6.99 ± 0.20 | 34.9 ± 0.30 |
| 64 | 7.83 ± 0.14 | 35.3 ± 0.18 |
| 65 | 7.81 ± 0.13 | 35 ± 0.22 |
| 72 | 8.21 ± 0.09 | 35.2 ± 0.22 |
| 74 | 6.54 ± 0.15 | 34.3 ± 0.35 |
| 80 | 12.78 ± 0.14 | 35.1 ± 0.17 |
| 81 | 8.76 ± 0.20 | 34.7 ± 0.21 |
| 82 | 15.2 ± 0.23 | 35.5 ± 0.28 |
| 84 | 16.42 ± 0.17 | 34.4 ± 0.33 |
The IC50 values are listed in Table 2.
2.4. Quinone Reductase 2 Inhibitory Activity and X-ray Structure Analysis
QR2 inhibition assays were performed on analogues of synthetic compound JH-2–29, as described in the Experimental Section. Compounds 32 and 44 exhibited potent QR2 inhibitory activity with IC50 values of 1.7 ± 0.06 µM and 0.27 ± 0.01 µM, respectively. Both of these substances are more potent than resveratrol, which has an IC50 value of 5.1 µM in the same assay.
To gain insight into the binding mode of these two inhibitors, the X-ray crystal structures of both compounds 32 and 44 in complex with human QR2 were determined. Complete X-ray data sets were collected and processed, and the final structural models were refined to 1.74 Å and 1.55 Å for compound 32 and compound 44 complexes, respectively. As revealed by the strong and well-defined electron density shown in Figure 7, both compounds 32 and 44 bind to QR2 in an orientation similar to that of resveratrol. However, these two compounds interact directly with the side chain of Thr71, whereas resveratrol binds to this residue indirectly through a water molecule. The direct hydrogen bond interaction between the amino group and the hydroxyl group of residue Thr71 is most likely responsible for enhancing the QR2 inhibitory activity of compounds 32 and 44 relative to resveratrol. The 3-methoxy groups on ring B of compounds 32 and 44 interact with the amide NH2 group of residue Asn161, and there is one more hydrogen bond interaction between this side chain and the 4-methoxy group of compound 44, which may contribute to the higher potency of compound 44 relative to 32. The X-ray structures of QR2 in complex with compounds 32 and 44 also indicate that these resveratrol analogues sit parallel to the plane of the isoalloxazine ring of the bound cofactor FAD, occupying a position that is similar to resveratrol in the QR2-resveratrol complex.37
3. Conclusions
A focused set of 45 resveratrol analogues have been designed, synthesized, and tested in an effort to maximize their aromatase and QR2 inhibitory activities. Compounds 82 and 84 exhibit excellent aromatase inhibitory activity, with IC50 values 70 nM and 36 nM, respectively. From the preliminary structure-activity relationships and molecular modeling results, it appears that the para amino group on the trans stilbene benzene ring is essential for aromatase inhibitory activity, and the introduction of an imidazole moiety improves the activity greatly. Compounds 32 and 44 also displayed potent QR2 inhibitory activity, and their crystal structures in complex with QR2 have been determined. They provide a detailed description of the enzyme active site, and can be utilized in the further modification for discovery of new QR2 inhibitors.
4. Experimental
4.1. General procedures
Melting points were determined in capillary tubes using a Mel-Temp apparatus and are not corrected. Infrared spectra were obtained as films on salt plates using CHCl3 as the solvent except where otherwise specified, using a Perkin-Elmer Spectrum One FT-IR spectrometer, and are baseline-corrected. 1H NMR spectra were obtained at 300 MHz (1H) and 75 MHz (13C) or 500 MHz (1H) and 125 MHz (13C), using Bruker ARX300 and Bruker Avance 500 (TXI 5 mm probe) spectrometers, respectively. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. Analytical thin-layer chromatography was performed on Baker-flex silica gel IB2-F plastic-backed TLC plates. Flash chromatography was performed with 230–400 mesh silica gel. Unless otherwise stated, chemicals and solvents were of reagent grade and used as obtained from commercial sources without further purification. Dichloromethane was distilled from CaH2 and THF from sodium prior to use.
4.1.1. General Procedure 1 (GP 1) for the Preparation of Aromatic Alkenes from Aromatic Aldehydes
Methyl triphenylphosphonium bromide (4.3 g, 12.05 mmol) and sodium amide (0.47 g, 12.05 mmol) in dry diethyl ether (15 mL) were stirred at room temperature under argon for 10 h. A solution of benzaldehyde (6 mmol) in diethyl ether (10 mL) was added to the reaction mixture dropwise under argon in 30 min at −10 °C. After 10 min, the reaction mixture was warmed up to room temperature and stirred for 2 h. The reaction mixture was then filtered and the solvent was removed in vacuo to yield yellow oil that was purified by silica gel column chromatography, eluting with hexane-dichloromethane mixture, to yield the pure product in good yield.
4.1.2. General Procedure 2 (GP 2) for the Heck Reactions
In a round-bottomed flask, iodobenzene (2.21 mmol) was added to a mixture of tetrabutylammonium bromide (1.1 g, 3.33 mmol), potassium acetate (350 mg, 3.57 mmol), palladium acetate (25 mg, 0.11mmol) and aromatic alkenes (2.44 mmol) stirred in DMF (20 mL) at room temperature under argon. The reaction mixture was warmed up to 80 °C for 3 h and then cooled to room temperature. Diethyl ether (25 mL) was added and the organic layer was washed with water (3 × 20 mL) and then dried over anhydrous sodium sulfate. The solution wa s concentrated and the residue was purified by silica gel column chromatography, eluting with hexane-dichloromethane mixture, to yield the pure product as a solid in moderate yield.
4.1.3. General Procedure 3 (GP 3) for the Hydrolysis of Acetyl Groups
NaOMe (2 mg, 0.04 mmol) was added to a solution of (E)-(styryl)phenyl acetate (0.5 mmol) in ethanol (15 mL). The reaction mixture was stirred for 10 h and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography, eluting with a solution of 1% methanol in dichloromethane, to yield the pure product in good yield.
4.1.4. General Procedure 4 (GP 4) for the Catalytic Hydrogenation of Nitro Groups and Stilbenes
Stilbene compound (0.35 mmol) was dissolved in ethyl acetate (15 mL) and stirred with palladium on carbon (10%, 10 mg) under H2 at room temperature for 24 h. The mixture was filtered, and the solvent was removed in vacuo to provide a solid that was recrystallized from dichloromethane and hexane to yield the pure product in good yield.
4.2. Syntheses
4.2.1. 1,2-Dimethoxy-3-vinylbenzene (11).38
This compound was prepared in 97% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.14-6.99 (m, 3 H), 6.82 (dd, J = 10.8 Hz, J = 1.2 Hz, 1 H), 5.77 (d, J = 17.7 Hz, 1 H), 5.30 (d, J = 11.1 Hz, 1 H), 3.84 (s, 3 H), 3.81 (s, 3 H).
4.2.2. 2,4-Dimethoxy-1-vinylbenzene (12).39
This compound was prepared in 82% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 1 H), 7.04–6.94 (m, 1 H), 6.51-6.45 (m, 2 H), 5.65 (d, J = 17.7 Hz, 1 H), 5.17 (d, J = 11.4 Hz, 1 H), 3.83 (s, 3 H), 3.81 (s, 3 H).
4.2.3. 1,4-Dimethoxy-2-vinylbenzene (13).40
This compound was prepared in 93% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) 7.12-7.01 (m, 2 H), 6.80 (s, 2 H), 5.75 (d, J = 17.7 Hz, 1 H), 5.29 (d, J = 8.4 Hz, 1 H), 3.80 (s, 3 H), 3.79 (s, 3 H).
4.2.4. 1,2-Dimethoxy-4-vinylbenzene (14).40
This compound was prepared in 87% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.95-6.93 (m, 2 H), 6.90 (d, J = 1.8 Hz, 1 H), 6.69 (dd, J = 17.4 Hz, J = 10.6 Hz, 1 H), 5.59 (d, J = 17.4 Hz, 1 H), 5.13 (d, J = 10.6 Hz, 1 H), 3.88 (s, 3 H), 3.85 (s, 3 H).
4.2.5. 1,3-Dimethoxy-5-vinylbenzene (15).41
This compound was prepared in 93% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) 6.59-6.54 (m, 3 H), 5.62 (d, J = 17.4 Hz, 1 H), 5.16 (d, J = 10.8 Hz, 1 H), 3.82-3.81 (m, 9 H).
4.2.6. 1,2,3-Trimethoxy-4-vinylbenzene (16).42
This compound was prepared in 58% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.18 (d, J = 8.7 Hz, 1 H), 6.95-6.86 (m, 1 H), 6.65 (d, J = 8.7 Hz, 1 H), 5.63 (dd, J = 18.0, 1.2 Hz, 1 H), 5.17 (dd, J = 18.0, 1.2 Hz, 1 H), 3.86 (s, 6 H), 3.84 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 153.2, 151.4, 142.2, 130.9, 124.7, 120.5, 113.0, 107.5, 61.1, 60.8, 55.9.
4.2.7. 1,2,5-Trimethoxy-3-vinylbenzene (17).43
This compound was prepared in 42% yield by following GP 1. White solid: mp 54–55 °C. 1H NMR (300 MHz, CDCl3) δ 7.02–6.92 (m, 2 H), 6.48 (s, 1 H), 5.57 (dd, J = 17.7, 1.2 Hz, 1 H), 5.13 (dd, J = 14.1, 1.2 Hz, 1 H), 3.88 (s, 3 H), 3.85 (s, 3 H), 3.81 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 151.3, 149.5, 143.2, 130.8, 118.4, 112.0, 109.3, 97.6, 56.6, 56.4, 56.0.
4.2.8. 5-Vinylbenzo[d][1,3]dioxole (18).44
This compound was prepared in 60% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.99 (s, 1 H), 6.85 (d, J = 8.1 Hz, 1 H), 6.78 (d, J = 8.1 Hz, 1 H), 6.64 (dd, J = 16.8 Hz, J = 10.8 Hz, 1 H), 5.95 (s, 2 H), 5.60 (d, J = 16.8 Hz, 1 H), 5.15 (d, J = 10.8 Hz, 1 H); 13C NMR (75 MHz, CDCl3) δ 147.9, 147.3, 136.3, 132.0, 120.9, 111.8, 108.0, 105.3, 100.9.
4.2.9. 1,2,3-Trimethoxy-5-vinylbenzene (19).41
This compound was prepared in 85% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.59-6.54 (m, 3 H), 5.62 (d, J = 17.8 Hz, 1 H), 5.16 (d, J = 10.8 Hz, 1 H), 3.82-3.81 (m, 9 H).
4.2.10. 1-Methoxy-3-vinylbenzene (20).45
This compound was prepared in 73% yield by following GP 1. Colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.95-6.93 (m, 2 H), 6.90 (d, J = 1.8 Hz, 1 H), 6.69 (dd, J = 17.4 Hz, J = 10.6 Hz, 1 H), 5.59 (d, J = 17.4 Hz, 1 H), 5.13 (d, J = 10.6 Hz, 1 H), 3.88 (s, 3 H), 3.85 (s, 3 H).
4.2.11. 3,5-Dimethoxy-4’-amino-trans-stilbene (32)
This compound was prepared in 64% yield by following GP 2. Yellow solid: mp 90–91 °C. IR (film) 3369, 1595, 1516, 1456, 1203, 1151, 1066, 962 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.31 (d, J = 8.4 Hz, 2 H), 6.99 (d, J = 16.2 Hz, 1 H), 6.83 (d, J = 16.2 Hz, 1 H), 6.65 (d, J = 8.4 Hz, 2 H), 6.62 (d, J = 2.1 Hz, 2 H), 6.34 (t, J = 2.1 Hz, 1 H ), 3.81 (s, 6 H), 3.75 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 160.8, 146.2, 139.9, 129.1, 127.7, 124.9, 115.1, 104.0, 99.2, 55.3; CIMS 256 (MH+); HRMS calcd for C16H17O2N m/z 255.1259, found: m/z 255.1261 (M+).
4.2.12. 2,4-Dimethoxy-4’-amino-trans-stilbene (33)
This compound was prepared in 59% yield by following GP 2. Yellow solid: mp 97–98 °C. IR (film) 3372, 1609, 1577, 1517, 1464, 1286, 1207, 968 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J = 8.4 Hz, 1 H), 7.34 (d, J = 8.4 Hz, 2 H), 7.22 (d, J = 16.5 Hz, 1 H), 6.93 (d, J = 16.5 Hz, 1 H), 6.66 (d, J = 8.4 Hz, 2 H), 6.49-6.46 (m, 2 H), 3.85 (s, 3 H), 3.82 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 159.9, 157.6, 145.3, 129.1, 127.4, 127.0, 126.6, 120.0, 119.6, 115.3, 104.8, 98.4, 55.4, 55.3; CIMS m/z 256 (MH+); HRMS calcd for C16H18O2N m/z 256.1338, found: m/z 256.1344 (MH+).
4.2.13. 2,4-Dimethoxy-3’-amino-trans-stilbene (34)
This compound was prepared in 42% yield by following GP 2. Yellow solid: mp 69–70 °C. IR (film) 3369, 1598, 1577, 1504, 1456, 1287, 1158, 971cm−1; 1H NMR (300 MHz, CDCl3) δ 7.49 (d, J = 8.4 Hz, 1 H), 7.35 (d, J = 16.5 Hz, 1 H), 7.11 (t, J = 7.8 Hz, 1 H), 6.94-6.86 (m, 3 H), 6.57 (dd, J = 2.1 Hz, 8.1 Hz, 1 H), 6.50 (dd, J = 2.4 Hz, 8.7 Hz, 1 H), 6.46 (d, J = 2.4 Hz, 1 H), 3.85 (s, 3 H), 3.82 (s, 3 H), 3.77 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 160.4, 157.9, 146.1, 139.3, 129.4, 127.1, 127.0, 123.0, 119.5, 117.4, 114.2, 112.8, 104.9, 98.4, 55.4, 55.3; ESIMS m/z 256 (MH+); HRMS calcd for C16H18O2N m/z 256.1338, found: m/z 256.1340 (MH+).
4.2.14. 2,5-Dimethoxy-4’-amino-trans-stilbene (35).46
This compound was prepared in 35% yield by following GP 2. Yellow solid: mp 91–92 °C. IR (film) 3370, 1601, 1516, 1493, 1282, 1215, 1046, 969 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.36 (d, J = 8.4 Hz, 2 H), 7.27 (d, J = 16.2 Hz, 1 H), 7.11 (d, J = 2.7 Hz, 1 H), 6.99 (d, J = 16.2 Hz, 1 H), 6.81 (d, J = 8.7 Hz, 1 H), 6.76 (d, J = 2.7 Hz, 1 H), 6.71 (d, J = 8.4 Hz, 2 H), 4.15 (br, 2 H), 3.82 (s, 3 H), 3.80 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 153.7, 151.1, 146.0, 129.3, 128.3, 127.8, 119.4, 115.1, 112.8, 112.2, 111.1, 56.2, 55.7; CIMS m/z 256 (MH+).
4.2.15. 2,3-Dimethoxy-4’-amino-trans-stilbene (36)
This compound was prepared in 75% yield by following GP 2. Yellow solid: mp 97–98 °C. IR (film) 3367, 2923, 1539, 1515, 1343, 961 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.37 (d, J = 8.1 Hz, 2 H), 7.29 (d, J = 16.8 Hz, 1 H), 7.21 (dd, J = 1.2 Hz, 7.8 Hz, 1 H), 7.07-7.02 (m, 2 H), 6.82-6.77 (m, 3 H), 3.88 (s, 3 H), 3.84 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 152.8, 146.3, 146.1, 131.9, 129.8, 128.0, 127.7, 123.9, 118.8, 117.3, 115.0, 110.4, 60.8, 55.6; ESIMS m/z 256 (MH+); HRMS calcd for C16H17O2N m/z 255.1259, found: m/z 255.1257 (M+).
4.2.16. (3,4-Dimethoxy-4’-amino-trans-stilbene (37).47
This compound was prepared in 75% yield by following GP 2. Yellow solid: mp 158–159 °C. IR (film) 3368, 1622, 1604, 1516, 1465, 1137, 1023, 965 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.32 (d, J = 8.4 Hz, 2 H), 7.04 (d, J = 1.8 Hz, 1 H), 7.00 (dd, J = 1.8 Hz, 8.1 Hz, 1 H), 6.88 (s, 1 H), 6.83 (d, J = 8.1 Hz, 1 H), 6.67 (d, J = 8.4 Hz, 2 H), 3.92 (s, 3 H), 3.87 (s, 3 H), 3.75 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 149.0, 148.3, 145.8, 131.0, 128.1, 127.4, 126.8, 124.8, 119.2, 115.2, 111.1, 108.3, 55.8, 55.7; CIMS m/z 256 (MH+).
4.2.17. 3,4-Dimethoxy-4’-cyano-trans-stilbene (38)
This compound was prepared in 61% yield by following GP 2. Yellow solid: mp 102–103 °C. IR (film) 3398, 2223, 1595, 1515, 1463, 1267, 1139, 964 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 8.4 Hz, 2 H), 7.53 (d, J = 8.4 Hz, 2 H), 7.14 (d, J = 16.5 Hz, 1 H), 7.06 (d, J = 6.3 Hz, 2 H), 6.93 (d, J = 16.5 Hz, 1 H), 6.86 (d, J = 9.0 Hz, 1 H), 3.93 (s, 3 H), 3.90 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 149.6, 149.1, 142.0, 132.4, 132.1, 129.2, 126.5, 124.6, 120.6, 119.0, 111.1, 110.0, 108.8, 55.9; CIMS m/z 266 (MH+); HRMS calcd for C17H15O2N m/z 265.1103, found: m/z 265.1100 (M+).
4.2.18. 3,4-Dimethoxy-4’-acetoxy-trans-stilbene (39).48
This compound was prepared in 59% yield by following GP 2. Yellow solid: mp 125–126 °C. IR (film) 3368, 2927, 1760, 1515, 1420, 1196, 964 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 8.4 Hz, 2 H), 7.07-6.97 (m, 5 H), 6.93 (d, J = 16.5 Hz, 1 H), 6.83 (d, J = 7.8 Hz, 1 H), 3.93 (s, 3 H), 3.89 (s, 3 H), 2.29 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 169.5, 149.7, 149.1, 135.3, 130.2, 128.7, 127.1, 125.7, 121.7, 119.9, 111.1, 108.6, 55.9, 55.8, 21.1; CIMS m/z 299 (MH+).
4.2.19. 3,4-Dimethoxy-4’-fluoro-trans-stilbene (40)
This compound was prepared in 57% yield by following GP 2. Yellow solid: mp 117–118 °C. IR (film) 3368, 1600, 1515, 1461, 1225, 1139, 1022, 964 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.45 (dd, J = 5.7 Hz, 8.4 Hz, 2 H), 7.07-7.02 (m, 4 H), 6.97 (d, J = 16.5 Hz, 1 H), 6.91 (d, J = 16.5 Hz, 1 H), 6.86 (d, J = 7.8 Hz, 1 H), 3.95 (s, 3 H), 3.90 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ148.9, 133.6, 130.2, 128.2, 127.7, 127.5, 125.5, 119.7, 115.6, 115.3, 111.1, 108.5, 55.8, 55.7; CIMS m/z 259 (MH+); HRMS calcd for C16H15O2F m/z 258.1056, found: m/z 258.1051 (M+).
4.2.20. 3,4,5-Trimethoxy-4’-nitro-trans-stilbene (41).49
This compound was prepared in 68% yield by following GP 2. Orange solid: mp 192–193 °C. IR (film) 1594, 1581, 1506, 1331, 1265, 1126,1108 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 8.7 Hz, 2 H), 7.61 (d, J = 8.7 Hz, 2 H), 7.18 (d, J = 16.2 Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.76 (s, 2 H), 3.91 (s, 6 H), 3.87 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 153.5, 146.7, 143.8, 133.3, 131.8, 126.7, 125.7, 124.2, 104.1, 61.0, 56.2; CIMS m/z 316 (MH+).
4.2.21. 3,4,5-Trimethoxy-4’-amino-trans-stilbene (42)
This compound was prepared in 52% yield by following GP 2. Yellow solid: mp 105–106 °C. IR (film) 3367, 2926, 1599, 1583, 1505, 1457, 1417, 1243, 1126, 959 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.13 (t, J = 7.5 Hz, 1 H), 7.11 (d, J = 16.2 Hz, 1 H), 6.92-6.87 (m, 2 H), 6.81 (s, 1 H ), 6.71 (s, 2 H), 6.57 (dd, J = 7.8, 1.8 Hz, 1 H), 3.89 (s, 6 H), 3.86 (s, 3 H), 3.66 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 153.3, 146.6, 138.1, 137.7, 133.1, 129.5, 128.3, 117.1, 114.6, 112.7, 103.4, 60.9, 56.0. ESIMS m/z 286 (MH+); HRMS calcd for C17H20O3N m/z 286.1443, found: m/z 286.1446 (MH+).
4.2.22. 3,4,5-Trimethoxy-4’-aminomethyl-trans-stilbene (43)
This compound was prepared in 57% yield by following GP 2. White solid: mp 139–140 °C. IR (film) 3341, 2937, 1672, 1583, 1508, 1462, 1421, 1237, 1125, 964 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.45 (d, J = 8.1 Hz, 2 H), 7.25 (d, J = 8.1 Hz, 2 H), 7.02 (d, J = 16.2 Hz, 1 H), 6.95 (d, J = 16.2 Hz, 1 H), 6.7 (s, 2 H), 4.47 (d, J = 5.7 Hz, 2 H), 3.90 (s, 6 H), 3.86 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 161.0, 153.3, 136.8, 136.6, 132.8, 128.8, 128.1, 127.4, 126.6, 103.4, 60.9, 56.0, 41.8; ESIMS m/z 283 (MH – NH3)+; HRMS calcd for C18H19O3 m/z 283.1334, found: m/z 283.1336 (MH – NH3)+.
4.2.23. 3,4,5-Trimethoxy-4’-amino-trans-stilbene (44)
This compound was prepared in 50% yield by following GP 2. Yellow solid: mp 135–136°C. IR (film) 3368, 1606, 1580, 1516, 1417, 1234, 1124, 959 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.33 (d, J = 8.4 Hz, 2 H), 6.92 (d, J = 16.2 Hz, 1 H), 6.83 (d, J = 16.2 Hz, 1 H), 6.73 (d, J = 8.4 Hz, 2 H), 6.68 (s, 2 H), 3.89 (s, 6 H), 3.86 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 153.3, 146.1, 137.2, 133.7, 128.2, 127.6, 124.9, 115.1, 114.5, 103.0, 60.9, 56.0; CIMS m/z 286 (MH+); HRMS calcd for C17H20O3N m/z 286.1443, found: m/z 286.1442 (MH+).
4.2.24. 2,3,4-Trimethoxy-4’-amino-trans-stilbene (45)
This compound was prepared in 31% yield by following GP 2. Yellow solid: mp 139–140 °C. IR (film) 3369, 1606, 1516, 1493, 1413, 1297, 1094, 969 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2 H), 7.21 (d, J = 1.2 Hz, 1 H), 7.09 (d, J = 16.5 Hz, 1 H), 6.88 (d, J = 16.5 Hz, 1 H), 6.65-6.61 (m, 3 H), 3.84 (s, 3 H), 3.84 (s, 3 H), 3.82 (s, 3 H), 3.69 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 152.6, 151.3, 145.7, 142.3, 128.6, 127.9, 127.5, 125.0, 120.1, 119.2, 115.1, 107.7, 61.2, 60.8, 56.0; ESIMS m/z 286 (MH+); HRMS calcd for C17H20O3N m/z 286.1443, found: m/z 286.1441 (MH+).
4.2.25. 2,3,5-Trimethoxy-4’-amino-trans-stilbene (46)
This compound was prepared in 38% yield by following GP 2. Yellow solid: mp 105–106 °C. IR (film) 3369, 1608, 1518, 1464, 1206, 1032, 965 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.33 (d, J = 8.4 Hz, 2 H), 7.21 (d, J = 16.2 Hz, 1 H), 7.08 (s, 1 H), 6.86 (d, J = 16.2 Hz, 1 H), 6.65 (d, J = 8.4 Hz, 2 H), 6.51 (s, 1 H), 3.89 (s, 6 H), 3.84 (s, 3 H), 3.69 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 151.2, 148.9, 145.6, 143.3, 128.8, 127.4, 126.9, 119.3, 118.9, 115.1, 109.0, 97.8, 56.7, 56.4, 56.0; ESIMS m/z 286 (MH+); HRMS calcd for C17H20O3N m/z 286.1443, found: m/z 286.1442 (MH+).
4.2.26. (E)-3-(2-(Benzo[d][1,3]dioxol-5-yl)vinyl)benzenamine (47)
This compound was prepared in 66% yield by following GP 2. Yellow solid: mp 86–87 °C. IR (film) 3377, 1599, 1500, 1486, 1444, 1251, 1035, 963 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.14 (t, J = 7.8 Hz, 1 H), 7.05 (d, J = 1.5 Hz, 1 H), 7.00, (d, J = 16.2 Hz, 1 H), 6.94-6.89 (m, 2 H), 6.85 (d, J = 16.2 Hz, 1 H), 6.80 (d, J = 8.1 Hz, 2 H ), 6.59 (dd, J = 7.8 Hz, 1.5 Hz, 1 H), 5.97 (s, 2 H), 3.69 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 148.0, 147.1, 146.5, 138.4, 131.8, 129.5, 128.1, 127.1, 121.3, 117.0, 114.3, 112.6, 108.3, 105.4, 101.0; CIMS m/z 240 (MH+); HRMS calcd for C15H13O2N m/z 239.0946, found: m/z 239.0945 (M+).
4.2.27. (E)-4-(2-(Benzo[d][1,3]dioxol-5-yl)vinyl)benzenamine (48)
This compound was prepared in 75% yield by following GP 2. Yellow solid: mp 132–133 °C. IR (film) 3368, 2924, 1606, 1516, 1489, 1446, 1253, 1038, 960 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.4 Hz, 2 H), 7.03 (d, J = 1.5 Hz, 1 H), 6.89 (dd, J = 8.4 Hz, 1.5 Hz, 1 H), 6.87 (d, J = 16.2 Hz, 1 H), 6.82 (d, J = 16.2 Hz, 1 H), 6.78 (d, J = 8.1 Hz, 1 H), 6.66 (d, J = 8.4 Hz, 2 H), 5.96 (s, 2 H), 3.73 (br, 2 H); 13C NMR (125 MHz, CDCl3) δ 148.0, 146.6, 145.8, 132.4, 128.0, 127.4, 127.0, 124.7, 120.7, 115.1, 108.3, 105.2, 100.9; ESIMS m/z 240 (MH+); HRMS calcd for C15H14O2N m/z 240.1025, found: m/z 240.1022 (MH+).
4.2.28. 3-Nitro-4’-nitro-trans-stilbene (49).50
This compound was prepared in 46% yield by following GP 2. Yellow solid: mp 224–226 °C. IR (film) 1593, 1524, 1514, 1356, 1340, 972, 960, 868 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.40 (t, J = 1.5 Hz, 1 H), 8.25 (d, J = 9.0 Hz, 2 H), 8.16 (d, J = 8.1 Hz, 1 H), 7.82 (d, J = 7.8 Hz, 1 H), 7.67 (d, J = 9.0 Hz, 2 H), 7.57 (t, J = 7.8 Hz, 1 H), 7.30 (d, J = 16.5 Hz, 1 H), 7.24 (d, J = 16.5 Hz, 1 H); 13C NMR (75 MHz, CDCl3) δ 148.7, 147.3, 142.5, 137.9, 132.6, 130.4, 129.8, 129.2, 127.2, 124.2, 123.0, 121.2; ESIMS m/z 271 (MH+).
4.2.29. 3-Nitro-4’-amino-trans-stilbene (50)
This compound was prepared in 39% yield by following GP 2. Yellow solid: mp 177–178 °C. IR (film) 3469, 3390, 1620, 1527, 1356, 970, 819 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.29 (s, 1 H), 8.01 (d, J = 6.6 Hz, 1 H), 7.72 (d, J = 7.8 Hz, 1 H), 7.46 (t, J = 7.8 Hz, 1 H), 7.34 (d, J = 8.4 Hz, 2 H), 7.12 (d, J = 16.5 Hz, 1 H), 6.91 (d, J = 16.5 Hz, 1 H), 6.67 (d, J = 8.4 Hz, 2 H), 3.81 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 150.4, 149.3, 141.1, 133.0, 132.6, 130.9, 129.2, 124.9, 121.6, 121.2, 120.7, 114.7; CIMS m/z 241 (MH+); HRMS calcd for C14H12O2N2 m/z 240.0899, found: m/z 240.0898 (M+).
4.2.30. 3-Methoxyl-4’-amino-trans-stilbene (51).51
This compound was prepared in 51% yield by following GP 2. Yellow solid: mp 70–71 °C. IR (film) 2917, 1666, 1601, 1518, 1289, 965 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.37 (d, J = 8.4 Hz, 2 H), 7.30 (t, J = 7.8 Hz, 1 H), 7.14-7.05 (m, 3 H), 6.95 (d, J = 16.2 Hz, 1 H), 6.83 (dd, J = 2.1 Hz, 7.8 Hz, 1 H), 6.69 (d, J = 7.8 Hz, 2 H), 3.87 (s, 3 H), 3.75 (br, 1 H); 13C NMR (75 MHz, CDCl3) δ 159.8, 146.3, 139.4, 129.5, 129.0, 127.8, 124.8, 118.8, 115.2, 112.5, 111.3, 55.2; ESIMS m/z 226 (MH+).
4.2.31. 3,5-Dinitro-4’-nitro-trans-stilbene (52)
This compound was prepared in 42% yield by following GP 2. Yellow solid: mp 281–282 °C. IR (film) 3368, 1533, 1506, 1343, 1331, 961 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.90 (d, J = 2.1 Hz, 2 H), 8.72 (t, J = 1.8 Hz, 1 H), 8.29 (d, J = 9.0 Hz, 2 H), 7.94-7.88 (m, 3 H), 7.81 (d, J = 16.5 Hz, 1 H); 13C NMR (75 MHz, DMSO-d6) δ 149.5, 147.9, 143.7, 141.0, 132.5, 130.0, 129.0, 127.6, 125.1, 118.4; negative ion ESIMS m/z 314 (M – H+)−; negative ion HRMS calcd for C14H8O6N3 m/z 314.0413, found: m/z 314.0418 (M – H+)−.
4.2.32. 3,5-Dimethoxyl-4’-amino-trans-stilbene (53).52
This compound was prepared in 67% yield by following GP 2. Yellow solid: mp 134–135 °C. IR (film) 3368, 1588, 1509, 1462, 1336, 1205, 1164, 1064, 952 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 8.7 Hz, 2 H), 7.61 (d, J = 8.7 Hz, 2 H), 7.18 (d, J = 16.2 Hz, 1 H), 7.09 (d, J = 16.2 Hz, 1 H), 6.68 (d, J = 1.8 Hz, 2 H), 6.43 (t, J = 2.1 Hz, 1 H), 3.83 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.7, 133.8, 129.7, 128.2, 126.8, 123.4, 106.6, 105.0, 100.9, 100.0, 55.2; CIMS m/z 286 (MH+).
4.2.33. 3,5-Dimethoxyl-3’-amino-trans-stilbene (54)
This compound was prepared in 44% yield by following GP 2. Yellow solid: mp 72–73 °C. IR (film) 3368, 1599, 1590, 1459, 1204, 1152, 1065, 960 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.16 (t, J = 7.8 Hz, 1 H), 7.00 (s, 2 H), 6.93 (d, J = 7.8 Hz, 1 H), 6.84 (t, J = 2.1 Hz, 1 H), 6.67 (d, J = 2.4 Hz, 2 H), 6.61 (dd, J = 2.4 Hz, 8.4 Hz, 1 H), 6.41 (t, J = 2.1 Hz, 1 H), 3.84 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.9, 146.6, 139.4, 138.1, 129.5, 129.3, 128.4, 117.3, 114.7, 112.8, 104.5, 99.8, 55.3; ESIMS m/z 256 (MH+). HRMS calcd for C16H17O2N m/z 256.1338, found: m/z 256.1341 (MH+).
4.2.34. 3,5-Dimethoxy-4’-acetoxy-trans-stilbene (55).53
This compound was prepared in 50% yield by following GP 2. White solid: mp 121–122 °C. IR (film) 3399, 1759, 1591, 1505, 1457, 1194, 1152, 968 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.49 (d, J = 8.7 Hz, 2 H), 7.09-7.02 (m, 3 H), 6.96 (d, J = 16.5 Hz, 1 H), 6.64 (d, J = 2.1 Hz, 2 H), 6.39 (t, J = 2.4 Hz, 1 H), 3.82 (s, 6 H), 2.29 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 169.4, 160.9, 150.1, 139.1, 134.9, 128.9, 128.1, 127.5, 121.8, 104.5, 100.0, 55.3, 21.1; CIMS m/z 299 (MH+).
4.2.35. 3,5-Dimethoxy-4’-aminomethyl-trans-stilbene (56)
This compound was prepared in 50% yield by following GP 2. White solid: mp 115–116 °C. IR (film) 3292, 2927, 1660, 1593, 1457, 1426, 1204, 1151, 1067, 963 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2 H), 7.26 (d, J = 8.4 Hz, 2 H), 7.06 (d, J = 16.5 Hz, 1 H), 6.99 (d, J = 16.5 Hz, 1 H), 6.65 (d, J = 2.1 Hz, 2 H), 6.39 (t, J = 2.1 Hz, 1 H), 5.92 (br, 1 H), 4.47 (d, J = 6.0 Hz, 2 H), 3.82 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.9, 139.0, 136.9, 136.6, 128.8, 128.4, 128.1, 126.8, 104.5, 100.0, 55.3, 41.8; ESIMS m/z 253 (MH – NH3)+; HRMS calcd for C17H17O2 m/z 253.1229, found: m/z 253.1228 (MH – NH3)+.
4.2.36. 3,5-Dimethoxy-4’-fluoro-trans-stilbene (57).54
This compound was prepared in 56% yield by following GP 2. White solid: mp 44–45 °C. IR (film) 1597, 1509, 1457, 1228, 1204, 1157, 1067, 962 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.48-7.43 (m, 2 H), 7.06–7.00 (m, 3 H), 6.93 (d, J = 16.2 Hz, 1 H), 6.64 (d, J = 2.4 Hz, 2 H), 6.39 (t, J = 2.1 Hz, 1 H), 3.82 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.9, 139.1, 133.2, 128.3, 128.0, 127.9, 115.7, 115.4, 104.4, 99.8, 55.3; ESIMS m/z 259 (MH+).
4.2.37. 3,5-Dimethoxyl-4’-chloro-trans-stilbene (58).55
This compound was prepared in 57% yield by following GP 2. White solid: mp 65–66 °C. IR (film) 3369, 1587, 1455, 1347, 1206, 1151, 1057, 956 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.41 (d, J = 8.7 Hz, 2 H), 7.30 (d, J = 8.7 Hz, 2 H), 7.02 (d, J = 16.8 Hz, 1 H), 6.97 (d, J = 16.8 Hz, 1 H), 6.64 (d, J = 2.4 Hz, 2 H), 6.39 (t, J = 2.1 Hz, 1 H), 3.81 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.9, 138.8, 135.5, 133.2, 129.2, 128.89, 127.8, 127.6, 104.5, 100.1, 55.3; ESIMS m/z 275 (MH+).
4.2.38. 3,5-Dimethoxy-4’-cyano-trans-stilbene (59)
This compound was prepared in 63% yield by following GP 2. White solid: mp 106–107 °C. IR (film) 3400, 2223, 1602, 1586, 1458, 1427, 1206, 1154, 1061, 948 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 8.1 Hz, 2 H), 7.53 (d, J = 8.1 Hz, 2 H), 7.11 (d, J = 16.2 Hz, 1 H), 7.02 (d, J = 16.2 Hz, 1 H), 6.65 (d, J = 2.1 Hz, 2 H), 6.42 (t, J = 2.1 Hz, 1 H), 3.81 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 161.0, 141.5, 138.1, 132.4, 132.3, 127.1, 126.8, 118.9, 110.6, 104.9, 100.7, 55.3; CIMS m/z 266 (MH+); HRMS calcd for C17H15O2N m/z 265.1103, found: m/z 265.1096 (M+).
4.2.39. 4-(3,5-Dimethoxyphenethyl)benzenamine (60)
This compound was prepared in 95% yield by following GP 4. Yellow solid: mp 41–42 °C. IR (film) 3368, 2927, 2870, 1597, 1517, 1456, 1105, 967 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.00 (d, J = 8.1 Hz, 2 H), 6.62 (d, J = 8.1 Hz, 2 H), 6.36 (d, J = 2.1 Hz, 2 H), 6.32 (t, J = 2.1 Hz, 1 H), 3.77 (s, 6 H), 2.82 (s, 4 H); 13C NMR (75 MHz, CDCl3) δ 160.6, 144.4, 144.3, 131.7, 129.1, 115.1, 106.4, 97.8, 55.2, 38.5, 36.8; CIMS m/z 258 (MH+); HRMS calcd for m/z C16H20O2N 258.1494, found: m/z 258.1495 (MH+).
4.2.40. 4-(3,4-Dimethoxyphenethyl)aniline (61)
This compound was prepared in 90% yield by following GP 4. Yellow solid: mp 107–108 °C. IR (film) 3363, 2920, 1625, 1515, 1453, 1232, 1144, 1026 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.96 (d, J = 8.4 Hz, 2 H), 6.75 (d, J = 8.1 Hz, 1 H), 6.71 (dd, J = 1.8 Hz, 8.4 Hz, 1 H ), 6.66 (d, J = 1.8 Hz, 1 H), 6.63 (d, J = 8.1 Hz, 2 H), 3.84 (s, 3 H), 3.82 (s, 3 H), 3.80 (br, 2 H), 2.79 (s, 4 H); 13C NMR (75 MHz, CDCl3) δ 148.5, 147.0, 144.2, 134.6, 131.8, 129.2, 120.1, 115.1, 111.7, 111.0, 55.8, 55.7, 37.8, 37.2; CIMS m/z 258 (MH+); HRMS calcd for C16H19O2N m/z 257.1416, found: m/z 257.1419 (M+).
4.2.41. 4-(3,4,5-Trimethoxyphenethyl)aniline (62).49
This compound was prepared in 90% yield by following GP 4. Yellow solid: mp 84–85 °C. IR (film) 3368, 1589, 1516, 1457, 1420, 1237, 1124 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.96 (d, J = 8.4 Hz, 2 H), 6.61 (d, J = 8.4 Hz, 2 H), 6.36 (s, 2 H), 3.82 (s, 6 H), 3.81 (s, 3 H), 3.50 (br, 2 H), 2.79 (s, 4 H); 13C NMR (75 MHz, CDCl3) δ 152.9, 144.4, 137.7, 135.9, 131.5, 129.2, 115.1, 105.3, 60.8, 55.9, 38.6, 37.1; ESIMS m/z 288 (MH+).
4.2.42. 3,4'-(Ethane-1,2-diyl)dibenzenamine (63)
This compound was prepared in 98% yield by following GP 4. White solid: mp 72–73 °C. IR (film) 3390, 3335, 3217, 2915, 1618, 1590, 1515 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.06 (dd, J = 7.8 Hz, 8.1 Hz, 1 H), 6.97 (d, J = 8.1 Hz, 2 H), 6.63-6.58 (m, 3 H), 6.52-6.50 (m, 2 H), 3.42 (br, 4 H), 2.77 (s, 4 H); 13C NMR (75 MHz, CDCl3) δ 146.2, 144.2, 143.3, 132.0, 129.1, 118.8, 115.2, 115.1, 112.6, 38.2, 36.9; ESIMS m/z 213 (MH+); HRMS calcd for C14H16N2 m/z 212.1313, found: m/z 212.1314 (M+).
4.2.43. 3-Amino-4’-amino-trans-stilbene (64)
Stannous chloride (435 mg, 2.3 mmol) was added to a solution of (E)-4-(3-nitrostyryl)benzenamine (50) (110 mg, 0.46 mmol) in ethanol (15 mL), and the reaction mixture was stirred under reflux for 2 h. The ethanol was removed, aq NaOH (1 N, 20 mL) was added to the residue and mixture was stirred for 0.5 h. The aqueous layer was then extracted by diethyl ether (3 × 15 mL) and the organic layer was dried over sodium sulfate. The solvent was evaporated in vacuo at room temperature to provide a yellow solid that was recrystallized from ethyl acetate and hexane to yield a white solid (62 mg, 64.3%): mp 149–150 °C. IR (film) 3392, 3325, 3207, 1598, 1515, 1450, 1291, 970 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 8.4 Hz, 2 H), 7.11 (t, J = 7.8 Hz, 1 H), 6.96 (d, J = 16.5 Hz, 1 H), 6.88 (d, J = 7.8, 1 H ), 6.81 (d, J = 16.5 Hz, 2 H), 6.65 (d, J = 8.4 Hz, 2 H), 6.54 (dd, J = 8.4 Hz, 1.8 Hz, 1 H), 3.68 (br, 4 H); 13C NMR (75 MHz, CDCl3) δ 146.5, 146.0, 129.4, 128.4, 128.0, 127.6, 125.2, 116.9, 115.1, 113.9, 112.5; ESIMS m/z 211 (MH+); HRMS calcd for C14H14N2 m/z 210.1157, found: m/z 210.1160 (M+).
4.2.44. 3,5-Diamino-4’-amino-trans-stilbene (65)
Stannous chloride (1.08 g, 5.7 mmol) was added to a solution of (E)-1,3-dinitro-5-(4-nitrostyryl)benzene (52) (100 mg, 0.32 mmol) in ethanol (15 mL) and the reaction mixture was stirred under reflux for 2 h. The ethanol was removed, aq NaOH (1 N, 20 mL) was added to the residue and the mixture was stirred for 0.5 h. The aqueous layer was then extracted by diethyl ether (3 × 15 mL) and the organic layer was dried over sodium sulfate. The solvent was evaporated in vacuo at room temperature to provide a yellow solid that was recrystallized from ethyl acetate and hexane to yield a yellow solid (43.9 mg, 61%): mp 173–174 °C. IR (film) 3338, 1592, 1515, 1462, 965 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2 H), 6.90 (d, J = 16.2 Hz, 1 H), 6.72 (d, J = 16.2 Hz, 1 H), 6.63 (d, J = 8.4 Hz, 2 H), 6.25 (d, J = 1.8 Hz, 2 H), 5.93 (t, J = 1.8 Hz, 1 H), 3.71 (br, 2 H), 3.55 (br, 4 H); 13C NMR (75 MHz, CDCl3) δ 147.5, 145.9, 139.9, 128.3, 128.0, 127.6, 125.3, 115.1, 104.0, 101.0; CIMS m/z 226 (MH+); HRMS calcd for C14H15N2 m/z 255.1266, found: m/z 255.1268 (M+).
4.2.45. 3,4-Dimethoxy-4’-hydroxyl-trans-stilbene (66).56
This compound was prepared in 86% yield by following GP 3. Yellow solid: mp 180–181 °C. IR (film) 3369, 2927, 1608, 1516, 1463, 1252, 964 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 8.7 Hz, 2 H), 7.05 (d, J = 2.1 Hz, 1 H), 7.02 (dd, J = 8.7 Hz, 2.1 Hz, 1 H), 6.92 (s, 2 H), 6.85 (d, J = 8.7 Hz, 1 H), 6.62 (d, J = 8.7 Hz, 2 H), 3.94 (s, 3 H), 3.90 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 154.9, 148.5, 130.6, 130.4, 127.5, 126.4, 126.2, 119.4, 115.5, 111.1, 108.4, 55.8, 55.7; CIMS m/z 257 (MH+).
4.2.46. 3,5-Dimethoxy-4’-hydroxyl-trans-stilbene (67).57
This compound was prepared in 81% yield by following GP 3. White solid: mp 87–88 °C. IR (film) 3368, 1590, 1513, 1455, 1204, 1149, 961 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.40 (d, J = 8.1 Hz, 2 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.89 (d, J = 16.2 Hz, 1 H), 6.82 (d, J = 8.1 Hz, 2 H), 6.65 (d, J = 2.1 Hz, 2 H), 6.39 (t, J = 2.1 Hz, 1 H), 3.84 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.8, 155.3, 139.6, 130.0, 128.6, 127.9, 126.5, 115.5, 104.3, 99.5, 55.3; CIMS m/z 257 (MH+).
4.2.47. (5-Vinyl-1,3-phenylene)bis(oxy)bis(tert-butyldimethylsilane) (69).58
Tert-butylchlorodimethylsilane (6.55 g, 43.4 mmol) was added to a well-stirred solution of 3,5-dihydroxybenzaldehyde (68) (2 g, 14.48 mmol) and DIPEA (5.56 g, 43.4 mmol) in DMF (20 mL) at 0 °C, and the reaction mixture was stirred for further 3 h at room temperature. The reaction mixture was then diluted with dichloromethane (30 mL), washed with sat aq NaCl (3 × 15 mL) and dried over anhydrous sodium sulfate. The solution was concentrated and the residue was purified by silica gel column chromatography, eluting with a gradient solution from hexane to dichloromethane, to yield 3,5-bis(tert-butyldimethylsilyloxy)benzaldehyde as a colorless oil. The solution of 3,5-bis(tert-butyldimethylsilyloxy)benzaldehyde in diethyl ether (15 mL) was then added dropwise to a well-stirred solution of methyl triphenylphosphonium bromide (7.1 g, 20 mmol) and sodium amide (780 mg, 20 mmol) in dry diethyl ether (50 mL) at 0 °C. After 10 min, the reaction mixture was warmed up to room temperature and stirred for 2 h. The reaction mixture was filtered and the solvent was removed in vacuo to yield yellow oil that was purified by silica gel column chromatography, eluting with 50% hexane in dichloromethane, to yield a colorless oil (2.7 g, 52%): 1H NMR (300 MHz, CDCl3) δ 6.61-6.51 (m, 1 H), 6.50 (d, J = 2.4 Hz, 2 H), 6.23 (t, J = 2.4 Hz, 1 H), 5.64 (d, J = 17.7 Hz, 1 H), 5.18 (d, J = 10.2 Hz, 1 H), 0.95 (s, 18 H), 0.18 (s, 12 H); 13C NMR (75 MHz, CDCl3) δ 156.5, 139.2, 136.6, 113.8, 111.6, 111.3, 25.6, 18.1, −4.5.
4.2.48. 5-Vinyl-1,3-phenylene Diacetate (71).59
TBAF (3.41 g, 13.1 mmol) was added dropwise to a solution of (5-vinyl-1,3-phenylene)bis(oxy)bis(tert-butyldimethylsilane) (69) (2.27 g, 6.22 mmol) in anhydrous THF (15 mL) at 0 °C, and the reaction mixture was then stirred for 3 h at room temperature. The reaction mixture was diluted with diethyl ether (20 mL), washed with sat aq NaCl (3 × 15 mL) and dried over anhydrous sodium sulfate. The solution was concentrated and the residue was purified by silica gel chromatography, eluting with CH2Cl2, to yield 5-vinylbenzene-1,3-diol (70) (846 mg, 100%) as a colorless oil. Acetic anhydride (1.83 g, 18 mmol) was added dropwise to a solution of compound 70 (800 mg, 6 mmol), pyridine (1.4 g, 18 mmol) and DMAP (7.32 mg, 0.06mmol) in CH2Cl2 (20 mL) at 0 °C. The reaction mixture was then stirred for 10 h at room temperature and the solution was concentrated to provide a residue that was purified by silica gel column chromatography, eluting with 50% hexane in dichloromethane, to yield 71 (1.17 g, 89%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.00 (d, J = 2.1 Hz, 2 H), 6.80 (t, J = 2.1 Hz, 1 H), 6.67-6.58 (m, 4 H), 5.71 (d, J = 17.4 Hz, 1 H), 5.29 (d, J = 10.2 Hz, 1 H), 2.27 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 168.9, 151.1, 139.8, 135.2, 116.7, 115.8, 114.5, 21.0; ESIMS m/z 242 (MNa+).
4.2.49. 3,5-Diacetoxy-4’-amino-trans-stilbene (72)
This compound was prepared in 40% yield by following GP 2. Yellow solid: mp 146–147 °C. IR (film) 3369, 1765, 1601, 1517, 1203, 1122, 964 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2 H), 7.05 (d, J = 1.8 Hz, 2 H), 6.97 (d, J = 16.5 Hz, 1 H), 6.80 (d, J = 16.5 Hz, 1 H), 6.74 (t, J = 2.1 Hz, 1 H), 6.64 (d, J = 8.4 Hz, 2 H), 3.87 (br, 2 H), 2.28 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 169.0, 151.1, 146.5, 130.6, 127.9, 127.1, 123.1, 116.3, 115.0, 113.4, 21.1; ESIMS m/z 312 (MH+); HRMS calcd for C18H18O4N m/z 312.1236, found: m/z 312.1239 (MH+).
4.2.50. 3,5-Diacetoxy-4’-nitro-trans-stilbene (73)
This compound was prepared in 43% yield by following GP 2. Yellow solid: mp 112–113 °C. IR (film) 3411, 1767, 1593, 1514, 1341, 1198, 1124 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 8.7 Hz, 2 H), 7. 58 (d, J = 8.7 Hz, 2 H), 7.16 (d, J = 16.5 Hz, 1 H), 7.15 (d, J = 2.1 Hz, 2 H), 7.08 (d, J = 16.5 Hz, 1 H), 6.87 (t, J = 2.1 Hz, 1 H), 2.30 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 168.9, 151.3, 147.0, 142.9, 138.3, 131.3, 128.1, 127.0, 124.1, 117.3, 115.4, 21.0; CIMS m/z 342 (MH+); HRMS calcd for C18H15O6N m/z 341.0899, found: m/z 341.0901 (M+).
4.2.51. 3,5-Dihydroxyl-4’-amino-trans-stilbene (74)
This compound was prepared in 70% yield by following GP 3. Yellow solid: mp 215–216 °C. IR (film) 3368, 1586, 1514, 1473, 1346, 1151, 964 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.15 (d, J = 8.4 Hz, 2 H), 6.80 (d, J = 16.5 Hz, 1 H), 6.62 (d, J = 16.5 Hz, 1 H), 6.57 (d, J = 8.4 Hz, 2 H), 6.31 (d, J = 2.1 Hz, 2 H), 6.02 (t, J = 2.1 Hz, 1 H); 13C NMR (75 MHz, CD3OD) δ 159.6, 148.7, 141.6, 129.8, 128.7, 128.6, 125.7, 116.5, 105.6, 102.4; CIMS m/z 228 (MH+); HRMS calcd for C14H13O2N m/z 227.0946, found: m/z 227.0944 (M+).
4.2.52. 3,5-Dihydroxyl-4’-nitro-trans-stilbene (75).60
This compound was prepared in 81% yield by following GP 3. Yellow solid: mp 256–257 °C. IR (film) 3392, 1600, 1592, 1513, 1334, 1166 cm−1; 1H NMR (300 MHz, CD3OD) δ 8.20 (d, J = 8.7 Hz, 2 H), 7.73 (d, J = 7.5 Hz, 2 H), 7.24 (d, J = 16.2 Hz, 1 H), 7.13 (d, J = 16.2 Hz, 1 H), 6.54 (d, J = 2.1 Hz, 2 H), 6.25 (t, J = 2.1 Hz, 1 H); 13C NMR (75 MHz, CD3OD) δ 159.9, 148.0, 145.6, 139.8, 134.8, 128.1, 127.1, 125.0, 106.6, 104.2; CIMS m/z 258 (MH+).
4.2.53. (3,5-Dimethoxybenzyl)triphenylphosphonium Bromide (78).61
NaBH4 (137 mg, 3.61 mmol) was added slowly to a solution of 3,5-dimethoxybenzaldehyde (5) (1.5 g, 9.04 mmol) in THF (20 mL). The reaction mixture was stirred for 2 h at room temperature. Water (0.5 mL) was added to the reaction mixture and stirred for 10 min, followed by concentrated HCl (0.29 mL, 3.61 mmol) to neutralize the base. Dichloromethane (25 mL) was added and the organic layer was washed with water (3 × 15 mL) and then dried over anhydrous sodium sulfate. The solvent was removed in vacuo to yield the crude product (3,5-dimethoxyphenyl)methanol (76) (1.3 g, 87%). Tribromophosphine (1.6 g, 5.9 mmol) was then added dropwise to a solution of 76 (0.99 g, 5.9 mmol) in dichloromethane (15 mL) at 0 °C. The reaction mixture was stirred for 2 h and then warmed up to room temperature. After 2 h, the organic layer was washed with aq NaHCO3 (3 × 10 mL) and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to yield the crude product 1-(bromomethyl)-3,5-dimethoxybenzene (77) (1.65 g, 83.6%), which was then added to a solution of triphenylphosphine (2.1 g, 8 mmol) in anhydrous toluene (20 mL). The resulting mixture was then stirred under reflux for 3 h. The reaction mixture was allowed to cool to room temperature, and the precipitate was filtered and washed with hexane to provide compound 78 (3.5 g, 95%) as white solid: mp 264–265 °C. 1H NMR (300 MHz, CDCl3) δ 7.75-7.66 (m, 9 H), 7.62-7.55 (m, 6 H), 6.28 (t, J = 2.4 Hz, 2 H), 6.24 (t, J = 2.4 Hz, 1 H), 5.22 (d, J = 14.4 Hz, 2 H), 3.48 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 160.5, 134.9, 134.4, 134.3, 130.1, 129.9, 118.2, 117.0, 109.1, 109.0, 101.2, 55.5.
4.2.54. 2-(1H-Imidazol-1-yl)-1-(4-nitrophenyl)ethanone (80)
Imidazole (223 mg, 3.28 mmol) was added to a solution of 2-bromo-1-(4-nitrophenyl)ethanone (79) (200 mg, 0.82 mmol) in THF (15 mL) and the reaction mixture was stirred for 15 h at room temperature. THF was then removed in vacuo, and the residue was dissolved in ethyl ether (25 mL). The organic layer was washed with sat aq NaCl (3 × 15 mL) and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to yield a brown solid (140 mg, 90%): mp 164–165 °C. IR (film) 3368, 2921, 1709, 1525, 1347, 1220 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 9.0 Hz, 2 H), 8.14 (d, J = 9.0 Hz, 2 H), 7.54 (s, 1 H), 7.15 (s, 1 H), 6.95 (s, 1 H), 5.50 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 190.3, 150.9, 138.3, 137.9, 129.8, 129.0, 124.2, 120.0, 52.8; CIMS m/z 232 (MH+); HRMS calcd for C11H9O3N3 m/z 231.0644, found: m/z 231.0648 (M+).
4.2.55. 1-(4-Aminophenyl)-2-(1H-imidazol-1-yl)ethanone (81)
Pd/C (10%, 10 mg) was added to a solution of 2-(1H-imidazol-1-yl)-1-(4-nitrophenyl)ethanone (80) (100 mg, 0.43 mmol) in methanol (20 mL). The suspension was stirred under H2 for 4 h at 1.5 atm hydrogen pressure. The mixture was filtered, and the solvent was removed in vacuo to provide a yellow solid that was purified by silica gel column chromatography, eluting with 1% mehanol in dichloromethane, to yield a yellow solid (69 mg, 79%): mp 201–202 °C. IR (film) 3440, 3427, 3326, 1672, 1655, 1603 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.80 (d, J = 8.7 Hz, 2 H), 7.62 (s, 1 H), 7.06 (t, J = 1.2 Hz, 1 H), 6.99 (s, 1 H), 6.66 (d, J = 8.7 Hz, 2 H), 5.52 (s, 2 H); 13C NMR (75 MHz, CD3OD) δ 192.1, 156.1, 139.7, 131.8, 128.4, 123.9, 122.3, 114.3, 52.9; ESIMS m/z 202 (MH+); HRMS calcd for C11H11ON3 m/z 201.0902, found: m/z 201.0904 (M+).
4.2.56. (Z)-1-(3-(3,5-Dimethoxyphenyl)-2-(4-nitrophenyl)allyl)-1H-imidazole (82)
Potassium carbonate (118.7 mg, 0.86 mmol) and a small amount of 18-crown-6 were added to a solution of compounds 80 (100 mg, 0.43 mmol) and 78 (213 g, 0.43 mmol) in anhydrous CH2Cl2 (15 mL), the resulting mixture was stirred under reflux for 24 h. The insoluble material was then filtered off and the filtrate was concentrated to provide the orange oil that was purified by flash column chromatography, eluting with a solution of 1% methanol in dichloromethane, to afford 82 (67.5 mg, 43%) as a yellow solid that was recrystallized from hexane and dichloromethane: mp 132–133 °C. IR (film) 3368, 1592, 1515, 1456, 1344, 1204, 1154, 1067, 856 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 8.7 Hz, 2 H), 7.52 (d, J = 8.7 Hz, 2 H), 7.42 (s, 1 H), 7.20 (s, 1 H), 6.98 (s, 1 H), 6.83 (s, 1 H), 6.47-6.43 (m, 3 H), 5.17 (s, 2 H), 3.78 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 161.0, 147.3, 145.9, 137.0, 136.7, 136.5, 134.1, 129.9, 127.1, 124.0, 118.5, 106.4, 100.3, 55.3, 45.8; ESIMS m/z 366 (MH+); HRMS calcd for C20H20O4N3 m/z 366.1454, found: m/z 366.1456 (MH+).
4.2.57. (Z)-4-(1-(3,5-Dimethoxyphenyl)-3-(1H-imidazol-1-yl)prop-1-en-2-yl)benzenamine (84)
Stannous chloride (259 mg, 1.37 mmol) was added to a solution of (Z)-1-(3-(3,5-dimethoxyphenyl)-2-(4-nitrophenyl)allyl)-1H-imidazole (82) (100 mg, 0.27 mmol) in ethanol (15 mL) and the reaction mixture was stirred under reflux for 2 h. The ethanol was removed, aq NaOH (1 N, 15 mL) was added to the residue and the mixture was stirred for 0.5 h. The aqueous layer was then extracted by diethyl ether (3 × 15 mL) and the organic layer was dried over sodium sulfate. The solvent was evaporated in vacuo at room temperature to provide a yellow solid that was purified by silica gel flash column chromatography, eluting with a solution of 2% methanol in dichloromethane, to provide a white solid (62.5 mg, 69%): mp 134–135 °C.IR (film) 3368, 1590, 1517, 1456, 1204, 1153, 1067, 830 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.43 (s, 1 H), 7.20 (d, J = 8.4 Hz, 2 H), 7.00 (s, 1 H), 6.96 (s, 1 H), 6.86 (s, 1 H), 6.61 (d, J = 8.4 Hz, 2 H), 6.40-6.37 (m, 3 H), 5.04 (s, 2 H), 3.73 (s, 6 H), 3.55 (br, 2 H); 13C NMR (75 MHz, CDCl3) δ 160.8, 146.6, 138.6, 136.8, 135.0, 130.8, 129.3, 129.1, 127.1, 118.6, 115.0, 106.3, 99.5, 55.2, 46.0; ESIMS m/z 336 (MH+); HRMS calcd for C20H22O2N3 m/z 336.1712, found: m/z 336.1714 (MH+).
4.2.58. 4-Amino-trans-stilbene (JH-2–29).62
Pd(OAc)2 (51 mg, 0.23 mmol) was added to a well-stirred mixture of compound 85 (0.78 mL, 6.85 mmol) and 24 (1.0 g, 4.57 mmol), Bu4NBr (2.94 g, 9.13 mmol), and K2CO3 (1.58 g, 11.4 mmol) in anhydrous DMF (20 mL) under argon atmosphere at room temperature. The resulting brown solution was stirred at 80 °C under argon atmosphere for 4 h, cooled down to room temperature, and diluted with H2O (100 mL). 1 M HCl was added to the reaction mixture until pH<5. The resulting suspension was extracted with EtOAc (3 × 100 mL). The combined organic layers were first washed with water (3 × 30 mL) and brine (1 × 30 mL) and dried over sodium sulfate. The solvent was removed under reduced pressure. The crude residue was purified by recrystallization from hexanes to afford JH-2–29 as a light yellow solid (0.35 g, 40%): mp 148–150 °C. 1H NMR (300 MHz, acetone-d6) δ 7.48 (d, J = 7.3 Hz, 2 H), 7.30-7.28 (m, 2 H), 7.17 (tt, J = 7.3 Hz, J = 1.0 Hz, 1 H), 7.09 (d, J = 16.2 Hz, 1 H), 6.93 (d, J = 16.2 Hz, 1 H), 6.67 (d, J = 8.4 Hz, 2 H), 4.82 (s, 2 H); ESIMS m/z 196 (MH+).
4.3. Molecular Modeling
The crystal structure of aromatase in complex with androgen was downloaded from the Protein Data Bank (PDB code 3eqm).63 Hydrogens were added and minimized using the Amber force field and the Amber charges. Modeled analogues were constructed in SYBYL 8.1,64 and energy was minimized with the Amber force field and Amber charges. Docking analogues into the binding site of aromatase was performed using the GOLD program. For the genetic algorithm (GA) runs, a maximum number of 100,000 GA operations were performed on a single population of 100 individuals. Operator weights for crossover, mutation, and migration were set to 95, 95, and 10, respectively, which are the standard default settings recommended by the authors. The maximum distance between hydrogen bond donors and acceptors for hydrogen bonding was set to 3.5 Å. After docking, the best docked conformation of compound 32 was merged into the ligand-free protein. The new ligand–protein complex was subsequently subjected to energy minimization using the Amber force field with Amber charges. During the energy minimization, the structure of the compound 32 and a surrounding 6 Å sphere were allowed to move, while the structures of the remaining protein were frozen. The energy minimization was performed using the Powell method with a 0.05 kcal/(mol Å) energy gradient convergence criterion and a distance-dependent dielectric function.
4.4. Aromatase Assay
Aromatase activity was assayed as previously reported, with the necessary modifications to assay in a 384-well plate.65 Briefly, the test compound (3.5 µL) was preincubated with 30 µL of NADPH regenerating system (2.6 mM NADP+, 7.6 mM glucose 6-phosphate, 0.8 U/mL glucose-6-phosphate dehydrogenase, 13.9 mM MgCl2, and 1 mg/mL albumin in 50 mM potassium phosphate buffer, pH 7.4) for 10 min at 37 °C. The enzyme and substrate mixture (33 µL of 1 µM CYP19 enzyme, BD Biosciences, 0.4 µM dibenzylfluorescein, 4 mg/mL albumin in 50 mM potassium phosphate, pH 7.4) was added, and the plate was incubated for 30 min at 37 °C before quenching with 25 µL of 2 N NaOH. After termination of the reaction and shaking for 5 min, the plate was further incubated for 2 h at 37 °C. This enhances the ratio of signal to background. Fluorescence was measured at 485 nm (excitation) and 530 nm (emission). IC50 values were based on three independent experiments performed in duplicate using five concentrations of test substance (Table 1).
Kinetic aromatase assays were carried out in order to determine inhibitor mechanism. Test compound was preincubated with 30 µL of NADPH regenerating system (2.6 mM NADP+, 7.6 mM glucose 6-phosphate, 0.8 U/mL glucose 6-phosphate dehydrogenase, 13.9 mM MgCl2, and 1 mg/mL albumin in 50 mM potassium phosphate buffer, pH 7.4) for 10 min at 37 °C. The IC50 was used as the final concentration for each inhibitor. Substrate was added at five concentrations: 16, 8, 4, 2, and 1 µM. CYP19 (1 µM) was added and fluorescence was measured at 485 nm (excitiation) and 530 nm (emission) every 10 seconds for at least five minutes. Error values represent three independent experiments for each compound.
4.5. Expression and Purification of Human QR2
Human QR2 was purified from 3 L of E. coli BL21(DE3) grown in Luria-Bertani medium supplemented with 100 µg/mL ampicillin (Fisher Scientific, Pittsburg h, PA) following our previously reported procedures.18,24 The concentration of purified QR2 was determined using the Bio-RAD Protein Assay. QR2 was concentrated to 24 mg/mL for crystallization and 4 mg/mL for all kinetics assays. Freshly prepared, non-frozen QR2 was used to set up crystallization experiments. Frozen QR2, previously stored at −80 °C, was thawed on ice and then adjusted to the desired concentration by diluting in storage buffer without glycerol for kinetic assays and inhibitor studies.
4.6. Steady-State Kinetic Assays and IC50 Value Determination
The catalytic activity of QR2 was determined at 23 °C using a SpectraMax Plus 384 UV/Vis microplate reader by monitoring the increase in absorbance at 612 nm, which is due to formation of the product formazan, the reduced form of the substrate MTT [3-(4,5-dimethyltiazol-2-yl)-2,5-diphenyltetrazolium bromide]. Assays were run in 96-well plates with a final assay volume of 200 µL. Each assay mixture contained 12 nM QR2, 17.5 µM NMeH, and 200 µM MTT in a reaction buffer containing 100 mM NaCl, 50 mM Tris, and 0.1% Triton-X100 (Fisher Biotech 93004). All reactions were initiated by the addition of QR2. The initial slopes of the reaction (ΔAbs/Δtime) were measured and were used to calculate the initial rates of the reaction using a value of 11,300 mM−1 cm−1 for the molar extinction coefficient.
IC50 values, the inhibitory concentration that produces 50% inhibition, were determined in 96-well plates at 23 °C using the same assay conditions described above except that inhibitor concentrations were varied from 0.2 to 100 µM. Assays at each inhibitor concentration were performed in triplicate, and the average and standard deviations in the rate values were used to determine the final IC50 values by calculating the percent inhibition (%I) at each inhibitor concentration versus the negative control with zero inhibitor. The percent inhibition data were then plotted as a function of inhibitor concentration, [I], and the data were then fit to the following equation: %I = %Imax/[(1 + [I]/ IC50)] using nonlinear regression describing a simple binding isotherm. All data were globally fit to the equation using the Enzyme Kinetics Module of the program SigmaPlot from SPSS Scientific. IC50 values are reported along with their standard error in the fitted parameter.
4.7. Crystallization and X-ray Structure Determination of Human QR2 Inhibitor Complexes
QR2 was crystallized using the hanging-drop, vapor-diffusion method by adding 1 µL of purified QR2 to 1 µL of reservoir solution which contained 1.7 M ammonium sulfate, 0.1 M BisTris, pH 6.5, 0.1 M NaCl, 5 mM DTT, and 12 µM FAD. Diffraction quality crystals grew within two weeks, and individual crystals (approximately 0.1 mm in length) were transferred to a 10 µL solution, prepared with 9 µL of reservoir solution and 1 µL of a stock solution of inhibitor (10 mM in 100% DMSO). The final inhibitor concentration was 0.4 mg/mL and the crystals soaked for 24 h. Crystals were retrieved with a nylon loop which was then used to swipe the crystals through the well solution supplemented with 20% glycerol. The crystals were immediately flash-frozen by plunging into liquid nitrogen. Crystals were stored in shipping dewars containing liquid nitrogen until X-ray data collection.
X-Ray diffraction data were collected at beamline 21-ID-F (λ = 0.978272 Å) at the Life Sciences-Collaborative Access Team (LS-CAT) at the Advanced Photon Source (APS), Argonne National Laboratories. Flash-frozen crystals were transferred from shipping dewars and then mounted on a goniostat under a stream of dry N2 at 100 °K. X-ray data were collected on a MarMosaic 225 mm CCD detector. Complete X-ray data sets were collected on QR2-Compund-32 and QR2-Compound-44 complexes. X-ray data were processed and scaled using the program HKL2000.66 The structures of both complexes belonged to space group P212121. Data on the QR2-compound 32 complex were collected to 1.74 Å (1.77-1.74 Å) where the numbers in parentheses represent the highest resolution shell. The overall completeness of the dataset was 91.0% (94.2%) and the average I/σI was 28.5 (4.3). Data on the QR2-compound 44 complex were collected to 1.55 Å (1.58-1.55 Å) resolution with a completeness of 99.7% (99.7%) with an overall average I/σI of 37.2 (5.3).
Intensities were converted to structure-factor amplitudes by the method of French and Wilson67 using the program TRUNCATE in the CCP4 program suite.68 The initial phases for the model were determined by the method of molecular replacement using the program PHASER69 in the CCP4 program suite. The search model used for molecular replacement consisted of the dimer of wild-type QR2 (PDB:2QX4)24 with all side chains intact and all waters and ligands removed with the exception of FAD. The final and optimal molecular replacement solution contained a single dimer in the asymmetric unit. An initial round of combined positional and B-factor refinement was performed with the program REFMAC70 in the CCP4 program suite using a maximum-likelihood target function and no sigma cutoff on structure factor amplitudes. Initial difference Fourier maps were calculated and visualized using the program COOT71 The initial Fo-Fc difference maps revealed strong (+4σ), residual electron density peaks in the active site for the inhibitors. Molecular models for the inhibitors were built using the Monomer Library Sketcher program in the CCP4 program suite, and monomer library descriptions were created for refinement. The inhibitors were manually built into density using the program COOT, and the structures were refined using REFMAC. Iterative rounds of refinement were performed, and water molecules were added manually into strong (+4σ) difference density peaks in the initial refinement stages, and into peaks of (+3σ) in the final stages of refinement. Iterative refinement using REFMAC was continued until the Rcryst and Rfree values reached their lowest plateau values. The final X-ray structural model of the QR2-compound 44 complex was refined to a value of 20.2% (25.6%) for Rwork and a value of 25.4% (35.2%) for Rfree. The X-ray structure of the QR2-compound 32 complex was refined to a value of 20.8% (25.5%) for Rwork and a value of 23.7% (28.6%) for Rfree. Structural models and electron density maps presented in the figures were produced using the program PyMOL.
Scheme 1.

Reagents and conditions: (i) CH3PPh3Br, NaNH2, Et2O, rt; (ii) KOAc, Pd(OAc)2, Bu4NBr, DMF, 50–80 °C.
Scheme 2.
Reagents and conditions: (i) H2, Pd/C, MeOH.
Scheme 3.

Reagents and conditions: (i) SnCl2, EtOH.
Scheme 4.

Reagents and conditions: (i) NaOMe, MeOH.
Scheme 5.

Reagents and conditions: (i) TBDMSCl, DIPEA, DMF; (ii) CH3PPh3Br, NaNH2, Et2O; (iii) TBAF, THF; (iv) Ac2O, DMAP, DCM; (v) KOAc, Pd(OAc)2, Bu4NBr, DMF, 70 °C, 10 h; (vi) NaOMe, MeOH.
Scheme 6.

Reagents and conditions: (i) NaBH4, THF; (ii) PBr3, DCM; (iii) PPh3, toluene, reflux; (iv) Imidazole, THF, rt; (v) H2, Pd/C, MeOH; (vi) K2CO3, 18-crown-6, DCM, reflux; (vii) SnCl2, EtOH, reflux.
Scheme 7.

Reagents and conditions: (i) K2CO3, Pd(OAc)2, Bu4NBr, DMF, 80 °C.
Acknowledgement
This work was supported by program project grant P01 CA48112 awarded by the National Cancer Institute. Some of this research was conducted in a facility constructed with the financial support of a Research facilities Improvement program grant C06-14499 from the National Institutes of Health. Bin Sun wishes to thank China Scholarship Council and Dr Hongxiang Lou (Shandong Uniersity, P. R. China) for the financial support and assistance. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor research program (Grant 085P1000817).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takaoka MJ. J. Faculty Sci. Hokkaido Imperial University. 1940;3:1–16. [Google Scholar]
- 2.Kopp P. Eur J Endocrinol. 1998;138:619–620. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]
- 3.Baur JA, Sinclair DA. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Zhou D, Okubo T, Kao YC, Eng ET, Grube B, Kwon A, Yang C, Yu B. Ann. N. Y. Acad. Sci. 2002;963:229–238. doi: 10.1111/j.1749-6632.2002.tb04115.x. [DOI] [PubMed] [Google Scholar]
- 6.Brueggemeier RW, Richards JA, Joomprabutra S, Bhat AS, Whetstone JL. J. Steroid Biochem. Mol.Biol. 2001;79:75–84. doi: 10.1016/s0960-0760(01)00127-3. [DOI] [PubMed] [Google Scholar]
- 7.Banting L, Nicholls PJ, Shaw MA, Smith HJ. Prog. Med. Chem. 1989;26:253–298. doi: 10.1016/s0079-6468(08)70242-x. [DOI] [PubMed] [Google Scholar]
- 8.Lonard DM, Smith CL. Steroids. 2002;67:15–24. doi: 10.1016/s0039-128x(01)00133-7. [DOI] [PubMed] [Google Scholar]
- 9.Park WC, Jordan VC. Trends Mol. Med. 2002;8:82–88. doi: 10.1016/s1471-4914(02)02282-7. [DOI] [PubMed] [Google Scholar]
- 10.Nicholls H. Trends Mol. Med. 2002;8:S12–S13. doi: 10.1016/s1471-4914(02)02304-3. [DOI] [PubMed] [Google Scholar]
- 11.Miller WR. Best. Pract. Res. Clin. Endocrinol Metab. 2004;18:1–32. doi: 10.1016/s1521-690x(03)00044-7. [DOI] [PubMed] [Google Scholar]
- 12.Miller WR. Semin. Oncol. 2003;30:3–11. doi: 10.1016/s0093-7754(03)00302-6. [DOI] [PubMed] [Google Scholar]
- 13.Buzdar AU. Clin. Breast Cancer. 2003;4 Suppl 2:S84–S88. doi: 10.3816/cbc.2003.s.020. [DOI] [PubMed] [Google Scholar]
- 14.Recanatini M, Cavalli A, Valenti P. Med. Res. Rev. 2002;22:282–304. doi: 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]
- 15.Maiti A, Cuendet M, Croy VL, Endringer DC, Pezzuto JM, Cushman M. J. Med. Chem. 2007;50:2799–2806. doi: 10.1021/jm070109i. [DOI] [PubMed] [Google Scholar]
- 16.Mokbel K. International Journal of Clinical Oncology. 2002;7:279–283. doi: 10.1007/s101470200040. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Lee KW, Chan FL, Chen S, Leung LK. Toxicol. Sci. 2006;92:71–77. doi: 10.1093/toxsci/kfj190. [DOI] [PubMed] [Google Scholar]
- 18.Maiti A, Reddy PVN, Sturdy M, Marler L, Pegan SD, Mescar AD, Pezzuto JM, Cushman M. J. Med. Chem. 2009;52:1873–1884. doi: 10.1021/jm801335z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Celli CM, Tran N, Knox R, Jaiswal AK. Biochem. Pharmacol. 2006;72:366–376. doi: 10.1016/j.bcp.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 20.Jamieson D, Tung AT, Knox RJ, Boddy AV. J.Cancer. 2006;95:1229–1233. doi: 10.1038/sj.bjc.6603414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knox RJ, Jenkins TC, Hobbs SM, Chen S, Melton RG, Burke PJ. Cancer Res. 2000;60:4179–4186. [PubMed] [Google Scholar]
- 22.Buryanovskyy L, Fu Y, Boyd M, Ma YL, Hsieh TC, Wu JM, Zhang ZT. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutin JA, Audinot V, Ferry G, Delagrange P. Trends Pharmacol. Sci. 2005;26:412–419. doi: 10.1016/j.tips.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Calamini B, Santarsiero BD, Boutin JA, Mesecar AD. Biochem. J. 2008;413:81–91. doi: 10.1042/BJ20071373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angela F, Carolina F, Carolina M, Marcella G, Giulia N. Nat. Prod. Res. 2007;21:564–573. Part A. [Google Scholar]
- 26.Sun B, Yuan HQ, Xi GM, Ma YD, Lou HX. Bioorg. Med. Chem. 2009;17:4981–4989. doi: 10.1016/j.bmc.2009.05.077. [DOI] [PubMed] [Google Scholar]
- 27.Eicher T, Fey S, Puhl W, Büchel E, Speicher A. Eur. J. Org. Chem. 1998;5:877. [Google Scholar]
- 28.Ghosh D, Griswold J, Erman M, Pangborn W. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 30.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 31.Wang R, Lu Y, Wang S. J. Med. Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 32.Recanatini M, Bisi A, Cavalli A, Belluti F, Gobbi S, Rampa A, Valenti P, Palzer M, Palusczak A, Hartmann RF. J. Med. Chem. 2001;44:672–680. doi: 10.1021/jm000955s. [DOI] [PubMed] [Google Scholar]
- 33.Leonetti F, Favia A, Rao A, Aliano R, Paluszcak A, Hartmann RF, Carotti A. J. Med. Chem. 2004;47:6792–6803. doi: 10.1021/jm049535j. [DOI] [PubMed] [Google Scholar]
- 34.Gobbi S, Cavalli A, Negri M, Schewe KE, Belluti F, Piazzi L, Hartmann RW, Recanatini M, Bisi A. J. Med. Chem. 2007;50:3420–3422. doi: 10.1021/jm0702938. [DOI] [PubMed] [Google Scholar]
- 35.Neves MAC, Dinis TCP, Colombo G, Melo MLS. J. Med. Chem. 2009;52:143–150. doi: 10.1021/jm800945c. [DOI] [PubMed] [Google Scholar]
- 36.Saberi MR, Vinh TK, Yee SW, Griffiths N, Evans PJ, Simons C. J. Med. Chem. 2006;49:1016–1022. doi: 10.1021/jm0508282. [DOI] [PubMed] [Google Scholar]
- 37.Buryanovskyy L, Fu Y, Boyd M, Ma YL, Hsieh TC, Wu JM, Zhang ZT. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carreno MC, Mahugo J, Urbano A. Tetrahedron Lett. 1997;38:3047–3050. [Google Scholar]
- 39.Takashima Y, Kaneko Y, Kobayashi Y. Tetrahedron. 2010;66:197–207. [Google Scholar]
- 40.Marx VM, Burnell DJ. J. Am. Chem. Soc. 2010;132:1685–1689. doi: 10.1021/ja909073r. [DOI] [PubMed] [Google Scholar]
- 41.Alonso F, Riente P, Yus M. Eur. J. Org. Chem. 2009;34:6034–6042. [Google Scholar]
- 42.Arrish JP, Jung YC, Shin S, Jung KW. J. Org. Chem. 2002;67:7127–7130. doi: 10.1021/jo020159p. [DOI] [PubMed] [Google Scholar]
- 43.Waterman PG. Phytochemistry. 1976;15:347–356. [Google Scholar]
- 44.Banerjee B, Capps SG, Kang J, Robinson JW, Castle SL. J. Org. Chem. 2008;73:8973–8978. doi: 10.1021/jo801721z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcoux D, Goudreau SR, Charette AB. J. Org. Chem. 2009;74:8939–8955. doi: 10.1021/jo902066y. [DOI] [PubMed] [Google Scholar]
- 46.Coe BJ, Hamor TA, Jones CJ, McCleverty JA, Bloor D, Cross GH, Axon TL. J. Chem. Soc. Dalton.Trans. 1995;4:673–684. [Google Scholar]
- 47.Motoshima K, Noguchi-Yachide T, Sugita K, Hashimoto Y, Ishikawa M. Bioorg. Med. Chem. 2009;17:5001–5014. doi: 10.1016/j.bmc.2009.05.066. [DOI] [PubMed] [Google Scholar]
- 48.Velder J, Ritter S, Lex J, Schmalz HG. Synthesis. 2006;2:273–278. [Google Scholar]
- 49.Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E. J. Med. Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- 50.Ling C, Minato M, Lahti PM, Van Willigen H. J. Am. Chem. Soc. 1992;114:9959–9969. [Google Scholar]
- 51.Iding H, Jolidon S, Krummenacher D, Rodriguez SRM, Thomas AW, Wirz B, Wostl W, Wyler R. PCT Int. Appl. 2004:55. [Google Scholar]
- 52.Liu H, Dong A, Gao C, Tan C, Liu H, Zu X, Jiang Y. Bioorg. Med. Chem. 2008;16:10013–10021. doi: 10.1016/j.bmc.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 53.Moro AV, Cardoso FSP, Correia CRD. Tetrahedron Lett. 2008;49:5668–5671. [Google Scholar]
- 54.Moran BW, Anderson FP, Devery A, Cloonan S, Butler WE, Varughese S, Draper SM, Kenny PTM. Bioorg. Med. Chem. 2009;17:4510–4522. doi: 10.1016/j.bmc.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno CS, Schrader KK, Rimando AM. J. Agric. Food Chem. 2008;56:9140–9145. doi: 10.1021/jf801988p. [DOI] [PubMed] [Google Scholar]
- 56.Kang SS, Cuendet M, Endringer DC, Croy VL, Pezzuto JM, Lipton MA. Bioorg. Med. Chem. 2009;17:1044–1054. doi: 10.1016/j.bmc.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 57.Suresh A, Tatsuya M, Zaw LT, Feng L, Nwet W, Yasuhiro T, Hiroyasu E, Shigetoshi K. J. Nat. Prod. 2009;72:1631–1636. [Google Scholar]
- 58.Angela F, Carolina F, Carolina M. Nat. Prod. Res. Part A: Structure and Synthesis. 2006;20:247–252. [Google Scholar]
- 59.Marcella G, Carolina M, Angela F. Tetrahedron Lett. 2002;43:597–598. [Google Scholar]
- 60.Michael M, Jill M, Johan A, Carmen A, Marie L, Michelle D. PCT Int. Appl. 2007:337. [Google Scholar]
- 61.Gabriele B, Benabdelkamel H, Plastina P, Fazio A, Sindona G, Donna LD. Synthesis. 2008;18:2953–2956. [Google Scholar]
- 62.Arvela RK, Leadbeater NE, Sangi MS, Williams VA, Granados P, Singer RD. J. Org. Chem. 2005;70:161–168. doi: 10.1021/jo048531j. [DOI] [PubMed] [Google Scholar]
- 63.www.rcsb.org.
- 64.SYBYL 8.1 for Linux; Tripos, Inc.: 2007. 1699 South Hanley Rd., St. Louis, MO 63144-2917, USA.
- 65.Maiti A, Cuendet M, Croy VL, Endringer DC, Pezzuto JM, Cushman M. J. Med. Chem. 2007;50:2799–2806. doi: 10.1021/jm070109i. [DOI] [PubMed] [Google Scholar]
- 66.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. Vol 276. New York: Academic Press; 1997. pp. 307–326. part A ed. [DOI] [PubMed] [Google Scholar]
- 67.French S, Wilson K. Acta Cryst. 1994;A34:517–525. [Google Scholar]
- 68.CCP4 Collaborative Project Number 4. Acta Cryst. 1994;D50:760–763. [Google Scholar]
- 69.Storoni LC, McCoy AJ, Read RJ. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 70.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr. D. Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 71.Emsley P, Cowtan K. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]