

Abstract
Purpose
Prostate stem cell antigen (PSCA), a cell surface glycoprotein expressed in normal human prostate and bladder, is over-expressed in the majority of localized prostate cancer and most bone metastases. We have previously shown that the hu1G8 minibody, a humanized anti-PSCA antibody fragment (single-chain Fv-CH3 dimer, 80 kDa), can localize specifically and image PSCA-expressing xenografts at 21 h post-injection. However, the humanization and antibody fragment reformatting decreased its apparent affinity. Here, we sought to evaluate PET imaging contrast with affinity matured minibodies.
Methods
Yeast scFv display, involving four rounds of selection, was used to generate the three affinity matured antibody fragments (A2, A11, and C5) that were reformatted into minibodies. These three affinity matured anti-PSCA minibodies were characterized in vitro, and following radiolabeling with 124I were evaluated in vivo for microPET imaging of PSCA-expressing tumors.
Results
The A2, A11, and C5 minibody variants all demonstrated improved affinity compared to the parental (P) minibody and were ranked as follows: A2 > A11 > C5 > P. The 124I-labeled A11 minibody demonstrated higher immunoreactivity than the parental minibody and also achieved the best microPET imaging contrast in two xenograft models, LAPC-9 (prostate cancer) and Capan-1 (pancreatic cancer), when evaluated in vivo.
Conclusion
Of the affinity variant minibodies tested, the A11 minibody that ranked second in affinity was selected as the best immunoPET tracer to image PSCA-expressing xenografts. This candidate is currently under development for evaluation in a pilot clinical imaging study.
Keywords: Imaging, Prostate, Cancer, PSCA, Antibody, PET
Introduction
New imaging modalities for prostate cancer are needed for the accurate detection, staging, and monitoring of prostate cancer. PET imaging using 18F-FDG, although widely embraced in oncology, frequently falls short in prostate cancer due to low and heterogeneous uptake of the tracer; furthermore, proximity to the bladder interferes with imaging primary disease. More recently, metabolic tracers have been successfully tested for prostate cancer, including 11C-choline, 11C-acetate, 18F-fluorocholine, and anti-18F-FACBC, a synthetic leucine analog [1, 2]. Although these tracers are able to quantify cellular processes which are modified in prostate cancer, they are not prostate cancer specific. Prostate cancer metastasizes primarily to lymph nodes and bones, with bone metastases being the predominant manifestation and major cause for mortality. Bone scintigraphy using 99mTc-methylene diphosphonate remains the primary clinical imaging tool for detection of metastases; however, it is not tumor specific. In addition, bone scans have limited utility for monitoring response to treatment, since signal can remain long after viable tumor is eliminated, and in many instances, a post-treatment flare reaction can further complicate interpretation.
To generate more specific probes for prostate cancer, cell surface markers and receptors can be targeted. One strategy comprises radiolabeling antibodies or antibody fragments, which recognize cell surface markers, with a radionuclide which can be detected in vivo by single photon emission computed tomography (immunoSPECT) or positron emission tomography (immunoPET) [3]. Capromab pendetide, or ProstaScint™ (Cytogen Corp., Princeton, NJ, USA), is an 111In-labeled murine anti-PSMA (prostate-specific membrane antigen) monoclonal antibody (mAb) that was approved by the US Food and Drug Administration (FDA) for the detection of recurrent local disease and lymph node metastases [4]. However, there are several limitations with this tracer including low imaging contrast, delayed imaging, and the induction of an immune response after the first injection [5].
Another promising cell surface target for molecular imaging of prostate cancer is the prostate stem cell antigen (PSCA), which is over-expressed in a majority of prostate cancers and has low levels of expression in a very limited number of normal tissues [6, 7]. Other than expression in normal prostate, the most noticeable PSCA expression detected by immunohistochemistry was in the bladder and neuroendocrine cells of the stomach and colon [8]. In the same study PSCA protein was shown to be over-expressed by a majority of localized prostate cancers and to correlate with Gleason score, pathologic stage and androgen independence. Furthermore, PSCA was detected in over 60% of lymph node and liver metastases and strong PSCA staining was observed in 87–100% of bone metastases [8, 9]. In addition, PSCA is also highly expressed in localized bladder and pancreatic carcinoma [10–12].
We have previously evaluated a humanized anti-PSCA intact antibody (hu1G8) as a molecular imaging probe for microPET imaging [13]. Hu1G8 was radiolabeled with the positron emitter 124I (T 1/2 4.2 days) and injected into SCID mice bearing LAPC-9 xenografts. Tumors were detected by microPET imaging, with the maximal target to background signal obtained 1 week after injection. In an effort to optimize PSCA-targeted prostate tumor imaging, we have engineered several hu1G8 antibody fragments to improve the pharmacokinetics of the immunoPET probe. One of these engineered proteins, the hu1G8 minibody (single-chain Fv-CH3 dimer, 80 kDa) shown in Fig. 1, demonstrated faster blood clearance kinetics resulting in clear image contrast of prostate cancer xenografts at 21 h [14].
Fig. 1.

Parental hu1G8 (P) and affinity variant minibodies A2, A11, and C5. a Sequence of CDRs affected by residue substitution(s). b Schematic presentations of minibodies and scFv fragments. *Random residue substitution, V L variable light, V H variable heavy, C H constant heavy
The apparent affinities of the intact hu1G8 antibody and minibody are 5 and 46 nM, respectively, which represents a ∼ninefold loss [14]. The effect of affinity on intact antibody and antibody fragments tumor targeting is a controversial subject. The mathematical model of intact antibody distribution of Fujimori et al. suggests that if one considers that a uniform repartition is desired for therapy, a lower affinity antibody might be preferable [15]. In animal models, a number of studies show increased delivery and/or therapeutic efficacy of higher affinity intact antibody [16–18]. In another study intact antibodies with KD above 1 nM (3.4 and 11) have shown better tumor delivery than antibodies with sub nM KD [19]. Tumor targeting of small antibody fragments such as single-chain Fv (scFv, 25 kDa) or diabody (scFv dimer, 50 kDa) has also suggested that the best tumor targeting would be obtained at close to nanomolar affinity for these smaller engineered antibody fragments [20, 21].
Our goal was to improve the affinity of the parental anti-PSCA hu1G8 minibody in an effort to generate a tracer with better PET imaging contrast. Molecular evolution associated with yeast display is a powerful strategy for antibody affinity maturation. It enables selection of higher affinity scFv from a yeast library generated from error-prone polymerase chain reaction (PCR) applied to the complete scFv, rather than focusing on mutation of the CDR loop residues [22]. Using this method, high antibody affinity can be achieved through the cumulative effect of many small structural alterations [23]. This strategy has been successfully used to improve the affinity and stability of a humanized antibody specific to carcinoembryonic antigen (CEA) which resulted in improved retention time into CEA-expressing xenografts [24]. Here, a similar strategy was used to improve the affinity of an anti-PSCA scFv fragment. Three minibody variants demonstrating higher affinity to PSCA were derived. Furthermore, to evaluate the potential utility of these affinity variants for imaging, the minibodies were radiolabeled with 124I for in vivo microPET imaging studies in two xenograft models: LAPC-9 (prostate cancer) and Capan-1 (pancreatic cancer).
Materials and methods
Cell lines, antibodies
Yeast strain EBY100 (GAL1-AGA1::URA3 ura3-52 trp1 leu2Δ1 his3Δ200 pep4:HIS2 prb1Δ1.6R can1 GAL) was grown in YPD medium, EBY100 transfected with expression vector pYD2 was selected on SD-CAA medium [25]. Hybridoma and mouse myeloma cells NS0 (ATCC, Manassas, VA, USA) were grown in Iscove’s modified Dulbecco’s medium with 5% fetal bovine serum (FBS). The prostate cancer LAPC-9 was isolated and maintained as previously described [26]. Capan-1 was obtained from ATCC. The B-cell lymphoma SKW6.4 and the PSCA-transduced SKW 6.4 cell lines were used to determine the immunoreactive fraction. They were maintained as previously described [14]. R-phycoerythrin (PE) AffiniPure F(ab′)2 anti-human or anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were used for flow cytometry detection. Alkaline phosphatase (AP)-conjugated streptavidin (Jackson ImmunoResearch Laboratories) was used for enzyme-linked immunosorbent assay (ELISA) detection. PSCA level of cell surface expression was determined using QIFIKIT (Dako North America Inc., Carpinteria, CA, USA).
Construction of mutant scFv yeast display library
The coding sequences of the VL and VH were extracted from the parental anti-PSCA huIG8 minibody, fused with an 18 residue GlySer-rich linker to form scFv in the VL-VH orientation [14, 27], and cloned into pYD2 expression vector at NcoI and NotI restriction sites. Random mutations were introduced into huIG8 scFv by error-prone PCR to construct a library of huIG8 scFv variants displayed on yeast surface as previously described [28]. Briefly, the anti-PSCA huIG8 scFv gene in the pYD2 expression vector was subjected to 20 cycles of PCR with Taq (Sigma-Aldrich Corporation, St. Louis, MO, USA) in a reaction mixture containing 0.5 mM MnCl2. The mutated scFv gene was re-amplified using a high-fidelity DNA polymerase for 35 cycles. Both PCR reactions used primers Gap5′ and Gap3′ [28]. The PCR product was electrophoresed on an agarose gel and purified using QIAquick gel extraction (Qiagen, Chatsworth, CA, USA). The mutated gene mixture was used to transform lithium acetate-treated EBY100 cells with NcoI/NotI-digested pYD2 vector, using the TRAFO method with gap repair [29]. Transformation mixes were cultured and sub-cultured in SD-CAA media (Current Protocols in Molecular Biology, Chap. 13). The library size was determined by plating serial dilutions of the transformation mixture on SD-CAA plates.
Selection of mutant scFvs yeast display library
The transformed culture was induced in SG-CAA medium (SD-CAA medium with glucose replaced by galactose) for 24 h at 20°C to express and display scFv on the surface of yeast. For the first round of selection, 2 × 107 yeast (more than 30 times the library size) were washed, resuspended in FACS buffer [phosphate-buffered saline (pH 7.4), 0.5% bovine serum albumin] containing 200 nM of PSCA fused to the human IgG1-Fc domain (PSCA-γ1Fc), and incubated for 1 h at room temperature. The concentrations of PSCA-γ1Fc used for rounds 2, 3, and 4 of sorting were 5, 2, and 1 nM, respectively. After incubation, cells were washed three times with ice-cold FACS buffer, resuspended in a 1:400 (v/v) dilution of 1 mg/ml SV5-Alexa647 and 1:500 (v/v) dilution of 1 mg/ml anti-human (Fc specific)-PE conjugate, and incubated for 30 min at 4°C. Before sorting, the stained cells were washed three times with FACS buffer, resuspended in 100 μl of FACS buffer, and sorted on a FACSAria (Becton Dickinson, San Jose, CA, USA). Typically 0.5–1% of the PSCA binding population was gated for collection under the yield mode for the first two rounds and the purity mode for the last two rounds. The collected cells were grown in SD-CAA media and used for the next round of sorting after induction in SG-CAA. Twenty yeast clones picked from the fourth round of sorting were analyzed for comparing the binding activity to the parental scFv by flow cytometry.
Cloning, expression, and selection of minibodies
To construct the minibodies, the scFv genes were each fused to a signal peptide upstream, and the human IgG1 hinge and CH3 region downstream [27]. Two million mouse myeloma cells were transfected with 10 µg of linearized (cut with Pvu II) plasmid DNA carrying the minibody construct by electroporation and selected in glutamine-deficient media. Clones were screened for expression by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. Supernatants of individual clones were electrophoresed on SDS-PAGE and transferred onto a nitrocellulose membrane for detection with an anti-human Fc alkaline phosphatase-conjugated antibody. The strength of the signals was compared and the highest producing clones were expanded.
Protein purification and characterization
Soluble minibodies were purified from cell culture supernatants by protein L chromatography (Poros 50 L, Applied Biosystems, Foster City, CA, USA) using an ÄKTA Purifier (GE Healthcare, Piscataway, NJ, USA). The protein was eluted using 0.1 M glycine pH 2.5 and immediately neutralized by 2 M Tris-HCl, pH 8 (10% v/v). The purified proteins were then dialyzed against phosphate-buffered saline (PBS) and concentrated with a Vivascience Vivaspin 20 (MWCO: 30,000). Final protein concentrations were determined by measuring UV absorbance at 280 nm (absorbance coefficient: 1.67). Purified proteins were analyzed by SDS-PAGE under non-reducing conditions. Native structural size was determined by size exclusion chromatography (Superdex G75, GE Healthcare).
Binding assays
The apparent affinity of intact antibodies for cells expressing PSCA was measured as previously described [28]. Briefly, a range of concentrations of intact antibody were incubated overnight at 4°C with 5 × 104 SKW 6.4-PSCA cells. Cell-bound antibodies were then detected with PE-conjugated anti-human or anti-mouse IgG antibody. Staining was analyzed by flow cytometry, and the median fluorescence intensity (MFI) was determined using the CellQuest software (Becton Dickinson). To determine the KD, MFI values were fitted to the equation: 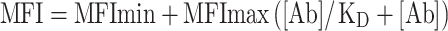 . For standard flow cytometry analysis, 5 × 105 cells were incubated for 30 min on ice with 100 µl of minibody at a 2 µg/ml concentration. Cells were washed and stained with a PE-conjugated goat-anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Competition ELISA assays were performed as previously described [13]. ELISA microtiter plates were coated overnight with 0.5 µg/ml of PSCA-mγ2a recombinant protein. A fixed amount of biotinylated parental minibody (0.3 nM) was mixed with non-biotinylated minibody competitor (parental, A2, A11, or C5) at a concentration ranging from 0.1 to 200 nM. Signal was monitored with AP-conjugated streptavidin. Phosphatase substrate tablets (Sigma-Aldrich Corporation) were dissolved in diethanolamine buffer to develop the assay. Assays were carried out in triplicate.
. For standard flow cytometry analysis, 5 × 105 cells were incubated for 30 min on ice with 100 µl of minibody at a 2 µg/ml concentration. Cells were washed and stained with a PE-conjugated goat-anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Competition ELISA assays were performed as previously described [13]. ELISA microtiter plates were coated overnight with 0.5 µg/ml of PSCA-mγ2a recombinant protein. A fixed amount of biotinylated parental minibody (0.3 nM) was mixed with non-biotinylated minibody competitor (parental, A2, A11, or C5) at a concentration ranging from 0.1 to 200 nM. Signal was monitored with AP-conjugated streptavidin. Phosphatase substrate tablets (Sigma-Aldrich Corporation) were dissolved in diethanolamine buffer to develop the assay. Assays were carried out in triplicate.
Radioiodination
Purified minibodies were radioiodinated using the iodogen method with the positron emitting isotope 124I (sodium iodide in 0.02 M NaOH) provided by IBA Molecular (Sterling, VA) as previously described [30]. The immunoreactive fraction was assayed by incubating the same amount of radioiodinated minibody with an excess amount of SKW6.4-PSCA or SKW6.4 cells or no cells for 1 h at room temperature in the same volume. Approximately 0.2 µCi (0.03 µg) of radiolabeled minibody was incubated with 20 million cells or no cells in 1 ml PBS/1% FBS. After incubation cells were pelleted by centrifugation and 0.7 ml of supernatant was counted in a Wallac WIZARD Automatic Gamma Counter (PerkinElmer Life and Analytical Sciences Inc., Boston, MA, USA). The specific immunoreactive fraction, which is the fraction that binds to the positive cells (SKW6.4-PSCA), was compared to the negative cells (SKW6.4). Experiments were carried out in duplicate.
MicroPET/CT imaging and biodistribution studies
All animal studies were conducted under protocols approved by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. LAPC-9 xenografts were established in 7- to 8-week-old male SCID mice (Charles River Laboratories, Wilmington, MA, USA) by s.c. inoculation of 1 × 106 LAPC-9 cells near the shoulder blade as described previously [13]. Capan-1 xenografts were established following the same protocol as described above except that 7- to 8-week-old male nude mice were injected near the hip. Mice were imaged when tumor masses were in the range of 100–300 mg. Thyroid uptake of radioiodine was blocked by adding 10 drops of Lugol (Sigma-Aldrich Corporation) per 100 ml of drinking water 24 h before injection of radioiodinated protein. In order to block stomach uptake, 1.5 mg of potassium perchlorate in 0.2 ml of PBS was administered by gastric lavage 30 min before injection. Each mouse was injected into the tail vein with 97–207 µCi of 124I minibody (30–50 µg of protein). Mice were serially imaged using a Focus 220 microPET scanner (Siemens Preclinical Solutions, Knoxville, TN, USA). To enable imaging, mice were anesthetized using 2% isoflurane, positioned in a prone position along the long axis of the microPET scanner, and imaged. Acquisition time was 10 min (one bed position), and images were reconstructed using a filtered backprojection (FBP) reconstruction algorithm. Directly after the final microPET scan a CT scan was performed using a microCAT II (Concorde Microsystems, Knoxville, TN, USA) for 10 min as described [31]. After the final CT scan, mice were euthanized and tumors, liver, spleen, kidney, lung, and blood were harvested, weighed, and counted in a Wallac WIZARD Automatic Gamma Counter (PerkinElmer Life and Analytical Sciences Inc., Boston, MA, USA). Results were calculated as percentage of injected dose per gram (%ID/g) of tissue.
MicroPET analysis
Cylindrical regions of interest (ROI) were drawn from three-dimensional FBP reconstructed PET/CT coregistered images using AMIDE as previously described [32]. From sagittal images, four ROIs were drawn within the tumor in high activity areas. From the coronal orientation, four ROIs were drawn in the arm muscle region in the low activity areas close to the tumors but not directly adjacent to the tumors and termed “background.” The appropriate positioning of ROIs was verified looking at each of the three orientations PET/CT images. Quantitative data were compared for significance using a one-tailed Student’s t test. All significance testing was determined at the p < 0.05 confidence level. Statistical analysis was performed using Excel 2000 (Microsoft, Redmond, WA, USA).
Results
Affinity maturation
In order to improve the affinity of hu1G8, random mutations were introduced into the hu1G8 scFv gene using error-prone PCR. The resulting gene repertoire was cloned into pYD2 that enables expression of scFv variants at the cell surface of yeasts. A yeast display library of 5.9 × 105 transformants was created. The library generated was subjected to four rounds of equilibrium-based selection using decreasing concentrations of purified recombinant PSCA protein. Twenty yeast clones from the fourth round of cell sorting were analyzed by flow cytometry. Eight of these clones showing strong staining by flow cytometry were selected and their scFv sequenced. When compared to the parental scFv sequence A2 DNA sequences were identical to five other clones with ten mutations, A11 had six mutations, and C5 had five mutations. These mutations translated into six, five, and four amino acid substitutions for A2, A11, and C5 scFv protein sequences, respectively. Two CDRs, namely VL CDR3 and VH CDR2, were affected by amino acid substitutions (Fig. 1a). In VL CDR3, at the same location, a serine was replaced by a glycine for both the A2 and A11 clones. In VH CDR2, an asparagine was replaced by a tyrosine and a threonine was replaced by a serine for the A2 clone. Introduction of a tyrosine could be more important as they are targeted by iodination. When compared to the parental scFv, two clones had a change in the number of tyrosine. In addition to the A2 clone that had an additional tyrosine introduced into VH CDR2, the A11 clone had an additional tyrosine introduced into its framework.
Biochemical characterization of the minibody variants
The parental minibody gene was used as a backbone to generate three minibody affinity variants by replacing the scFv insert with each of the three scFv affinity variants: A2, A11, and C5 (Fig. 1b). The A2, A11, C5, and parental minibody expression yield levels ranged from ∼6 to 20 mg/l of terminal culture. Minibodies were purified by protein L chromatography and analyzed on SDS-PAGE (Fig. 2a). The A2, A11, C5, and parental minibodies migrated as molecular weight species corresponding to an ∼80 kDa covalently linked minibody [14]. More significant for this study, SDS-PAGE analysis also showed that the four purified minibody variants had similar size and purity. The A11 minibody variant which has been selected for an exploratory imaging clinical trial was subsequently the subject of extended protein expression development, stability assays, and further protein characterization. The best yield obtained for one A11 minibody clone was 30 mg/l when expanded to terminal cultures into 2% FBS, as determined by ELISA (data not shown). The purified A11 minibody eluted at ∼29 min (Fig. 2b), the same time as the parental minibody on a size exclusion chromatography [14]. Five-day stability experiments at 4°C in PBS and at 37°C in 5% FBS demonstrated a 12 and 13% decrease in activity, respectively, compared to frozen sample (Fig. 2c).
Fig. 2.

Biological characterization. a SDS-PAGE analysis in non-reducing conditions of the four minibodies purified by protein L chromatography: parental (P), A11, A2, and C5. lane-M, molecular weight marker. b Size exclusion chromatography profile of A11 minibody purified by protein L chromatography on calibrated Superdex 200 Column. c A11 minibody stability; 1 mg/ml stock solution was stored at −20°C in 20% glycerol, or incubated at 4°C in PBS or 37°C in PBS/5% FBS for 5 days. Binding activity was determined by ELISA. Each dilution from 3,200 to 12,800 was assayed in triplicate
Affinity ranking
Competition ELISA demonstrated that all three minibody variants had higher affinity than the parental hu1G8 minibody (Fig. 3a). The apparent affinity as measured by competition ELISA was estimated to be 11, 16, and 26 nM for A2, A11, and C5, respectively, compared to 50 nM for the parental minibody. However, none of the minibodies was able to compete as well as the mouse monoclonal antibody 1G8. The four minibodies were also evaluated for their ability to bind to PSCA-expressing cells by flow cytometry (Fig. 3b). Flow cytometry data resulted in the same ranking as for the competition ELISA targeting recombinant PSCA. In conclusion, the apparent affinity of A2 minibody to cell surface PSCA was the strongest closely followed by A11, then C5 with the parental minibody showing the weakest affinity to PSCA: A2 > A11 > C5 > parental.
Fig. 3.

Affinity ranking. a By competitive ELISA binding assay. Plates were coated with PSCA-mγ2a recombinant protein and biotinylated parental minibody was used as probe and mixed with different concentrations of non-labeled competitor: intact monoclonal antibody 1G8, minibody: parental (P), A2, A11, and C5. b By flow cytometry 50 nM of each minibody was incubated with PSCA-expressing cells. Cells were then stained with anti-human Fc PE-conjugated antibody. Negative control, A2, A11, C5, and P minibody as are indicated
Evaluation of the minibody variants for microPET imaging of LAPC-9 xenografts
The parental hu1G8 minibody labeled with 124I has previously been evaluated for microPET imaging of a variety of prostate cancer xenograft models [14]. SCID mice bearing LAPC-9 tumors is a model for which we previously demonstrated specific tumor targeting of minibody, as an irrelevant minibody of the same isotype labeled with 124I did not localize to the xenograft [14]. We used this model to initially evaluate the affinity matured minibodies for microPET imaging efficiency. The A2, A11, and C5 minibodies were all purified by identical procedure. On the same day, each minibody affinity variant labeled with the same 124I batch was injected intravenously into mice bearing PSCA-expressing tumors. In this experiment radiolabeling was consistent among the three variants at 88.6% (±1.5%). However, A11 showed better immunoreactive fraction (60.2%) compared to C5 (49.3%) and A2 (43.9%). The best images obtained for each affinity matured minibody are shown in Fig. 4a. The A11 minibody exhibited the best biodistribution and imaging results with a tumor to blood ratio of 1.1 and a tumor to background ratio of 4.8 (Fig. 4b). In subsequent studies, A11 was then compared directly to the parental minibody. Labeling efficiencies and immunoreactivities of the parental and A11 minibody were determined after labeling with different batches of 124I. The labeling efficiencies for parental and A11 minibodies were 89.9% (±2.7; n = 4) and 86.6% (±4; n = 4), respectively (Fig. 5). However, the immunoreactive fraction of the A11 minibody was 66% (±6; n = 4) and statistically better (p < 0.05) than the immunoreactive fraction of the parental minibody [45% (±14; n = 4)]. Comparison of biodistribution studies between the two minibodies showed similar tumor uptake for parental and A11 minibodies at 21 h (3.9%ID/g). However, a difference between A11 and the parental minibody was observed when tumor was compared to background values, i.e., blood activity in the biodistribution, and soft tissue activity in the images. The A11 minibody demonstrated overall better biodistribution and imaging results than the parental hu1G8 minibody in the LAPC-9 model with a tumor to blood ratio of 1.1 vs 0.8 and a tumor to background ratio of 5.7 vs 2.4 (Table 1). In conclusion, the tumor image contrast was more than twofold better with A11 than parental minibody in this experiment.
Fig. 4.

In vivo evaluation in SCID mice bearing LAPC-9 tumors that were injected with 124I-labeled minibodies (A2, A11, and C5). a Coregistered microPET/microCT scan of SCID mice bearing LAPC-9 (PSCA-positive human prostate cancer) xenografts. The mice were injected with radiolabeled minibody variants (A2, A11, and C5) and scanned at 21 h post-injection. Coronal projections are presented that are adjusted to the same scale. b Biodistribution, Tu/Bl (tumor/blood) and imaging data Tu/Ba (tumor/background) at 21 h post-injection. Tumor and normal organ uptakes are expressed as percent injected dose per gram (%ID/g ± SE)
Fig. 5.

Labeling of the parental and A11 minibodies. Labeling efficiency and immunoreactivity were determined for each minibody (n = 4). *The difference between the parental (P) (grey bars) and A11 (black bars) minibodies immunoreactivity was significant (p < 0.05)
Table 1.
Biodistribution (%ID/g ± SE) and uptake ratios of 124I-labeled parental (P) and A11 minibodies at 27 h in mice (n = 3) bearing LAPC-9AD tumors
| Organ | P | A11 |
|---|---|---|
| Tumor | 3.9 ± 0.26 | 3.8 ± 0.95 |
| Blood | 4.9 ± 0.56 | 3.4 ± 0.28 |
| Liver | 1.3 ± 0.41 | 0.8 ± 0.04 |
| Spleen | 0.8 ± 0.33 | 0.7 ± 0.2 |
| Kidney | 1.9 ± 0.41 | 1.3 ± 0.02 |
| Ratio | ||
| Tu/Bl | 0.8 ± 0.08 | 1.1 ± 0.15 |
| Tu/Ba | 2.4 ± 0.24 | 5.7 ± 0.56 |
Tu/Bl (tumor/blood) and imaging data (Tu/Ba, tumor/background)
Evaluation of A11 for microPET imaging of Capan-1 xenografts
A11 was also compared to the parental minibody for targeting and imaging of PSCA-expressing tumors in a second model: nude mice carrying Capan-1 xenografts (human pancreatic cancer). We determined that the Capan-1 cells express ∼30,000 PSCA molecules on their cell surface using QIFIKIT. The labeling efficiency was again very similar for the parental hu1G8 (88.4%) and A11 (84.7%) minibodies for this experiment. The immunoreactive fraction was 48.9% for the parental minibody and 68.4% for the A11 minibody. We obtained similar imaging contrast with the A11 minibody in both LAPC-9 and Capan-1 models at 21 h with tumor to background ratios of 4.8 in the LAPC-9 model and 4.2 in the Capan-1 model. Although the tumor uptakes in the Capan-1 model at 21 h of both parental and A11 were similar, A11 exhibited a slightly better tumor to blood ratio than the parental minibody (Fig. 6). ROI analysis resulted in a tumor to background ratio of 2.2 and 4.2 (n = 5, p < 0.001) for parental hu1G8 and A11 minibody, respectively. Thus, 124I-labeled A11 minibody was a significantly better microPET probe for imaging PSCA-expressing Capan-1 tumors compared to the parental minibody.
Fig. 6.

In vivo evaluation in nude mice bearing Capan-1 xenografts. a Coregistered microPET/microCT scan of ten nude mice bearing Capan-1 (PSCA-positive human pancreatic cancer) xenografts. The mice (n = 5) were injected with 124I-radiolabeled minibody variants: parental (P) minibody top row, A11 minibody bottom row. Mice were scanned and biodistribution performed at 21 h post-injection. Coronal projections are presented that are adjusted to the same scale. b Biodistribution (n = 5) and Tu/Bl (tumor/blood). Tumor and normal organ uptakes are expressed as percent injected dose per gram (%ID/g ± SE)
Discussion
We previously demonstrated specific detection of PSCA-expressing xenografts with the parental hu1G8 minibody at 21 h using microPET imaging [14]. Here, we describe how affinity variant minibodies were generated and a better microPET probe was selected. Molecular evolution and yeast display technology was used to generate three affinity matured scFvs: A2, A11, and C5 that translated into three minibodies with better apparent affinity than the parental minibody. The in vivo evaluation resulted in the selection of A11 as the best affinity variant to be labeled with 124I to detect PSCA-expressing xenografts by microPET imaging. Finally, comparisons of A11 with the parental minibody in two different xenograft models showed that the A11 affinity matured minibody resulted in a tumor to soft tissue contrast twofold higher than what was obtained with the parental minibody.
The three minibody variants generated, A2, A11, and C5 when analyzed by competition ELISA, all demonstrate apparent affinity improvement on the parental minibody. Thus, in these three cases the selection of affinity matured scFv using yeast display technology translated into corresponding minibodies with improved affinity. In addition, the competition ELISA results show that A2, A11, and C5 were able to compete with the parental minibody which suggests that they all recognize the same epitope on PSCA as the parental minibody. In conclusion we have generated four minibodies: A2, A11, C5, and P that recognize the same epitope on PSCA with apparent affinity measured at 11, 16, 26, and 50 nM, respectively.
The effect of radioiodination of minibody on PSCA binding was evaluated using SKW6.4 and SKW6.4-PSCA cells. With consistent labeling efficiency ∼88% from one 124I batch to the next for both the parental and A11 minibodies, we observed that the A11 minibody immunoreactive fraction 66% (±6; n = 4) was statistically better (p < 0.05) than the parental minibody immunoreactive fraction 45% (±14; n = 4). Smaller standard deviation values were obtained when comparing the difference in immunoreactivity for the same two minibodies on the same day with the same 124I batch. Thus, variations in the 124I batch seem to affect immunoreactivity.
The biodistribution data in both LAPC-9 and Capan-1 xenograft models only indicate a small improvement in tumor to blood ratio (1.3 and 1.2 times, respectively) that was not significantly different between the parental and A11 minibodies. Although tumor and blood activities as well as tumor to blood ratios are important pharmacokinetic measurements, they are not directly related to imaging measurements. Here, we measured the contrast between the tumor and the soft tissue around the tumor to quantify imaging results. Ratios would be much different if the tumor signal was compared to the strongest background activity such as the blood (heart) or the bladder, which are visible in the microPET images presented. However, the comparison between the parental and A11 minibody in the low PSCA-expressing Capan-1 xenograft model is more relevant as the sample size in the same experiment was bigger (n = 5) which allows statistical analysis of smaller variations. In this model we obtained a tumor to soft tissue ratio of 4.2 for A11 which is almost twofold higher (p < 0.001) than the ratio obtained for the parental minibody (2.2).
In vivo, the minibody that ranked with the intermediate apparent affinity (A11) demonstrated best microPET imaging results, with the best affinity minibody (A2) showing the worst results. The poor in vivo result of A2 is unlikely a consequence of higher affinity since the difference between A2 and A11 KD was determined to be only around 5 nM. One possible reason for the poor performance of A2 is the presence of a new tyrosine in the VH CDR2. Iodination of this tyrosine during labeling might affect binding to PSCA. Consistent with an effect of iodination with the poor imaging results obtained for A2 imaging is that the A2 minibody had the lowest immunoreactivity of the three affinity variants. However, A2 immunoreactivity was still close to 40%, and other parameters such as protein stability in vivo could also be involved. The overall observation is that different affinity variants with similar apparent affinity exhibited different microPET imaging properties. Comparison between in vitro and in vivo characterization of peptide probes to select the best microPET tracers has recently been carried out [33]. In this study, 43 peptides were evaluated as microPET tracers to detect αvβ6-expressing tumors. This in vivo selection resulted in the selection of four peptides that would not have been selected on the bases of their in vitro characterization including their affinity. Thus, the best strategy to develop new microPET tracers seems to be the development of high-throughput technology for the generation and testing of new protein-based microPET tracers rather than extensive in vitro studies of many different parameters.
The A11 minibody has been selected for evaluation in a clinical setting for a number of reasons. We generated clones that express high yields of minibody that was stable at 37°C. A11 has higher affinity to PSCA and higher immunoreactivity after 124I labeling than the parental minibody. Finally, A11 antibody showed highest microPET imaging contrast in two different xenograft models.
Acknowledgments
The expert technical support of Dr. David Stout, Waldemar Ladno and Judy Edwards at the Crump Institute for Molecular Imaging at UCLA is gratefully acknowledged. Appreciation is also extended to the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility at UCLA. This work was supported by NIH Grants R25T CA098010, P50 092131, P50 CA086306, U54 CA119367, and Department of Defense Grant W81WXH-08-1-0442. RER and AMW are members of Jonsson Comprehensive Cancer Center (CA 16042).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Emonds KM, Swinnen JV, Mortelmans L, Mottaghy FM. Molecular imaging of prostate cancer. Methods. 2009;48:193–199. doi: 10.1016/j.ymeth.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Zaheer A, Cho SY, Pomper MG. New agents and techniques for imaging prostate cancer. J Nucl Med. 2009;50:1387–1390. doi: 10.2967/jnumed.109.061838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu AM. Antibodies and antimatter: the resurgence of immuno-PET. J Nucl Med. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- 4.Haseman MK, Rosenthal SA, Polascik TJ. Capromab Pendetide imaging of prostate cancer. Cancer Biother Radiopharm. 2000;15:131–140. doi: 10.1089/cbr.2000.15.131. [DOI] [PubMed] [Google Scholar]
- 5.Seltzer MA, Barbaric Z, Belldegrun A, Naitoh J, Dorey F, Phelps ME, et al. Comparison of helical computerized tomography, positron emission tomography and monoclonal antibody scans for evaluation of lymph node metastases in patients with prostate specific antigen relapse after treatment for localized prostate cancer. J Urol. 1999;162:1322–1328. doi: 10.1016/S0022-5347(05)68277-8. [DOI] [PubMed] [Google Scholar]
- 6.Raff AB, Gray A, Kast WM. Prostate stem cell antigen: a prospective therapeutic and diagnostic target. Cancer Lett. 2009;277:126–132. doi: 10.1016/j.canlet.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 1998;95:1735–1740. doi: 10.1073/pnas.95.4.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu Z, Thomas G, Yamashiro J, Shintaku IP, Dorey F, Raitano A, et al. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene. 2000;19:1288–1296. doi: 10.1038/sj.onc.1203426. [DOI] [PubMed] [Google Scholar]
- 9.Lam JS, Yamashiro J, Shintaku IP, Vessella RL, Jenkins RB, Horvath S, et al. Prostate stem cell antigen is overexpressed in prostate cancer metastases. Clin Cancer Res. 2005;11:2591–2596. doi: 10.1158/1078-0432.CCR-04-1842. [DOI] [PubMed] [Google Scholar]
- 10.Amara N, Palapattu GS, Schrage M, Gu Z, Thomas GV, Dorey F, et al. Prostate stem cell antigen is overexpressed in human transitional cell carcinoma. Cancer Res. 2001;61:4660–4665. [PubMed] [Google Scholar]
- 11.Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4324. [PubMed] [Google Scholar]
- 12.McCarthy DM, Maitra A, Argani P, Rader AE, Faigel DO, Van Heek NT, et al. Novel markers of pancreatic adenocarcinoma in fine-needle aspiration: mesothelin and prostate stem cell antigen labeling increases accuracy in cytologically borderline cases. Appl Immunohistochem Mol Morphol. 2003;11:238–243. doi: 10.1097/00129039-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Olafsen T, Gu Z, Sherman MA, Leyton JV, Witkosky ME, Shively JE, et al. Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J Immunother. 2007;30:396–405. doi: 10.1097/CJI.0b013e318031b53b. [DOI] [PubMed] [Google Scholar]
- 14.Leyton JV, Olafsen T, Lepin EJ, Hahm S, Bauer KB, Reiter RE, et al. Humanized radioiodinated minibody for imaging of prostate stem cell antigen-expressing tumors. Clin Cancer Res. 2008;14:7488–7496. doi: 10.1158/1078-0432.CCR-07-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–1198. [PubMed] [Google Scholar]
- 16.Colcher D, Minelli MF, Roselli M, Muraro R, Simpson-Milenic D, Schlom J. Radioimmunolocalization of human carcinoma xenografts with B72.3 second generation monoclonal antibodies. Cancer Res. 1988;48:4597–4603. [PubMed] [Google Scholar]
- 17.Schlom J, Eggensperger D, Colcher D, Molinolo A, Houchens D, Miller LS, et al. Therapeutic advantage of high-affinity anticarcinoma radioimmunoconjugates. Cancer Res. 1992;52:1067–1072. [PubMed] [Google Scholar]
- 18.Velders MP, van Rhijn CM, Briaire IH, Fleuren GJ, Warnaar SO, Litvinov SV. Immunotherapy with low and high affinity monoclonal antibodies 17-1A and 323/A3 in a nude mouse xenograft carcinoma model. Cancer Res. 1995;55:4398–4403. [PubMed] [Google Scholar]
- 19.Verel I, Heider KH, Siegmund M, Ostermann E, Patzelt E, Sproll M, et al. Tumor targeting properties of monoclonal antibodies with different affinity for target antigen CD44V6 in nude mice bearing head-and-neck cancer xenografts. Int J Cancer. 2002;99:396–402. doi: 10.1002/ijc.10369. [DOI] [PubMed] [Google Scholar]
- 20.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61:4750–4755. [PubMed] [Google Scholar]
- 21.Nielsen UB, Adams GP, Weiner LM, Marks JD. Targeting of bivalent anti-ErbB2 diabody antibody fragments to tumor cells is independent of the intrinsic antibody affinity. Cancer Res. 2000;60:6434–6440. [PubMed] [Google Scholar]
- 22.Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Midelfort KS, Hernandez HH, Lippow SM, Tidor B, Drennan CL, Wittrup KD. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J Mol Biol. 2004;343:685–701. doi: 10.1016/j.jmb.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Graff CP, Chester K, Begent R, Wittrup KD. Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37 degrees C. Protein Eng Des Sel. 2004;17:293–304. doi: 10.1093/protein/gzh038. [DOI] [PubMed] [Google Scholar]
- 25.Razai A, Garcia-Rodriguez C, Lou J, Geren IN, Forsyth CM, Robles Y, et al. Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J Mol Biol. 2005;351:158–169. doi: 10.1016/j.jmb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, et al. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999;59:5030–5036. [PubMed] [Google Scholar]
- 27.Wu AM, Yazaki PJ, Tsai S, Nguyen K, Anderson AL, McCarthy DW, et al. High-resolution microPET imaging of carcinoembryonic antigen-positive xenografts by using a copper-64-labeled engineered antibody fragment. Proc Natl Acad Sci U S A. 2000;97:8495–8500. doi: 10.1073/pnas.150228297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, et al. Impact of single-chain Fv antibody fragment affinity on nanoparticle targeting of epidermal growth factor receptor-expressing tumor cells. J Mol Biol. 2007;371:934–947. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 30.Olafsen T, Kenanova VE, Wu AM. Tunable pharmacokinetics: modifying the in vivo half-life of antibodies by directed mutagenesis of the Fc fragment. Nat Protoc. 2006;1:2048–2060. doi: 10.1038/nprot.2006.322. [DOI] [PubMed] [Google Scholar]
- 31.Kenanova V, Olafsen T, Crow DM, Sundaresan G, Subbarayan M, Carter NH, et al. Tailoring the pharmacokinetics and positron emission tomography imaging properties of anti-carcinoembryonic antigen single-chain Fv-Fc antibody fragments. Cancer Res. 2005;65:622–631. [PMC free article] [PubMed] [Google Scholar]
- 32.Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131–137. doi: 10.1162/153535003322556877. [DOI] [PubMed] [Google Scholar]
- 33.Gagnon MK, Hausner SH, Marik J, Abbey CK, Marshall JF, Sutcliffe JL. High-throughput in vivo screening of targeted molecular imaging agents. Proc Natl Acad Sci U S A. 2009;106:17904–17909. doi: 10.1073/pnas.0906925106. [DOI] [PMC free article] [PubMed] [Google Scholar]