

Abstract
The rapid spread on multi-drug resistant strains of Staphylococcus aureus requires not just novel treatment options, but the development of faster methods for the identification of new hits for drug development. The exponentially increasing speed of computational methods makes a more extensive use in the early stages of drug discovery attractive if sufficient accuracy can be achieved. Computational target identification using systems-level methods suggested the histidine biosynthesis pathway as an attractive target against S. aureus. Potential inhibitors for the pathway were identified through docking, followed by ensemble rescoring that is sufficiently accurate to justify immediate testing of the identified compounds by whole cell assays, avoiding the need for time-consuming and often difficult intermediary enzyme assays. This novel strategy is demonstrated for three key enzymes of the S. aureus histidine biosynthesis pathway, which is predicted to be essential for bacterial biomass productions. Virtual screening of a library of ~106 compounds identified 49 potential inhibitors of three enzymes of this pathway. 18 representative compounds were directly tested on three S. aureus-and two E. coli strains in standard disc inhibition assays. 13 compounds are inhibitors of some or all of the S. aureus strains, while 14 compounds weakly inhibit growth in one or both E. coli strains. The high hit rate obtained from a fast virtual screen demonstrates the applicability of this novel strategy to the histidine biosynthesis pathway.
Keywords: Antibiotics, systems biology, virtual screening, histidine biosynthesis
Introduction
Staphylococcus aureus is a rapidly growing problem for modern society. From 1999 to 2005, the number of S. aureus related hospitalizations increased by 62%.1 The treatment of the infections is complicated by the bacteria’s ability to develop resistance towards methicillin and the other commonly used antibiotics, necessitating the use of drugs such as vancomycin, that are both costly and difficult to administer to patients. Methicillin-resistant S. aureus (MRSA) was responsible for 43% of all the S. aureus-related hospitalizations in 1999. By 2005 the percentage had increased to 58%, leading to a mortality rate of ~6.3 per 10,000.2 In recent years vancomycin-resistant S. aureus (VRSA) strains have appeared.3 It is therefore of great importance to develop new antibiotics with new targets for the treatment of S. aureus, including its multidrug-resistant strains.
The pressing need for new antibiotics and the economic realities of drug discovery necessitate faster and more efficient methods for the development of new antibiotics. Given the exponentially increasing speed of computers, the use of computational methods in the early stages of target identification and drug discovery is particularly attractive. To this end, the combination of genomic information with systems biology provides a novel tool for target identification. We have previously reconstructed the metabolic networks of thirteen fully sequenced S. aureus strains and used flux balance analysis to identify their unconditionally essential enzymes as well as their synthetic lethal pairs.4 One of the families of targets identified in these studies is the histidine biosynthesis pathway, an unbranched pathway consisting of 10 enzymatic reactions with no routes to bypass any of the enzymes (Fig. 1). 6
Figure 1.

Histidine biosynthesis pathway
Although virtual screening has become an established tool for computer aided molecular design and frequently reproduces experimentally observed binding poses, there is usually no good correlation between docking scores and experimentally observed binding constants. Therefore, a significant number of compounds from virtual screens are usually selected for experimental confirmation by enzyme assays early in the hit discovery process. This requires significant effort in the acquisition and screening of the compounds and typically results in varying enrichment factors that depend on the scoring function and the enzyme studied. It would therefore be desirable to further refine the scoring to increase enrichment and possibly bypass the biochemical assay in favor of whole cell assays. As a result, several rescoring procedures have been proposed to improve the accuracy of the computational predictions. In a recent ipso facto study of a large dataset MM-PBSA rescoring of docking complexes increased the percentage of correctly docked poses (within 2Å of the X-ray position) from 56% (found in the initial docking) to 76%.5 A study of the related MM-GBSA rescoring method led to correlation coefficients between predicted and experimental binding constants ranging from R2= 0.64 to R2=0.81.5, 6 This is in line with our findings on the FAS II pathway,7 where MM-PBSA rescoring of ensembles of snapshots from MD simulations (ensemble rescoring) led to improved compound selection. Specifically, 19 of 41 compounds selected this way were shown to be active in enzyme assays and 14 were active in subsequent whole cell assays. This suggested that the computational predictions can be sufficiently accurate to be tested directly in in vivo disk inhibition assays, which would accelerate the process.
Here, we report the results of a study of inhibitors of the histidine biosynthesis pathway, where ensemble rescoring was used to select compounds that were then directly tested in whole-cell assays. To demonstrate this novel strategy to identify potential inhibitors of the histidine biosynthesis, we chose three enzymes from the pathway as targets for antibiotic hit identification based on the availability of crystal structures and established biochemical assays: Phosphoribosyl-AMP Cyclohydrolase (HisI),8, 9 Imidazoleglycerol Phosphate Dehydratase (IGPD),10 and Histidinol Phosphate Aminotransferase (HisC).11–15 The efficacy of the identified hits will then be tested in whole-cell assays.
Materials and Methods
Computational methods
Homology models of the S. aureus enzymes were built in Prime16 using comparative modeling using the template structures discussed in the text. The docking experiments were performed in Glide,17, 18 and using the “Lead” subset of the ZINC database19 of commercially available compounds. This subset was obtained from the complete dataset by applying filters20 to have good drug potential, resulting in ~106 small molecules docked to the enzyme of interest using Glide’s high throughput mode. The highest scoring 100,000 hits were saved and docked to the enzyme again, this time using Glide’s standard precision mode. The highest scoring 10,000 hits were then saved, and docked to the enzyme using the extra precision mode. The highest scoring 2,000 hits were saved, and by manual inspection we selected a small number of potential inhibitors representative of the chemical space covered by the best scored docking hits for ensemble rescoring. In this procedure, side chain flexibility is introduced through 8 ns MD simulations, allowing the active site residues to move, and to test how these movements affect the binding mode of the ligand. Desolvation and hydrophobic effects are included through MM-PBSA/GBSA calculations of the binding energy on snapshots extracted from the last 4 ns of the MD run. These scores were then used to select compounds for experimental testing. A separate analysis showed that for the cases studied here, conformational sampling and MM-PBSA/GBSA calculations of the separate structures had a minor effect on the relative ordering of the binding energies of the ligands.
Experimental protocols
For the experiments two E. coli (MG1655 and a random patient isolate) and three S. aureus (MU50, USA300 and a random patient isolate) and one P. ariginosa (PA01) strains were used. The propagation of bacteria and their experimental manipulation were performed as previously described.7 In brief, sterile filter discs soaked with 10µl of 10mg/ml of the different inhibitors were applied to the surface of the evenly-inoculated LB plates, minimal medium agar plates, or minimal medium agar plates complemented by 0.2mM L-histidine and incubated for 16–20h at 37 °C. 10µl 1mg/ml Ampicillin (to demonstrate the effect of resistance strains) and DMSO were utilized as positive and negative controls, respectively. To test for potential human cell toxicity, human foreskin fibroblast cells were treated with 100 µg/ml of selected compounds or DMSO and their cell viabilities were determined by MTT (3-[4,5-dimethyl-triazolyl-2]2,5-diphenyl tetrazolium bromide) viability assay and trypan blue exclusion assay, as previously described.7 Further details are available in the Supporting Information.
Results and Discussion
The histidine biosynthesis pathway in S. aureus
The first step in the biosynthesis of histidine is catalyzed by the enzyme ATP phosphoribosyltransferase (HisG) and results in the condensation of ATP and 5-phosphoribosyl 1-pyrophosphate to form N’-5’-phosphoribosyl-ATP. The triphosphate group of the product is then hydrolyzed by the enzyme phophoribosyl-ATP pyrophosphohydrolase (HisE) to form N’-5’-phosphoribosyl-AMP, and subsequently the purine ring is hydrolyzed by phosphoribosyl-AMP cyclohydrolase (HisI) to form N’-[(5’-phosphoribosyl)-formimino]-5-aminoimidazole-4 carboxamide-ribonucleotide. In the fourth step of the pathway, the product is converted to N’-[(5’-phosphoribulosyl)-formimino]-5-aminoimidazole-4 carboxamide-ribonucleotide via an Amadori rearrangement. This product is then cleaved into imidazole glycerol phosphate (IGP) and 5-aminoimidazole-4-carboxamide 1-beta-D ribofuranosyl 5’-monophosphate (AICAR). The first of these is used in the further synthesis of histidine, while the latter is used in the de novo synthesis of purine.7 In the sixth step of the pathway IGP is dehydrated by imidazole glycerole phosphate dehydratase (IGPD). This gives the product imidazole acetol phosphate, which in the seventh step of the biosynthesis is transaminated by the enzyme histidinol phosphate aminotransferase (HisC) to give L-histidinol phosphate. This product is then converted into L-histidinol, which is oxidized to L-histidine in two steps via the L-histidinal intermediate by a single enzyme (HisD).
Based on our analyses of the metabolic network of thirteen S. aureus strains by flux balance analysis,4 inhibition of a single enzyme of the pathway is expected to stop the production of histidine, an essential biomass component. Evolutionarily, the pathway is highly conserved among different bacterial species, which makes the pathway a potential target for the treatment of bacterial infections other than caused by S. aureus. The histidine pathway has been extensively studied in the past, and these studies have contributed to the development of the operon theory of gene clustering and function.21 More recently, the histidine biosynthesis in plants has received a lot of attention due to its vital role for plant growth.22 Particularly the sixth enzyme of the pathway, IGPD, has been used as a target for the design of herbicides.23–29
Identification of inhibitor scaffolds through computational chemistry
Phosphoribosyl-AMP Cyclohydrolase
(HisI) (EC 3.5.4.19) catalyzes the third step of the histidine biosynthetic pathway, the hydrolysis of phosphoribosyl-AMP (Fig. 1). The crystal structure of HisI for S. aureus has not yet been determined, but the structure of HisI from Methanobacterium thermoautotrophicum (PDB ID: 1zps), which has a 51% homology to HisI from S. aureus, has been solved to a resolution of 1.70 Å. We have built a homology model of the S. aureus MRSA252 enzyme and compared it to the structure of the M. thermoautotrophicum enzyme to validate its use in the docking experiments. HisI is a homodimeric metalloenzyme dependent on both Zn2+ and Mg2+. Two pairs of metal ions are bound on the interface between the two subunits, and make up two active sites, each containing one zinc and one magnesium ion, respectively. The active sites are solvent exposed. The magnesium binding site consists of three aspartate residues from one subunit (Asp85, Asp87, and Asp89) while the zinc ion is coordinated by one cysteine residue from the same subunit (Cys86), and two cysteine residues from the other subunit (Cys109 and Cys102). EDTA inhibits the enzyme by chelation of Mg2+, and exogenous Zn2+ also reduces enzyme function, but no small molecule inhibitors of this enzyme have previously been reported.
Application of the virtual screening procedure to the lead library from ZINC yielded 12 small molecules that have good poses in both the docking and the subsequent MD simulations (for a full listing, compare Table S1 in the Supporting Information). Six of these molecules, shown in Table 1 together with their G-scores from Glide XP as well as predicted binding energies from MM-PBSA and MM-GBSA calculations, were selected based on their structural diversity. It should be noted that the absolute binding energies calculated by either method are not meaningful.5 Unfortunately, no known inhibitors are available for referencing the values and that the scores can only be used for a relative ranking of the compounds.
Table 1.
Selected Inhibitors for HisI
| ID | Structure | G-score kcal/mol |
MM- PBSA kcal/mol |
MM- GBSA kcal/mol |
|---|---|---|---|---|
| HisI1 | 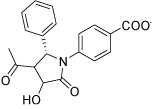 |
−15.23 | −55.73 | −22.50 |
| HisI2 | 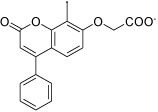 |
−14.44 | −60.15 | −29.58 |
| HisI4 | 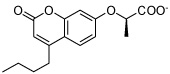 |
−14.08 | −60.16 | −25.46 |
| HisI8 | 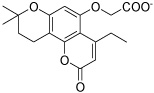 |
−13.80 | −22.33 | −22.28 |
| HisI9 | 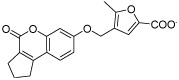 |
−13.69 | −35.48 | −30.28 |
| HisI11 | 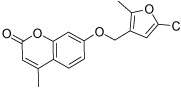 |
−13.42 | −45.83 | −19.40 |
Figure 2 shows two representative examples of the binding modes observed after 8 ns MD simulation (for representations of other poses, see the Supporting Information). The HisI active site is solvent exposed and contains a zinc and a magnesium ion, which explains the high abundance of carboxylate derivatives among the docking hits. Inhibitor HisI2 is a typical case for the bidentate coordination of the zinc ion by the carboxylate group. This zinc coordination is always preferred over the coordination of the magnesium ion (only one of the selected docking poses, HisI10, showed the carboxylate group coordinating to both metal ions). The magnesium ion is surrounded by three aspartate residues from one side and a hydrophobic pocket consisting of the residues Ile25, Ile25’, Val27, Val27’, Leu37, Leu37’, Val39, Val39’, Tyr41 and Tyr41’ from both subunits (shown as four blue colored β-sheets in Figure 2). Several docking poses place a hydrophobic group pointing into this pocket (HisI2, 3, 4, 6, and 8), and most of these allow a cation-π interaction with the magnesium ion and an aromatic moiety.
Figure 2.

Active site of homology model of S. aureus with HisI2 (left) and HisI9 (right) in the active site after 8ns MD.
Even though several of the potential HisI inhibitors share a chromene-2-one core, different poses in the active site were obtained in the docking calculations. For the chromene-2-one derivative HisI9 (shown in Figure 2 on the right) a monodentate coordination to the zinc is observed in all cases. The main interaction appears to be a π-stacking with Trp67 that is only observed after the MD simulation. In addition, Lys69 acts a hydrogen bond donor to the furan moiety of HisI9.
Imidazoleglycerol Phosphate Dehydratase
IGPD (EC 4.2.1.19) catalyzes the dehydration of imidazoleglycerol phosphate to imidazoleacetol phosphate, which is the sixth step of histidine biosynthesis (Fig. 1). A crystal structure of the S. aureus N315 IGPD with a resolution of 2.01 Å (PDB ID: 2ae8) is available. However, many moieties close to the active site are not resolved, making this structure unsuitable for virtual screening approaches. We have therefore chosen the well characterized Aridopsis thaliana IGPD (PDB ID: 2f1d, 42% homology) to build a homology model of the S. aureus MRSA252 IGPD. IGPD is a manganese dependent metalloenzyme consisting of 24 identical subunits. Each active site consists of three subunits and two manganese ions. Each manganese ion is coordinated by a glutamate and three histidine residues from two different subunits (Mn2+1: His47A, His169A, Glu173A, and His74B; Mn2+2: His170A, His73B, Glu77B, and His145B). The IGPD enzyme has been used as target for the development of herbicides, and several inhibitors of this enzyme are known from these efforts.23–29 The known inhibitors resemble the natural substrate imidazole-glycerol phosphate (IGP). They typically contain a phosphonate group instead of the phosphate group present in IGP, a triazole group instead of imidazole, and a number of hydroxy groups. Known inhibitors are reported to have IC50 values in the range from low µM to nM – the most potent inhibitor having an IC50 value of 20 nM.11,13
The selected potential IGPD inhibitors and their G-scores are listed in Table 2. It is evident that the MM-PBSA and MM-GBSA scores obtained for all inhibitors as well as for the known inhibitors (data not shown) are relatively unfavorable. This might be a consequence of an underestimation of binding to the manganese ions, possibly due to the Mn2+ parameters employed for the MM-PBSA/GBSA calculations (see the Supporting Information for further details). Structurally, there is a high abundance of carboxylate derivatives among the best docking hits. In the majority of these, the carboxylate group is placed between the two manganese ions coordinating both of them. The key interactions are exemplified by the structures of IGPD13, 14, 17, and 18 in the IGPD active site after 4ns of MD simulation shown in Figure 3.
Table 2.
Selected Inhibitors of IGPD
| ID | Structure | G-score kcal/mol |
MM- PBSA kcal/mol |
MM- GBSA kcal/mol |
|---|---|---|---|---|
| IGPD1 | 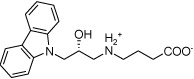 |
−8.61 | −17.22 | −36.66 |
| IGPD6 | 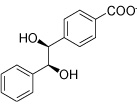 |
−8.05 | 12.28 | −1.75 |
| IGPD10 | 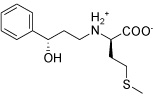 |
−7.78 | −5.53 | −24.67 |
| IGPD13 | 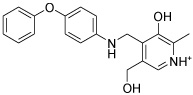 |
−7.70 | −1.30 | −20.34 |
| IGPD14 | 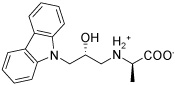 |
−7.68 | −0.30 | −20.53 |
| IGPD16 | 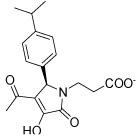 |
−7.62 | 10.30 | −13.25 |
| IGPD17 | 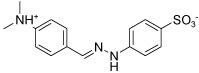 |
−7.32 | 5.21 | −12.98 |
Figure 3.

Structures of potential inhibitors a) IGPD13, b) IGPD14, c) IGPD17, and d) IGPD18 bound to IGPD active site from 4 ns MD simulations.
Most of the potential inhibitors interact with Asp69, either forming a salt bridge through an ammonium group like in the case of IGPD1, 2, and 5 or by hydrogen bond donating, either through a hydroxy group (IGPD6, 8, 10, 11, 12, 14, and 16) or a NH functionality (IGPD17 and 18), as shown in Figure 3 b–d. In accordance with the negatively charged substrate, the active site is highly positively charged with the presence of three arginines (Arg8 and Arg295, with an additional arginine residue Arg268 in the periphery of the active site) and Lys534. As a result, all selected potential inhibitors posses a negative charge, either as a carboxylate or a sulfonate. The sole exception, IGPD13, has an acidic phenol hydroxyl group that is likely deprotonated upon binding to the manganese, as shown in Figure 3a. Five of the selected poses interact with Lys534 (IGPD3, 5, 9, 10, and 18), and four interact with Glu12 (IGPD2, 6, 13, and 18). The rest of the active site pocket is filled out by aromatic groups stacking against the α-helix forming one wall of the active site (IGPD13, 17 and 18) or the β-sheet forming the other (IGPD14). As mentioned before, all known inhibitors of the IGPD enzyme are small, polar compounds. We have therefore chosen fewer hits containing large, hydrophobic groups and more relatively small, polar compounds for further evaluation than is actually representative of the abundance with which they appear among the docking hits (e.g., IGPD2, 3, 5, and 6). We have also included two sulfonate derivatives (IGPD7 and 17) in the compound set to be evaluated experimentally, since a sulfonate group is a better bioisostere for the phosphonate group than the carboxylate group.
Histidinol Phosphate Aminotransferase
(HisC). The seventh step of the histidine biosynthesis in eubacteria is the transamination between imidazoleacetol phosphate and glutamate to form L-histidinol phosphate (LHP) and α-ketoglutarate (Fig. 1), which is catalyzed by the enzyme HisC (EC 2.6.1.9). HisC uses the cofactor pyridoxal-5’-phosphate (PLP) to transfer the amino group from glutamate, forming pyridoxamine-5’-phosphate (PMP) that is subsequently transferred to the substrate. Except for the metal ions Cu2+ and Co2+ and hydroxyl amine, iodoacetate and semicarbazide, no HisC inhibitors are currently known. In our docking calculations, we have used the crystal structure of HisC complexed with PMP from E. coli (PDB ID: 1fg7, 29% homology), which has been solved to a resolution of 1.5 Å.14 HisC from S. aureus MRSA252 has a high sequence similarity with its E. coli ortholog, making the latter a suitable model. One important difference is that the E. coli residue Tyr110 is substituted to a Phe in the S. aureus ortholog. Although the active enzyme functions as a homodimer, all relevant residues are contained in the monomer that was used in the docking calculations. In the crystal structure, the phosphate group of PMP forms a salt bridge with Arg222 and hydrogen bonds to Thr211, Ser213, and to the backbone amide groups of Ala84 and Asp85. The amino group forms a hydrogen bond to Lys214 and the hydroxy group hydrogen bonds to Tyr187 and Asn157. Asp184 forms a salt bridge with the protonated pyridinium nitrogen and Tyr110 interacts with the pyridinium ring via π-stacking. The substrate, LHP, is known to form hydrogen bonds to Tyr20 and Asn157 and salt bridges to Arg322 and Arg335 via its phosphate group. Furthermore, the LHP imidazole ring forms hydrogen bonds to the residues Tyr20, Asp85, and Tyr110 in the E. coli HisC.22 Based on the sequence alignment MRSA252 HisC cannot form the latter hydrogen bond.
The docking hits for the HisC enzyme are shown in Table 3 and are again negatively charged or polar compounds for the same reasons as discussed for IGPD. The affinity of carboxylate derivatives to the HisC enzyme can be ascribed to the phosphate binding site of the PMP cofactor (Arg222, Thr209, and Ser211) and to the residues Asn157 and Arg335 which are part of the phosphate binding site of the substrate LHP. In all the docking hits that are carboxylate derivatives, the carboxylate functionality binds to either of the two phosphate binding sites.
Table 3.
Selected Inhibitors of HisC
| ID | Structure | G-score kcal/mol |
MM- PBSA kcal/mol |
MM- GBSA kcal/mol |
|---|---|---|---|---|
| HisC5 | 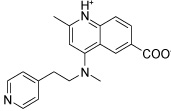 |
−15.02 | −40.92 | −49.55 |
| HisC9 | 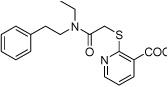 |
−14.50 | −38.74 | −43.95 |
| HisC11 | 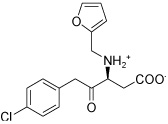 |
−14.25 | −28.80 | −35.94 |
| HisC14 | 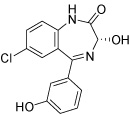 |
−13.84 | −12.95 | −35.13 |
| HisC16 | 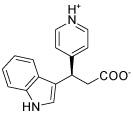 |
−13.70 | −29.43 | −37.33 |
| HisC19 | 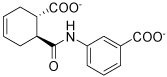 |
−13.53 | −13.80 | −26.48 |
The two main binding modes where the carboxylate group occupies the PMP phosphate binding site and its pyridine ring interacting with the LHP binding site and where the carboxylate group binds to the LHP phosphate binding site are exemplified by the calculated docking poses of HisC5 and HisC9 and are shown in Figure 4a and b, respectively. Other structures that do not contain a carboxylate functionality have other polar groups that follow the same motifs and bind to at least one of the phosphate binding sites, as for example the azepine moiety of HisC14 shown in Figure 4c. Other key interactions include π-stacking with Tyr110 and hydrogen bonding or salt bridges with Lys214 and Asp184, respectively. Overall, the calculated binding energies, shown in Table 3, are strongly favorable, but the lack of known inhibitors prevents the referencing to experimental data and allows only a relative ranking of the compounds.
Figure 4.

Structures of potential inhibitors a) HicC5, b) HisC9, c) HisC14, and d) HisC19, bound to HisC active site from 8 ns MD simulations.
Several hits with good G-scores contain two carboxylate groups, which would seem favorable, since both phosphate binding sites then can interact with a carboxylate group. This is demonstrated in Figure 4d, which shows the docking pose of HisC19 where both carboxylate groups interact with the two phosphate binding sites of PMP and LHP. However, since the active site is buried deep within the enzyme, the cost of desolvation of these bis-anions is likely to decrease the binding of very polar ligands, as is indicated by the much lower binding energies calculated by MM-PBSA and MM-GBSA. We therefore selected only HisC19 as one example of this class of compounds for further experimental evaluation. Instead, we have chosen several structures where other polar groups are able to interact with the second phosphate binding site, e.g. the pyridine group of HisC5 and the amide group of HisC9 (Figure 4a, b, respectively).
Biological validation of potential inhibitors of the histidine pathway
A recent study of inhibitors of the FASII biosynthetic pathway7 demonstrated that 42% of the inhibitors predicted to be good binders by the MM-PBSA ensemble rescoring method were subsequently confirmed experimentally in enzyme- and whole cell assays. These results encouraged us to use a disk inhibition assay as a primary screen for the computationally predicted compounds. Although this approach can potentially yield both false positive results due to inhibition of targets other than the computational predicted ones and false negative results due to limited transport across the cell membrane, the simplicity of the assay and the fact that the results are more relevant for the ultimate goal of bacterial growth inhibition made this approach attractive.
We therefore selected several representative structures with good calculated MM-PBSA binding energies, structural features representative of the diversity of the larger dataset, and reasonable interactions to the active site of the respective enzymes to be tested in disk inhibition assays. The goal of the selection was to identify the maximum number of active compounds with the minimum size of the total dataset. Of the potential HisI inhibitors, we selected HisI1, 2, 4, 8, 9, and 11 to be biologically tested. These compounds have been selected due to good calculated binding energies, and because they represent both of the two different binding modes discussed above. For IGPD, we have included the potential inhibitors IGPD1, 6, 10, 13, 14, 16, and 17. For HisC, we chose the potential inhibitors HisC9, 11, 14, 16, and 19 for biological evaluation. These 18 compounds were obtained commercially and tested in a standard bacterial growth assay30 in two MRSA strains (Mu50 and USA300) as well as a random patient isolate from the University of Pittsburgh Medical Center. As example of a gram-negative bacterium, we also tested the E. coli MG1655 strain and an E. coli random patient isolate. 1 mg/ml Ampicillin and 10 µl DMSO were used as positive and negative controls, respectively. Figure 5 shows some representative plates for t E. coli and S. aureus strains (for complete results, see Figure S4 in the Supporting Information) and the results for active compounds are summarized in Table 4.
Figure 5.

Bacterial disc inhibition assays and human fibroblast toxicity assay. The inhibitory effect of selected small molecules against a./ E. coli MG1655 strain, b./ S. aureus Mu50 strain
Table 4.
Summary of results of growth inhibition and toxicity assays for 18 selected small molecules
| ID |
S. aureus patient strain |
S. aureus Mu50 |
S. aureus USA300 |
E. coli patient strain |
E. coli 1655 |
MTT activity |
Trypan blue |
|---|---|---|---|---|---|---|---|
| HisI1 | − | ±/− | − | − | + | N.D. | N.D. |
| HisI2 | + | + | + | + | + | − | − |
| HisI4 | + | + | − | ± | ± | ± | ± |
| HisI8 | − | + | ± | − | + | N.D. | N.D. |
| HisI9 | + | +/± | − | − | − | N.D. | N.D. |
| HisI11 | ± | ± | − | ± | + | ± | − |
| IGPD1 | +++ | + | +++ | ± | ± | ± | − |
| IGPD6 | + | + | + | ± | + | ± | + |
| IGPD10 | − | − | − | − | − | N.D. | N.D. |
| IGPD13 | ++ | ++ | ++ | +/− | + | ± | + |
| IGPD14 | − | − | − | − | − | N.D. | N.D. |
| IGPD16 | − | + | − | + | + | N.D. | N.D. |
| IGPD17 | − | − | − | − | ± | N.D. | N.D. |
| HisC9 | ± | ± | ± | ± | + | N.D. | N.D. |
| HisC11 | − | − | − | − | − | N.D. | N.D. |
| HisC14 | ± | + | +/± | +/− | + | ± | ± |
| HisC16 | + | +/± | − | + | ± | N.D. | N.D. |
| HisC19 | − | − | − | − | ± | N.D. | N.D. |
| Amp | +++ | + | + | +++ | + | ± | − |
| DMSO | − | − | − | − | − | − | − |
Summary of bacterial disk inhibition and human fibroblast toxicity assay results. +: weak inhibition; ++: weak inhibition plus zone of partial inhibition; +++: strong inhibition; ±: partial inhibition; −: no inhibition; N.D. not determined
Out of 18 tested compounds 10 compounds have different levels of activity against the S. aureus patient strain, 13 against S. aureus Mu50, and 7 against S. aureus USA300 as shown in Table 4. Especially encouraging is the fact that several of the compounds show significant activity towards the drug resistant strains of S. aureus, Mu50 and USA300 that show only weak susceptibility towards the well-established antibiotic ampicillin.
It is interesting to note that of the six tested HisI ligands, HisI1 and HisI9 were predicted to bind in a different orientation than the rest. These two ligands show the least activity of the potential HisI inhibitors in the disc inhibition assay, which suggests that their mode of binding is not favorable. The compound HisI8 seems to be the third least active compound, which is in accordance with a lower calculated binding energy for this ligand than for the rest.
Out of the seven tested IGPD ligands, three show activity. This is the small polar compound IGPD6, the compound with a pyridinol group instead of a carboxylate group (IGPD13) and the carbazolyl derivative IGPD1. It is noteworthy that while IGPD1 shows strong inhibition of two S. aureus strains, the very similar compound IGPD14 shows no inhibitory effect at all. Glide does rank IGPD1 higher than IGPD14, but both poses are given high G-scores. However, MD and MM-PBSA/GBSA calculations are able to identify IGPD14 as a false positive. Two especially promising compounds are IGPD1 and IGPD13. The first shows strong inhibition against the S. aureus patient strain and the USA300 strain and it shows weak inhibition against the Mu50 strain. The second shows a clear zone of growth inhibition plus partial inhibition of the S. aureus strains Mu50 and USA300 respectively, and a weak inhibition of growth in the patient strain.
Three out of the five tested potential HisC ligands show an inhibitory effect. These three ligands all interact with one of the phosphate binding sites and with Tyr110, indicating the importance of these interactions for inhibitory activity on this enzyme. The positive results for HisC14 indicates that other groups than carboxylate can interact with the phosphate binding sites of this enzyme. The lack of activity of the dicarboxylate HisC19 might be a consequence of too high a desolvation cost for binding of this very polar compound to the deeply buried HisC active site pocket. It might also be due to HisC19 being too polar to effectively penetrate the cell wall.
Activity against E. coli is generally much lower, which is possibly a result of both the selection of the compounds towards the S. aureus orthologs of the three enzymes studied as well as the different properties of the cell wall in gram negative bacteria, which could make it difficult for the mostly negatively charged inhibitors to pass the outer membrane. Nevertheless, 14 compounds were found to be weakly active or give partial inhibition for the E. coli 1655 strain.
To check the computational prediction that the small molecules exert their effect through binding the the enzymes in the histidine biosynthesis pathway, we performed a complementation assay (see Table S4 in the Supporting Information) where the bacteria were grown on minimal medium agar plates, where the effect of the potential inhibitors is very similar to the LB plates. Upon addition to 0.2mM L-histidine, five of ten inhibitors selected showed complete complementation of growth of the S. aureus, while the other five showed partial complementation in at least one of the strains tested. These results are consistent with the hypothesis that the small molecules indeed inhibit whole cell growth through interference with the histidine biosynthesis.
Finally, we tested the toxicity of seven of the more active potential inhibitors in mammalian cells for off-target effects using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability and trypan blue exclusion assays. For this purpose, we used normal BJ foreskin fibroblast cells immortalized by hTERT, the catalytic subunit of human telomerase.31 The results of these studies are presented in Figure S5 (in the Supporting Information) and summarized in Table 4. Again, IGPD1 is the most promising compound for further development, since it has slightly lower human cell toxicity than the known antibiotic ampicillin. In comparison, the second best inhibitor, IGPD13, is significantly more cytotoxic (according to the trypan blue exclusion assay). Overall, the human cell toxicity of the selected compounds in these assays is fairly limited, indicating that off-target activity of the selected compounds is relatively low.
Conclusions
The development of novel compounds with antibiotic activity is a pressing need in view of the rapidly expanding resistance of pathogens towards known antibiotics. In the ongoing race between drug development and new resistance, not only new compounds but more importantly, new methods for the discovery of new hits are needed. In the present study, the application of virtual screening and MM-PBSA ensemble rescoring to three enzymes in the histidine biosynthetic pathway, which was predicted to be essential for growth of S. aureus by systems-level approaches,4 yielded 49 potential inhibitors. Based on structural diversity and physicochemical properties, 18 compounds were selected for experimental tests in a whole cell growth inhibition assay. We found 13 and 14 compounds to be active in different strains of S. aureus and E. coli, respectively. Although the observed growth inhibitions are in many cases weak or only partial, two compounds (IGPD1 and IGPD13) show very promising activity comparable to the effects of the well-known antibiotic ampicillin.
Several conclusions can be drawn from these studies. First, the scalable and fast computer-based methods of flux balance analysis and virtual screening used for target and hit identification, respectively, is an effective strategy for the early stages of antibiotic drug discovery. Secondly, refinement of the docking results by ensemble rescoring together with judicious compound selection leads at least in the examples present here and in our earlier studies7 to excellent hit rates and justifies the immediate use of cell-based assays, which in many respects have a higher information content for drug discovery. Thirdly, at least two of the compounds disclosed here merit further development as inhibitors of the histidine biosynthesis pathway that is unconditionally essential for growth in S. aureus even though the possibility of transport of histidine or one of the relevant biosynthetic intermediates from external sources has to be, in analogy to studies of the FASII pathway,32 investigated in future studies.
Supplementary Material
Acknowledgements
We gratefully acknowledge support of this research by the National Institutes of Health (NIAID U01-0700499 to Z.O and O. W.) and by travel grants from The American-Scandinavian Foundation, The Danish Chemical Society, The Otto Mønsted Foundation, The Knud Højgaard Foundation, The Augustinus Foundation, and The Oticon Foundation to S.T.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Procedures for computational and biological experiments, additional calculated G-scores and MM-PBSA/GBSA binding energies, as well as additional biological data. This material is available free of charge via the Internet at http://pubs.acs.org. PyMOL sessions with both docking poses and enzyme-ligand complexes after 8 ns MD runs are available from the authors upon request.
References
- 1.Klein E, Smith DL, Laxminarayan R. Hospitalizations and Deaths Caused by Methicillin-Resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 2007;13:1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman J. Vancomycin-Resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis. 2008;46:668–674. doi: 10.1086/527392. [DOI] [PubMed] [Google Scholar]
- 4.Lee D-S, Burd H, Liu J, Almaas E, Wiest O, Barabási A-L, Oltvai ZN, Kapatral V. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel anti-microbial drug targets. J Bacteriol. 2009;191:4015–4024. doi: 10.1128/JB.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson DC, Humblet C, Joseph-McCarthy D. Investigation of MM-PBSA Rescoring of Docking Poses. J. Chem. Inf. Model. 2008;48:1081–1091. doi: 10.1021/ci700470c. [DOI] [PubMed] [Google Scholar]
- 6.Guimarães CRW, Cardozo M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- 7.Shen Y, Liu J, Estiu G, Isin B, Ahn Y-Y, Lee D-S, Barabasi A-L, Kapatral V, Wiest O, Oltvai ZN. A blueprint for antimicrobial hit discovery targeting metabolic networks. Proc. Natl. Acad. Sci. 2010;107:1082–1087. doi: 10.1073/pnas.0909181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sivaraman J, Myers RS, Boju L, Sulea T, Cygler M, Davisson VJ, Schrag JDI. Crystal Structure of Methanobacterium thermoautotrophicum Phosphoribosyl-AMP Cyclohydrolase His. Biochemistry. 2005;44:10071–10080. doi: 10.1021/bi050472w. [DOI] [PubMed] [Google Scholar]
- 9.D’Ordine RL, Klem TJ, Davisson VJ. N1-(5’-Phophoribosyl)adenosine-5’-Monophosphate Cyclohydrolase: Purification and Characterization of a Unique Metalloenzyme. Biochemistry. 1999;38:1537–1546. doi: 10.1021/bi982475x. [DOI] [PubMed] [Google Scholar]
- 10.Glynn SE, Baker PJ, Sedelnikova SE, Davies CL, Eadsforth TC, Levy CW, Rodgers HF, Blackburn GM, Hawkes TR, Viner R, Rice DW. Structure and Mechanism of Imidazoleglycerol-Phosphate Dehydratase. Structure. 2005;13:1809–1817. doi: 10.1016/j.str.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez FJ, Vega MC, Lehmann F, Sandmeier E, Gehring H, Christen P, Wilmanns M. Structural Studies of the Catalytic Reaction Pathway of a Hyperthermophilic Histidinol-phosphate Aminotransferase. J. Biol. Chem. 2004;279:21478–21488. doi: 10.1074/jbc.M400291200. [DOI] [PubMed] [Google Scholar]
- 12.Marienhagen J, Kennerknecht N, Sahm H, Eggeling LJB. Functional Analysis of All Aminotransferase Proteins Inferred from the Genome Sequence of Corynebacterium glutamicum. J. Bacteriol. 2005;187:7639–7646. doi: 10.1128/JB.187.22.7639-7646.2005. 2005, 187, 7639–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haruyama K, Nakai T, Miyahara I, Hirotsu K, Mizuguchi H, Hayashi H, Kagamiyama H. Structures of Escherichia Coli Histidinol-Phosphate Aminotransferase and Its Complexes with Histidinol-Phosphate and N-(5’-Phosphopyridoxyl)-L-Glutamate: Double Substrate Recognition of the Enzyme. Biochemistry. 2001;40:4633–4644. doi: 10.1021/bi002769u. [DOI] [PubMed] [Google Scholar]
- 14.Sivaraman J, Li Y, Larocque R, Schrag JD, Cygler M, Matte A. Crystal Structure of Histidinol Phosphate Aminotransferase (HisC) from Escherichia Coli, and its Covalent Complex with Pyridoxal-5’-phosphate and L-Histidinol Phosphate. J. Mol. Biol. 2001;311:761–776. doi: 10.1006/jmbi.2001.4882. [DOI] [PubMed] [Google Scholar]
- 15.Ames BN, Horecker BN. The Biosynthesis of Histidine: Imidazoleacetol Phosphate Transaminase. J. Biol. Chem. 1956;220:113–128. [PubMed] [Google Scholar]
- 16.Prime 1.6. New York, NY: Schrödinger, LLC; 2007. [Google Scholar]
- 17.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring.1.Method and Assessment of Docking Accuracy. J.Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 18.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J.Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 19.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teague SJ, Davis AM, Leeson PD, Oprea T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. Intl. Ed. Engl. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 21.Alifano P, Fani R, Liò P, Lazcano A, Bazzicalupo M, Carlomagno MS, Bruni CB. Hisitidine Biosynthetic Pathway and Genes: Structure, Regulation, and Evolution. Microbiol. Rev. 1996;60:44069. doi: 10.1128/mr.60.1.44-69.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stepansky A, Leustek T. Histidine biosynthesis in plants. Amino Acids. 2006;30:127–142. doi: 10.1007/s00726-005-0247-0. [DOI] [PubMed] [Google Scholar]
- 23.Mori I, Iwasaki G, Kimura Y, Matsunaga S, Ogawa A, Nakano T, Buser H-P, Hatano M, Tada S, Hayakawa K. Synthesis of Inhibitors of Imidazole Glycerol Phosphate Dehydratase. J. Am. Chem. Soc. 1995;117:4411–4412. [Google Scholar]
- 24.Gohda K, Kimura Y, Mori I, Ohta D, Kikuchi T. Theoretical evidence of the existence of a diazafulvene intermediate in the reaction pathway of imidazoleglycerol phosphate dehydratase: design of a novel and potent heterocycle structure for the inhibitor on the basis of the electronic structure-activity relationship study. Biochim. Biophys. Acta. 1998;1385:107–114. doi: 10.1016/s0167-4838(98)00049-1. [DOI] [PubMed] [Google Scholar]
- 25.Gohda K, Mori I, Ohta D, Kikuchi T. A CoMFA analysis with conformational propensity: An attempt to analyze the SAR of a set of molecules with different conformational flexibility using a 3D-QSAR method. J. Comput.-Aided Mol. Design. 2000;14:265–275. doi: 10.1023/a:1008193217627. [DOI] [PubMed] [Google Scholar]
- 26.Schweitzer BA, Loida PJ, Jacob CA, Chott RC, Collantes EM, Hegde SG, Mosier PD, Profeta S. Discovery of Imidazole Glycerol Phosphate Dehydratase Inhibitors through 3-D Database Searching. Bioorg. Med. Chem. Lett. 2002;12:1743–1746. doi: 10.1016/s0960-894x(02)00283-4. [DOI] [PubMed] [Google Scholar]
- 27.Schweitzer BA, Loida PJ, Thompson-Mize RL, Jacob CA, Hegde SG. Design and synthesis of β-carboxamido phosphonates as potent inhibitors of imidazole glycerol phosphate dehydratase. Bioorg. Med. Chem. Lett. 1999;9:2053–2058. doi: 10.1016/s0960-894x(99)00338-8. [DOI] [PubMed] [Google Scholar]
- 28.Ohta D, Mori I, Ward E. Inhibitors of imidazoleglycerolphosphate dehydratase as herbicides. Weed Science. 1997;45:610–620. [Google Scholar]
- 29.Cox JM, Hawkes TR, Bellini P, Ellis RM, Barrett R, Swanborough JJ, Russell SE, Walker PA, Barnes NJ, Knee AJ, Lewis T, Davies PR. The Design and Synthesis of Inhibitors of Imidazole glycerol Phosphate Dehydratase as Potential Herbicides. Pestic. Sci. 1997;50:297–311. [Google Scholar]
- 30.Woods GL, Washington JA. Antibacterial susceptibility tests: dilution and disk diffusion methods. In: Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH, editors. Manual of Clinical Microbiology. Washington, DC: ASM Press; 1995. [Google Scholar]
- 31.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 32.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.