

We provide evidence that Scyl1 is also a cytoplasmic component of the nuclear aminoacylation-dependent tRNA export pathway. Scyl1, like the Saccharomyces cerevisiae Cex1p, may collect aminoacyl-tRNAs from the nuclear tRNA export receptors at the cytoplasmic side of the NPC and channel them to eEF-1A for use in protein synthesis.
Abstract
Scyl1 is an evolutionarily conserved N-terminal protein kinase-like domain protein that plays a role in COP1-mediated retrograde protein trafficking in mammalian cells. Furthermore, loss of Scyl1 function has been shown to result in neurodegenerative disorders in mice. Here, we report that Scyl1 is also a cytoplasmic component of the mammalian nuclear tRNA export machinery. Like exportin-t, overexpression of Scyl1 restored export of a nuclear export-defective serine amber suppressor tRNA mutant in COS-7 cells. Scyl1 binds tRNA saturably, and associates with the nuclear pore complex by interacting, in part, with Nup98. Scyl1 copurifies with the nuclear tRNA export receptors exportin-t and exportin-5, the RanGTPase, and the eukaryotic elongation factor eEF-1A, which transports aminoacyl-tRNAs to the ribosomes. Scyl1 interacts directly with exportin-t and RanGTP but not with eEF-1A or RanGDP in vitro. Moreover, exportin-t containing tRNA, Scyl1, and RanGTP form a quaternary complex in vitro. Biochemical characterization also suggests that the nuclear aminoacylation-dependent pathway is primarily responsible for tRNA export in mammalian cells. These findings together suggest that Scyl1 participates in the nuclear aminoacylation-dependent tRNA export pathway and may unload aminoacyl-tRNAs from the nuclear tRNA export receptor at the cytoplasmic side of the nuclear pore complex and channels them to eEF-1A.
INTRODUCTION
Cell division is triggered when a cell has reached a critical size, and attainment of this size is highly dependent on the overall protein translation rate (for review, see Jorgensen and Tyers, 2004). An important contributor to the translation rate is nuclear tRNA export, as evidenced by increased transcription of tRNA genes in cells lacking the tumor suppressor protein p53 or RB (for reviews, see White, 2004a,b, 2005; Ernens et al., 2006), and increased expression of the mammalian nuclear tRNA export receptor exportin-t (Xpo-t) in rapidly dividing cells (Kruse et al., 2000). Nuclear tRNA export also plays an important role in cell cycle regulation in Saccharomyces cerevisiae, because cells arrest in G1 upon depletion of Utp8p (Bernstein and Baserga, 2004), an essential intranuclear component of the nuclear tRNA export machinery (Steiner-Mosonyi et al., 2003; Strub et al., 2007), and by redistribution of the nuclear tRNA export receptor Los1p to the cytoplasm when the DNA is damaged (Ghavidel et al., 2007). More recent studies have shown that nuclear import of tRNAs also plays an important role in nutrient-related regulation of cell growth (Shaheen and Hopper, 2005; Shaheen et al., 2007; Hurto et al., 2007; Whitney et al., 2007), and to facilitate transport of the human immunodeficiency virus-1 replication complexes into the nucleus of HeLa cells (Zaitseva et al., 2006). Thus, tRNA trafficking between the nucleus and cytoplasm is of fundamental importance to cell biology, and disruption of this process is likely to have grave consequences on cell viability and ultimately survival of the organism.
Many studies reported to date have revealed the complexity of the life cycle of a tRNA molecule. Eukaryotic tRNA genes are transcribed by RNA polymerase III as precursor tRNAs (pre-tRNAs). The pre-tRNAs then undergo trimming of the 5′ and 3′ ends, various base modifications, addition of the CCA trinucleotide to the 3′ end, and in some cases removal of an intron (for review, see Hopper and Phizicky, 2003). In mammals and plants, processing of intronless and intron-containing pre-tRNAs occurs in the nucleus (Lund and Dahlberg, 1998; Hopper and Phizicky, 2003; Paushkin et al., 2004; Englert et al., 2007). The maturation of pre-tRNAs also occurs in the nucleus of Saccharomyces cerevisiae (Hopper and Phizicky, 2003). However, removal of the intron from pre-tRNAs in S. cerevisiae takes place in the cytoplasm, and the spliced tRNAs are imported back into the nucleus for reasons that are not known (Huh et al., 2003; Yoshihisa et al., 2003, 2007; Shaheen and Hopper, 2005; Takano et al., 2005). The fully processed tRNAs from both classes of precursor tRNAs are then subjected to an aminoacylation quality control step in the nucleus to establish that they are functional before export to the cytoplasm (Lund and Dahlberg, 1998; Sarkar et al., 1999; Grosshans et al., 2000a; Azad et al., 2001; Steiner-Mosonyi and Mangroo, 2004). In S. cerevisiae, this proofreading step occurs in the nucleolus (Steiner-Mosonyi and Mangroo, 2004). Nuclear tRNA aminoacylation, however, is not the only mechanism used to inspect the functionality of processed tRNAs, because it is not absolutely required for tRNA export in both Xenopus laevis and S. cerevisiae (Arts et al., 1998b; Azad et al., 2001). Consequently, nuclear tRNA export is thought to occur by two pathways referred to as aminoacylation dependent and aminoacylation independent. However, findings reported suggest that the nuclear tRNA aminoacylation-dependent pathway is primarily used to export mature tRNAs from the nucleus in S. cerevisiae (Steiner-Mosonyi and Mangroo, 2004). tRNAs that are deemed functional in S. cerevisiae are transported out of the nucleolus by Utp8p and delivered to carrier proteins for export to the cytoplasm (Steiner-Mosonyi et al., 2003; Strub et al., 2007). Whether such a protein is required in mammalian cells is not known.
The nuclear envelope provides a selectively permeable barrier between the cytoplasm and nucleus due to the presence of large proteinaceous assemblies known as nuclear pore complexes (NPCs). Although small molecules (<25 kDa) can diffuse freely through the NPC, translocation of large macromolecules such as proteins and nucleic acids is dependent on carrier proteins and components of the NPC known as nucleoporins (Nups). The carrier proteins that facilitate nuclear import and export of macromolecules are referred to as import/export receptors. Many of these proteins have been identified in both mammalian cells and S. cerevisiae. The majority of nuclear import/export receptors belong to a family of soluble transport proteins known as β-karyopherins. The function of these proteins is regulated by the GTPase Ran in mammalian cells and Gsp1p in S. cerevisiae (for reviews, see Wozniak et al., 1998; Gorlich and Kutay, 1999; Rodriguez et al., 2004). The β-karyopherins that facilitate translocation of tRNAs across the NPC are Xpo-t and exportin-5 (Xpo-5) (Kutay et al., 1998; Arts et al., 1998a; Bohnsack et al., 2002; Calado et al., 2002) in mammalian cells, and Los1p and Msn5p (Hellmuth et al., 1998; Takano et al., 2005; Eswara et al., 2009), the orthologues of Xpo-t and Xpo-5, respectively, in S. cerevisiae. Furthermore, PAUSED, the orthologue of Xpo-t, is required for movement of tRNAs across the NPC in plants (Hunter et al., 2003).
Like other nuclear export β-karyopherins, the nuclear tRNA export receptors bind the tRNA cargo and RanGTP/Gsp1pGTP and move across the NPC; upon arrival in the cytoplasm RanBP1/Yrb1p binds to RanGTP/Gsp1pGTP and RanGAP/Rna1p activates the GTPase activity of Ran/Gsp1p. Hydrolysis of guanosine triphosphate (GTP) to guanosine diphosphate (GDP) by Ran/Gsp1p allows the receptor–cargo complex to dissociate (Izaurralde et al., 1997; Hellmuth et al., 1998; Sarkar and Hopper, 1998; Kutay et al., 1998; Arts et al., 1998a; Bohnsack et al., 2002; Calado et al., 2002). Recent studies have shown that in S. cerevisiae Cex1p also participates in this step of the nuclear aminoacylation-dependent tRNA export pathway (McGuire and Mangroo, 2007). Cex1p has been proposed to collect aminoacyl-tRNAs from the nuclear tRNA export receptors at the cytoplasmic side of the NPC, and transfer them to the eukaryotic elongation factor eEF-1A by using a channelling mechanism (McGuire and Mangroo, 2007). However, it is not known whether unloading of the nuclear tRNA export receptors in mammalian cells involves a similar mechanism.
Cex1p belongs to an evolutionarily conserved protein family with a characteristic protein kinase-like domain at their N termini. The mammalian member of this family is Scyl1, which plays a role in COP1-mediated retrograde protein trafficking in HeLa cells (Burman et al., 2008). Furthermore, loss of Scyl1 function has been shown to result in neurodegenerative disorders in mice (Schmidt et al., 2007). YATA, the Drosophila orthologue of Scyl1, also functions in protein trafficking, and null mutants of YATA exhibit several abnormalities, including deterioration of neural tissues (Sone et al., 2009). Here, we investigated whether Scyl1 plays a role in nuclear tRNA export in HeLa cells. The data suggest that Scyl1 is also a cytoplasmic component of the nuclear aminoacylation-dependent tRNA export pathway. Scyl1, like Cex1p, may collect aminoacyl-tRNAs from the nuclear tRNA export receptors at the cytoplasmic side of the NPC, and channel them to eEF-1A for use in protein synthesis. It is possible that this mechanism is conserved throughout eukarya, because an uncharacterized orthologue of Cex1p and Scyl1 is found in all eukaryotic genomes sequenced to date. In addition, this study is the first to show that a protein implicated in the development of neurodegenerative disorders participates in nuclear tRNA export.
MATERIALS AND METHODS
Plasmids, Antibodies, and Oligonucleotides
The pGEX2T-TEV plasmid was obtained from Dr. D. Heinrichs (University of Western Ontario, London, ON, Canada). The pET23d and pYX242 plasmids were purchased from Novagen (San Diego, CA). pCDNA 6.2-GW/EmGFP-miR was purchased from Invitrogen (Carlsbad, CA). The pCMV-Tag2A plasmid was purchased from Stratagene (La Jolla, CA). pQE32-Ran was provided by Dr. D. Gorlich (University of Heidelberg, Heidelberg, Germany). The pEGFP-NUP98 plasmid was obtained from Dr. R. Truant (McMaster University, Hamilton, ON, Canada). The pEGFP-N1 plasmid was purchased from (Clonetech, Mountain View, CA). The pYX242-LOS1 and pYX242-CEX1 plasmids have been described previously (Cleary and Mangroo, 2000). pET23a-RanGAP was provided by Dr. V. Gerke (University of Muenster, Muenster, Germany). The pUC13-tRNAamSer plasmid was obtained from Dr. J. Capone (McMaster University). The pSVBpUC plasmid was provided by Dr. U. Rajbhandary (Massachusetts Institute of Technology, Cambridge, MA). pYX242-SCYL1 was constructed by polymerase chain reaction (PCR) amplification of the HeLa Scyl1 cDNA (IMAGE ID# 6581110) from Open Biosystems (Huntsville, AL), and ligation into the EcoRI and AvrII sites of pYX242. The pET23d-SCYL1 plasmid was prepared by amplification of the Scyl1 cDNA, and ligation into the NcoI and EcoRI sites in pET23d. The pGEX2T-TEV-SCYL1 plasmid was generated by PCR amplification of the Scyl1 cDNA, and cloning into the EcoRI site in pGEX2T-TEV. The pCMV-Tag2A-SCYL1 plasmid was constructed by PCR amplification of the Scyl1 cDNA, and cloning into the EcoRI and EcoRV sites in pCMV-Tag2A. The pGEX2T-TEV-NUP98 plasmid was made by PCR amplification of the NUP98 open reading frame (ORF) from pEGFP-NUP98, and cloning into the BamHI and EcoRI sites in pGEX2T-TEV. The pGEX2T-TEV-XPOT plasmid was constructed by PCR amplification of the Xpo-t ORF from pYES2-XPOT, and cloning into the BamHI and EcoRI sites in pGEX2T-TEV. The pCMV-Tag2A-XPOT plasmid was generated by PCR amplification of the Xpo-t ORF from pYES2-XPOT, and cloning into the EcoRI and SalI sites in pCMV-Tag2A. The pSVBpUC-tRNAamSer plasmids were constructed by excising the wild-type and G11:C24 tRNAamSer genes from pUC13, and ligating them into the BglII site in pSVBpUC. pET23d-XPOT was constructed by PCR amplification of the Xpo-t ORF from pYES2-XPOT, and cloning into the NcoI and EcoR1 sites in pET23d. A fragment containing the promoter, the EGFPam29,78 ORF and the transcription termination sequences was removed from pEGFPam29,78 with AseI and AflII; the ends of the excised fragment were blunted and inserted into the SmaI site in pSVBpUC-tRNAamSer, or the pSVBpUC-G11:C24 tRNAamSer vectors. The sources of the antibodies used for Western blot analyses and immunoprecipitation are provided in Supplemental Data. The sequences of the oligonucleotide probes used for fluorescence in situ hybridization (FISH) and Northern blot analysis are given in the Supplemental Data. Site-directed mutagenesis is performed as described in Supplemental Data.
Cell Culture and Transfections
HeLa and COS-7 cells were maintained in DMEM containing 10% fetal bovine serum and penicillin-streptomycin at 37°C in 5% CO2. Transfections were carried out with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as described by the manufacturer.
S. cerevisiae In Vivo Nuclear tRNA Export Assay
The S. cerevisiae HEY301-129 strain has been reported previously (Cleary and Mangroo, 2000). The strain harboring yEPLAC195 containing the G11:C24 tRNAamTyr mutant gene was transformed with pYX242, pYX242-LOS1, pYX242-CEX1, or pYX242-SCYL1, and transformants were selected on synthetic dextrose medium lacking uracil and leucine (SD-Ura-Leu). Transformants were grown overnight at 30°C in SD-Ura-Leu medium and tested for suppression of the amber mutation in the trp1 gene of HEY301-129 by growth on SD-Ura-Leu-Trp medium.
Immunofluorescence Microscopy
HeLa cells grown on coverslips were washed with 1× phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in 1× PBS for 20 min at room temperature. The cells were permeabilized in 1× PBS containing 0.5% Triton X-100 on ice for 10 min, rinsed with 1× PBS, and incubated in blocking buffer (1× PBS containing 5% skim milk and 0.1% Tween 20) for 1 h. The cells were incubated with monoclonal antibody mAb414 (1:500), α-Nup98 (1:50), α-Scyl1 (1:500), and α-calnexin (1:500) in 1× PBS containing 0.1% Tween 20 (PBS-T) for 1 h at 37°C in a humid chamber. The cells were washed with PBS-T and incubated with Alexa Fluor (488 or 595)-conjugated secondary antibodies for 1 h at room temperature, washed with PBS-T, and mounted using DAKO Cytomation (Roche Applied Science, Indianapolis, IN). The cells were viewed with an upright DM RE scanning confocal laser microscope (Leica Microsystems, Deerfield, IL) using a 63× oil immersion objective. The images were processed using MBFImageJ (McMaster Biophotonics Facility, McMaster University) or the MetaMorph imaging software (Molecular Devices, Sunnyvale, CA).
Analysis of Scyl1–tRNA Interaction by Substrate-induced Intrinsic Fluorescence Quenching
Substrate-induced intrinsic fluorescence quenching of tryptophan residues was used to determine whether Scyl1 binds tRNA as described previously (Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007). Scyl1 was expressed and purified as outlined in Supplemental Data. Reactions were set up in 20 mM HEPES, pH 7.4, containing 120 mM NaCl, 0.25 μM Scyl1, and various concentrations of a mixture of yeast mature tRNA, Escherichia coli 5S rRNA, or a single-stranded 76-mer DNA oligonucleotide (0.25, 0.5, 1, 2, 4, 8, and 16 μM) and incubated for 1 h at 4°C. Control reactions lacking Scyl1 were prepared as described above. Tryptophan fluorescence was measured using a spectrofluorometer (Photon Technology International, Lawrenceville, NJ) with excitation and emission slits set to 4 nm, and excitation and emission wavelengths set to 295 and 340 nm, respectively. The fluorescence intensity of each control containing only tRNA/DNA was subtracted from the appropriate reaction and is expressed as a percent decrease in fluorescence intensity obtained with Scyl1 alone.
In Vitro Protein Binding Assays
The proteins were expressed and purified as described in Supplemental Data. Glutathione transferase (GST)-Xpo-t (20 μg; 158 pmol), GST-Scyl1 (20 μg; 178 pmol), GST-Nup98 (20 μg; 161 pmol), or GST (158–178 pmol) was incubated with glutathione-Sepharose 4B (GT-Sepharose) in IPP150 buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% NP-40) containing PIN protease inhibitor cocktail (Roche Applied Science) and 1 mM dithiothreitol in the presence or absence of an excess of tRNA (6 μM; Xpo-t; 70 μM, Scyl1) for 2 h at 4°C. The resins were washed with IPP150 containing PIN protease inhibitor cocktail and incubated with a molar excess of prey proteins (Scyl1, Ran-GTP/GDP/5′-guanylyl imido diphosphate (GppNHp) [the nonhydrolyzable analogue of GTP], or eEF-1A) for 2 h at 4°C. Ran was loaded with 100 μM GTP, GDP, or GppNHp as described (Lee and Aitchison, 1999). The resins were washed with IPP150 buffer, and boiled in 1× LDS sample buffer (Invitrogen) to elute the bound proteins. The proteins were separated by electrophoresis on 4–12% Novex Bis-Tris gels and subjected to Western blot analysis.
Immunoprecipitations and Western Blot Analysis
HeLa cells were harvested and lysed at 4°C in NP-40 buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1% NP-40) containing protease inhibitor cocktail, 1 mM Na3VO4, and 10 mM NaF. Immunoprecipitations were performed using α-Scyl1 antibodies covalently linked to CNBr-activated Sepharose 4B. Total cell lysates were incubated with the resin overnight at 4°C in NP-40 lysis buffer containing protease inhibitors. The resin was washed with NP-40 buffer and eluted with 0.2 M glycine, pH 2.65. The proteins in the eluate were precipitated with 25% trichloroacetic acid for 30 min on ice and centrifuged for 30 min at 4°C. The protein pellet was washed with acetone containing 0.05 N HCl, followed by a wash with ice cold acetone. The protein pellet was air dried, solubilized in LDS sample buffer, and loaded onto a 4–12% Novex Bis-Tris gel. The separated proteins were transferred to Immbilon-P polyvinylidene difluoride membranes, and probed with mAb414 (1:1000), α-Nup98 (1:1000), α-Xpo-5 (1:1000), α-Xpo-t (1:1000), α-Ran (1:1000), α-Nup107 (1:1000), or α-eEF-1A (1:1000) in TBS-T with 0.25% skim milk. The bound antibodies were detected using horseradish peroxidase-conjugated α-rabbit and α-mouse secondary antibodies (1:5000) and the enhanced chemiluminescence chemiluminescent detection system.
Northern Blot Analysis of Nuclear and Cytoplasmic RNA from COS-7 Cells
COS-7 cells were transfected with pSVBpUC containing the wild-type or the G11:C24 mutant tRNAamSer gene. At 48 h after transfection, the cells were washed with 1× PBS containing 5 mM EDTA and lifted in 1× PBS. Cell fractionation was carried out using the ProteoJet cytoplasmic and nuclear fractionation kit as instructed by the manufacturer (Fermentas, Burlington, ON, Canada). Total RNA was isolated from each fraction using TriZol (Invitrogen) as specified by the manufacturer, and separated by electrophoresis on a 10% polyacrylamide gel containing 8 M urea. The RNA was transferred electrophoretically to NyTran Super Charge positively charged membrane (Whatman Schleicher and Schuell, Keene, NH) for 2 h using 40 V at 4°C in 1× TAE (40 mM Tris, 40 mM sodium acetate, and 2 mM EDTA, pH 8.0). The membrane was baked at 80°C for 2 h and incubated for 4 h at 37°C in 4× SET (1× SET = 0.03 M Tris-HCl, pH 8.0, containing 0.15 M NaCl and 2 mM EDTA) containing 250 μg/ml sheared salmon sperm DNA, 0.1% SDS, and 10× Denhardt's solution. Hybridization was carried out overnight at 37°C in the same buffer containing a 5′-32P-labeled oligonucleotide probe (∼1–2 × 106 cpm/ml). The membranes were washed four times at room temperature with 3× SET containing 0.1% SDS for 30 min each and subjected to autoradiography.
Analysis of the Aminoacylation Status of Mammalian tRNAs Isolated from Nuclear and Cytosolic Fractions
HeLa cells grown on 10-cm2 dishes were lifted using TrypLE Express and pelleted by centrifugation. The cells were lysed using the cell lysis buffer from the ProteoJet cytoplasmic and nuclear fractionation kit. A brief low-speed centrifugation was used to separate the cytosolic and nuclear fractions. The pellet containing nuclei was washed twice with the nuclei wash buffer provided by the fractionation kit and resuspended in 300 mM sodium acetate, pH 5.0. Total RNA was then isolated from the cytosolic and nuclear fractions under acidic conditions using TRIzol as prescribed by the manufacturer. The RNA pellets were resuspended in 10 mM sodium acetate, pH 5.0, and stored at −80°C. To deacylate the tRNAs, total RNA from each fraction was incubated in 0.2 M Tris-HCl, pH 9.5, for 1 h at 37°C. Aliquots corresponding to 0.5 A260 of RNA from the nuclear and cytosolic fractions treated with or without base were subjected to electrophoresis on a 6.5% acid-urea polyacrylamide gel (6.5% polyacrylamide [19:1 acrylamide/bisacrylamide] in 0.1 M sodium acetate, pH 5.0, containing 8 M urea) for 18 h at 4°C. RNA was transferred electrophoretically to NyTran Super Charge positively charged membrane for 2 h at 4°C in 1× TAE, pH 8.0. Northern analysis was performed as described above.
Fluorescence In Situ Hybridization
FISH was carried out essentially as described (http://www.singerlab.org/protocols). Cells grown on coverslips were fixed for 1 h at room temperature in 1× PBS containing 4% formaldehyde. The cells were then rinsed with 1× PBS and permeabilized in 1× PBS containing 0.5% Triton X-100 for 10 min on ice. The cells were rehydrated for 5 min in 30 μl of 2× SSC containing 50% formamide, and hybridized overnight at 37°C with 5 pmol of Cy3- or fluorescein-labeled probe in 2× SSC containing 50% formamide, 10% dextran sulfate, 0.02% bovine serum albumin, 2 mM vanadyl ribonucleoside complex, and 40 μg of E. coli tRNA in a volume of 30 μl. The cells were washed with 2× SSC containing 50% formamide and 1× SSC containing 50% formamide at room temperature, followed by 1× SSC containing 50% formamide at 37°C. The cells were stained with 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI), washed three times with 1× PBS and mounted using DAKO Cytomation.
The Mammalian in Vivo Nuclear tRNA Export Assay Based on Amber Suppression
COS-7 cells grown on coverslips were transfected with pEGFPam29,78 alone, pSVBpUC-tRNAamSer-EGFPam29,78, or pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78. COS-7 cells were also doubly transfected with the pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78 and either pCMV-XPOT, pCMV-SCYL1, or pCMVTag2A. Twenty-four hours after transfection, the cells were fixed with 4% paraformaldehyde, rinsed with 1× PBS containing 0.1 M glycine, and stained with DAPI. The cells were then rinsed 3× with 1× PBS and mounted with DAKO fluorescence mounting medium, and enhanced green fluorescent protein (EGFP) expression was monitored with an Elcipse E600 fluorescent microscope (Nikon, Tokyo, Japan) with a 63× oil immersion objective. Images were captured using a CoolSNAPFX charge-coupled device camera and analyzed using the MetaMorph imaging software.
Knockdown of Scyl1 by MicroRNA (miRNA) Expression
HeLa cells were grown on 10-cm2 dishes until 80% confluent before they were transfected with 24 μg of the miRNA expression plasmid pCDNA 6.2-GW/mRFP-Scyl1 (sense oligonucleotide, 5′-TGCTGCATGTCTGCTGACCACTTCTCGTTTT GGCCACTGACTGACGAGAAGTGCAGCAGACATG-3′) or the pCDNA 6.2/mRFP-control (sense oligonucleotide, 5′-TGCTGACGTGACACGTTCGGAGAATTGTTTTGGCCACTGACT GACAATTCTCCACGTGTCACGT-3′) (generous gifts from Dr. P. S. McPherson, McGill University, Montreal, QC, Canada; Burman et al., 2008) using Lipofectamine 2000. Forty-eight hours after transfection, the cells were plated on coverslips and grown for 24 h, and immunofluorescence microscopy was used to monitor the knockdown of Scyl1. Alternatively, the transfected cells were plated on coverslips and grown for 24 h, and FISH was performed to detect tRNATyr by using the procedure described above.
Knockdown of Nup98 by miRNA Expression
HeLa cells were grown on 10-cm2 dishes until 80% confluent before they were transfected with 24 μg of the miRNA expression plasmid pCDNA 6.2-GW/EmGFP-Nup98 (sense oligonucleotide, 5′-TGCTGTAGTGACTTGCTTTCATATTCGTTTTGGCCACTGACTGACGAATATGAGCAAGTCACTA-3′) by using the small interfering RNA sequence reported previously (Ebina et al., 2004) or the pCDNA 6.2/EmGFP-control (sense oligonucleotide, 5′-TGCTGACGTGACACGTTCGGAGAATTG TTTTGGCCACTGACTGACAATTCTCCACGTGTCACGT-3′) by using Lipofectamine 2000. Forty-eight hours after transfection, the cells were plated on coverslips and grown for 24 h, and immunofluorescence microscopy and Western blot analyses were used to monitor the knockdown of Nup98. Alternatively, the transfected cells were plated on coverslips, grown for 24 h, and then FISH was performed to detect tRNALys by using the procedure described above.
RanGTPase Protection Assay
Ran [γ-32P]GTP (1 μM) was added to PBSM (10 mM Na2HPO4, 1.75 mM KH2PO4, 137 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, pH 7.4, and 10% glycerol), containing 0.5 nM RanGAP, which was purified as described previously (Haberland and Gerke, 1999), 2 μM Xpo-t with or without 6 μM tRNA in a final volume of 500 μl. At the specified time, a 50-μl aliquot of the incubation was added to 200 μl of 20 mM phosphoric acid containing 5% activated charcoal (wt/vol). The charcoal mixtures were then centrifuged for 10 min in 1.5-ml tubes, and 100 μl was removed for scintillation counting to determine the amount of GTP hydrolyzed.
RESULTS
Development of an In Vivo Mammalian Nuclear tRNA Export Assay Based on Amber Suppression
Amber suppression and nuclear-cytoplasmic distribution analyses established that mutation of the C11:G24 base pair of the yeast tyrosine amber suppressor tRNA to G11:C24 resulted in nuclear retention of the tRNA in S. cerevisiae (Cleary and Mangroo, 2000). This defect can be rescued by overexpression of tRNA-binding proteins directly involved in nuclear tRNA export such as the nuclear tRNA export receptors Los1p and Xpo-t (Cleary and Mangroo, 2000). Furthermore, this in vivo nuclear tRNA export assay has been used to show that Utp8p and Cex1p, identified during a three-hybrid interaction screen for tRNA-binding proteins, participate in nuclear tRNA export in S. cerevisiae (Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007). To develop a similar nuclear tRNA export assay for mammalian cells, we investigated whether mutation of the C11:G24 base pair of the human serine amber suppressor tRNA to G11:C24 (Supplemental Figure S1) renders the tRNA nuclear export defective. COS-7 cells were transfected with pSVBpUC-tRNAamSer or pSVBpUC-G11:C24 tRNAamSer, and the cellular location of the tRNA was monitored by FISH by using a Cy3-labeled oligonucleotide complementary to the anticodon stem-loop and variable region of tRNAamSer (Figure 1A). The wild type (WT) tRNAamSer (top row) was detected primarily in the cytoplasm, whereas the G11:C24 tRNAamSer mutant (second row) was found in the nucleus. The G11:C24 tRNAamSer mutant seems to be distributed throughout the nucleoplasm, because very little of the RNA could be detected in nucleoli, which are visualized as punctuate dots when FISH is used to detect the U18 small nucleolar RNA (U18 snoRNA) (third row). Retention of the G11:C24 tRNAamSer mutant in the nucleus was not due to a maturation defect, as only the mature form of the wild type (Figure 1B, lane 1) and mutant tRNA (lane 2) was detected by Northern blot analysis of total RNA isolated from COS-7 cells transfected with pSVBpUC-tRNAamSer or pSVBpUC-G11:C24 tRNAamSer.
Figure 1.

The G11:C24 mutation affects the efficiency of nuclear export of tRNAamSer. (A) FISH analysis of the nuclear-cytoplasmic distribution of the WT and G11:C24 mutant amber suppressor tRNA. COS-7 cells were transfected with pSVBpUC containing the WT tRNAamSer or G11:C24 tRNAamSer gene. The subcellular distribution of the wild-type tRNAamSer and G11:C24 tRNAamSer mutant and the location of U18 snoRNA were detected using Cy3- and fluorescein-labeled oligonucleotides, respectively. The values presented are averages of three independent experiments. DNA was visualized by staining with DAPI. Bar, 10 μm. (B) The G11:C24 mutation did not affect maturation of tRNAamSer. Total RNA was isolated from COS-7 cells transfected with pSVBpUC carrying the WT (lane 1) or G11:C24 mutant tRNAamSer gene (lane 2). An aliquot containing 20 μg of total RNA was subjected to PAGE under denaturing conditions, and tRNAamSer was detected by Northern blot analysis. (C) Analyses of the purity of nuclei isolated from COS-7 cells. Nuclear (N) and cytoplasmic (C) fractions were prepared from COS-7 cells, and Western blot analysis was used to monitor the amount of the cytoplasmic markers tubulin (left, first row) and actin (second row) associated with nuclei, and the amount of the nucleolar marker fibrillarin present in the cytoplasmic fraction (third row). The amount of protein in the two fractions was assessed by Coomassie Blue staining of the blot (right). (D) Northern blot analysis of the nuclear-cytoplasmic distribution of the WT and G11:C24 mutant tRNAamSer. N and C fractions were isolated from COS-7 cells transfected with pSVBpUC carrying the G11:C24 mutant or WT suppressor tRNA gene. Total RNA as isolated from each fraction and subjected to PAGE under denaturing conditions. The RNA was transferred electrophoretically to membrane, and Northern analysis was performed to detect tRNAamSer.
To verify that the G11:C24 tRNAamSer mutant is export-defective, nuclear-cytoplasmic distribution analysis was conducted. Total RNA was isolated from nuclear and postnuclear fractions obtained from COS-7 cells transfected with pSVBpUC-tRNAamSer or pSVBpUC-G11:C24 tRNAamSer, and the cellular location of the suppressor tRNA was monitored by Northern blot analysis (Figure 1D). The extent of lysis of the nucleus, and the purity of the nuclei preparation was assessed by monitoring the location of fibrillarin, and actin and tubulin, respectively (Figure 1C, left). Western blot analysis of the same amount of nuclear (N) and postnuclear (C) fractions followed by densitometric analysis of the blots showed that the nuclear fraction contained approximately <0.5% of the tubulin (top row) and essentially no actin (second row), indicating that the nuclei preparation was essentially free of cytoplasmic contaminants. Approximately 0.5% of the fibrillarin was detected in the cytoplasmic fraction (third row), suggesting that the separation method resulted in a small amount of lysis of the nuclei. Proteins in the nuclear and cytoplasmic fractions also were visualized directly by Coomassie Blue staining of the blot (Figure 1C, right). Northern analysis of the same amount of total RNA from each fraction followed by densitometric analyses of the autoradiogram showed that ∼3% of the wild-type tRNAamSer (Figure 1D, top row) was present in the nucleus. The G11:C24 tRNAamSer mutant was found in both the nuclear and cytoplasmic fractions (bottom row). However, unlike the wild type tRNA ∼45% of the G11:C24 tRNAamSer mutant was detected in the nucleus, suggesting that the efficiency of nuclear export of the G11:C24 tRNAamSer mutant was significantly affected.
To detect nuclear export of the G11:C24 tRNAamSer mutant by overexpression of a protein that participates in the export process, an amber mutant of EGFP was used as the reporter. The EGFPam29,78 mutant gene was made by mutating the codons coding for serine at positions 29 and 78 of EGFP to amber termination codons. These mutations blocked synthesis of EGFP, because no fluorescence signal was detected by microscopy in COS-7 cells transfected with pEGFPam29,78 (data not shown). To avoid the necessity to transfect three plasmids at once, the mutant EGFPam29,78 gene was introduced into the pSVBpUC-G11:C24 tRNAamSer and pSVBpUC-tRNAamSer plasmids. When COS-7 cells were transfected with pSVBpUC-tRNAamSer-EGFPam29,78 fluorescence emission was observed in 68% of the cells analyzed, indicating synthesis of the full-length EGFP (Figure 2A, first row). In contrast, no signal was detected in COS-7 cells transfected with pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78 and pCMV-Tag 2A (Figure 2A, second row) or in cells transfected with pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78 alone (data not shown). Furthermore, the G11:C24 tRNA mutant was found primarily in the nucleus of transfected cells (Figure 2A). However, EGFP was produced in cells transfected with pCMV-Tag 2A-XPOT and pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78 (Figure 2A, fourth row). In addition, FISH analyses of the same cells demonstrated that overexpression of Xpo-t shifted accumulation of G11:C24 tRNAamSer mutant toward the cytoplasm. These results suggest that overexpression of Xpo-t increased the efficiency of nuclear export of the mutant tRNA. In addition, the data indicate that the amber suppression nuclear tRNA export assay can be used to test whether a protein is involved in nuclear tRNA export in mammalian cells.
Figure 2.

Overexpression of Scyl1 rescues export of the yeast and human mutant amber suppressor tRNAs defective in nuclear export. (A) Scyl1 increased the efficiency of nuclear export of the export-defective human serine amber suppressor tRNASer mutant. COS-7 cells grown on coverslips were transfected with pSVBpUC-tRNAamSer-EGFPam29,78, pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78, and pCMVTag2A, pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78, and pCMV-XPOT, or pSVBpUC-G11:C24 tRNAamSer-EGFPam29,78 and pCMV-SCYL1, and allowed to express for 24 h. The subcellular distribution of the WT tRNAamSer and the G11:C24 tRNAamSer was detected by FISH using Cy3-labeled oligonucleotides. Expression of EGFP was monitored by fluorescent microscopy. The DNA was visualized by DAPI staining. The asterisk (*) denotes the percentage of cells expressing EGFP, and the values presented are averages of three independent experiments. For each experiment, 100 cells were analyzed for EGFP expression. Bar, 10 μm. (B) Scyl1 increased the efficiency of nuclear export of the export-defective S. cerevisiae tyrosine amber suppressor tRNA mutant. The HEY301-129 strain containing the yEPLAC195-G11:C24 tRNAamTyr plasmid was transformed with pYX242-LOS1, pYX242-CEX1, pYX242-SCYL1, or pYX242 and streaked on CSD-Ura-Leu or CSD-Ura-Leu-Trp to select for amber suppression of the trp1 allele.
Overexpression of Scyl1 Facilitates Nuclear Export of the Export-defective Human Amber Suppressor tRNASer in COS-7 Cells and the Yeast Amber Suppressor tRNATyr in S. cerevisiae
Cex1p is a cytoplasmic tRNA-binding protein that facilitates nuclear tRNA export in S. cerevisiae (McGuire and Mangroo, 2007). PSI-BLAST search indicated that Cex1p is structurally homologous to proteins in higher eukaryotes, including the human Scyl1. These proteins possess an N-terminal kinase-like domain (Supplemental Figure S2). However, Cex1p, Scyl1 and their orthologues lack the characteristic catalytic residues found in protein kinases. Furthermore, studies reported indicate that Scyl does not possess kinase activity (Kato et al., 2002). Cex1p and its orthologues also contain a HEAT repeat domain, which is involved in protein-protein interactions. ClustalW analysis indicated that the amino acid sequences of Cex1p and Scyl1 are 23% identical and 41% similar. The sequence and structural similarities between Scyl1 and Cex1p suggest that Scyl1 could be involved in nuclear tRNA export in mammalian cells. Thus, the mammalian nuclear tRNA export assay was used to determine whether Scyl1 from HeLa cells plays a role in nuclear tRNA export. Like Xpo-t, overexpression of Scyl1 increased the efficiency of nuclear export of the G11:C24 tRNAamSer mutant based on expression of EGFP and accumulation of the mutant tRNA in the cytoplasm (Figure 2A, third row). EGFP expression and cytoplasmic accumulation of the G11:C24 tRNAamSer mutant were observed in 10% of the cells analyzed by microscopy.
The ability of Scyl1 to restore export of the nuclear export-defective yeast tyrosine amber suppressor G11:C24 tRNA mutant was also investigated (Figure 2B). Export of the tRNA mutant was monitored by growth of HEY301-129 with a chromosomal trp1am gene on amber suppression medium. Transformants expressing the G11:C24 tRNAamTyr mutant and Scyl1, Los1p or Cex1p grew on Trp amber suppression medium (Figure 2B). In contrast, no growth was observed for the transformant expressing the tRNA mutant and harboring the empty plasmid. This strain, however, grew on nonamber suppression medium containing tryptophan. These data also suggest that Scyl1 is involved in nuclear tRNA export and may be functionally equivalent to Cex1p.
Scyl1 Binds tRNA Directly and Saturably In Vitro
Substrate-induced intrinsic fluorescence quenching of tryptophan residues was used to determine whether Scyl1 binds tRNA. The data indicate that Scyl1 bound mature yeast tRNA directly and saturably, with a Kd value of 7 μM (Figure 3). Scyl1 also was found to interact with a DNA oligonucleotide that is 76 nucleotides in length, but this binding was not saturable using the same concentrations used for tRNA binding (data not shown). Moreover, Scyl1 interacted nonsaturably with the E. coli 5S RNA, with a Kd value of 5 M (Figure 3). This behavior is not surprising, because a large number of tRNA-binding proteins, including aminoacyl-tRNA synthetases have been shown to interact nonspecifically with noncognate RNA substrates in vitro (Gite and RajBhandary, 1997; Wang and Schimmel, 1999). Interaction with noncognate RNA in vitro also was observed for proteins involved in nuclear tRNA export, including Utp8p and Cex1p (Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007; McGuire et al., 2009). These results suggest that tRNA is probably the substrate for Scyl1 in vivo.
Figure 3.

Scyl1 binds tRNA directly and saturably. Substrate induced intrinsic fluorescence quenching of tryptophan residues was used to determine whether purified Scyl1 binds tRNA (•) or E. coli 5S RNA (■) directly in vitro as described in Materials and Methods.
Scyl1 Associates with the NPC
Scyl1 has been shown by cell fractionation to be located in the cytosol and with low density microsomal membranes (Liu et al., 2000). Furthermore, fluorescence microscopy localized Scyl1 to the endoplasmic reticulum (ER)–Golgi intermediate compartment and cis-Golgi (Burman et al., 2008). To understand the role of Scyl1 in nuclear tRNA export, we investigated whether it associates with the NPC of the nuclear envelope (Figure 4). HeLa cells were fixed first and then permeabilized followed by incubation with anti-Scyl1 and mAb414, a mAb that recognizes the FXFG-repeat domains of a subset of vertebrate Nups of the NPC. Fluorescence microscopy showed that the majority of Scyl1 (A, first row) is located in the cytoplasm and associated with cytoplasmic structures, which is consistent with previous reports. Interestingly, overlay analysis of the Scyl1 signal with that of mAb414 (B, first row) showed that a small amount of Scyl1 is associated with the NPC (C and inset, first row).
Figure 4.

Scyl1 associates specifically with the NPC in HeLa cells. HeLa cells were fixed with 4% paraformaldehyde in PBS and then permeabilized with 0.5% Triton X-100 in PBS (first and third rows) or fixed and permeabilized simultaneously in 2% paraformaldehyde and 0.5% Triton X-100 (second and fourth rows). The location of Scyl1, the NPC and Calnexin was detected by immunofluorescence microscopy. Localization of Scyl1 with the NPC or calnexin was determined by overlay analyses of the micrographs. Insets represent zoomed in regions outlined with white rectangles. Bars, 10 μm.
To test further whether Scyl1 association with the NPC is authentic, HeLa cells were permeabilized and fixed simultaneously to remove Scyl1 that is not bound to any subcellular structures (A′, second row). Immunofluorescence microscopy showed that the treatment resulted in removal of a large amount of Scyl1 (compare A and A′). Furthermore, overlay analysis indicated that Scyl1 colocalized with FG repeat Nups (B′, second row) of the NPC (C′ and inset, second row). Colocalization of Scyl1 with FG repeat Nups was also observed when HeLa cells were permeabilized and then fixed (data not shown). These data show that while Scyl1 is predominantly cytoplasmic, a small amount of the protein is located at the NPC.
To verify that this association with the NPC is specific, we investigated whether Scyl1 colocalizes with the ER marker calnexin. The cells were fixed first and then permeabilized followed by incubation with anti-Scyl1 (Figure 4D, third row) and anti-calnexin (E). Overlay analysis indicated that Scyl1 did not colocalize with calnexin (F and inset). Colocalization of Scyl1 with calnexin (F′ and inset, fourth row) was also not observed when the cells were fixed and permeabilized simultaneously, or permeabilized and then fixed (data not shown).
Scyl1 Associates with the NPC by Interacting Directly with Nup98
Cex1p has been shown previously to associate with the NPC by interacting directly with Nup116p (McGuire and Mangroo, 2007). The human orthologue of Nup116p is Nup98, and like Nup116p, Nup98 is found on the cytoplasmic and nuclear faces of the NPC in mammalian cells (Griffis et al., 2003). Scyl1 also was shown previously to copurify with Nup98 (Schmidt et al., 2007). Moreover, immunofluorescence confocal scanning microscopy established that Scyl1 colocalizes with Nup98 (Supplemental Figure S3). To ascertain whether Scyl1 associates with Nup98 in vivo, immunoprecipitation was used to determine whether Nup98 copurifies with Scyl1. Sepharose or α-Scyl1 covalently bound to Sepharose was incubated with total cell lysates prepared from HeLa cells. After the resins were washed bound proteins were eluted and subjected to Western blot analysis (Figure 5A). Nup98 (first row) and Scyl1 (third row) were detected in total cell lysate (lane 1), but not in the eluate from the Sepharose matrix alone (lane 2). However, Nup98 (first row, lane 3) and Scyl1 were detected (third row, lane 3) in the eluate from the α-Scyl1-Sepharose matrix. SYPRO Ruby staining of the blot (fourth row) shows that proteins present in the total cell lysates (lane 1) were not present in the eluate from the control matrix (lane 2), but a number of them were in the eluate from the α-Scyl1-Sepharose matrix (lane 3). The complex protein mixture associated with Scyl1 is related to Scyl1 being part of protein complexes involved in retrograde protein trafficking and clathrin-mediated endocytosis (Burman et al., 2008). Nup98 has been shown to interact with Nup107 found on the cytoplasmic and nuclear sides of the NPC in mammalian cells (Belgareh et al., 2001). Western blot analysis shows that Nup107 also copurified with Scyl1 (second row, compare lanes 2 and 3). Nup358, which is found on the cytoplasmic side of the NPC did not copurify with Scyl1, indicating that the copurification of Nup98 and Nup107 with Scyl1 is specific (data not shown). These data together with the cytoplasmic location of Scyl1 suggest that Scyl1 probably associates with Nup98 and Nup107 on the cytoplasmic side of the NPC.
Figure 5.

Scyl1 associates with the NPC by interacting directly with Nup98. (A) Nup98 copurifies with Scyl1. Total cell lysates prepared from HeLa cells (lane 1) were incubated with Sepharose 4B (lane 2) or α-Scyl1 coupled to Sepharose 4B (lane 3). The washed resins were boiled in sample buffer, and the eluates were subjected to Western blot analysis to detect Nup98 (first row), Nup107 (second row), and Scyl1 (third row). The proteins were visualized directly by SYPRO Ruby staining of the blot (fourth row). (B) Scyl1 interacts directly with Nup98 in vitro. Bound GST-Nup98 (20 μg; 161 pmol) (lane 3) or GST (lane 4) was incubated with a twofold molar excess of Scyl1 (28 μg; 322 pmol). The washed resins were boiled in sample buffer, and the eluates and purified Scyl1 (lane 1) and GST-Nup98 (lane 2) were subjected to SDS-PAGE. Scyl1 (first row) and GST-Nup98 (second row) were detected by Western blot analysis followed by Coomassie Blue staining of the blot (third row).
Protein binding assays were performed in vitro to test whether Scyl1 interacts directly with Nup98 (Figure 5B). GST-Nup98 (20 μg; 161 pmol) or GST bound to GT-Sepharose was incubated with a twofold molar excess of Scyl1 (28 μg; 322 pmol). The resins were washed and the bound proteins were eluted by boiling in 1× LDS sample buffer. Scyl1 (top row) and GST-Nup98 (middle row) were detected by Western blot analysis. Scyl1 was found to interact with bound Nup98 (top row, lane 3) and GST alone (lane 4). However, densitometric analysis of the blots indicated that the amount of Scyl1 bound to Nup98 was approximately fivefold higher compared with that bound to GST (top row, compare lanes 3 and 4). Coomassie Blue staining of the blot (bottom row) shows that the interaction between the two proteins was not stoichiometric. This finding suggests that the two proteins are interacting very weakly with each other. Nevertheless, the data imply that Scyl1 associates with the NPC by interacting, in part, directly with Nup98 in vivo.
To test further whether Scyl1 associates with the NPC by interacting directly with Nup98 in vivo, the effect of depletion of Nup98 on the localization of Scyl1 to the NPC was investigated by miRNA-mediated knockdown of Nup98 expression (Ebina et al., 2004) (Figure 6). HeLa cells were transfected with a miRNA expression plasmid carrying the emerald GFP (EmGFP) gene and a control miRNA or the Nup98 miRNA gene, and Western blot analyses (A) and immunofluorescence microscopy (B) were used to monitor the level of Nup98. The expression of the EmGFP gene was used to identify cells transfected with the miRNA expression plasmid. The level of Nup98 in cell lysate prepared from cells transfected with the Nup98 miRNA gene (lane 2) was significantly reduced compared with that in cell lysate prepared from cells transfected with the control miRNA gene (lane 1). Western blot analysis indicated that the level of actin was about the same in cell extract prepared from cells transfected with the control (lane 1) or Nup98 miRNA (lane 2) gene, indicating that approximately the same amount of cell extract was used. The ratio of the intensities of Nup98 to actin, ascertained by densitometric analyses of the blots, indicated that the level of Nup98 in cell extract prepared from cells transfected with the Nup98 miRNA gene was ∼45% of that in cell extract prepared from cells transfected with the control miRNA gene. However, the transfection efficiency obtained with the plasmid with the Nup98 miRNA gene is 30–35%, indicating that the 55% reduction in the level of Nup98 is an underestimation. Fluorescence microscopy also indicated that the level of Nup98 in all cells transfected with the Nup98 miRNA gene (T) was significantly reduced compared with that in cells that were not transfected (NT) with the Nup98 miRNA gene (B, compare bottom), and in cells transfected with the control miRNA gene (T) (top). Analyses of the fluorescence intensity indicate that the level of Nup98 in cells transfected with the Nup98 miRNA gene is 10–20% of that in cells transfected with the control miRNA gene, indicating very efficient knockdown of Nup98 expression (Ebina et al., 2004).
Figure 6.

Knockdown of Nup98 expression affects the localization of Scyl1 to the NPC in HeLa cells. (A) Nup98 level is reduced upon expression of Nup98 miRNA. HeLa cells were transfected with either the control (lane 1) or Nup98 miRNA (lane 2) expression plasmid. Cell lysates were prepared 72 h after transfection, and Western blot analysis was performed to monitor Nup98 (top row) or actin (bottom row) level. (B) Reduction in the level of Nup98 is detected in vivo by fluorescence microscopy. HeLa cells were transfected with either the control (top) or Nup98 (bottom) miRNA expression plasmid. After 72 h, the cells were fixed with 4% paraformaldehyde and the level of Nup98 (left column) was monitored by immunofluorescence microscopy. Transfected cells were identified by the expression of EmGFP (right column). T, transfected cells; NT, untransfected cells. The transfection efficiency was determined by analyzing 100 cells from three independent transfections. Twenty-five transfected cells in two independent transfections were analyzed for the level of Nup98 by measuring the fluorescence intensity using the MetaMorph software. (C) A decrease in the level of Nup98 affects the localization of Scyl1 to the NPC. HeLa cells were transfected with either the control (top) or Nup98 (bottom) miRNA expression plasmid. After 72 h, the cells were fixed with 2% paraformaldehyde and permeabilized simultaneously with 0.5% Triton X-100; localization of Scyl1 (left column) was monitored by immunofluorescence microscopy. The NPC was visualized by staining with mAb414 (middle). Overlay analysis was performed to monitor the level of Scyl1 at the NPC (right) in 25 transfected cells (left box and inset) and untransfected cells (right box and inset). Insets represent zoomed in regions outlined with white rectangles. T, transfected cells; NT, untransfected cells. Bars, 10 μm.
To investigate whether depletion of Nup98 affects Scyl1 localization at the NPC, colocalization analysis was conducted by immunofluorescence microscopy using mAb414 and anti-Scyl1 (C). We found that the level of Scyl1 at the NPC in all cells transfected with the Nup98 miRNA gene (T) (left inset) was reduced considerably compared with that in cells that were not transfected (NT) with the Nup98 miRNA gene (right inset), and in cells transfected with the control miRNA gene (inset). These data also suggest that interaction of Scyl1 with Nup98 is responsible for the association of Scyl1 with the NPC. However, a defect in nuclear export of tRNALys was not observed when Nup98 was depleted (data not shown). This finding is consistent with previous studies showing that inhibition of Nup98 function by microinjection of anti-Nup98 antibodies into X. laevis oocytes had no effect on nuclear tRNA export (Powers et al., 1997).
Scyl1 Interacts with Components of the Mammalian Nuclear tRNA Export Machinery
To elucidate further the role of Scyl1 in the nuclear tRNA export process, HeLa cell lysate was used to test whether proteins known to participate in nuclear tRNA export copurify with Scyl1. Total cell lysate was incubated with α-Scyl1-Sepharose or Sepharose alone and washed to remove unbound proteins. Bound proteins were eluted and subjected to Western blot analysis to detect copurification of the nuclear tRNA export receptors Xpo-t (Figure 7, first row) and Xpo-5 (second row), Ran (third row), and Crm1 (fourth row), which is a β-karyopherin that is not involved in nuclear tRNA export. Xpo-t (lane 1), Xpo-5 (lane 1), Ran (lane 1), Scyl1 (fifth row, lane 1), and Crm1 (lane 1) were detected in the cell lysates, but not in the eluate from the resin alone (lane 2). Xpo-t, Xpo-5 and Ran, but not Crm1, were found in the eluate from the α-Scyl1-resin (lane 3). SYPRO Ruby staining of the blot (sixth row) showed that proteins in the cell lysate (lane 1) were not present in the eluate from the resin alone (lane 2), but many of them copurified with Scyl1 (lane 3). The results indicate that Xpo-t, Xpo-5 and Ran copurified specifically with Scyl1.
Figure 7.
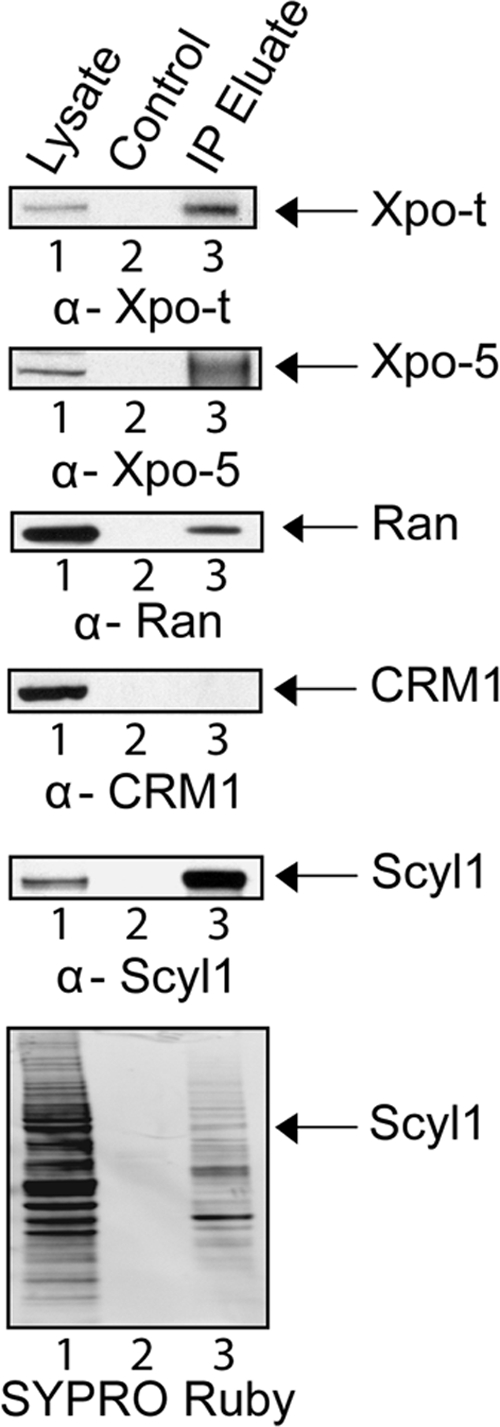
Xpo-t, Xpo-5, and Ran but not Crm1 copurify with Scyl1. Total cell lysate from HeLa cells (lane 1) was incubated with Sepharose 4B (lane 2) or α-Scyl1 coupled to Sepharose 4B (lane 3). The resins were washed and boiled in sample buffer to release bound proteins. Western blot analysis was used to detect Xpo-t (first row), Xpo-5 (second row), Ran (third row), Crm1 (fourth row), and Scyl1 (fifth row) in the eluates. The proteins were visualized directly by SYPRO Ruby staining of the blot (sixth row).
In vitro protein binding studies were performed to test whether Scyl1 interacts directly with Xpo-t (Figure 8A) and Ran (B), because the roles of these two proteins in tRNA export are better characterized than that of Xpo-5. GST-Xpo-t (20 μg; 158 pmol) was bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of saturating amounts of mature yeast tRNAs (6 μM). The reported Kd value of Xpo-t in the absence of RanGTP is 0.6 μM (Kutay et al., 1998). The resins were washed to remove unbound protein and tRNA, and incubated with a twofold molar excess of Scyl1 (27 μg; 316 pmol) (lanes 3 and 4). As a control, the same amount of Scyl1 was incubated with GST bound to GT-Sepharose (lane 5). The washed resins were boiled in 1× LDS sample buffer to release the bound proteins, and the eluates were subjected to Western blot analysis to detect Scyl1 (top row) and GST-Xpo-t (middle row). Scyl1 was found to interact with Xpo-t in the presence (lane 3) or absence of tRNA (lane 4). Scyl1 also interacted with GST (lane 3), but the amount bound, based on densitometric analysis of the blots, was ∼1% of that observed with Xpo-t. Coomassie Blue staining of the blot (bottom row) detected both Scyl1 and GST-Xpo-t and shows that the interaction between the two proteins was not stoichiometric (compare lanes 3 and 4). This suggests that the interaction between the two proteins is very weak.
Figure 8.
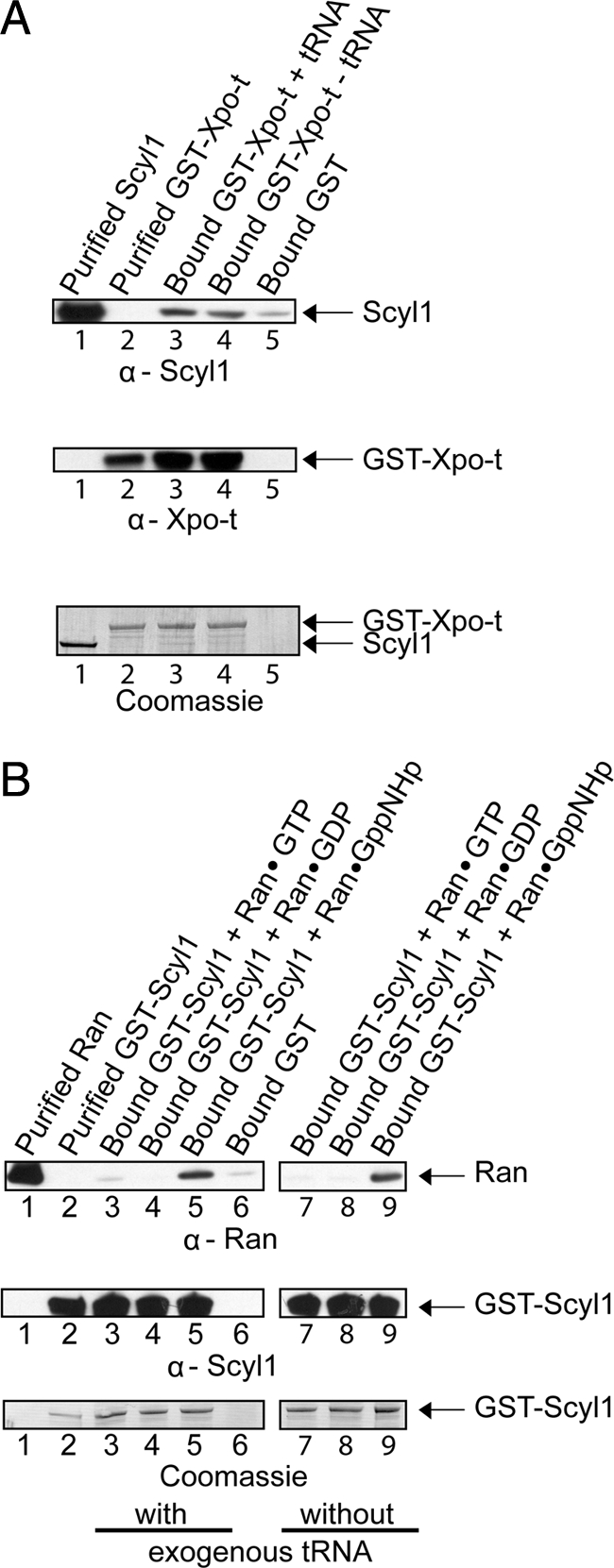
Scyl1 interacts directly with Xpo-t and RanGTP but not RanGDP in vitro. (A) Scyl1 interacts with Xpo-t in a tRNA-independent manner. GST bound to GT-Sepharose (lane 5) or GST-Xpo-t (20 μg; 158 pmol) bound to GT-Sepharose with (lane 3) or without (lane 4) 6 μM yeast mature tRNA was incubated with a twofold molar excess of Scyl1 (27 μg; 316 pmol). The washed resins were boiled in sample buffer, and the eluates and purified Scyl1 (lane 1) and GST-Xpo-t (lane 2) were separated by SDS-PAGE. Scyl1 (first row) and GST-Xpo-t (second row) were detected by Western blot analysis followed by Coomassie Blue staining of the blot (third row). (B) Scyl1 interacts with RanGTP but not RanGDP. GST-Scyl1 (20 μg; 178 pmol) bound to GT-Sepharose in the presence (lanes 3–5) or absence (lanes 7–9) of 70 μM yeast mature tRNA and with a twofold molar excess of Ran (9 μg, 356 pmol) loaded with 100 μM GTP (lanes 3 and 7), GDP (lanes 4 and 8), or GppNHp (lanes 5 and 9). The same amount of Ran was incubated with bound GST (lane 6). The resins were washed and boiled in sample buffer to elute bound proteins. Ran (lane 1), GST-Scyl1 (lane 2), and the eluates were separated by SDS-PAGE, followed by Western blot analyses to detect Ran (first row) and GST-Scyl1 (second row). Coomassie Blue staining of the blot was used to detect the protein directly (third row).
To test the interaction with Ran, GST-Scyl1 (20 μg; 178 pmol) bound to GT-Sepharose was incubated with (Figure 8B, lanes 3–5) or without an excess of mature yeast tRNA (70 μM) (lanes 7–9) and a twofold molar excess of Ran (9 μg; 356 pmol) loaded with GTP (lanes 3 and 7), GDP (lanes 4 and 8) or the nonhydrolyzable GTP analogue GppNHp (lanes 5 and 9). The same amount of Ran was also incubated with GST bound to GT-Sepharose (lane 6). The resins were washed and eluted by boiling in 1× LDS sample buffer. The eluates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blot analysis was performed to detect Ran (top row) and GST-Scyl1 (middle row). An interaction was observed with Ran (top row) loaded with GppNHp (lanes 5 and 9), but not with Ran loaded with GTP (lanes 3 and 7) or GDP (lanes 4 and 8). Furthermore, a negligible amount of Ran was found to interact with GST alone (lane 6). The interaction between Scyl1 and Ran was not dependent on tRNA, because the same amount of RanGppNHp was detected in the presence (lane 5) or absence (lane 9) of tRNA. Coomassie Blue staining of the blot detected GST-Scyl1 (bottom row); however, the amount of Ran present was too low to be detected (data not shown), indicating that the interaction between Scyl1 and Ran is very weak. The RanQ69L mutant protein, defective in hydrolysis of GTP, also was found to interact much more strongly with Scyl1 than wild-type Ran loaded with GTP or GDP (data not shown). This suggests that the lack of interaction between Scyl1 and Ran in the GTP form is likely due to Ran hydrolyzing the GTP and dissociating from the protein. The preference for the GTP bound form of Ran suggests that the interaction between Scyl1 and Ran is specific. Furthermore, the data suggest that Scyl1, Xpo-t and RanGTP may be part of a complex in vivo. The finding that Scyl1 interacts with Ran is interesting, as Scyl1 does not possess a typical Ran-binding domain motif found in known Ran-binding proteins. Although the RanGTPase Gsp1p was shown to copurify with Cex1p (McGuire and Mangroo, 2007), it is not known whether they interact directly with each other.
Scyl1 Forms a Complex with Xpo-t-tRNA-RanGTP In Vitro
Ran bound to a β-karyopherin is not accessible to RanGAP activation of its GTPase activity in vitro (Lounsbury and Macara, 1997; Hellmuth et al., 1998; Kutay et al., 1998). This enzymatic activity-based assay has been used to show that Xpo-t binds tRNA in a RanGTP-dependent manner (Kutay et al., 1998). To verify that Xpo-t interacts with Ran, protein binding studies were conducted in vitro (Figure 9). GST-Xpo-t (20 μg; 158 pmol) bound to GT-Sepharose was incubated with (lanes 3 and 4) or without (lanes 5 and 6) 6 μM yeast mature tRNA and with Ran (8 μg, 316 pmol) in the GDP (lanes 3 and 5) or GppNHp (lanes 4 and 6) form. The washed resins were boiled in 1× LDS buffer to elute bound proteins, and Western blot analysis was used to detect GST-Xpo-t (top row) and Ran (bottom row). RanGDP did not interact with Xpo-t in the presence (lane 3) or absence (lane 5) of tRNA. However, an interaction was detected between Xpo-t and RanGppNHp in the presence (lane 4) or absence (lane 6) of tRNA. Although the interaction between Xpo-t and Ran was not dependent on tRNA, the two proteins seem to be interacting specifically as binding was dependent on the GTP bound form of Ran.
Figure 9.
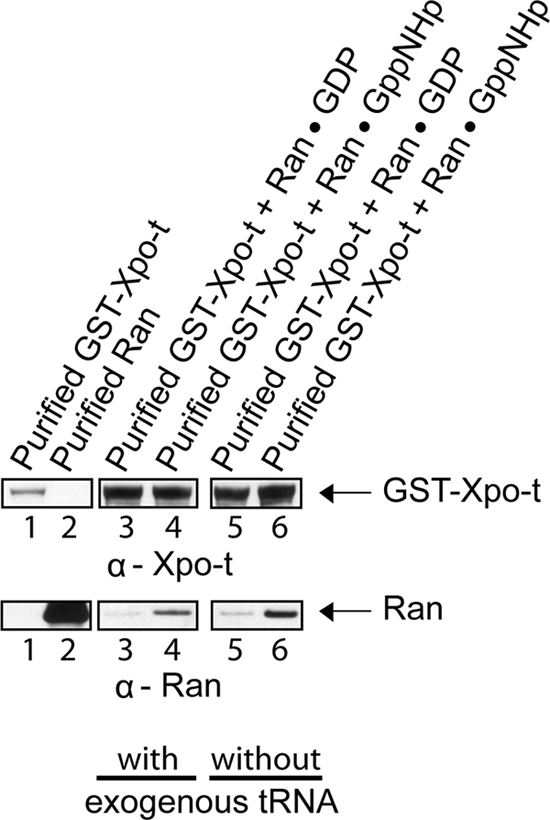
Xpo-t interacts directly with RanGTP but not RanGDP in a tRNA-independent manner. GST-Xpo-t (20 μg; 158 pmol) bound to GT-Sepharose in the presence (lanes 3 and 4) or absence (lanes 5 and 6) of an excess of yeast mature tRNA (6 μM) was incubated with Ran (8 μg; 316 pmol) in the GDP (lanes 3 and 5) or RanGppNHp (lanes 4 and 6) form. The resins were washed and boiled in sample buffer to elute bound proteins. GST-Xpo-t (lane 1), Ran (lane 2), and the eluates were subjected to SDS-PAGE, followed by Western blot analyses to detect GST-Xpo-t (top row) and Ran (bottom row).
Previous studies have shown that Xpo-t interacts with RanGTP only in the presence of tRNA (Kutay et al., 1998). The affinity of Xpo-t for tRNA in the absence of RanGTP is reported to be 0.6 μM (Kutay et al., 1998). Therefore, an explanation why we were not able to observe a tRNA-dependent interaction between Xpo-t and RanGTP is that GST-Xpo-t purified from E. coli by using GT-Sepharose is loaded with endogenous E. coli tRNA. To test this possibility, 500 μg of GST-Xpo-t purified from E. coli by affinity chromatography using GT-Sepharose was subjected to RNA extraction. Analysis of the extract by electrophoresis on an agarose gel indicated that tRNA was bound to the protein (data not shown). To remove tRNA from purified GST-Xpo-t, we used ion-exchange chromatography or ammonium sulfate precipitation (Kutay et al., 1998) after purification by GT-Sepharose chromatography. These treatments removed the tRNA from GST-Xpo-t; however, the protein bound Ran in the GTP and GDP forms irrespective of the absence or presence of tRNA (data not shown). Furthermore, purified GST-Xpo-t treated with ribonuclease A bound RanGTP but not RanGDP, indicating that tRNA is bound to GST-Xpo-t (data not shown). Analysis of the RNA established that GST-Xpo-t remained bound to fragmented tRNA, which was verified by the RanGTPase protection assay discussed below (data not shown).
To understand the significance of the association observed between Scyl1 and Xpo-t and Ran, Xpo-t free of tRNA is required to test whether Scyl1 only interacts with the Xpo-t-RanGTP-tRNA export complex. Thus, to obtain Xpo-t free of E. coli tRNA, Xpo-t tagged at the C terminus with a (His)6 tag was expressed in E. coli and purified by Ni2+-chelation chromatography followed by ammonium sulfate precipitation as described previously (Kutay et al., 1998). The Xpo-t-(His)6 fusion protein was found to be free of tRNA after the ammonium sulfate precipitation. In addition, we tested whether the Xpo-t-(His)6 fusion protein has the ability to bind RanGTP in the presence of tRNA by using the in vitro enzymatic activity assay based on receptor protection of the RanGAP-induced GTPase activity of Ran (Lounsbury and Macara, 1997; Hellmuth et al., 1998; Kutay et al., 1998) (Figure 10). Incubation of RanGTP with RanGAP results in rapid hydrolysis of GTP by Ran, and essentially no RanGTPase activity was detected in the absence of RanGAP. The presence of Xpo-t in the absence of yeast mature tRNA did not block RanGAP-induced Ran catalyzed hydrolysis of GTP. In contrast, the presence of Xpo-t and 6 μM yeast mature tRNA blocked RanGAP-induced GTP hydrolysis by Ran (Kutay et al., 1998). Addition of Scyl1 with Xpo-t and tRNA also resulted in a block of RanGAP activation of the GTPase activity of Ran. These data indicate that Xpo-t subjected to ammonium sulfate precipitation interacted with RanGTP in a tRNA-dependent manner. However, Xpo-t-(His)6 could not be used to test whether Xpo-t interacts with RanGTP in a tRNA-dependent manner in vitro, as Xpo-t bound to Ni2+-Sepharose was released during incubation with tRNA and/or RanGTP. Thus, for the in vitro protein binding analyses described below we used affinity purified GST-Xpo-t complexed to endogenous E. coli tRNA. In addition, these studies were supplemented with or without exogenous yeast mature tRNA to verify that GST-Xpo-t retained the tRNA during purification.
Figure 10.

Xpo-t blockage of RanGAP stimulation of the GTPase activity of Ran is dependent on the presence of tRNA and is not alleviated by the inclusion of Scyl1. Ran-[γ-32P]GTP (1 μM) was incubated with (•) or without (▴) 0.5 nM RanGAP. Ran also was incubated with RanGAP in the presence of 2 μM Xpo-t with (▾) or without (■) 6 μM yeast mature tRNA, or with 2 μM Xpo-t, 2 μM Scyl1, and 6 μM yeast mature tRNA (♦). At the specified times, hydrolysis of Ran-bound GTP was determined by counting released [32P]phosphate by using the charcoal method.
To ascertain whether Xpo-t loaded with tRNA, Ran and Scyl1 form a complex in vitro, GST-Xpo-t (20 μg; 158 pmol) bound to GT-Sepharose was incubated with (Figure 11, lanes 4 and 5) or without (lanes 6 and 7) yeast mature tRNA and with Ran (4 μg, 158 pmol) loaded with GDP (lanes 4 and 6) or GppNHp (lanes 5 and 7). The resins were washed and incubated with Scyl1 (14 μg; 158 pmol). The same amounts of Ran and Scyl1 were incubated with bound GST (lane 8). After the resins were washed the bound proteins were eluted by boiling in 1× LDS buffer, and Western blot analyses were performed to detect GST-Xpo-t (first row), Ran (second row), and Scyl1 (third row). Scyl1 bound to Xpo-t in the presence (lanes 4 and 5) or absence (lanes 6 and 7) of exogenous tRNA, and a negligible amount of Scyl1 was detected in the eluate from the bound GST resin (lane 8). The protein also interacted with Xpo-t in the presence of Ran loaded with GDP (lanes 4 and 6) or GppNHp (lanes 5 and 7). However, Scyl1 interacted better with Xpo-t in the presence of RanGppNHp (compare lanes 4 with 5 and lanes 6 with 7) than in the presence of RanGDP. The ratio of the intensities of Scyl1 to Xpo-t, quantified by densitometry, indicated that the amount of Scyl1 bound to Xpo-t in the presence of RanGppNHp was approximately threefold higher compared with that bound to Xpo-t in the presence of RanGDP. RanGppNHp (lanes 5 and 7) but not RanGDP (lanes 4 and 6) was found in the complex irrespective of whether exogenous tRNA is present (lane 5) or absent (lane 7), which is consistent with studies published (Kutay et al., 1998), and the finding that GST-Xpo-t purified from E. coli contains tRNA. Ran also interacted with GST, but densitometric analyses of the blots indicated that the amount bound is ∼5% of that found with Xpo-t and Scyl1 (compare lanes 8 with 7 and 5). Thus, the finding that Ran loaded with GDP did not form a complex with Xpo-t containing tRNA and Scyl1 indicated that the complex formed between Ran loaded with GppNHp, Xpo-t containing tRNA and Scyl1 was not due to Ran interacting with the GST portion of the GST-Xpo-t fusion protein. To verify that the complex contains tRNA, the complex formed in the absence of yeast mature tRNA was eluted using glutathione to release GST-Xpo-t from the resin and analyzed for tRNA. The result of this analysis indicated the presence of tRNA in the complex (data not shown). Together, the data show that Xpo-t bound to endogenous E. coli tRNA, Scyl1 and RanGTP form a quaternary complex in vitro. Although it was not possible to test whether Scyl1 is capable of forming a complex with Xpo-t and Ran in the absence of tRNA, the in vitro protein binding data suggest that Scyl1 probably interacts with the Xpo-t-tRNA-RanGTP ternary complex upon reaching the cytoplasmic side of the NPC in vivo.
Figure 11.

Scyl1, Xpo-t containing endogenous E. coli tRNA and RanGTP form a complex. GST-Xpo-t (20 μg; 158 pmol) bound to GT-Sepharose was incubated in the presence (lanes 4 and 5) or absence (lanes 6 and 7) of an excess of exogenous yeast mature tRNA (6 μM) and Ran (4 μg; 158 pmol) in the GDP (lanes 4 and 6) or GppNHp (lanes 5 and 7) bound form. The resins were washed and incubated with Scyl1 (14 μg; 158 pmol). The same amount of Ran and Scyl1 was incubated with bound GST (lane 8). The resins were washed and boiled in sample buffer to elute bound proteins. GST-Xpo-t (lane 1), Ran (lane 2), Scyl1 (lane 3) and the eluates were subjected to SDS-PAGE, followed by Western blot analyses to detect GST-Xpo-t (first row), Ran (second row), and Scyl1 (third row). The proteins also were detected directly by SYPRO Ruby staining of the blot (fourth row).
Aminoacylated tRNAs Are Found in the Nucleus of HeLa Cells
tRNAs can be exported out of the nucleus of X. laevis and S. cerevisiae in the aminoacylated (Lund and Dahlberg, 1998; Sarkar et al., 1999; Grosshans et al., 2000a; Azad et al., 2001; Steiner-Mosonyi and Mangroo, 2004) or nonaminoacylated form (Arts et al., 1998b; Azad et al., 2001). However, it has been suggested that tRNAs are primarily exported out of the nucleus in the aminoacylated form in S. cerevisiae (Steiner-Mosonyi and Mangroo, 2004). For mammalian cells, it is not known whether the nuclear tRNA aminoacylation-dependent pathway is principally used to export tRNAs. This information is necessary to understand the role of Scyl1 in nuclear tRNA export. To determine whether the nuclear aminoacylation-dependent pathway is mainly used to export tRNAs from the nucleus in mammalian cells, a biochemical approach capable of separating the aminoacylated and nonaminoacylated forms of tRNAs was used to test whether aminoacyl-tRNAs are present in the nucleus of HeLa cells (Varshney et al., 1991; Steiner-Mosonyi et al., 2003; Steiner-Mosonyi and Mangroo, 2004). The ester bond linking the amino acid to the tRNA was preserved during isolation of nuclei and extraction of total nuclear RNA by using acidic conditions. We have shown that nuclei isolated by the fractionation procedure used are intact and essentially free of cytosolic contaminants (Figure 1C).
To assess the aminoacylation status of nuclear tRNA at steady state, total nuclear RNA was isolated from nuclei prepared from HeLa cells grown to 60–70% confluence, and then separated by PAGE under acidic conditions to prevent deacylation of the tRNAs; this electrophoretic system has been used extensively to monitor the aminoacylation status of tRNAs in yeast, mammalian cells, and bacteria (Mangroo and RajBhandary, 1995; Drabkin and RajBhandary, 1998; Steiner-Mosonyi et al., 2003; Kohrer et al., 2003; Steiner-Mosonyi and Mangroo, 2004). The aminoacylated and nonaminoacylated forms of specific tRNA isoacceptors were detected by Northern blot analysis. Total nuclear and cytosolic RNA treated with base to cleave the ester bond linking the amino acid and tRNA served as the source of deacylated tRNA markers (Figure 12, lane 1). The analyses indicated that cytosolic tRNALys (A) and nuclear tRNALys, tRNAAsn, tRNAGly, tRNATyr and tRNAVal (B) that were not treated with base (lane 2) migrated more slowly than the deacylated tRNAs (lane 1). The data show that the tRNAs are present mainly in the aminoacylated form (90–98%) in the nucleus, and suggest that these tRNAs are exported from the nucleus in the aminoacylated form. Moreover, these findings suggest that under normal conditions the nuclear aminoacylation-dependent pathway may be primarily used to export tRNAs in mammalian cells. Thus, it is possible that Scyl1 is a cytoplasmic component of this pathway.
Figure 12.
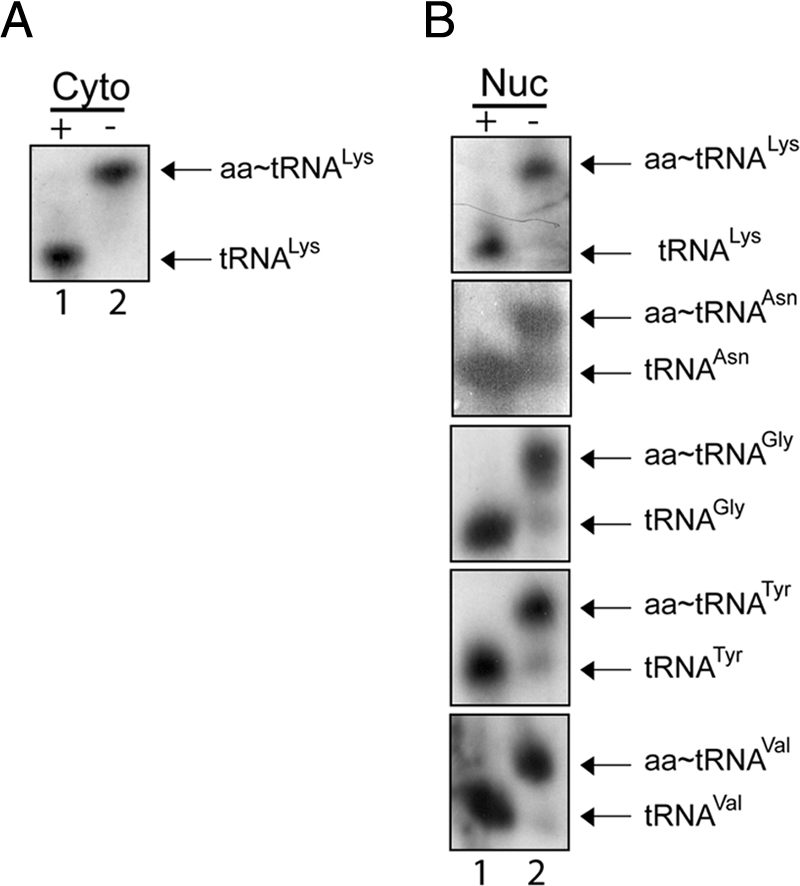
Aminoacylated tRNAs are present in the nucleus of HeLa cells. Total RNA was isolated from nuclear and cytosolic fractions prepared from HeLa cells under acidic conditions. RNA treated with (+) or without (−) base was separated on a 6.5% acid-urea polyacrylamide gel. Northern blot analysis was performed to monitor the aminoacylation status of tRNALys in the cytoplasmic (A) and various tRNA isoacceptors in the nuclear (B) fractions using 32P-labeled oligonucleotide probes.
eEF-1A Copurifies with Scyl1 from Cell Extract but They Do Not Interact Directly In Vitro
To test further whether Scyl1 may be involved in the nuclear aminoacylation-dependent tRNA export pathway, we investigated whether eEF-1A copurifies with Scyl1, because eEF-1A transports aminoacylated tRNAs to the ribosomes for use in protein synthesis. HeLa cell lysate was incubated with α-Scyl1-Sepharose or Sepharose, and the resins were washed to remove unbound proteins. Bound proteins were released and subjected to Western blot analyses to detect eEF-1A (Figure 13A, top row) and Scyl1 (bottom row). eEF-1A (lane 1) and Scyl1 (lane 1) were detected in the cell lysate, but not in the eluate from the resin alone (lane 2). However, Scyl1 (lane 3) and a small amount of eEF-1A (lane 3) were observed in the eluate from the α-Scyl1 resin, suggesting that Scyl1 associates with eEF-1A in vivo.
Figure 13.
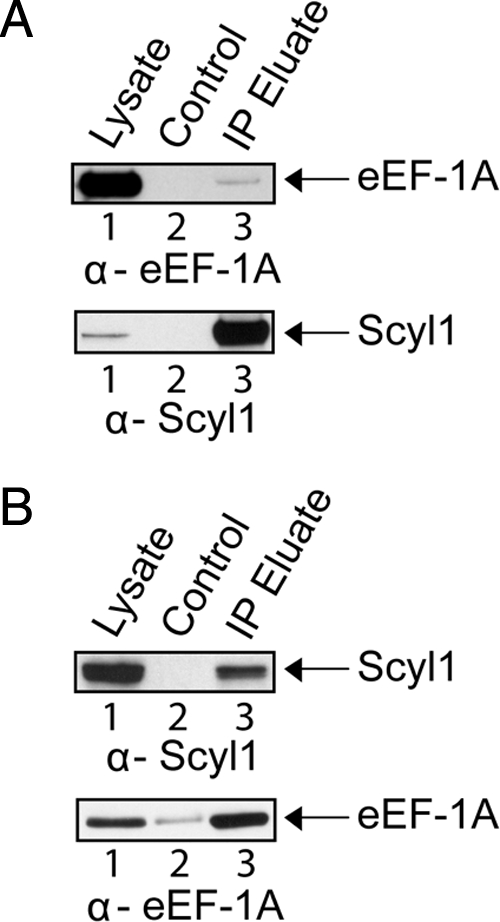
eEF-1A and Scyl1 copurify with each other. (A) eEF-1A copurifies with Scyl1. HeLa cell lysates (lane 1) were incubated with Sepharose (lane 2) or α-Scyl1-Sepharose (lane 3). The resins were washed and boiled in sample buffer to elute bound proteins. Western blot analysis was used to detect eEF-1A (first row) and Scyl1 (second row). (B) Scyl1 copurifies with eEF-1A. HeLa cell lysates (lane 1) were incubated with Sepharose (lane 2) or α-eEF-1A-Sepharose (lane 3). The resins were washed and boiled in sample buffer to elute bound proteins. Western blot analysis was used to detect Scyl1 (first row) and eEF-1A (second row).
To test whether Scyl1 copurifies with eEF-1A, cell lysate from HeLa cells was incubated with α-eEF-1A-Sepharose or Sepharose, and the resins were washed to remove unbound proteins. Bound proteins were eluted and subjected to Western blot analysis to detect Scyl1 (Figure 13B, top row) and eEF-1A (bottom row). Scyl1 (top) and eEF-1A (bottom) were detected in the cell lysate (lane 1), and a negligible amount of eEF-1A was detected in the eluate from the Sepharose resin control (lane 2). However, both Scyl1 (lane 3) and eEF-1A (lane 3) were detected in the eluate from the α-eEF-1A-Sepharose resin. Together, the data indicate that the association of Scyl1 with eEF-1A in vivo is authentic. However, in vitro protein binding studies show that eEF-1A does not interact directly with Scyl1 (Supplemental Figure S4). It is possible that the interaction detected between Scyl1 and eEF-1A in vivo is mediated by another protein.
miRNA-mediated Knockdown of Scyl1 Did Not Affect Nuclear tRNA Export in HeLa Cells
Previous studies have shown that knockdown of Scyl1 by using miRNA resulted in a block in retrograde protein transport from the Golgi to the ER (Burman et al., 2008). Thus, miRNA-mediated knockdown of Scyl1 was used to ascertain the importance of Scyl1 function in nuclear tRNA export in mammalian cells (Figure 14). HeLa cells were transfected with a plasmid carrying the red fluorescent protein (RFP) gene and a control miRNA or the Scyl1 miRNA gene, and Western blot analysis (A) and immunofluorescence microscopy (B) were used to monitor the level of Scyl1. The RFP served as a marker to identify cells transfected with the miRNA containing plasmid. The level of Scyl1 in cell lysate prepared from cells transfected with the Scyl1 miRNA gene (A, top row, lane 2) was significantly lower than that observed in cell lysate prepared from cells transfected with the control miRNA gene (lane 1). The amount of actin in cell extract prepared from cells transfected with the control miRNA gene was lower (A, bottom, lane 1) compared with that in cell extract prepared from cells transfected with the Scyl1 miRNA gene (lane 2). The ratio of the intensities of Scyl1 to actin, determined by densitometric analyses of the blots, indicated that the amount of Scyl1 in cell extract prepared from cells transfected with the Scyl1 miRNA gene was ∼20% of that in cell extract prepared from cells transfected with the control miRNA gene. The 80% reduction in the level of Scyl1 observed by Western analysis is an underestimation, because the efficiency of transfection with the plasmid carrying the Scyl1 miRNA gene is ∼45–50%. Fluorescence microscopy shows that the level of Scyl1 in all cells transfected with the Scyl1 miRNA gene (T) (B, bottom row) was substantially reduced compared with that in cells that were not transfected with the Scyl1 miRNA gene (NT), and in cells transfected with the control miRNA gene (T) (top row). Knockdown of Scyl1 expression was very efficient, because fluorescence intensity analyses indicate that the level of Scyl1 in cells transfected with the Scyl1 miRNA gene is 7–10% of that in cells transfected with the control miRNA gene.
Figure 14.

Knockdown of Scyl1 expression does not affect nuclear tRNA export. (A) Scyl1 levels are reduced upon expression of a Scyl1 miRNA. HeLa cells were transfected with either the control or Scyl1 miRNA expression plasmid. Cell lysates were prepared 72 h after transfection and Western blot analysis was performed to monitor Scyl1 (top row) and actin (bottom) levels. (B) A reduction in Scyl1 levels can be detected in vivo. HeLa cells were transfected with either a control (top) or Scyl1 (bottom) miRNA expression plasmid. After 72 h, the cells were fixed and the levels of Scyl1 (left column) were monitored by immunofluorescence microscopy. Transfected cells were identified by expression of RFP (middle column), and the efficiency of transfection was determined by analyzing 100 cells from three independent transfections. Twenty-five transfected cells in two independent transfections were analyzed for the level of Scyl1 by measuring the fluorescence intensity by using the MetaMorph software. (C) Decreased Scyl1 expression did not affect the nuclear-cytoplasmic distribution of tRNA. HeLa cells were transfected with either a control or Scyl1 miRNA expression plasmid. The cellular location of tRNATyr in 25 transfected cells was detected using an oligonucleotide probe conjugated to fluorescein. Cells expressing the control (first row) and Scyl1 (second and third rows) miRNAs were visualized by confocal laser scanning microscopy. T, transfected cells; NT, untransfected cells. Bars, 10 μm.
FISH was performed to monitor the effect of decreasing the level of Scyl1 on nuclear tRNA transport. HeLa cells were transfected with the miRNA expression plasmid containing the control or Scyl1 miRNA gene. tRNATyr in cells expressing RFP was visualized using an oligonucleotide probe complementary to the mature tRNA sequence that is conjugated to fluorescein (Figure 14C). Cells expressing the control (T) or the Scyl1 miRNA (T) were found to have a similar tRNATyr localization pattern (C, compare top and bottom). In addition, the tRNATyr nuclear-cytoplasmic distribution in cells that were not transfected with the control (NT) or the Scyl1 (NT) miRNA gene was comparable with that in cells transfected (T) with the control or Scyl1 miRNA gene. These data indicate that depletion of Scyl1 had no noticeable effect on nuclear tRNA export and suggest that the function of Scyl1, like the S. cerevisiae Cex1p, in the mammalian nuclear tRNA export process is not absolutely required.
DISCUSSION
A yeast tRNA three-hybrid interaction screen combined with an in vivo nuclear tRNA export assay based on amber suppression has been used previously to identify components of the S. cerevisiae nuclear tRNA export machinery. This strategy resulted in the identification of Cex1p, a redundant cytoplasmic factor of the nuclear aminoacylation-dependent tRNA export pathway (Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007). Cex1p, an uncharacterized gene found in all eukaryotic genomes sequenced to date, mammalian Scyl1 and Drosophila YATA constitute a protein family with a characteristic N-terminal protein kinase-like domain (Supplemental Figure S2). Both Scyl1 and YATA have been shown to participate in intracellular protein trafficking (Burman et al., 2008; Sone et al., 2009). However, based on the function of Cex1p we surmise that members of the protein kinase-like domain family in higher eukaryotes also may be involved in nuclear tRNA export. To test this possibility, we assess whether Scyl1 plays a role in nuclear tRNA export in HeLa cells. The results of this investigation show that Scyl1 is also a cytoplasmic component of the mammalian nuclear tRNA export machinery that plays a redundant role in the export process. Thus, it is possible that orthologues of Scyl1 in other eukaryotes also are involved in nuclear tRNA export.
Studies conducted in X. laevis and S. cerevisiae have shown that tRNAs can be exported from the nucleus in the aminoacylated or nonaminoacylated form (Lund and Dahlberg, 1998; Arts et al., 1998b; Sarkar et al., 1999; Grosshans et al., 2000a; Azad et al., 2001; Steiner-Mosonyi and Mangroo, 2004). However, genetic and biochemical analyses established that the nuclear aminoacylation-dependent pathway is mainly responsible for nuclear tRNA export in S. cerevisiae (Steiner-Mosonyi and Mangroo, 2004). Xpo-t, which is the main tRNA export receptor in mammalian cells, has been shown to have the capacity to export both forms of tRNA (Arts et al., 1998b). Xpo-5 also binds aminoacylated and nonaminoacylated tRNAs, but it has a minor role in nuclear tRNA export (Bohnsack et al., 2002; Calado et al., 2002); Xpo-5 is mainly responsible for nuclear export of pre-microRNA to the cytoplasm for maturation and regulation of gene expression (Lund et al., 2003). Therefore, in contrast to S. cerevisiae, it is unclear which pathway is primarily used for nuclear tRNA export in mammalian cells.
To ascertain whether the nuclear aminoacylation-dependent or -independent pathway is mainly used to export tRNAs in mammalian cells, the aminoacylation status of nuclear tRNAs was evaluated using an electrophoretic method to monitor the state of aminoacylation of endogenous tRNA extracted from HeLa nuclei. This strategy was used previously to show that the major tRNA species of at least 19 families were aminoacylated in the nucleus of S. cerevisiae (Steiner-Mosonyi and Mangroo, 2004). We found that all of the tRNA isoacceptors studied were present primarily in the aminoacylated form in nuclei obtained from HeLa cells (Figure 12). This finding suggests that the nuclear aminoacylation-dependent tRNA export pathway is principally responsible for nuclear export of tRNA in mammalian cells. This inference is consistent with the finding that a number of aminoacyl-tRNA synthetases are found in the nucleus (Nathanson and Deutscher, 2000). Like in S. cerevisiae nuclear tRNA aminoacylation may occur in the nucleolus, because methionyl-tRNA synthetase has been localized to this compartment (Ko et al., 2000). It is, therefore, possible that this pathway is the major one used for nuclear export of tRNA in other eukaryotes. More importantly, the data suggest that Scyl1 most likely participates in the nuclear aminoacylation-dependent tRNA export pathway. This supposition is consistent with the finding that eEF-1A and Scyl1 copurify with each other (Figure 13), and with genetic and biochemical results showing that Cex1p is a component of the nuclear aminoacylation-dependent tRNA export pathway in S. cerevisiae (McGuire and Mangroo, 2007). The data, however, do not exclude the possibility that Scyl1 also may participate in the nuclear aminoacylation-independent tRNA export pathway.
Scyl1 is a cytosolic protein that has been shown to be associated with ER–Golgi intermediate compartments and cis-Golgi (Liu et al., 2000; Burman et al., 2008). We also established that Scyl1 is primarily located in the cytoplasm (Figure 4). Nevertheless, colocalization studies showed that a small percentage of the protein is found at the NPC (Figure 4 and Supplemental Figure S3). This property of Scyl1 is specific, because it was not detected at the ER by colocalization analysis with the ER marker calnexin (Figure 4). Scyl1 also was found to associate with Nup98 in vivo by copurification analyses (Figure 5). Nup98 is the orthologue of the S. cerevisiae Nup116p, which has been shown to interact directly and specifically with Cex1p at the NPC, and to be involved in nuclear tRNA export (Sarkar and Hopper, 1998; McGuire and Mangroo, 2007). Like Nup116p, Nup98 is located on the nucleoplasmic and cytoplasmic sides of the NPC (Griffis et al., 2003). Furthermore, Nup98 on the cytoplasmic side of the NPC has been shown to form a subcomplex with Nup214, Nup62, and Nup88 (Griffis et al., 2003). Nup98 also has been shown to be part of an independent subcomplex by interacting with Nup107 on the cytoplasmic and nuclear sides of the NPC. Nup107 but not Nup62, Nup214, or Nup88 copurified with Scyl1 (Figure 5; data not shown). These data indicate that Scyl1 is associating with a specific Nup subcomplex consisting of Nup98 and Nup107, and they suggest that Scyl1 facilitates nuclear tRNA export by acting at the cytoplasmic side of the NPC. The association of Scyl1 with the NPC is, in part, mediated by Scyl1 binding directly to Nup98, because the two proteins were found to interact directly by in vitro protein binding studies (Figure 5), and knockdown of Nup98 expression resulted in a significant decrease in the level of Scyl1 associated with the NPC (Figure 6C). These results combined with the finding that Scyl1 associates with the tRNA export receptors Xpo-t and Xpo-5, and Ran, but not Crm1 in vivo (Figure 7) suggest that Scyl1 locates to the NPC to participate in a step after the nuclear export receptor–tRNA–RanGTP complex has reached the cytoplasmic side of the NPC. This conclusion is further supported by data showing that Scyl1, Xpo-t containing endogenous E. coli tRNA and RanGTP, but not RanGDP, form a complex in vitro (Figure 11). Although the function of Scyl1 is not understood, the finding that it interacts directly with tRNA and Xpo-t in vitro strongly suggests that Scyl1 interacts with the export receptors at the NPC to collect aminoacyl-tRNAs. Cex1p also has been proposed to have a similar function in S. cerevisiae, because it copurifies with Gsp1p and binds tRNA and Los1p directly in vitro (McGuire and Mangroo, 2007). However, further studies are required to understand fully how Scyl1 and Cex1p retrieve the tRNA cargo from the export receptors.
The GTPase Ran/Gsp1p plays an essential role in both nuclear import and export processes facilitated by β-karyopherins. For nuclear export, interaction of the export receptor with the GTP bound form of Ran/Gsp1p facilitates binding of the cargo to the receptor. The resulting ternary complex moves across the NPC and once in the cytoplasm, RanBP1/Yrb1p binds Ran/Gsp1p and the GTPase activity of Ran/Gsp1p is activated by the RanGTPase activating protein RanGAP/Rna1p. Hydrolysis of GTP to GDP by Ran/Gsp1p facilitates dissociation of the receptor-cargo-Ran/Gsp1p complex (for reviews, see Gorlich and Kutay, 1999; Rodriguez et al., 2004). Xpo-t, Xpo-5, Msn5p, and Los1p have been shown to interact with RanGTP/Gsp1pGTP in a tRNA-dependent manner in vitro (Hellmuth et al., 1998; Kutay et al., 1998; Arts et al., 1998a; Bohnsack et al., 2002; Calado et al., 2002; Eswara et al., 2009; Figure 10). This finding led to the suggestion that loading of Xpo-t, Xpo-5, Msn5p, and Los1p with tRNA is dependent on RanGTP/Gsp1pGTP in vivo. Moreover, in vitro and in vivo studies in S. cerevisiae and X. laevis suggest that unloading of the tRNA cargo from Los1p and Xpo-t after translocation across the NPC requires GTP hydrolysis by Gsp1p/Ran (Izaurralde et al., 1997; Sarkar and Hopper, 1998). Ran also copurified with Scyl1 by immunoprecipitation with α-Scyl1 (Figure 7). Furthermore, immunoprecipitation resulted in coisolation of Scyl1 and Xpo-t or Xpo-5 (Figure 7). Scyl1 also interacted directly with Xpo-t and Ran in vitro (Figure 8). The interaction observed between Ran and Scyl1 is dependent on GTP but not GDP. Moreover, Scyl1, Xpo-t containing endogenous E. coli tRNA and RanGTP form a quaternary complex in vitro (Figure 11). These data combined with finding that Scyl1 associates with the NPC suggest that in vivo Scyl1 interacts with the Xpo-t–tRNA–RanGTP complex at the NPC before RanBP1 interaction with Ran and activation of its GTPase activity by RanGAP. This interpretation is consistent with the finding that in vitro Scyl1 and Xpo-t containing tRNA did not form a complex with RanGDP (Figure 8B), and with the data showing that the presence of Scyl1 did not affect Xpo-t protection of RanGTP from RanGAP in the presence of tRNA (Figure 10). It is therefore likely that Scyl1 retrieves the tRNA from the export receptor once Ran hydrolyzes GTP to GDP. Similarly, Cex1p has been proposed to collect aminoacyl-tRNAs from the export receptor after GTP hydrolysis by Gsp1p (McGuire and Mangroo, 2007). However, a detailed investigation is required to determine whether the GTPase activity of Ran is necessary to facilitate unloading of aminoacyl-tRNAs from the nuclear tRNA export receptors by Scyl1 and to understand the significance of the interaction between Scyl1 and RanGTP. Regardless, the data together clearly demonstrate that Scyl1 is directly involved in the nuclear tRNA export process in HeLa cells.
Channeling is a mechanism used to segregate biochemical processes and involves the direct transfer of a substrate from one component to another in a multistep pathway. Studies in S. cerevisiae established that channeling is used in tRNA maturation, delivery of tRNAs from aminoacyl-tRNA synthetases in the nucleolus to the nuclear tRNA export receptors, delivery of cytoplasmic tRNAs to the aminoacyl-tRNA synthetases, and transfer of aminoacyl-tRNAs from aminoacyl-tRNA synthetases to the ribosomes (Simos et al., 1996; Wolin and Matera, 1999; Grosshans et al., 2000a,b; McGuire and Mangroo, 2007). Aminoacyl-tRNAs exiting the nucleus also are channeled to the ribosomes by eEF-1A in S. cerevisiae, because disruption of the genes for Los1p and eEF-1A affected cell growth due to nuclear accumulation of tRNA (Grosshans et al., 2000a). Cex1p is thought to be involved in channeling aminoacyl-tRNAs to eEF-1A in S. cerevisiae (McGuire and Mangroo, 2007). It has been proposed to deliver the tRNAs to eEF-1A after retrieving them from the tRNA export receptor at the cytoplasmic side of the NPC (McGuire and Mangroo, 2007). This is based on the finding that eEF-1A and Cex1p copurify with each other by tandem affinity purification, and on the observation that disruption of the Cex1p and eEF-1A genes affected the efficiency of nuclear tRNA export and cell growth (McGuire and Mangroo, 2007). Although it is not known whether a channeling mechanism is involved in the various aspects of the life cycle of tRNAs in mammalian cells, it seems that Scyl1 may be involved in transferring aminoacyl-tRNAs from the receptors to eEF-1A. This is, in part, based on the finding that eEF-1A and Scyl1 copurify with each other and the detection of aminoacylated tRNAs in nuclei of HeLa cells (Figures 12 and 13). However, Scyl1, like Cex1p, did not interact directly with eEF-1A in vitro (Supplemental Figure S4) (McGuire and Mangroo, 2007). Thus, it is possible that the interaction observed between Scyl1 and eEF-1A in vivo is mediated by an unidentified protein. However, a more detailed analysis is required to test this possibility.
The mammalian Scyl1 and the Drosophila YATA have been shown to participate in intracellular protein trafficking (Burman et al., 2008; Sone et al., 2009). Moreover, loss of the function of Scyl1 and YATA results in neurodegenerative disorders in mice and Drosophila. The results of the present study suggest that Scyl1 is also a cytoplasmic component of the mammalian nuclear tRNA export process. The biochemical characterization suggests that Scyl1 collects aminoacyl-tRNAs from the nuclear tRNA export receptors on the cytoplasmic side of the NPC, probably after Ran hydrolyzes GTP, and channels them to eEF1A with the assistance of an unidentified protein. Finally, this study is the first to show that a protein implicated in the development of neurodegenerative disorders participates in nuclear tRNA export.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. M. G. Coppolino for use of the mammalian cell culture facility and advice on culturing various cell lines and Drs. D. Gorlich, R. Truant, J. P. Capone, U. L. RajBhandary, M. A. Powers, V. Doye, V. Gerke, and P. S. McPherson for their generous gifts of antibodies and plasmids. This work was supported by an operating grant from the Canadian Institutes of Health Research.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-03-0176) on May 26, 2010.
REFERENCES
- Arts G. J., Fornerod M., Mattaj I. W. Identification of a nuclear export receptor for tRNA. Curr. Biol. 1998a;8:305–314. doi: 10.1016/s0960-9822(98)70130-7. [DOI] [PubMed] [Google Scholar]
- Arts G. J., Kuersten S., Romby P., Ehresmann B., Mattaj I. W. The role of exportin-t in selective nuclear export of mature tRNAs. EMBO J. 1998b;17:7430–7441. doi: 10.1093/emboj/17.24.7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad A. K., Stanford D. R., Sarkar S., Hopper A. K. Role of nuclear pools of aminoacyl-tRNA synthetases in tRNA nuclear export. Mol. Biol. Cell. 2001;12:1381–1392. doi: 10.1091/mbc.12.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgareh N., et al. An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J. Cell Biol. 2001;154:1147–1160. doi: 10.1083/jcb.200101081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein K. A., Baserga S. J. The small subunit processome is required for cell cycle progression at G1. Mol. Biol. Cell. 2004;15:5038–5046. doi: 10.1091/mbc.E04-06-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack M. T., Regener K., Schwappach B., Saffrich R., Paraskeva E., Hartmann E., Gorlich D. Exp5 exports eEF1A via tRNA from nuclei and synergizes with other transport pathways to confine translation to the cytoplasm. EMBO J. 2002;21:6205–6215. doi: 10.1093/emboj/cdf613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman J. L., Bourbonniere L., Philie J., Stroh T., Dejgaard S. Y., Presley J. F., McPherson P. S. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J. Biol. Chem. 2008;283:22774–22786. doi: 10.1074/jbc.M801869200. [DOI] [PubMed] [Google Scholar]
- Calado A., Treichel N., Muller E. C., Otto A., Kutay U. Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J. 2002;21:6216–6224. doi: 10.1093/emboj/cdf620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J. D., Mangroo D. Nucleotides of the tRNA D-stem that play an important role in nuclear-tRNA export in Saccharomyces cerevisiae. Biochem. J. 2000;347:115–122. [PMC free article] [PubMed] [Google Scholar]
- Drabkin H. J., RajBhandary U. L. Initiation of protein synthesis in mammalian cells with codons other than AUG and amino acids other than methionine. Mol. Cell. Biol. 1998;18:5140–5147. doi: 10.1128/mcb.18.9.5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina H., Aoki J., Hatta S., Yoshida T., Koyanagi Y. Role of Nup98 in nuclear entry of human immunodeficiency virus type 1 cDNA. Microbes and Infection. 2004;6:715–724. doi: 10.1016/j.micinf.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Englert M., Latz A., Becker D., Gimple O., Beier H., Akama K. Plant pre-tRNA splicing enzymes are targeted to multiple cellular compartments. Biochimie. 2007;89:1351–1365. doi: 10.1016/j.biochi.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Ernens I., Goodfellow S. J., Innes F., Kenneth N. S., Derblay L. E., White R. J., Scott P. H. Hypoxic stress suppresses RNA polymerase III recruitment and tRNA gene transcription in cardiomyocytes. Nucleic Acids Res. 2006;34:286–294. doi: 10.1093/nar/gkj402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswara M.B.K., McGuire A. T., Pierce J. B., Mangroo D. Utp9p Facilitates Msn5p-mediated Nuclear Reexport of Retrograded tRNAs in Saccharomyces cerevisiae. Mol. Biol. Cell. 2009;20:5007–5025. doi: 10.1091/mbc.E09-06-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavidel A., Kislinger T., Pogoutse O., Sopko R., Jurisica I., Emili A. Impaired tRNA nuclear export links DNA damage and cell-cycle checkpoint. Cell. 2007;131:915–926. doi: 10.1016/j.cell.2007.09.042. [DOI] [PubMed] [Google Scholar]
- Gite S., RajBhandary U. L. Lysine 207 as the site of cross-linking between the 3′-end of Escherichia coli initiator tRNA and methionyl-tRNA formyltransferase. J. Biol. Chem. 1997;272:5305–5312. doi: 10.1074/jbc.272.8.5305. [DOI] [PubMed] [Google Scholar]
- Gorlich D., Kutay U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- Griffis E. R., Xu S. L., Powers M. A. Nup98 localizes to both nuclear and cytoplasmic sides of the nuclear pore and binds to two distinct nucleoporin subcomplexes. Mol. Biol. Cell. 2003;14:600–610. doi: 10.1091/mbc.E02-09-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H., Hurt E., Simos G. An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev. 2000a;14:830–840. [PMC free article] [PubMed] [Google Scholar]
- Grosshans H., Simos G., Hurt E. Review: transport of tRNA out of the nucleus-direct channeling to the ribosome? J. Struct. Biol. 2000b;129:288–294. doi: 10.1006/jsbi.2000.4226. [DOI] [PubMed] [Google Scholar]
- Haberland J., Gerke V. Conserved charged residues in the leucine-rich repeat domain of the Ran GTPase activating protein are required for Ran binding and GTPase activation. Biochem. J. 1999;343:653–662. [PMC free article] [PubMed] [Google Scholar]
- Hellmuth K., Lau D. M., Bischoff F. R., Kunzler M., Hurt E., Simos G. Yeast Los1p has properties of an exportin-like nucleocytoplasmic transport factor for tRNA. Mol. Cell. Biol. 1998;18:6374–6386. doi: 10.1128/mcb.18.11.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper A. K., Phizicky E. M. tRNA transfers to the limelight. Genes Dev. 2003;17:162–180. doi: 10.1101/gad.1049103. [DOI] [PubMed] [Google Scholar]
- Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hunter C. A., Aukerman M. J., Sun H., Fokina M., Poethig R. S. PAUSED encodes the Arabidopsis exportin-t ortholog. Plant Physiol. 2003;132:2135–2143. doi: 10.1104/pp.103.023309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurto R. L., Tong A.H.Y., Boone C., Hopper A. K. Inorganic phosphate deprivation causes tRNA nuclear accumulation via retrograde transport in Saccharomyces cerevisiae. Genetics. 2007;176:841–852. doi: 10.1534/genetics.106.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E., Kutay U., von Kobbe C., Mattaj I. W., Gorlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P., Tyers M. How cells coordinate growth and division. Curr. Biol. 2004;14:R1014–R1027. doi: 10.1016/j.cub.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Kato M., Yano K., Morotomi-Yano K., Saito H., Miki Y. Identification and characterization of the human protein kinase-like gene NTKL: mitosis-specific centrosomal localization of an alternatively spliced isoform. Genomics. 2002;79:760–767. doi: 10.1006/geno.2002.6774. [DOI] [PubMed] [Google Scholar]
- Ko Y. G., Kang Y. S., Kim E. K., Park S. G., Kim S. Nucleolar localization of human methionyl-tRNA synthetase and its role in ribosomal RNA synthesis. J. Cell Biol. 2000;149:567–574. doi: 10.1083/jcb.149.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrer C., Yoo J. H., Bennett M., Schaack J., RajBhandary U. L. A possible approach to site-specific insertion of two different unnatural amino acids into proteins in mammalian cells via nonsense suppression. Chem. Biol. 2003;10:1095–1102. doi: 10.1016/j.chembiol.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Kruse C., Willkomm D. K., Grunweller A., Vollbrandt T., Sommer S., Busch S., Pfeiffer T., Brinkmann J., Hartmann R. K., Muller P. K. Export and transport of tRNA are coupled to a multi-protein complex. Biochem. J. 2000;346:107–115. [PMC free article] [PubMed] [Google Scholar]
- Kutay U., Lipowsky G., Izaurralde E., Bischoff F. R., Schwarzmaier P., Hartmann E., Gorlich D. Identification of a tRNA-specific nuclear export receptor. Mol. Cell. 1998;1:359–369. doi: 10.1016/s1097-2765(00)80036-2. [DOI] [PubMed] [Google Scholar]
- Lee D. C., Aitchison J. D. Kap104p-mediated nuclear import. Nuclear localization signals in mRNA-binding proteins and the role of Ran and Rna. J. Biol. Chem. 1999;274:29031–29037. doi: 10.1074/jbc.274.41.29031. [DOI] [PubMed] [Google Scholar]
- Liu S.C.H., Lane W. S., Lienhard G. E. Cloning and preliminary characterization of a 105 kDa protein with an N-terminal kinase-like domain. Biochim. Biophys. Acta. 2000;1517:148–152. doi: 10.1016/s0167-4781(00)00234-7. [DOI] [PubMed] [Google Scholar]
- Lounsbury K. M., Macara I. G. Ran-binding protein 1 (RanBP1) forms a ternary complex with Ran and karyopherin beta and reduces Ran GTPase-activating protein (RanGAP) inhibition by karyopherin beta. J. Biol. Chem. 1997;272:551–555. doi: 10.1074/jbc.272.1.551. [DOI] [PubMed] [Google Scholar]
- Lund E., Dahlberg J. E. Proofreading and aminoacylation of tRNAs before export from the nucleus. Science. 1998;282:2082–2085. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- Lund E., Guttinger S., Calado A., Dahlberg J. E., Kutay U. Nuclear export of microRNA precursors. Science. 2003;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- Mangroo D., RajBhandary U. L. Mutants of Escherichia coli initiator tRNA defective in initiation. Effects of overproduction of methionyl-tRNA transformylase and the initiation factors IF2 and IF3. J. Biol. Chem. 1995;270:12203–12209. doi: 10.1074/jbc.270.20.12203. [DOI] [PubMed] [Google Scholar]
- McGuire A. T., Keates R.A.B., Cook S., Mangroo D. Structural modeling identified the tRNA-binding domain of Utp8p, an essential nucleolar component of the nuclear tRNA export machinery of Saccharomyces cerevisiae. Biochem. Cell Biol. 2009;87:431–443. doi: 10.1139/o08-145. [DOI] [PubMed] [Google Scholar]
- McGuire A. T., Mangroo D. Cex1p is a novel cytoplasmic component of the Saccharomyces cerevisiae nuclear tRNA export machinery. EMBO J. 2007;26:288–300. doi: 10.1038/sj.emboj.7601493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson L., Deutscher M. P. Active aminoacyl-tRNA synthetases are present in nuclei as a high molecular weight multienzyme complex. J. Biol. Chem. 2000;275:31559–31562. doi: 10.1074/jbc.C000385200. [DOI] [PubMed] [Google Scholar]
- Paushkin S. V., Patel M., Furia B. S., Peltz S. W., Trotta C. R. Identification of a human endonuclease complex reveals a link between tRNA splicing and Pre-mRNA 3′ end formation. Cell. 2004;117:311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Powers M. A., Forbes D. J., Dahlberg J. E., Lund E. The vertebrate GLFG nucleoporin, Nup98, is an essential component of multiple RNA export pathways. J. Cell Biol. 1997;136:241–250. doi: 10.1083/jcb.136.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. S., Dargemont C., Stutz F. Nuclear export of RNA. Biol. Cell. 2004;96:639–655. doi: 10.1016/j.biolcel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Sarkar S., Azad A. K., Hopper A. K. Nuclear tRNA aminoacylation and its role in nuclear export of endogenous tRNAs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1999;96:14366–14371. doi: 10.1073/pnas.96.25.14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Hopper A. K. tRNA nuclear export in Saccharomyces cerevisiae: in situ hybridization analysis. Mol. Biol. Cell. 1998;9:3041–3055. doi: 10.1091/mbc.9.11.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt W. M., et al. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–697. doi: 10.1038/sj.embor.7401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen H. H., Hopper A. K. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2005;102:11290–11295. doi: 10.1073/pnas.0503836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen H. H., Horetsky R. L., Kimball S. R., Murthi A., Jefferson L. S., Hopper A. K. Retrograde nuclear accumulation of cytoplasmic tRNA in rat hepatoma cells in response to amino acid deprivation. Proc. Natl. Acad. Sci. USA. 2007;104:8845–8850. doi: 10.1073/pnas.0700765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simos G., Segref A., Fasiolo F., Hellmuth K., Shevchenko A., Mann M., Hurt E. C. The yeast protein Arc1p binds to tRNA and functions as a cofactor for the methionyl- and glutamyl-tRNA synthetases. EMBO J. 1996;15:5437–5448. [PMC free article] [PubMed] [Google Scholar]
- Sone M., et al. Loss of yata, a novel gene regulating the subcellular localization of APPL, induces deterioration of neural tissues and lifespan shortening. PLoS ONE. 2009;4:e4466. doi: 10.1371/journal.pone.0004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner-Mosonyi M., Leslie D. M., Dehghani H., Aitchison J. D., Mangroo D. Utp8p is an essential intranuclear component of the nuclear tRNA export machinery of Saccharomyces cerevisiae. J. Biol. Chem. 2003;278:32236–32245. doi: 10.1074/jbc.M302779200. [DOI] [PubMed] [Google Scholar]
- Steiner-Mosonyi M., Mangroo D. The nuclear tRNA aminoacylation-dependent pathway may be the principal route used to export tRNA from the nucleus in Saccharomyces cerevisiae. Biochem. J. 2004;378:809–816. doi: 10.1042/BJ20031306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub B. R., Eswara M.B.K., Pierce J. B., Mangroo D. Utp8p is a nucleolar tRNA-binding protein that forms a complex with components of the nuclear tRNA export machinery in Saccharomyces cerevisiae. Mol. Biol. Cell. 2007;18:3845–3859. doi: 10.1091/mbc.E06-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano A., Endo T., Yoshihisa T. tRNA actively shuttles between the nucleus and cytosol in yeast. Science. 2005;309:140–142. doi: 10.1126/science.1113346. [DOI] [PubMed] [Google Scholar]
- Varshney U., Lee C. P., RajBhandary U. L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- Wang C. C., Schimmel P. Species barrier to RNA recognition overcome with nonspecific RNA binding domains. J. Biol. Chem. 1999;274:16508–16512. doi: 10.1074/jbc.274.23.16508. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerase III transcription—a battleground for tumour suppressors and oncogenes. Eur. J. Cancer. 2004a;40:21–27. doi: 10.1016/j.ejca.2003.09.027. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerase III transcription and cancer. Oncogene. 2004b;23:3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell. Biol. 2005;6:69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- Whitney M. L., Hurto R. L., Shaheen H. H., Hopper A. K. Rapid and reversible nuclear accumulation of cytoplasmic tRNA in response to nutrient availability. Mol. Biol. Cell. 2007;18:2678–2686. doi: 10.1091/mbc.E07-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin S. L., Matera A. G. The trials and travels of tRNA. Genes Dev. 1999;13:1–10. doi: 10.1101/gad.13.1.1. [DOI] [PubMed] [Google Scholar]
- Wozniak R. W., Rout M. P., Aitchison J. D. Karyopherins and kissing cousins. Trends Cell Biol. 1998;8:184–188. doi: 10.1016/s0962-8924(98)01248-3. [DOI] [PubMed] [Google Scholar]
- Yoshihisa T., Ohshima C., Yunoki-Esaki K., Endo T. Cytoplasmic splicing of tRNA in Saccharomyces cerevisiae. Genes Cells. 2007;12:285–297. doi: 10.1111/j.1365-2443.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol. Biol. Cell. 2003;14:3266–3279. doi: 10.1091/mbc.E02-11-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva L., Myers R., Fassati A. tRNAs promote nuclear import of HIV-1 intracellular reverse transcription complexes. PLoS Biol. 2006;4:1689–1706. doi: 10.1371/journal.pbio.0040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.