

Abstract
The Fanconi anaemia (FA) FANCG protein is an integral component of the FA nuclear core complex that is required for monoubiquitylation of FANCD2. FANCG is also part of another protein complex termed D1-D2-G-X3 that contains FANCD2 and the homologous recombination repair proteins BRCA2 (FANCD1) and XRCC3. Formation of the D1-D2-G-X3 complex is mediated by serine-7 phosphorylation of FANCG and occurs independently of the FA core complex and FANCD2 monoubiquitylation. FANCG contains seven tetratricopeptide repeat (TPR) motifs that mediate protein-protein interactions and here we show that mutation of several of the TPR motifs at a conserved consensus residue ablates the in vivo binding activity of FANCG. Expression of mutated TPR1, TPR2, TPR5 and TPR6 in Chinese hamster fancg mutant NM3 fails to functionally complement its hypersensitivities to mitomycin C (MMC) and phleomycin and fails to restore FANCD2 monoubiquitylation. Using co-immunoprecipitation analysis, we demonstrate that these TPR-mutated FANCG proteins fail to interact with BRCA2, XRCC3, FANCA or FANCF. The interactions of other proteins in the D1-D2-G-X3 complex are also absent, including the interaction of BRCA2 with both the monoubiquitylated (FANCD2-L) and non-ubiquitylated (FANCD2-S) isoforms of FANCD2. Interestingly, a mutation of TPR7 (R563E), that complements the MMC and phleomycin hypersensitivity of human FA-G EUFA316 cells, fails to complement NM3, despite the mutated FANCG protein co-precipitating with FANCA, BRCA2 and XRCC3. Whilst interaction of TPR7-mutated FANCG with FANCF does appear to be reduced in NM3, FANCD2 is monoubiquitylated suggesting that sub-optimal interactions of FANCG in the core complex and the D1-D2-G-X3 complex are responsible for the observed MMC- and phleomycin-hypersensitivity, rather than a defect in FANCD2-monoubiquitylation. Our data demonstrates that FANCG functions as a mediator of protein-protein interactions and is vital for the assembly of multi-protein complexes including the FA core complex and the D1-D2-G-X3 complex.
Keywords: Fanconi anemia, FANCG, Tetratricopeptide motif, homologous recombination, radiomimetic, SH3 domain
1. Introduction
The FANCG gene was first cloned as a human cDNA that complemented the mitomycin C (MMC) hypersensitivity of the Chinese hamster mutant cell line UV40 and was therefore originally termed XRCC9 [1-3]. FANCG is one of twelve genes (FANCA, B, C, D1, D2, E, F, G, I, J, L and N), defects in which result in Fanconi anaemia (FA), a syndrome characterised by haematological and developmental defects, an increased incidence of cancer (particularly acute myeloid leukaemia in childhood), and cellular hypersensitivity to interstrand cross-linking agents [4-6]. A further three genes encoding FANCM/FAAP250, FAAP100 and FAAP24 (FAAP for Fanconi anaemia associated protein) related to FA have also been described [7-9].
FANCG is a component of a protein sub-complex comprising six other FA proteins (FANCA, B, C, E, F, and L) and FAAP100, which is recruited to chromatin by the FANCM-FAAP24 heterodimer to form the active FA nuclear core complex [10,11]. FANCG interacts directly with FANCA and FANCF [12,13], mediates the interaction of FANCA-FANCF [14] and is required for in vivo interactions between several of the core complex proteins [15,16]. An intact FA core complex is required for the interdependent monoubiquitylation of the FANCD2 and FANCI proteins and cells lacking FANCG (or other FA core sub-complex proteins) fail to express the monoubiquitylated form of FANCD2 [17-19].
The remaining three FA proteins, encoded by the breast cancer susceptibility genes FANCD1/BRCA2, FANCN/PALB2 and FANCJ/BRIP1, are not required for the monoubiquitylation of FANCD2 [20-22]. BRCA2 and PALB2 (partner and localiser of BRCA2) act together in homologous recombination repair (HRR) and function either downstream of, or in parallel with, FANCD2 monoubiquitylation [23]. Altogether, FANCM-FAAP24, the FA nuclear core complex, FANCD2-FANCI, BRCA2-PALB2 and FANCJ form the spine of a multifaceted response to DNA damage that has become known as the FANC/BRCA pathway or network [6,24-27].
In addition to being a vital component of the FA core complex, FANCG has a direct link with proteins that operate in HRR. It co-localises with RAD51 and BRCA2 in nuclear foci in response to DNA damage [28] and has been shown to directly interact with BRCA2 and the RAD51 paralog XRCC3 [28,29]. We recently demonstrated [30] that phosphorylation of FANCG at its serine-7 residue [31] is required, not only for its own in vivo interactions with BRCA2 and XRCC3, but also for the interaction of BRCA2-FANCD2 and the assembly of a protein complex comprising BRCA2/FANCD1, FANCD2, FANCG and XRCC3 (D1-D2-G-X3). Formation of D1-D2-G-X3 does not require core complex proteins other than FANCG and is independent of the monoubiquitylation of FANCD2.
The requirement of FANCG for mediating protein interactions in both the FA core complex and the D1-D2-G-X3 complex is consistent with the presence of at least seven tetratricopeptide repeat (TPR) motifs located throughout its length [32]. TPRs comprise degenerate 34 amino acid motifs, usually present in tandem arrays of 3-14 motifs that fold into a superhelical structure, forming a scaffold for protein-protein interactions [33,34]. TPR motifs contain 8 consensus residues (–W-LG-Y-A-F-A-P-) with amino acids at 4,7,8 and 11 (W-LG-Y) representing a pouched domain into which a second domain fits (formed by A-F-A at residues 20, 24 and 27). There is considerable sequence diversity within TPR motifs, and it is speculated that there may be specialisation within the motifs of individual proteins that allow binding to different substrates [33,35,36]. FANCG's TPR motifs were identified by comparison of human and fish orthologs [32], and the mammalian (human, hamster and mouse) proteins [2,37,38] show a high degree of conservation (Fig. 1). Human FANCG cDNA with mutations at the conserved 8th position of TPR1, TPR2, TPR5 or TPR6 (glycine to glutamine) fail to complement the MMC-hypersensitivity of lymphoblastoid cell line EUFA316 (FA-G), and the TPR-disrupted proteins do not co-immunoprecipitate with FANCA [32]. Analysis of the same mutants for direct interactions using a yeast two hybrid system showed that FANCG-FANCA interaction was severely reduced by TPR5 and TPR6 mutants; FANCG-FANCF by TPR1, TPR2, TPR5 and TPR6; FANCG-XRCC3 by TPR5 and TPR6; FANCG-BRCA2 N-terminus by TPR5 and TPR6 and FANCG-BRCA2 C-terminus by TPR1 and TPR2 [29]. This differential requirement for the TPR motifs by its binding partners may provide a mechanism by which FANCG could mediate the assembly of multi-protein complexes. For example, as TPR5 and TPR6 are required for the interaction of XRCC3 with FANCG, the remaining motifs could potentially be used to bind the C-terminus of BRCA2, thus bringing together XRCC3 and BRCA2 in the D1-D2-G-X3 complex [29].
Fig. 1.

The tetratricopeptide (TPR) repeat motifs of FANCG/XRCC9. Sequences are shown for human (Homo sapiens), Chinese hamster (Cricetulus griseus) and mouse (Mus musculus), with identical residues shown in black and similar residues shaded in grey. The upper arrow indicates the position of the conserved 8th residue (glycine or alanine) that was changed to glutamine to create the following mutant FANCG proteins: TPR1 (G216Q), TPR2 (G253Q), TPR4 (A401Q), TPR5 (G460Q) and TPR6 (G521Q). As the 8th residue of TPR7 is neither Gly nor Ala, this motif was mutated at another residue that is highly conserved. The lower arrow indicates the position of the arginine (R) residue that is mutated to glutamic acid (E) in TPR7 (R563E).
To test this idea and to further define the requirement of the TPR motifs for the in vivo interactions of FANCG, we transfected the Chinese hamster fancg mutant NM3 with constructs containing human FANCG cDNA and established cell lines expressing mutant forms of TPR1, TPR2, TPR4, TPR5, TPR6 and TPR7. The capacity of the mutated FANCG proteins to mediate interactions in the D1-D2-G-X3 complex were then determined using co-immunoprecipitation analysis. Complementation of the MMC and phleomycin hypersensitivities of NM3, and the ability to restore monoubiquitylation of FANCD2 were used to test the functionality of the TPR mutants.
2. Materials and Methods
2.1. Cell lines and culture conditions
The Chinese hamster ovary (CHO) wild type cell line AA8 and its derived mutant NM3 (which has a frameshift mutation in exon 3 of FancG) have been described previously [17,38]. Derivatives of the human FA-G lymphoblastoid cell line EUFA316 transfected with TPR-mutated human FANCG cDNA in the mammalian expression vector pMEP4 were those previously constructed by Blom et al. [32] and used with the kind permission of Hans Joenje. BD180 is a wild type lymphoblastoid cell line [30]. The FA-D2 cell line PD20, and its human cDNA complemented derivative, were described previously [39]. Fibroblast cell lines (CHO and PD20) were routinely maintained in Dulbecco's modified Eagle's media (D-MEM, Cambrex, Belgium) with ultraglutamine, supplemented with 10% foetal calf serum (Harlan Sera Laboratories), 1% non-essential amino acids, and either 100 units/ml penicillin and 100 mg/ml streptomycin sulphate or 50 μg/ml gentamicin [40]. Trypsinization was performed with 0.12% trypsin and 0.008% EDTA. BD180 and EUFA316 cell lines were grown in RPMI1640 medium supplemented as for D-MEM except 15% FCS was used. All cell lines were grown at 37°C under 5% CO2.
2.2 Transfection of NM3 cells with mutant-TPR FANCG cDNA
Mutant human FANCG cDNAs (TPR1, G216Q; TPR2, G253Q; TPR4, A401Q; TPR5, G460Q; TPR6, G521Q; TPR7, R563E) in the mammalian expression vector pCEP4 (Invitrogen) were generated using standard PCR mutagenesis methods as described previously and sequenced in full-length to confirm the presence of the intended mutation and the absence of other mutations [32]. For transfection, a 75 cm2 flask was seeded with 2 × 106 cells and incubated overnight. Transfection was with 1 μg of vector DNA and 50 μl of lipofectamine (Invitrogen, UK) according to the manufacturers specifications and as previously described [17]. Following overnight incubation, cells were harvested, replated and grown in medium containing 500 μg/ml hygromycin (Sigma-Aldrich, UK) for 10 days to allow the selection of stably transfected cells. Single colonies were isolated and grown to bulk culture in the presence of hygromycin.
2.3 Clonal survival and growth inhibition assays
Functional complementation of the TPR-mutated FANCG constructs in NM3 was determined by performing clonal survival experiments with MMC and phleomycin as previously described [17]. Each survival curve represents the mean of 2-4 experiments and the data were fitted on a semi-log plot for each cell line. For the EUFA316 cell lines, 5 × 105 cells in 2ml of medium were seeded into each well of a 6-well culture dish and phleomycin added at the appropriate doses. Following 96 h incubation, the resulting cell densities in each well were determined using a haemocytometer and normalized to the cell number obtained in untreated wells.
2.4 Immunoblotting, immunoprecipitation and antibodies
Western blotting was performed essentially as described previously [17]. Co-immunoprecipitation was performed using Sigma's (Poole, UK) EZview red protein affinity gel system with the same antibodies for FANCA, FANCF, FANCG, BRCA2 and XRCC3 as previously used [30]. The FANCD2 antibody (ab2187) used for immunoprecipitation and β-actin antibody (ab6276) was from Abcam Limited (Cambridge, UK), whilst anti-FANCD2 (H-300) from Santa Cruz Biotechnology (USA) was used for western blotting [39,41]. Total cell extracts were prepared from 1 to 2 × 107 exponentially growing cells treated with mitomycin C for 18h.
3. Results
3.1 Several mutated TPR motifs fail to complement the mitomycin C hypersensitivity of NM3
To investigate the role of the TPR motifs of FANCG in mediating in vivo interactions in the BRCA2/D1-D2-G-X3 protein complex, we cloned sequences encoding mutated TPR-motifs into the pCEP4 vector [32], transfected them into the CHO fancg mutant NM3 and selected stable transfectants. TPR3 was not tested as it was identified subsequent to the 6 other motifs, and a mutant of this motif had not been constructed [32]. Individual clones which expressed similar levels of FANCG were chosen (Fig. 2A) and these NM3-mTPR (mutated tetratricopeptide motif) cell lines were used in all subsequent experiments. We first determined whether the mutant motifs behaved similarly in these cell lines as had previously been reported for human cells [32], in order to establish whether CHO was a suitable model system for studying the interactions of FANCG. The mutants of TPR1 (G216Q), TPR2 (G253Q), TPR5 (G460Q), TPR6 (G521Q) and TPR7 (R563E) failed to complement the MMC-sensitivity of NM3, whilst TPR4 (A401Q) and wild-type FANCG did complement (Fig. 2B). These results are broadly similar to those obtained in the human cell line EUFA316, with the exception that mutant TPR7 failed to complement NM3 cells [32].
Fig. 2.
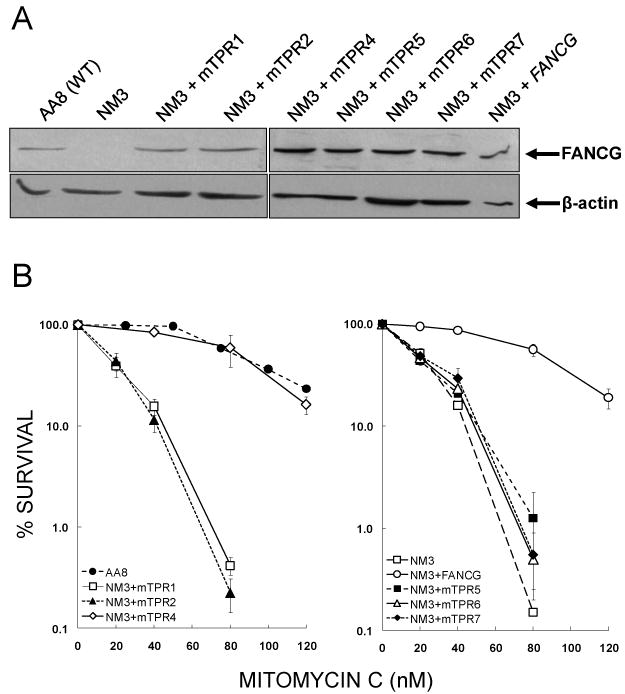
Validation of NM3 cell lines used to study FANCG and FANCG-mediated protein interactions. (A) Expression of mutated human FANCG protein in NM3 cell lines. Western blotting was used to determine expression of FANCG protein in NM3 cell lines transfected with human cDNA mutated at the indicated TPR motif. Clones that expressed FANCG at similar levels to each other and at a similar level to the endogenous expression of wild type hamster protein (AA8) were selected for further study. β-actin was used as a loading control. (B) MMC survival responses of NM3 cell lines expressing human FANCG with mutated TPR motifs. Clonogenic survival assays were performed with curves shown representing the means of 2-4 experiments and error bars showing the standard error of the mean (SEM). The responses of NM3 cell lines with mutated TPR1, TPR2, TPR5, TPR6 and TPR7 (respective D37 values = 22, 25, 29, 29 and 32 nM MMC) were similar to NM3 (D37 = 26 nM MMC), whilst the response of NM3+mTPR4 (D37 = 101 nM MMC) was similar to the wild type cell line AA8 (D37 = 99 nM MMC) and NM3 complemented with wild type human FANCG cDNA (D37 = 100 nM MMC).
Several of the TPR mutants fail to support the interaction of FANCG with its core complex partner FANCA in human EUFA316 cells [32], and we wished to confirm this observation in the NM3 lines. Using anti-FANCA antibody to immunoprecipitate, the mutant TPR1, TPR2, TPR5 and TPR6 proteins showed no detectable interaction with FANCA in NM3, whilst they did co-precipitate in AA8, NM3-FANCG, NM3-mTPR4 and NM3-mTPR7 (Fig. 3A). These results were confirmed in several repeat experiments (e.g. Fig. 3B) and in a reciprocal immunoprecipitation experiment (Fig. 3C). These observations are identical to those seen in EUFA316 cells [32]. Therefore, TPR7 (R563E) corrects the MMC sensitivity of EUFA316, buts fails to correct NM3 (Fig. 2B) despite the mutant protein interacting with FANCA in the hamster cells. Having established that most of the mutated TPRs behaved similarly in NM3 as they previously had in human EUFA316 cells, we next examined protein interactions in the D1-D2-G-X3 complex.
Fig. 3.
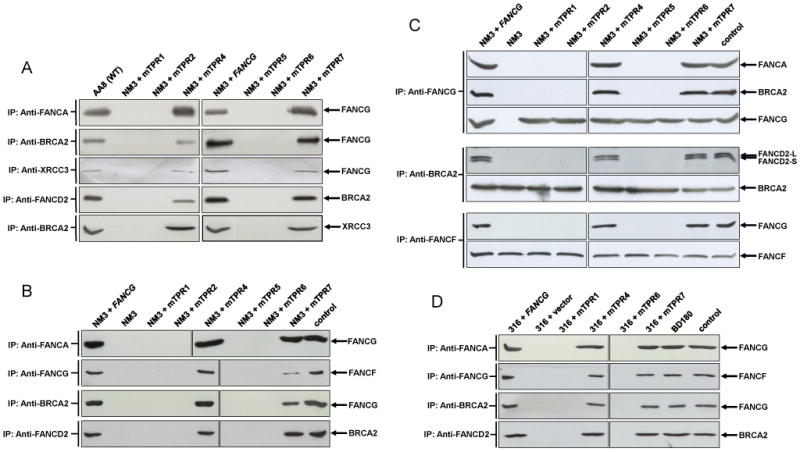
Interactions of TPR-mutated FANCG proteins in hamster NM3 cells (A, B and C) and human EUFA316 cells (D) as determined by co-immunoprecipitation analysis. Whole cell lysates from cultures treated with 50 nM MMC for 18 h were immunoprecipitated with the indicated anti-body and samples were loaded onto a SDS-PAGE gel. Following electrophoresis and membrane transfer, the blot was probed for the indicated protein. (A) The upper panel shows that mutations at TPR1, TPR2, TPR5 and TPR6 fail to support the interaction of FANCG with FANCA in NM3 cells. FANCG proteins with the same mutations were previously shown not to interact with FANCA in human FA-G EUFA316 cells (Blom et al. 2004), thereby confirming the suitability of the NM3 cell lines to analyse protein interactions in the D1-D2-G-X3 complex. The lower panel of (A) shows that FANCG mutated at TPR1, TPR2, TPR5 and TPR6 fails to co-precipitate with its direct binding partners BRCA2 and XRCC3 in the D1-D2-G-X3 complex and these same mutations fail to support the indirect protein interactions between BRCA2-FANCD2 and BRCA2-XRCC3. (B) Immunoprecipitation between FANCG-FANCF in NM3 cells and repeats for FANCA-FANCF, BRCA2-FANCG and BRCA2-FANCD2. Mutated TPR1, 2, 5 and 6 fail to support the interaction between FANCG-FANCF and appears to be reduced in NM3+mTPR7. The lane labelled input was loaded with1/50 of the protein extract used for immunoprecipitation in NM3+FANCG. (C) Reciprocal immunoprecipitations for FANCG-FANCA, FANCG-BRCA2, BRCA2-FANCD2 and FANCF-FANCG in NM3 cells. The lane labelled input was loaded with1/50 of the protein extract used for immunoprecipitation in NM3+FANCG. When immunoprecipitated with anti-FANCG, no interaction is detected between FANCG-FANCA and FANCG-BRCA2 in NM3, NM3+mTPR1, +mTPR2, +mTPR5 or +mTPR6, whilst blotting for FANCG acts as a control for the immunoprecipitation and indicates the expression of FANCG in these cell lines. Immunoprecipitation with anti-BRCA2 shows the interaction of BRCA2 with both isoforms of FANCD2 in NM3+FANCG, NM3+mTPR4 and +TPR7, with no interaction detected with either isoform of FANCD2 in NM3, NM3+mTPR1, +TPR2, +TPR5 or +TPR6. Immunoprecipitation with anti-FANCF confirms the lack of interaction between FANCF-FANCG in NM3, NM3+TPR1, +TPR2, +TPR5 and +TPR6 and interaction in NM3+FANCG and NM3+TPR4. No reduction in the interaction between FANCF-FANCG in NM3+TPR7 was evident in this experiment. (D) Protein interactions in selected human EUFA316 cells. Immunoprecipitations were the same as those shown for NM3 in Fig. 3B. The lane labelled input was loaded with1/50 of the protein extract used for immunoprecipitation in EUFA316+FANCG. Co-precipitation of FANCA-FANCG, FANCG-FANCF, BRCA2-FANCG and FANCD2-BRCA2 was observed in BD180 (wild type), EUFA316+FANCG, +mTPR4 and +mTPR7, whilst it was absent in EUFA316+vector, +TPR1 and +TPR6.
3.2 The TPR motifs of FANCG are required for in vivo protein-protein interactions of the D1-D2-G-X3 complex and the core complex
FANCG directly interacts with both BRCA2 [28] and XRCC3 [29] and is required for in vivo interactions amongst the components of the D1-D2-G-X3 complex [30], including the co-precipitation of the directly interacting BRCA2 and FANCD2 proteins [42]. Here we show that mutations in TPR1, TPR2, TPR5 and TPR6 of FANCG eradicates its ability to interact with both BRCA2 and XRCC3 in NM3 cells, whilst mutant TPR4, TPR7 and wild type FANCG do co-precipitate with the two recombination proteins (Figs.3A+B). Similarly, co-precipitation of FANCD2-BRCA2 and BRCA2-XRCC3 was retained in TPR4, TPR7 and wild type expressing cells, but interaction in the TPR1, TPR2, TPR5 and TPR6 mutants was not detected (Figs. 3A+B). The lack of interaction between FANCD2-BRCA2 and BRCA2-XRCC3 in these cell lines indicates that the motifs (TPR1, 2, 5 and 6) are not only required for the direct interactions of FANCG, but are also important for mediating the interaction of other proteins and the formation of the D1-D2-G-X3 complex.
To verify these results, we performed reciprocal experiments for the key interactions between FANCG and BRCA2 and between BRCA2 and FANCD2 (Fig. 3C). In these experiments we also probed for the protein that was used for the immunoprecipitation as a control for loading and expression. The reciprocal immunoprecipitations confirmed that FANCG-BRCA2 and BRCA2-FANCD2 co-precipitate in NM3-FANCG, NM3-mTPR4 and NM3-mTPR7, but fail to interact in NM3-mTPR1, NM3-mTPR2, NM3-mTPR5 and NM3-mTPR6 (Fig. 3C). The pull down with anti-BRCA2 antibody also confirmed our previous observation [30] that BRCA2 interacts with both the monoubiquitylated and non-ubiquitylated isoforms of FANCD2 (Fig. 3C), and demonstrates that FANCG, and several of its TPR motifs, are required for interaction of both BRCA2-FANCD2-L and BRCA2-FANCD2-S.
In addition to directly interacting with FANCA in the core complex, a direct interaction between FANCG and FANCF has been demonstrated [13]. Here we show that, as for FANCA-FANCG, no co-immunoprecipitation was seen between FANCG and FANCF in NM3+mTPR1, NM3+mTPR2, NM3+mTPR5 and NM3+mTPR6 (Fig. 3B), indicating that these TPR motifs are required for the interactions of FANCG in the core complex. However, whilst the interaction was clearly observed in NM3+FANCG and NM3+mTPR4, it appeared to be reduced in NM3+mTPR7 (Fig. 3B). We found the co-precipitation of FANCG-FANCF to be variable in NM3+mTPR7. In some instances the interaction appeared to be almost completely absent (data not shown) or reduced (Fig. 3B), whilst in other immunoprecipitations no reduction in the interaction was apparent (Fig. 3C).
In order to further validate our interaction data, we examined co-precipitation of several pairs of proteins in representative mTPR-expressing EUFA316 cells. TPR4/7 and TPR1/6 were selected as mutants, respectively complementing or failing to complement the MMC-hypersensitivity of EUFA316 [32]. Essentially, the interactions in the human cells were the same as observed in the NM3 cell lines (Fig. 3C). FANCA-FANCG, FANCG-FANCF, BRCA2-FANCG and FANCD2-BRCA2 did not co-precipitate in EUFA316+vector, EUFA316+mTPR1 and EUFA316+mTPR6 but did interact in BD180 (wild type), EUFA316+FANCG, EUFA316+mTPR4 and EUFA316+mTPR7 (Fig. 3D). No reduction in the interaction between FANCG-FANCF was evident in EUFA316+mTPR7 (Fig. 3D and data not shown).
3.3 Phleomycin hypersensitivity of NM3, EUFA316 and the TPR mutants
We previously demonstrated that NM3 shows sensitivity to DNA damaging agents other than interstrand cross-linking agents [17]. Indeed, Chinese hamster FancG mutants exhibit hypersensitivity to a broad spectrum of genotoxins including the radiomimetic compound bleomycin [1,17,43]. Given that FANCG interacts directly with the homologous recombination repair proteins XRCC3 and BRCA2, and hamster mutants of Xrcc3 and Brca2 are also sensitive to radiomimetic agents [44,45], we investigated whether the TPR motifs were required for resistance to a compound related to bleomycin. Phleomycin is a glycopeptide antibiotic of the bleomycin family; it binds and intercalates DNA, induces strand breaks and is a potent inhibitor of DNA replication [46]. As with MMC, NM3+mTPR1, 2, 5 and 6 exhibited sensitivity to phleomycin similar to that observed in NM3, whilst NM3+mTPR4 was as resistant as NM3+FANCG (Fig. 4A). Perplexingly, NM3+mTPR7 was significantly hypersensitive to phleomycin (Fig. 4A), even though co-precipitation of BRCA2-FANCG, XRCC3-FANCG, BRCA2-FANCD2 and BRCA2-XRCC3 was observed in this cell line (Fig. 3).
Fig. 4.

Phleomycin survival responses of NM3 and EUFA316 cell lines expressing FANCG with mutated TPR motifs. (A) Clonogenic survival assays were performed for hamster cell lines with curves shown representing the means of 2-4 experiments and error bars showing the standard error of the mean (SEM). The responses of NM3 cell lines with mutated TPR1, TPR2, TPR5, TPR6 and TPR7 (respective D37 values = 3.4, 3.4, 3.4, 3.7 and 4.3 μg/ml phleomycin) were similar to NM3 (D37 = 3.1 μg/ml phleomycin), whilst the response of NM3+mTPR4 (D37 = 8.9 μg/ml phleomycin) was similar to NM3 corrected with wild type FANCG cDNA (D37 = 8.0 μg/ml phleomycin). (B) Growth inhibition by MMC and phleomycin in human EUFA316 (FA-G) cell lines expressing the TPR-mutated forms of FANCG. Exponentially growing cells (106) were plated into 75 cm2 flasks, grown until near-confluence was reached in control cultures, trypsinised and cell numbers determined by haemocytometer. Data shown represents the minimum of 3 independent experiments. The dose of MMC required to inhibit growth by 50% was 23 nM for EUFA316+vector, 40 nM for EUFA316+mTPR1 & EUFA316+mTPR6, 69 nM for EUFA316+mTPR7, 81 nm for EUFA316+mTPR4 and 85 nM for EUFA316+FANCG. Doses of phleomycin reducing growth by 50% were 0.28 μg/ml for EUFA316+vector, 0.31 μg/ml for EUFA316+mTPR6, 0.33 μg/ml for EUFA316+mTPR1, 0.56 μg/ml for EUFA316+mTPR7, 0.61 μg/ml for EUFA316+mTPR4 and 0.63 μg/ml for EUFA316+FANCG.
The failure of mTPR7 to correct the MMC and phleomycin hypersensitivities of NM3 is also contradictory to its previously reported complementation of EUFA316 [32]. Using a growth inhibition assay (Fig. 4B), we show that mTPR4 and mTPR7 significantly complement the MMC sensitivity of EUFA316, whilst mTPR1 and mTPR6 only slightly increased resistance to MMC, confirming the observations of Blom et al. [32]. EUFA316 cells, like NM3, were hypersensitive to phleomycin. On the basis of the dose required to reduce cell division by 50%, EUFA316+vector was 2.3-fold sensitive compared to EUFA316+FANCG (Fig. 4B). Correction of this phleomycin-hypersensitivity by the mutant TPRs was the same as for MMC, in that significant complementation was seen with mTPR4 and mTPR7, but not with mTPR1 and mTPR6. Thus mTPR7 corrected the hypersensitivities of human EUFA316 cells but not NM3 hamster cells.
3.4 The TPR motifs are required for efficient monoubiquitylation of FANCD2
Given that TPR1, TPR2, TPR5 and TPR6 are required for interaction of FANCG with FANCA and FANCF, and NM3+mTPR7 shows variable interaction of FANCG-FANCF, we assayed the ability of the NM3 cell lines to monoubiquitylate FANCD2 (Fig. 5A). As might be expected, monoubiquitylation was very severely reduced in NM3+mTPR1, +mTPR2, +mTPR5 and +mTPR6 (close inspection of the film does suggest a very low level of FANCD2-L expression in these cell lines). However, the relative amount of FANCD2-L expression was not reduced in comparison to FANCD2-S in NM3+mTPR7, although the total amount of FANCD2 does appear to be slightly reduced. In human cells, EUFA316+vector, +mTPR1 and +mTPR6 did not support the monoubiquitylation of FANCD2, whilst normal monoubiquitylation was observed in EUFA316+FANCG, +mTPR4 and +mTPR7 (Fig. 5B), consistent with the interactions of FANCA-FANCG and FANCG-FANCF in these cells (Fig. 3D).
Fig. 5.
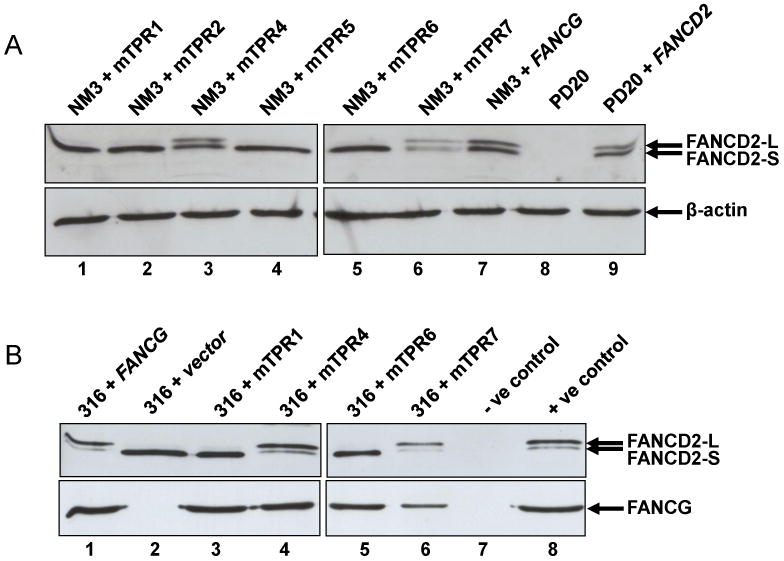
Expression of FANCD2 in NM3 and EUFA316 cell lines with mutated TPR motifs. Whole cell extracts were prepared from NM3 or EUFA316 cells treated with 50 nM MMC for 18 h. (A) NM3+mTPR1, +TPR2, +TPR5, and +TPR6 lack significant expression of the monoubiquitylated isoform (FANCD2-L), whilst NM3+TPR4 and +TPR7 and NM3+FANCG express both monoubiquitylated and non-ubiquitylated (FANCD2-S) isoforms. Human PD20 (FA-D2) cells and a cDNA-corrected counterpart (PD20+FANCD2) were used as negative and positive controls for FANCD2 detection by the antibody (lanes 8 & 9 respectively). β-actin was used as a loading control. (B) EUFA316+vector, +mTPR1, and +TPR6 lack significant expression of FANCD2-L, whilst EUFA316+mTPR4, +TPR7 and EUFA316+FANCG express both isoforms. Expression of FANCG in the same cell extracts are shown in the lower panel. As negative and positive controls for the detection of FANCD2 by the antibody, PD20 and PD20+FANCG were used (lanes 7 & 8 upper panel), whilst NM3 and NM3+FANCG were used as controls for FANCG (lanes 7 & 8 lower panel).
4. Discussion
We previously demonstrated that phosphorylation of FANCG at serine-7 was required for its in vivo interaction with the homologous recombination repair proteins BRCA2 (FANCD1) and XRCC3 [30]. Furthermore, FANCG Ser7-phosphorylation is required for the critical interaction between FANCD2 and BRCA2 and for co-precipitation of the component parts of the D1-D2-G-X3 complex. Here we show that several of the TPR repeat motifs are also required for these interactions in NM3 cells (Fig. 3). When the conserved 8th residue glycine was substituted with glutamine in TPR1, TPR2, TPR5 and TPR6, the resulting mutated FANCG proteins did not co-precipitate with either BRCA2 or XRCC3. The in vivo interactions between BRCA2-FANCD2-L, BRCA2-FANCD2-S and BRCA2-XRCC3 were also removed by these mutations, indicating that the TPR motifs of FANCG are required to mediate the interactions between these pairs of proteins. Similarly, interactions of proteins in the FA core complex were disrupted by the mutation of TPR1, TPR2, TPR5 and TPR6, with no co-precipitation detected between FANCG-FANCA, FANCG-FANCF (Fig. 3) and FANCA-FANCF (data not shown). This requirement for the TPR motifs, not only for the direct interactions of FANCG (BRCA2, XRCC3, FANCA and FANCF), but also for mediating and stabilising other protein-protein interactions in both the D1-D2-G-X3 and the FA core complexes (BRCA2-FANCD2, BRCA2-XRCC3 and FANCA-FANCF), is consistent with the role of other TPR-containing proteins in the assembly of multiprotein complexes [47-49]. For example, the molecular chaperone HSP90 is bound by several different TPR-containing proteins, such as protein phosphatase 5, Hop, FKBP52, and Cyp40, which then facilitate the formation of distinct TPR-containing HSP90 complexes [50-53].
The lack of interaction of TPR1 and TPR2 mutants with FANCA and XRCC3 is in apparent contradiction with the yeast-2-hybrid data which showed that TPR1 and TPR2 were not important for FANCA-FANCG or FANCG-XRCC3 interactions [29]. Nevertheless, mutations of these two motifs results in hypersensitivity to MMC and phleomycin in human [32] and hamster cells (Figs. 2 and 4). This discrepancy suggests that the interactions in yeast may not fully reflect the situation in mammalian cells, where other proteins or post-translational modifications may affect the strength and stability of the interactions. A patient derived missense mutation in FANCG, located at one end of TPR6 (amino acid 546, glycine to arginine) was found to interact with both FANCA and FANCF in the yeast 2-hybrid system [14], even though the G546R protein failed to correct the MMC-hypersensitivity of an FA-G indicator (fibroblast FAG326SV) cell line [54]. This pathogenic mutation at a residue conserved in human, hamster, mouse (Fig. 1) and zebrafish [32] further demonstrates the vital role of the TPR motifs in the activity of the FANCG protein and in FA pathway function.
Mutations of TPR4 at A401Q and TPR7 at R563E appeared not to disrupt the interactions of FANCG in human cells (Fig. 3D) and their expression in EUFA316 complemented both MMC and phleomycin hypersensitivity and restored FANCD2 monoubiquitylation (Figs. 4B and 5B). In contrast, mutated TPR7 failed to correct the sensitivities of NM3 (Figs. 2 and 4A) and whilst interactions between FANCG and BRCA2, XRCC3 and FANCA were detected by co-precipitation, the interaction between FANCG and FANCF appeared to be reduced in some experiments (Fig. 3B). It is not immediately evident why mutations at these residues/motifs should differ from mutations in the four other TPR motifs. The mutation of TPR4 is also at the 8th residue of the motif (although in this case alanine is conserved not glycine), and whilst the TPR7 mutation is located elsewhere, both substituted amino acids are highly conserved and present in the FANCG proteins of mammals (Fig. 1), fish, Xenopus laevis and chickens [32,37,55,56]. It may be that TPR4 and TPR7 are less critical for binding to FANCA, FANCF, BRCA2 and XRCC3 than TPRs 1, 2, 5 and 6, or that they are required for interactions with other proteins. The mutated residue in TPR7 (at position 16) is not one of the eight TPR consensus residues, although amino acids other than these eight may determine the specificity of a TPR domain for its target protein [33,36]. The lack of complementation by mTPR7 in NM3 cells does suggest that this motif is functionally important. It is possible that the R563E mutation is insufficient to completely disrupt the protein interactions of FANCG in human and hamster cells, as detected here by co-immunoprecipitation. However, mutation of this motif, which is the least conserved of the seven TPRs (Fig. 1), may be enough to impair the function of the human FANCG protein in CHO cells., Thus, whilst human FANCG and the hamster proteins bind and the interactions are detectable by immunoprecipitation, they may not be in a normal configuration so that the resulting protein complexes perform sub-optimally. The variable interaction of the human mTPR7-FANCG protein with hamster FANCF suggests the core complex is unstable in NM3+mTPR7. Nevertheless, these cells do express monoubiquitylated FANCD2 (Fig. 5A), suggesting that the observed MMC sensitivity is a result of an impairment of a core complex function other than FANCD2 monoubiquitylation.
We previously demonstrated that 3 independent CHO fancg mutants (NM3, UV40 and KO40) were hypersensitive to the anti-cancer glycopeptide bleomycin [17,43]. In addition, Carreau et al. [57] showed that EUFA316 and another FA-G lymphoblastoid cell line EUFA143 were also significantly hypersensitive to this drug. Here we show that NM3 and EUFA316 are also sensitive to the related chemical phleomycin and that several TPR mutants fail to complement this phenotype (Fig. 4A). We suggest that hypersensitivity to bleomycin and phleomycin in FA-G cell mutants is related to FANCG's role in mediating the formation of the D1-D2-G-X3 protein complex. Certainly, hamster brca2 and xrcc3 cell mutants [44,45] are hypersensitive to bleomycin and phleomycin (data not shown), and the extreme bleomycin sensitivity seen in the FA-D2 cell line EUFA202 also supports this hypothesis [57]. Bleomycin and phleomycin are regarded as X-ray mimetic compounds and human FA cell lines from most core complex complementation groups, including FA-G, are not generally perceived as exhibiting cellular hypersensitivity to ionising radiation [58]. However, the precise nature of the DNA damage that ionising radiations and these so-called mimetic drugs induce does significantly differ, including the types of DNA strand breaks they generate [46]. NM3 and another isogenic CHO fancg mutant KO40 exhibit little or no sensitivity to γ-rays despite their marked hypersensitivity to bleomycin and phleomycin [38,43]. We are currently examining the response of human cell lines from other FA complementation groups to further define the role of FA proteins in the repair of phleomycin/bleomycin-induced DNA damage.
Defective homologous recombination repair of I-SceI induced DNA double strand breaks (DSBs) of an artificial direct-repeat substrate has been reported for both a chicken DT40 fancg mutant and a human FA-G fibroblast cell line [55,59]. Whilst these defects are relatively modest (9-fold in the DT40 fancg mutant and 2-fold in human EUFA326 cells) it should be remembered that the assay measures the repair of a single artificial frank DSB that is quite different to the DNA strand breaks induced by bleomycin and phleomycin. Given the hypersensitivity observed in hamster and human FA-G mutants, and the requirement of the protein for the assembly of the D1-D2-G-X3 complex, we propose that FANCG is required for the efficient repair of phleomycin-induced DNA damage by homologous recombination, and this is currently under investigation.
TPR repeats are found in archaeal, bacterial and eukaryotic proteins and comprise 34 amino acids organised in two anti-parallel α-helices that pack together in a knobs in holes arrangement [33,35]. Multiple TPRs fold into a right-handed superhelical structure containing a ligand-binding grove that forms a scaffold for protein-protein interactions [35,60]. Secondary structure predications suggest that several FA proteins (including A, C, E, F, and G) are largely α-helical, and FANCE and FANCF have been shown to possess α-helical repeat domains [26,61,62]. These non-canonical helical repeats are related to TPR, ARM and HEAT repeats, structures that mediate protein-protein interactions [61,62]. Mutations of these “FANC” repeats (including patient-derived mutations) disrupt the interaction of FANCE with FANCD2 [61] and compromise the interaction of FANCF with other core complex proteins [62]. Here we have demonstrated that several of the TPR motifs of FANCG are required for its functionality and for mediating protein-protein interactions in both the FA core complex and the recently described D1-D2-G-X3 complex. In contrast to the phosphorylation of FANCG at Ser7, that is only required for formation of the latter complex, the motifs TPR1, TPR2, TPR5 and TPR5 do not differentiate for the interaction of FANCG with either complex. Mutation of the conserved 8th residue of TPR4 did not appear to disrupt the interactions of FANCG with either complex, and we cannot rule out the possibility that this particular motif may be required for interactions with other proteins and complexes. FANCG has been reported to functionally interact with several non-FA proteins including cytochrome p450 CYP2E1 [63], PKR kinase [64] and nonerythroid α spectrin (αIISp) [65]. It would be of interest to determine whether TPR4 and the other TPR motifs of FANCG are required for interaction with these proteins, particularly αIISp, a structural protein hypothesised to play a role in DNA repair, chromatin remodelling, transcription and RNA processing [66].
Our data indicate that FANCG, like many other TPR-containing proteins, functions as a mediator of protein-protein interactions and is important for the assembly of multi-protein complexes. Consistent with such a role for FANCG is the recent identification of two motifs either side of TPR4 in FANCG (amino acids 380-388 PRFSPPPSP and 443-449 KELPYCP) that bind to Src-homology 3 (SH3) domains such as that found in αIISp [67]. SH3-domains are protein modules that act as sites for protein complex formation via binding to these proline-rich consensus motifs [68]. TPR motifs are often found in combination with other protein binding sites such as SH2, SH3 and J domains [36] and it is tempting to speculate that TPR4 acts in conjunction with the two adjacent SH3-interacting domains to bind other proteins such as αIISp.
Acknowledgments
This work was funded by a grant provided by the North West Cancer Research Fund (NWCRF grant number CR715) to NJJ and an NIH grant R01 HL063776 to GMK. RC was supported by a BBSRC research studentship. We thank Larry Thompson for his comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Busch DB, Zdzienicka MZ, Natarajan AT, Jones NJ, Overkamp WJI, Collins A, Mitchell DL, Stefanini M, Botta E, Albert RB, Liu N, White DA, vanGool AJ, Thompson LH. A CHO mutant, UV40, that is sensitive to diverse mutagens and represents a new complementation group of mitomycin C sensitivity. Mutation Research-DNA Repair. 1996;363:209–221. doi: 10.1016/0921-8777(96)00014-6. [DOI] [PubMed] [Google Scholar]
- 2.Liu N, Lamerdin JE, Tucker JD, Zhou ZQ, Walter CA, Albala JS, Busch DB, Thompson LH. The human XRCC9 gene corrects chromosomal instability and mutagen sensitivities in CHO UV40 cells. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1997;94:9232–9237. doi: 10.1073/pnas.94.17.9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CGM, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, Pronk JC, Arwert F, Hoehn H, Digweed M, Buchwald M, Joenje H. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nature Genetics. 1998;20:281–283. doi: 10.1038/3093. [DOI] [PubMed] [Google Scholar]
- 4.Auerbach AD. Fanconi anemia and its diagnosis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003;97:425–440. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 6.Wang WD. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature Reviews Genetics. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 7.Ali AM, Singh TR, Meetei AR. FANCM-FAAP24 and FANCJ: FA proteins that metabolize DNA. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2009;668:20–26. doi: 10.1016/j.mrfmmm.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan ZJ, Xue YT, Oostra AB, Auerbach AD, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Takata M, Meetei AR, Wang WD. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO Journal. 2007;26:2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciccia A, Ling C, Coulthard R, Yan ZJ, Xue YT, Meetei AR, Laghmani EH, Joenje H, McDonald N, de Winter JP, Wang WD, West SC. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Molecular Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Kim JM, Kee YH, Gurtan A, D'Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: Mechanistic insights. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber PAJ, Medhurst AL, Youssoufian H, Mathew CG. Investigation of Fanconi anemia protein interactions by yeast two-hybrid analysis. Biochemical and Biophysical Research Communications. 2000;268:73–77. doi: 10.1006/bbrc.1999.2055. [DOI] [PubMed] [Google Scholar]
- 13.Medhurst AL, Huber PAJ, Waisfisz Q, de Winter JP, Mathew CG. Direct interactions of the five known Fanconi anaemia proteins suggest a common functional pathway. Human Molecular Genetics. 2001;10:423–429. doi: 10.1093/hmg/10.4.423. [DOI] [PubMed] [Google Scholar]
- 14.Gordon SM, Buchwald M. Fanconi anemia protein complex: mapping protein interactions in the yeast 2- and 3-hybrid systems. Blood. 2003;102:136–141. doi: 10.1182/blood-2002-11-3517. [DOI] [PubMed] [Google Scholar]
- 15.Medhurst AL, Laghmani EH, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang WD, Joenje H, de Winter JP. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 2006;108:2072–2080. doi: 10.1182/blood-2005-11-008151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Winter JP, van der Weel L, de Groot J, Stone S, Waisfisz Q, Arwert F, Scheper RJ, Kruyt FAE, Hoatlin ME, Joenje H. The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Human Molecular Genetics. 2000;9:2665–2674. doi: 10.1093/hmg/9.18.2665. [DOI] [PubMed] [Google Scholar]
- 17.Wilson JB, Johnson MA, Stuckert AP, Trueman KL, May S, Bryant PE, Meyn RE, D'Andrea AD, Jones NJ. The Chinese hamster FANCG/XRCC9 mutant NM3 fails to express the monoubiquitinated form of the FANCD2 protein, is hypersensitive to a range of DNA damaging agents and exhibits a normal level of spontaneous sister chromatid exchange. Carcinogenesis. 2001;22:1939–1946. doi: 10.1093/carcin/22.12.1939. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 19.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nature Genetics. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 21.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, de Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 22.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature Genetics. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 23.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu JM, Christ N, Liu XG, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Thompson LH, Hinz JM, Yamada NA, Jones NJ. How Fanconi anemia proteins promote the four Rs: Replication, recombination, repair, and recovery. Environmental and Molecular Mutagenesis. 2005;45:128–142. doi: 10.1002/em.20109. [DOI] [PubMed] [Google Scholar]
- 25.de Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2009;668:11–19. doi: 10.1016/j.mrfmmm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Gari K, Constantinou A. The role of the Fanconi anemia network in the response to DNA replication stress. Critical Reviews in Biochemistry and Molecular Biology. 2009;44:292–325. doi: 10.1080/10409230903154150. [DOI] [PubMed] [Google Scholar]
- 27.Thompson LH, Jones NJ. Stabilizing and remodeling the blocked DNA replication fork: Anchoring FANCM and the Fanconi anemia damage response. Molecular Cell. 2010;37:748–751. doi: 10.1016/j.molcel.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Hussain S, Witt E, Huber PAJ, Medhurst AL, Ashworth A, Mathew CG. Direct interaction of the Fanconi anaemia protein FANCG with BRCA2/FANCD1. Human Molecular Genetics. 2003;12:2503–2510. doi: 10.1093/hmg/ddg266. [DOI] [PubMed] [Google Scholar]
- 29.Hussain S, Wilson JB, Blom E, Thompson LH, Sung P, Gordon SM, Kupfer GM, Joenje H, Mathew CG, Jones NJ. Tetratricopeptide-motif-mediated interaction of FANCG with recombination proteins XRCC3 and BRCA2. DNA Repair. 2006;5:629–640. doi: 10.1016/j.dnarep.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, Jones NJ. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene. 2008;27:3641–3652. doi: 10.1038/sj.onc.1211034. [DOI] [PubMed] [Google Scholar]
- 31.Qiao FY, Mi J, Wilson JB, Zhi G, Bucheimer NR, Jones NJ, Kupfer GM. Phosphorylation of Fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. Journal of Biological Chemistry. 2004;279:46035–46045. doi: 10.1074/jbc.M408323200. [DOI] [PubMed] [Google Scholar]
- 32.Blom E, van de Vrugt HJ, de Vries Y, de Winter JP, Arwert F, Joenje H. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair. 2004;3:77–84. doi: 10.1016/j.dnarep.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions - to TPR or not to TPR. Trends in Biochemical Sciences. 1995;20:257–259. doi: 10.1016/s0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- 34.Groves MR, Barford D. Topological characteristics of helical repeat proteins. Current Opinion in Structural Biology. 1999;9:383–389. doi: 10.1016/s0959-440x(99)80052-9. [DOI] [PubMed] [Google Scholar]
- 35.Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: Structures, functions, and evolution. Journal of Structural Biology. 2001;134:117–131. doi: 10.1006/jsbi.2001.4392. [DOI] [PubMed] [Google Scholar]
- 36.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 37.van de Vrugt HJ, Koomen M, Berns MAD, de Vries Y, Rooimans MA, van der Weel L, Blom E, de Groot J, Schepers RJ, Stone S, Hoatlin ME, Cheng NC, Joenje H, Arwert F. Characterization, expression and complex formation of the murine Fanconi anaemia gene product FancG. Genes to Cells. 2002;7:333–342. doi: 10.1046/j.1365-2443.2002.00518.x. [DOI] [PubMed] [Google Scholar]
- 38.Lamerdin JE, Yamada NA, George JW, Souza B, Christian AT, Jones NJ, Thompson LH. Characterization of the hamster FancG/Xrcc9 gene and mutations in CHOUV40 and NM3. Mutagenesis. 2004;19:237–244. doi: 10.1093/mutage/geh019. [DOI] [PubMed] [Google Scholar]
- 39.Zhi G, Wilson JB, Chen X, Krause DS, Xiao Y, Jones NJ, Kupfer GM. Fanconi Anemia complementation group FANCD2 protein serine 331 phosphorylation is important for Fanconi Anemia pathway function and BRCA2 interaction. Cancer Res. 2009;69:8775–8783. doi: 10.1158/0008-5472.CAN-09-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu JH, Wilson JB, Wolfreys AM, Scott A, Jones NJ. Optimization of the comet assay for the sensitive detection of PUVA-induced DNA interstrand crosslinks. Mutagenesis. 2009;24:173–181. doi: 10.1093/mutage/gen068. [DOI] [PubMed] [Google Scholar]
- 41.Rudland PS, Platt-Higgins AM, Davies LM, De Silva Rudland S, Wilson JB, Aladwani A, Winstanley JHR, Barraclough DL, Barraclough BR, West CR, Jones NJ. Significance of the Fanconi anemia FANCD2 protein in sporadic and metastatic human breast cancer. American Journal of Pathology in press. 2010 doi: 10.2353/ajpath.2010.090779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Human Molecular Genetics. 2004;13:1241–1248. doi: 10.1093/hmg/ddh135. [DOI] [PubMed] [Google Scholar]
- 43.Tebbs RS, Hinz JM, Yamada NA, Wilson JB, Salazar EP, Thomas CB, Jones IM, Jones NJ, Thompson LH. New insights into the Fanconi anemia pathway from an isogenic FancG hamster CHO mutant. DNA Repair. 2005;4:11–22. doi: 10.1016/j.dnarep.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 44.Fuller LF, Painter RB. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutation Research. 1988;193:109–121. doi: 10.1016/0167-8817(88)90041-7. [DOI] [PubMed] [Google Scholar]
- 45.Overkamp WJI, Rooimans MA, Neuteboom I, Telleman P, Arwert F, Zdzienicka MZ. Genetic diversity of mitomycin C-hypersensitive Chinese hamster cell mutants - a new complementation group with chromosomal instability. Somatic Cell and Molecular Genetics. 1993;19:431–437. doi: 10.1007/BF01233248. [DOI] [PubMed] [Google Scholar]
- 46.Chen JY, Ghorai MK, Kenney G, Stubbe J. Mechanistic studies on bleomycin-mediated DNA damage: multiple binding modes can result in double-stranded DNA cleavage. Nucleic Acids Research. 2008;36:3781–3790. doi: 10.1093/nar/gkn302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demonacos C, Krstic-Demonacos M, La Thangue NB. A TPR motif cofactor contributes to p300 activity in the p53 response. Molecular Cell. 2001;8:71–84. doi: 10.1016/s1097-2765(01)00277-5. [DOI] [PubMed] [Google Scholar]
- 48.Kuraoka I, Ito S, Wada T, Hayashida M, Lee L, Saijo M, Nakatsu Y, Matsumoto M, Matsunaga T, Handa H, Qin J, Nakatani Y, Tanaka K. Isolation of XAB2 complex involved in pre-mRNA splicing, transcription, and transcription-coupled repair. Journal of Biological Chemistry. 2008;283:940–950. doi: 10.1074/jbc.M706647200. [DOI] [PubMed] [Google Scholar]
- 49.Nakatsu Y, Asahina H, Citterio E, Rademakers S, Vermeulen W, Kamiuchi S, Yeo JP, Khaw MC, Saijo M, Kodo N, Matsuda T, Hoeijmakers JHJ, Tanaka K. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. Journal of Biological Chemistry. 2000;275:34931–34937. doi: 10.1074/jbc.M004936200. [DOI] [PubMed] [Google Scholar]
- 50.Hinds TD, Sanchez ER. Protein phosphatase 5. International Journal of Biochemistry & Cell Biology. 2008;40:2358–2362. doi: 10.1016/j.biocel.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Odunuga OO, Longshaw VM, Blatch GL. Hop: more than an Hsp70/Hsp90 adaptor protein. Bioessays. 2004;26:1058–1068. doi: 10.1002/bies.20107. [DOI] [PubMed] [Google Scholar]
- 52.Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cellular Signalling. 2004;16:857–872. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Riggs DL, Cox MB, Cheung-Flynn J, Prapapanich V, Carrigan PE, Smith DF. Functional specificity of co-chaperone interactions with Hsp90 client proteins. Critical Reviews in Biochemistry and Molecular Biology. 2004;39:279–295. doi: 10.1080/10409230490892513. [DOI] [PubMed] [Google Scholar]
- 54.Nakanishi K, Moran A, Hays T, Kuang Y, Fox E, Garneau D, de Oca RM, Grompe M, D'Andrea AD. Functional analysis of patient-derived mutations in the Fanconi anemia gene, FANCG/XRCC9. Experimental Hematology. 2001;29:842–849. doi: 10.1016/s0301-472x(01)00663-4. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Molecular and Cellular Biology. 2003;23:5421–5430. doi: 10.1128/MCB.23.15.5421-5430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stone S, Sobeck A, van Kogelenberg M, de Graaf B, Joenje H, Christian J, Hoatlin ME. Identification, developmental expression and regulation of the Xenopus ortholog of human FANCG/XRCC9. Genes to Cells. 2007;12:841–851. doi: 10.1111/j.1365-2443.2007.01096.x. [DOI] [PubMed] [Google Scholar]
- 57.Carreau M, Alon N, Bosnoyan-Collins L, Joenje H, Buchwald M. Drug sensitivity spectra in Fanconi anemia lymphoblastoid cell lines of defined complementation groups. Mutation Research-DNA Repair. 1999;435:103–109. doi: 10.1016/s0921-8777(99)00041-5. [DOI] [PubMed] [Google Scholar]
- 58.Kalb R, Duerr M, Wagner M, Herterich S, Gross M, Digweed M, Joenje H, Hoehn H, Schindler D. Lack of sensitivity of primary Fanconi's anemia fibroblasts to UV and ionizing radiation. Radiation Research. 2004;161:318–325. doi: 10.1667/rr3138. [DOI] [PubMed] [Google Scholar]
- 59.Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das AK, Cohen PTW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. Embo Journal. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nookala RK, Hussain S, Pellegrini L. Insights into Fanconi Anaemia from the structure of human FANCE. Nucleic Acids Research. 2007;35:1638–1648. doi: 10.1093/nar/gkm033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kowal P, Gurtan AM, Stuckert P, D'Andrea AD, Ellenberger T. Structural determinants of human FANCF protein that function in the assembly of a DNA damage signaling complex. Journal of Biological Chemistry. 2007;282:2047–2055. doi: 10.1074/jbc.M608356200. [DOI] [PubMed] [Google Scholar]
- 63.Futaki M, Igarashi T, Watanabe S, Kajigaya S, Tatsuguchi A, Wang JX, Liu JM. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis. 2002;23:67–72. doi: 10.1093/carcin/23.1.67. [DOI] [PubMed] [Google Scholar]
- 64.Zhang XL, Li J, Sejas DP, Rathbun KR, Bagby GC, Pang QS. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR) Journal of Biological Chemistry. 2004;279:43910–43919. doi: 10.1074/jbc.M403884200. [DOI] [PubMed] [Google Scholar]
- 65.McMahon LW, Sangerman J, Goodman SR, Kumaresan K, Lambert MW. Human alpha spectrin II and the FANCA, FANCC, and FANCG proteins bind to DNA containing psoralen interstrand cross-links. Biochemistry. 2001;40:7025–7034. doi: 10.1021/bi002917g. [DOI] [PubMed] [Google Scholar]
- 66.Sridharan DM, McMahon LW, Lambert MW. alpha II-Spectrin interacts with five groups of functionally important proteins in the nucleus. Cell Biology International. 2006;30:866–878. doi: 10.1016/j.cellbi.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Lefferts JA, Wang C, Sridharan D, Baralt M, Lambert MW. The SH3 Domain of alpha II Spectrin Is a Target for the Fanconi Anemia Protein, FANCG. Biochemistry. 2009;48:254–263. doi: 10.1021/bi801483u. [DOI] [PubMed] [Google Scholar]
- 68.Kaneko T, Li L, Li SSC. The SH3 domain - a family of versatile peptide- and protein-recognition module. Frontiers in Bioscience. 2008;13:4938–4952. doi: 10.2741/3053. [DOI] [PubMed] [Google Scholar]