

Abstract
Glutamate racemase is an attractive antimicrobial drug target. Virtual screening using a transition-state conformation of the enzyme resulted in the discovery of several μM competitive inhibitors, dissimilar from current amino acid-like inhibitors, providing novel scaffolds for drug discovery. The most effective of these competitive inhibitors possesses a very high ligand efficiency value of −0.6 kcal/mol/heavy atom, and is effective against three distinct glutamate racemases representing two species of Bacillus. The benefits of employing the transition-state conformation of the receptor in virtual screening are discussed.
Keywords: Glutamate racemase; virtual screening; inhibitor, protein dynamics
Glutamate racemase (GR) catalyzes stereoinversion at the Cα of glutamate1 and is a source of d-glutamate in bacteria. d-Glutamate is an essential component of the peptidoglycan layer of bacterial cell walls and is a target for antibacterial drug development.2−4 GR possesses an exquisite substrate specificity, catalyzing stereoinversion of Cα via a 1,1 proton transfer, utilizing a “two-base” mechanism consisting of two cysteine residues that flank the Cα.5 GR-catalyzed racemization proceeds without the assistance of a cofactor, which has generated intense interest in its mechanism, as well as the related enzymes such as diaminopimelate (DAP) epimerase.6,7 GR−ligand cocrystal structures do not provide a reasonable explanation, a posteriori, for how the Cα proton is abstracted by the catalytic cysteine base, without invoking a reorganization of the active site.8,9 However, recent molecular dynamics−quantum mechanical/molecular mechanical studies on the Bacillus subtilis GR have yielded a “reactive” conformation resulting from a repositioning of active site moieties such that the carbanionic transition state is saturated with hydrogen bond donors.9
The plasticity of GR poses immense challenges for classic structure-based drug design.10 The current study seeks to surmount these obstacles by employing a virtual screening regime, in which the receptor is the “reactive” form of B. subtilis GR, as described in Spies et al.9 This methodology is summarized in the flowchart in Figure 1. The reactive conformation of GR, characterized in Spies et al.,9 was employed in the present study as a receptor for virtual screening of a large leadlike library of compounds. This approach is appealing in the sense that the reactive form of the enzyme is characterized by a compressed active site that exhibits dramatically enhanced protein−ligand interaction energy.9 After compounds were virtually screened and scored, approximately 1 million compounds, the docked enzyme−ligand complexes that yielded the greatest computationally determined pKi values (where Ki is the dissociation constant) or the most negative interaction energies, were slated for experimental investigation (Table 1). The various types of outcomes are also listed in Figure 1.
Figure 1.
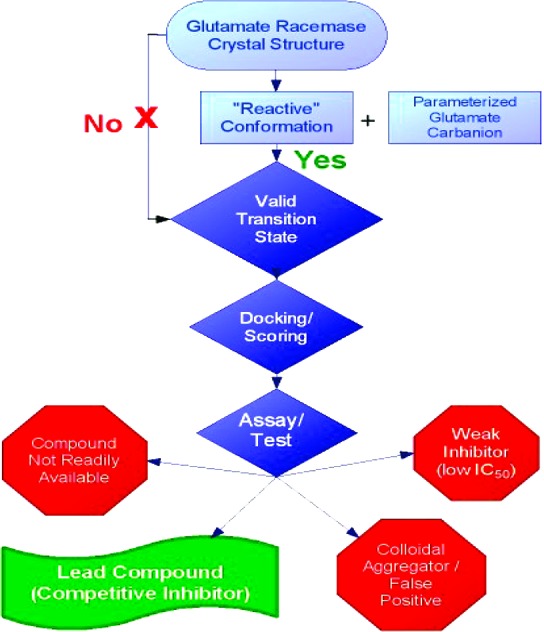
Flowchart for inhibitor discovery using a conformationally active form of GR.
Table 1. Highest-Ranking Hits Emerging from Virtual Screening of a Leadlikea Library of ∼1 Million Compounds to the Reactiveb (That Is, Transition State) Form of GR Versus the Outcome of Experimental GR Assays.
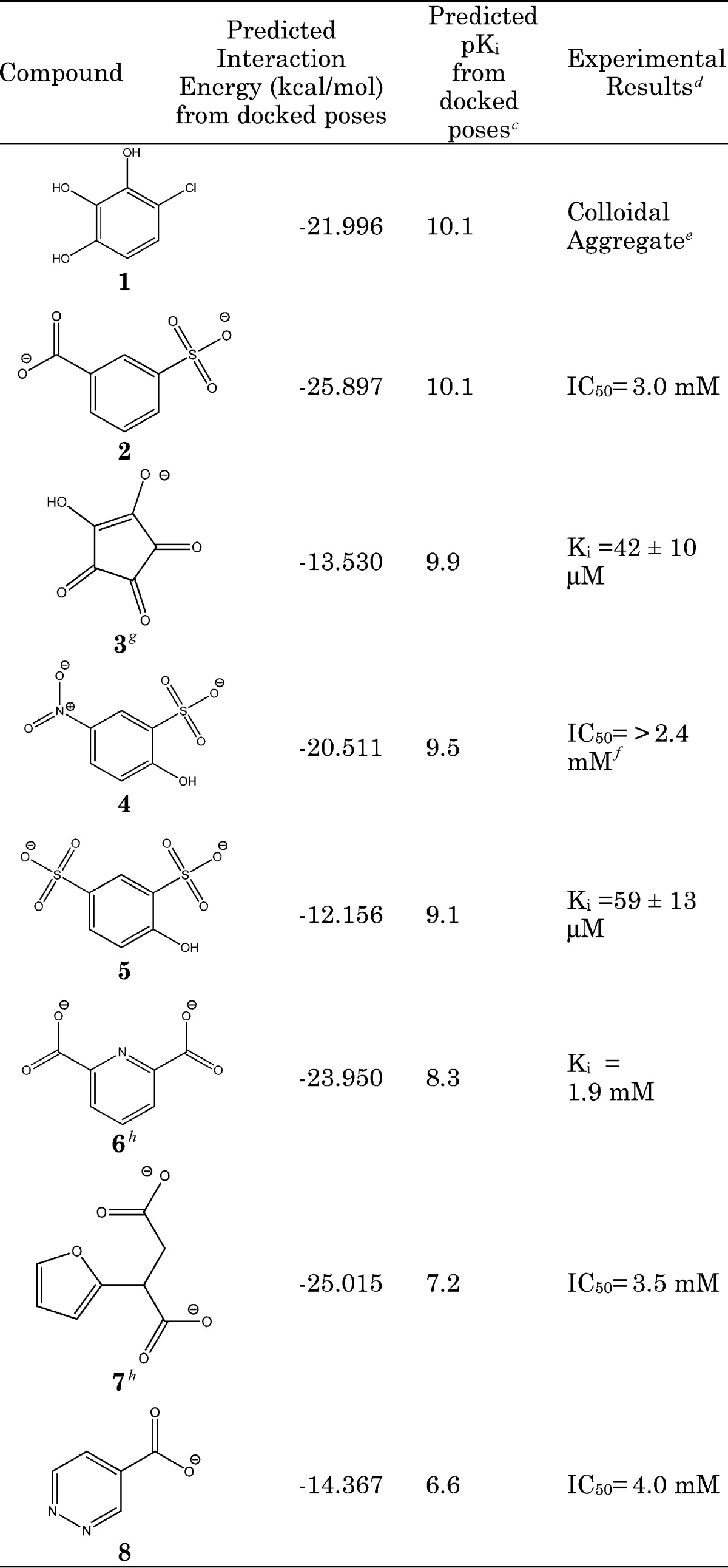 |
The Chemical Computing Group Conformational Database Version 2007, where lead-likeness is based on Oprea's parameters;14 see Computational Procedures in the Supporting Information.
The reactive form of GR, as characterized by Spies et al.9
As determined by the London dG scoring function, implemented within the LigX utility of MOE (v2008.10);15 values above pKi = 9.0 were considered for experimental investigation.
All Ki or IC50 values determined by the circular dichroism assay are described in the Supporting Information, except for 1 and 4.
Compound 1 appeared to display noncompetitive inhibition with an apparent Ki = 90 ± 7 μM but was found to inhibit GR through colloidal aggregation.
Inhibition by 4 was measured using the coupled enzyme assay, and the actual IC50 is expected to be higher due to partial inhibition of the coupled enzyme, l-glutamate dehydrogenase (see the Supporting Information for details).
Compounds 6 and 7 were chosen for their high interaction energies, while 8 was a readily available fragment of 2, 4, 5, and 6.
Two lead compounds were identified (where “lead compound” refers to a bona fide competitive inhibitor with a micromolar range Ki). Table 1 provides a full account of all of the tested compounds with the greatest predicted pKi values and their experimental Ki values. Compounds 1−5 have dG scores in the top 1% of the originally retained 500 compounds from the virtual screen. All of these compounds were found to be inhibitors (two in the micromolar range and two in the millimolar range), while 1 was a colloidal aggregator (vide infra). Scoring with dG was notably superior to simply using ligand interaction energies, as performed in Spies et al.,9 which generated only a few millimolar inhibitors. Furthermore, in the current study, interaction energies for the two best inhibitors, 3 and 5, were very low, underscoring the poor utility of this metric.
Figure 2 shows the initial rate kinetic data for GR inhibition by 3 and 5 (obtained by a circular dichroism assay,9 described in the Supporting Information), which was globally fitted to a competitive inhibition model (fitted parameters located in Table 2), respectively. To our knowledge, these are the first micromolar competitive inhibitors for GR that are not amino acids. Importantly, the more effective of these, 3, also inhibits GR isozymes from Bacillus anthracis with and without the so-called “valine bridge”, which has been shown to confer species specificity against the only other class of effective competitive inhibitors for GR3,11 (Figure S2 of the Supporting Information), suggesting that 3 may be developed as a broad antimicrobial chemotherapeutic.
Figure 2.
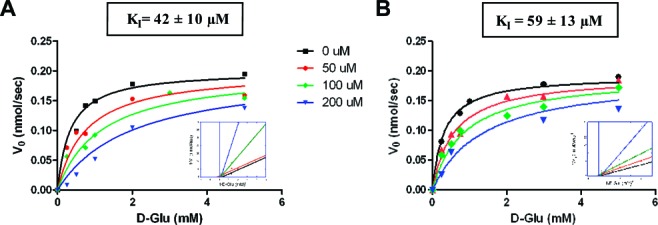
Experimental results for competitive inhibition of GR by compounds 3 (A) and 5 (B). Michaelis−Menten curves are globally fit to competitive inhibition models. See the Supporting Information for experimental methods described in full.
Table 2. Optimized Parameters from Global Nonlinear Regression (Competitive Inhibition Model).
| inhibitor | 3 | 5 |
|---|---|---|
| KM (mM) | 0.34 ± 0.08 | 0.30 ± 0.05 |
| Vmax (nmol/s) | 0.20 ± 0.01 | 0.19 ± 0.01 |
| KI (μM) | 42 ± 10 | 59 ± 13 |
| R2 | 0.97 | 0.98 |
Surprisingly, 1 was found to exhibit apparent noncompetitive inhibition against GR (Figure 3A). Shoichet and co-workers have thoroughly documented the pitfalls of hits, from virtual (and high-throughput) screening, that show apparent noncompetitive inhibition.12 It is often the case that such inhibitors form colloidal aggregates, which lead to local protein denaturation of the target enzyme and, thus, apparent noncompetitive inhibition. Colloidal aggregators can easily be identified by performing inhibition assays in the presence of increasing concentrations of mild detergents.13 This procedure was performed on 1, which showed almost total alleviation of inhibition in the presence of detergent, clearly identifying it as a colloidal aggregator and, thus, a false positive lead (Figure 3B).
Figure 3.
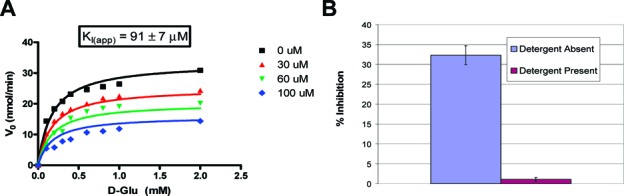
Compound 1 yields apparent noncompetitive inhibition using global kinetic analysis (A) but is shown to actually be a colloidal aggregator using the detergent assay of Shoichet and co-workers13 (B); the presence of 0.01% Triton X-100 in the assay mixture almost fully alleviates the enzyme inhibition (see the Supporting Information for a full description of this method).
It is instructive to examine the docked poses of 3 into GR using a more thorough approach than was used in the high-throughput docking that was employed in the initial screen. Docking into both the reactive form of GR and the crystal structure (PDB 1ZUW) were performed to compare the benefit of employing the reactive form in the original virtual screening. In these additional docking studies, 3 is minimized for each docked pose, followed by rescoring with the London dG scoring function (the computational details are described in the Supporting Information). Two salient features emerge from this docking study: (1) The shape of the active site pocket for the reactive form of GR enforces a very tight distribution of possible complex structures with 3 (Figure 4A,C), while the crystal structure (Figure 4B,D) has a wide range of distributions (the differential in tightness of placement is quantified below), and (2) calculated binding affinities between 3 and the reactive form of GR are uniformly stronger than those of 3 and the crystal structure (ΔG = −13.0 ± 0.80 kcal/mol for the reactive complex versus −11.1 ± 0.80 kcal/mol for the crystal complex). Free energy values obtained from the dG scoring function are not meant to provide a rigorous estimate of the actual free energy of binding but rather to provide a metric for ranking the affinity of a ligand for its receptor in an aqueous environment (see the Supporting Information for a full discussion of the dG scoring function). The averaged calculated dG value for the transition state complex and the crystal structure complex with 3, together with the ligand maps in Figure 4A,C, indicate a more favorable binding to the transition state receptor.
Figure 4.
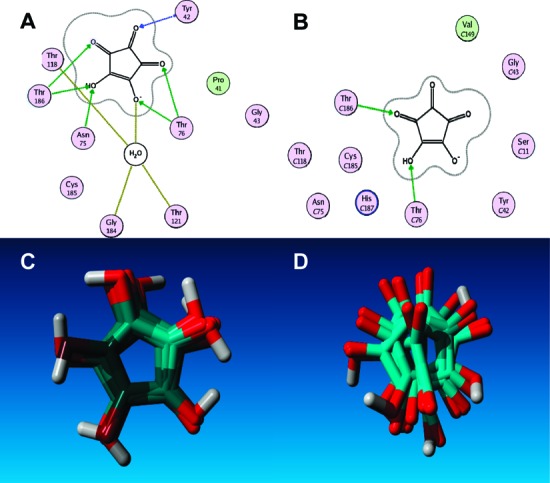
Ligand interaction maps for compound 3 docked in the reactive conformation (A) and crystal structure (B). Superposed renderings of the top-ranked docking positions of compound 3 in the reactive conformation (C) and crystal structure (D).
One may quantify the tightness in the distribution of the docked poses in the reactive complex with 3 versus the crystal structure by calculating the overlap in molecular volume between the docked poses, such that a high degree of overlap (i.e., a perfect overlap in which a compound is always docked to the exact volume) has a value of 1, while no overlap at all between docked poses yields a value of 0. Calculation of the Tanimoto volume (Tvol), which is described in the Supporting Information, has been shown to be a superior descriptor of similarity between the docked complexes and the more often used root-mean-square value.18 Here, we calculated a Tvol of 0.79 ± 0.03 for 3 docked into the reactive form of GR versus a value of 0.64 ± 0.17 for 3 docked into the crystal structure. This disparity is graphically illustrated in the overlap of docked poses for both complexes in Figure 4C,D, respectively.
The reactive form of GR tightly places docked poses of 3 into highly similar positions, as supported by the high Tvol value. However, the crystal structure receptor yields a wide range of positions for 3 with consistently lower calculated pKi values. Taken together, these volumes overlap, and calculated binding studies establish that the selection of the reactive form of GR for virtual screening is an attractive alternative approach for finding high-affinity lead compounds, as described in the current study.
Although 3 is not, a priori, a substrate analogue, it is, in many ways, a conformational analogue to the cyclic glutamate carbanion, the reactive intermediate described in Spies et al.,9 which is evidenced by calculating the Tvol of the glutamate carbanion versus the docked poses of 3 (Tvol = 0.55), as illustrated in Figure S3 of the Supporting Information. However, as Figure S3 of the Supporting Information indicates, the overlap between the cyclic glutamate carbanion and 3, while reasonable in terms of molecular volume, is not electrostatically optimal, due primarily to the region around the Cβ and Cγ positions of the glutamate carbanion. One may contrast the binding of 3 versus glutamine, a substrate analogue, which has a Ki of 50 mM. It is interesting to note that glutamine lacks the ionic and hydrogen bonding complementarity for forming intramolecular cyclized species as observed with glutamate. It may be that GR's affinity for substrate and transition state is partially derived from these cyclized forms.
The active site configuration of the complex between GR and 3 is structurally reminiscent of proline racemase (a structurally related family of cofactor independent racemases) with the transition state analogue pyrrole-2-carboxylic acid (PYC),19 which exhibits two binding states with Ki1 = 4.6 μM and Ki2 = 30 μM. The crystal structure of proline racemase reveals that PYC is compressed between two Cys residues, and the carboxylate receives five hydrogen bonds from the active site residues. The transition to the active form of proline racemase involves a conformation change that accommodates the planarity of the proline ring carbanionic intermediate, which is highly analogous to the case of the cyclic glutamate carbanion described in Spies et al.,9 as well as the nature of the lead compounds identified in the current study. In all of these systems, the plane of the ring is perpendicular to the axis connecting the two flanking Cys sulfur atoms. A superpose of reactive and crystal structure forms of the docked GR complexes illustrates that this perpendicularity between the plane of the ring and the sulfur atoms is only present in the reactive form, while the docked crystal structure complex is tilted and possesses significant ring strain (Figure 5). A characteristic feature of all of the docked crystal structure complexes with 3 is ring strain, with O−C−C−O carbonyl−carbonyl dihedral angles of approximately ∼25° versus ∼5° for complexes with the reactive form as the receptor. The differential hydrogen-bonding pattern in the docked crystal structure complex, relative to the reactive conformation, is responsible for the suboptimal ligation with 3. Additionally, there is a significant reduction in the volume of the active site pocket in the reactive form versus the crystal structure (from 188 Å3 in the crystal structure to 159 Å3 in the reactive form).
Figure 5.
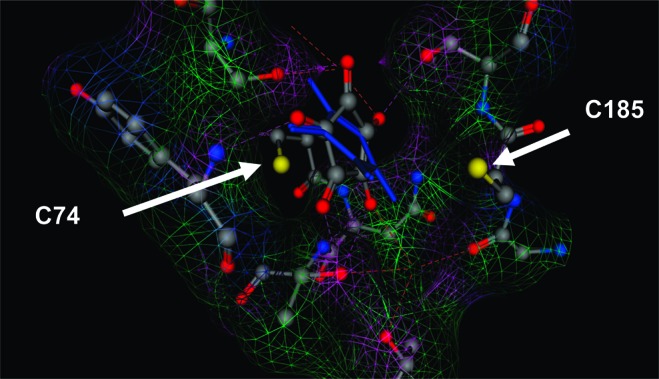
Superpose of the docked complex of GR with 3 from the reactive form (elemental color/ball and stick rendering) and GR with 3 from the crystal structure (cobalt blue/stick rendering). Each complex is from the top scoring docked configuration (see the Supporting Information for further details). Only the residues of the reactive form are shown for clarity. The Connolly (solvent-excluded) surface of the pocket of the reactive form of GR is rendered in colored mesh (purple is hydrogen bonding, green is hydrophobic, and blue is polar).
The most significant property of 3 is its very large ligand efficiency (LE) value of −0.6 kcal/mol/heavy atom. LE values are of paramount importance in fragment-based drug discovery (FBDD), which has been a highly successful and emerging approach for identifying promising drug candidates.20,21 The FBDD approach aims to grow small, weak-binding (yet possessing high LE value) inhibitors into nanomolar inhibitors, which retain Lipinski-like rule-of-five compliance throughout the process. LE values of −0.3 kcal/mol/heavy atom or greater are considered to be good, since careful optimization from MW ∼150 to >400 should yield a compound in the ∼tens of nanomolar range.21 However, 3 possesses an LE value far surpassing that of a standard fragment, such that the addition of a modest four heavy atoms places it in the nanomolar range. Thus, 3 is an excellent point from which to begin an optimization campaign, particularly in light of the fact that it is effective against three GRs, each unique in their oligomeric equilibrium as well as the presence or absence of the valine bridge (Val149 in RacE2; Figure S2 in the Supporting Information).
There are two widely accepted models that account for protein flexibility in small molecule binding, which may be of use when considering the preferential docking of lead compounds into the reactive form of an enzyme versus the corresponding crystal structure. The Monod−Wyman−Changeux (MWC) model describes an unliganded form of the enzyme, which may be in equilibrium with numerous conformations.22−25 The ligand may preferentially bind to one of these sampled receptor conformations. Alternatively, the Koshland−Nemethy−Filmer (KNF) model describes an “induced-fit” sequence, in which the ligand promotes a series of conformational changes in the receptor.26 The virtual screening methodology presented in the current work is appropriate for systems that exhibit behavior similar to the MWC model, since receptor conformational sampling may be determined a priori (i.e., the reactive conformation is simply an additional, albeit rare, form that may be elucidated by any number of methods, such as those described in Spies et al.9). However, systems exhibiting KNF behavior assume conformational states that may be unique for a particular protein−ligand complex, suggesting ambiguity in the receptor target.
Supporting Information Available
Experimental and computational methods, detailed results, and controls. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH AI076830 (M.A.S.) and NIH AI057156 (S.R.B.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gallo K. A.; Tanner M. E.; Knowles J. R. Mechanism of the Reaction Catalyzed by Glutamate Racemase. Biochemistry 1993, 32, 3991–7. [DOI] [PubMed] [Google Scholar]
- Bugg T. D.; Walsh C. T. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: Enzymology, antibiotics, and antibiotic resistance. Nat. Prod. Rep. 1992, 9, 199–215. [DOI] [PubMed] [Google Scholar]
- de Dios A.; Prieto L.; Martin J. A.; Rubio A.; Ezquerra J.; Tebbe M.; Lopez de Uralde B.; Martin J.; Sanchez A.; LeTourneau D. L.; McGee J. E.; Boylan C.; Parr T. R. Jr.; Smith M. C. 4-Substituted d-Glutamic Acid Analogues: The First Potent Inhibitors of Glutamate Racemase (MurI) Enzyme with Antibacterial Activity. J. Med. Chem. 2002, 45, 4559–70. [DOI] [PubMed] [Google Scholar]
- Lundqvist T.; Fisher S. L.; Kern G.; Folmer R. H.; Xue Y.; Newton D. T.; Keating T. A.; Alm R. A.; de Jonge B. L. Exploitation of structural and regulatory diversity in glutamate racemases. Nature 2007, 447, 817–22. [DOI] [PubMed] [Google Scholar]
- Tanner M. E.; Gallo K. A.; Knowles J. R. Isotope Effects and the Identification of Catalytic Residues in the Reaction Catalyzed by Glutamate Racemase. Biochemistry 1993, 32, 3998–4006. [DOI] [PubMed] [Google Scholar]
- Koo C. W.; Blanchard J. S. Chemical Mechanism of Haemophilus influenzae Diaminopimelate Epimerase. Biochemistry 1999, 38, 4416–22. [DOI] [PubMed] [Google Scholar]
- Pillai B.; Cherney M. M.; Diaper C. M.; Sutherland A.; Blanchard J. S.; Vederas J. C.; James M. N. Structural insights into stereochemical inversion by diaminopimelate epimerase: an antibacterial drug target. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzheinikov S. N.; Taal M. A.; Sedelnikova S. E.; Baker P. J.; Rice D. W. Substrate-induced conformational changes in Bacillus subtilis glutamate racemase and their implications for drug discovery. Structure 2005, 13, 1707–13. [DOI] [PubMed] [Google Scholar]
- Spies M. A.; Reese J. G.; Dodd D.; Pankow K. L.; Blanke S. R.; Baudry J. Determinants of Catalytic Power and Ligand Binding in Glutamate Racemase. J. Am. Chem. Soc. 2009, 131, 5274–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez J.; Shoichet B.. Virtual Screening in Drug Discovery; Taylor & Francis: Boca Raton, 2005; p 470. [Google Scholar]
- Dodd D.; Reese J. G.; Louer C. R.; Ballard J. D.; Spies M. A.; Blanke S. R. Functional comparison of the two Bacillus anthracis glutamate racemases. J. Bacteriol. 2007, 189, 5265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B. K. Screening in a spirit haunted world. Drug Discovery Today 2006, 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I. Property distribution of drug-related chemical databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (/MOE/), version 2008.10. Chemical Computing Group, Inc.,Montreal, Quebec, Canada, 2005. [Google Scholar]
- Bou A.; Pericas M. A.; Serratosa F. Synthetic applications of di-tert-butoxyethyne, II: New syntheses of squaric, semisquaric, and croconic acids. Tetrahedron Lett. 1982, 23, 361–364. [Google Scholar]
- Fatiadi A. J. Pseudooxocarbons. Synthesis of 1,2,3-Tris(dicyanomethylene)croconate Salts. A New Bond-Delocalized Dianion, Croconate Blue. J. Org. Chem. 1980, 45, 1338–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren G. L.; Andrews C. W.; Capelli A. M.; Clarke B.; LaLonde J.; Lambert M. H.; Lindvall M.; Nevins N.; Semus S. F.; Senger S.; Tedesco G.; Wall I. D.; Woolven J. M.; Peishoff C. E.; Head M. S. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [DOI] [PubMed] [Google Scholar]
- Buschiazzo A.; Goytia M.; Schaeffer F.; Degrave W.; Shepard W.; Gregoire C.; Chamond N.; Cosson A.; Berneman A.; Coatnoan N.; Alzari P. M.; Minoprio P. Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc. Natl. Acad. Sci. U.S.A. 2005, 103, 1705–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard S.; Berdini V.; Boulstridge J. A.; Carr M. G.; Cross D. M.; Curry J.; Devine L. A.; Early T. R.; Fazal L.; Gill A. L.; Heathcote M.; Maman S.; Matthews J. E.; McMenamin R. L.; Navarro E. F.; O'Brien M. A.; O'Reilly M.; Rees D. C.; Reule M.; Tisi D.; Williams G.; Vinkovic M.; Wyatt P. G. Fragment-Based Discovery of the Pyrazol-4-yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent Aurora Kinase Activity. J. Med. Chem. 2009, 52, 379–388. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- Tsai C. J.; Kumar S.; Ma B.; Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999, 8, 1181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. Ligand Binding and Internal Equilibria in Proteins. Biochemistry 1972, 11, 864–878. [DOI] [PubMed] [Google Scholar]
- Lee G. M.; Craik C. S. Trapping moving targets with small molecules. Science 2009, 324, 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. Jr.; Nemethy G.; Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.