

Abstract
Retinal degenerative diseases result in retinal pigment epithelial (RPE) and photoreceptor cell loss. These cells are continuously exposed to the environment (light) and to potentially pro-oxidative conditions, as the retina's oxygen consumption is very high. There is also a high flux of docosahexaenoic acid (DHA), a PUFA that moves through the blood stream toward photoreceptors and between them and RPE cells. Photoreceptor outer segment shedding and phagocytosis intermittently renews photoreceptor membranes. DHA is converted through 15-lipoxygenase-1 into neuroprotectin D1 (NPD1), a potent mediator that evokes counteracting cell-protective, anti-inflammatory, pro-survival repair signaling, including the induction of anti-apoptotic proteins and inhibition of pro-apoptotic proteins. Thus, NPD1 triggers activation of signaling pathway/s that modulate/s pro-apoptotic signals, promoting cell survival. This review provides an overview of DHA in photoreceptors and describes the ability of RPE cells to synthesize NPD1 from DHA. It also describes the role of neurotrophins as agonists of NPD1 synthesis and how photoreceptor phagocytosis induces refractoriness to oxidative stress in RPE cells, with concomitant NPD1 synthesis.
Keywords: age-related macular degeneration, retinal pigment epithelial cells, oxidative stress, neurotrophins, 15-lipoxygenase-1
RETINAL DEGENERATIVE DISEASES
Retinal degenerative diseases are a complex group of conditions with different etiologies that result in a common outcome: photoreceptor apoptotic cell death (1–5). Accordingly, there are differences in how these conditions evolve. For instance, in retinitis pigmentosa (RP), rod photoreceptor death initially occurs in the periphery, whereas in age-related macular degeneration (AMD), death is initiated in the macular zone and spreads in later phases throughout the retina (2, 5). RP is a collection of inherited blinding diseases caused by the mutation of a wide variety of genes resulting in more than 150 abnormalities of photoreceptor-specific proteins, including mutations of rhodopsin, peripherin, the β-subunit of cGMP phosphodiesterase, and retinal outer-segment membrane protein 1 (6–8).
Conversely, the etiology of AMD, which is the leading cause of blindness over the age of 65, is not as clear as that of RP. AMD is also a heterogeneous group of disorders, but the causes are proposed to be multifactorial, and the main known risk factors are both genetic and environmental (2, 5). There are two forms of AMD: the dry and the wet form. In the dry form, photoreceptors degenerate slowly and progressively, producing a thinning in the retinal layers and leaving deposits known as drusen. In the wet form, the less common of the two AMDs, the predominant feature is invasive choroidal neovascularization, which leads to severe vision loss (9).
AMD also affects the choriocapillaris and Bruch's membrane, which separates the retinal pigment epithelium (RPE) from the blood vessels (10). The results from the Age-Related Eye Disease Study (NEI) show that the intake of high amounts of antioxidants and zinc can reduce the risk of developing advanced AMD by about 25%, implying an indirect role for oxidative stress in the pathogenesis. More specifically, the presence of oxidative damage markers in postmortem retinas of patients with geographic atrophy shows, at least in the dry form, oxidative stress is involved in the pathogenic mechanisms of AMD (11). In this manner, oxidative stress is enhanced and exaggerated and mitochondrial function is compromised (8–13), which leads to apoptotic cell death. Tunel studies performed on postmortem human retinas of patients presenting geographic atrophy and exudative forms of the disease show that apoptosis is the main mechanism of degeneration, not only for photoreceptors, but also for the RPE and inner retinal layers (12).
Initiation and progression of AMD involves the unsuccessful resolution of the inflammatory response. Single nucleotide polymorphisms occurring in the gene encoding factor H (CFH/HF1) (14–17) were proposed to be a major risk factor for AMD. Factor H is an inhibitor of the alternative pathway of complement system activation that, as a result, has the ability to limit cell injury and inflammation (18, 19). Conversely, studies that focused on other regulatory proteins of the complement pathway, such as factor B and complement component 2 (C2) (18, 20), exhibit protective effects and reduce the risk of AMD to some extent. For example, the E318D variant of C2 (H10) as well as a variant in intron 10 of C2 and the R32Q variant of factor B (H7) confer a reduced risk of AMD.
Therefore, the identification of early pro-survival, anti- inflammatory signaling critical for the maintenance of photoreceptor cell integrity may be applicable for novel therapeutic intervention/s for slowing or halting disease progression.
THE PROTECTIVE ROLE OF THE RETINAL PIGMENT EPITHELIUM
As in the brain, retinal function relies on glycolysis and oxidative phosphorylation, which are coupled to the citric acid cycle. These processes require a sustained delivery of oxygen and glucose as well as adequate control of the enzymes that allow for equilibrated formation and consumption of ATP. The blood-retinal barrier actively promotes homeostasis by tightly controlling the stoichiometry and activity of a network of proteins that maintain ionic gradients and metabolic transport systems and preserve the environment through detoxifying mechanisms that scavenge and remove toxic molecules (Fig. 1) (21).
Fig. 1.

RPE/photoreceptor interactions and NPD1 bioactivity. A: RPE control of the permeability of the outer blood-retinal barrier involving remodeling of the blood vessels and selective flux of nutrients and catabolites. B: NPD1 pro-survival enhances the expression of Bcl-2 proteins and decreases COX-2 expression. C: Shedding and phagocytosis of the photoreceptor outer segments.
The outer blood-retinal barrier is composed of three distinctive structures: the fenestrated endothelium of the choriocapillaris, Bruch's membrane, and the RPE (21). The outer blood-retinal barrier mediates the exchange of small molecules and solutes and other metabolites from the blood stream to the photoreceptor layer (22). RPE cells are the most restrictive layer of the three components of the outer blood-retinal barrier, preventing the passage of biomolecules based on size and charge and thus preserving a controlled environment for photoreceptors. The retinal pigment epithelium, like other epithelia, is a compact structure where cells communicate laterally through tight junctions. In addition, the retinal pigment epithelium presents an elaborate transcellular transport system and a high polarization, allowing it to have different functions (22). The selective permeability of the outer blood-retinal barrier depends on RPE integrity. RPE cells are involved in the preservation of these structures by interacting reciprocally in their formation and maintenance.
While the retinal pigment epithelium plays a role in regulating nutrition, intercellular and intra/inter-tissue communication and remodeling, scavenging of sub-products, retinal functions, and promotion of neurotrophic signaling, it is also responsible for the recycling of the photobleached pigments produced by the photoreceptor activity of rods and, to some extent, the cones (23). Rhodopsin is a light-sensitive molecule that transduces the photon into a biochemical signal in rods. It is composed of the G-coupled receptor protein opsin and of the chromophore 11-cis-retinal. When rhodopsin interacts with a photon, 11-cis-retinal is isomerized to all-trans-retinal. The regeneration of 11-cis-retinal from all-trans-retinal takes place between the photoreceptor outer segment and the RPE (24). All-trans-retinol is esterified with a fatty acid to be isomerized to 11-cis-retinol and hydrolyzed from the ester bond in RPE cells. Then 11-cis retinol is oxidized to 11-cis-retinal, and the active chromophore then leaves the RPE apical surface and moves to the photoreceptor to regenerate rhodopsin; afterward, the cycle begins again (reviewed in 25 and 26). Failure of the retinal pigment epithelium to accomplish its function in the retinoid cycle leads to retinal degeneration. For instance, autosomal-recessive inherited retinitis punctata albescens has been associated with mutations in the RLBP1 gene that encodes the cellular retinaldehyde-binding protein, which carries 11-cis-retinol and 11-cis-retinaldehyde in the RPE and Müller cells. When cellular retinaldehyde-binding protein is mutated, it loses the ability to bind the second ligand, and, as a consequence, all-trans-retinyl esters accumulate and evolve to produce RPE atrophy, retinal pigmentary changes, and decreased blood vessel development (27).
Integral to the fragility of photoreceptor cells is their close relationship with RPE cells. In Stargardt's disease, a juvenile form of AMD, RPE cell functional integrity is initially compromised, and in turn photoreceptors are damaged. Once RPE cells die, photoreceptor cells then succumb (28). In other words, similar to what was proposed to occur in the blood-brain barrier in the neurovascular hypothesis of Alzheimer's disease (AD) (29), defective clearance of certain molecules across the blood-retinal barrier may initiate a series of faulty maintenance functions that could lead to a retino-vascular inflammatory response, contributing to the development of AMD.
RPE cells and vascular remodeling
The significance of RPE cells in vascular remodeling is highlighted in several studies. For example, absence of RPE cells in mice expressing fibroblast growth factor (FGF) 9 directed by the tyrosinase-related protein 2 promoter (FGF9 transgenic mice) is due to forcing embryonic RPE cells to become neural retinal cells through the ectopic expression of FGF9 (30). These mice fail to form blood vessels in the choroidal layers adjacent to regions where RPE cells are absent; however, vessels are found near the patch where RPE cells are present at postnatal day 7, indicating the importance of these cells in vessel formation. Moreover, dependency between RPE and endothelial vascular cells continues during adulthood through regulation of neovascularization. Compelling evidence links RPE cells with the secretion of angiogenic-related factors. In particular, RPE from transgenic apolipoprotein E2 mice, which express human apolipoprotein E2 protein and whose eyes present features common to AMD patients, shows reciprocal unbalanced expression of pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF), indicating that neovascularization may be increased (31). Furthermore, autocrine VEGF signaling in RPE cells stimulates VEGF-related gene expression as well as PEDF modulation (32), which is a potent angiogenic inhibitor (33). Taken together, the balanced production and secretion of these factors contribute to the formation, maintenance, and remodeling of cells that surround the RPE layer and/or are in their vicinity.
The maintenance of photoreceptor cell integrity
Photoreceptor cells shed outer segment tips, which are then phagocytized by RPE cells in a daily, intermittent, and circadian fashion in mammals (34–36). RPE cells are the most active phagocytes of the body. In mammals, the circadian shedding and phagocytosis has been calculated to be complete after 10 days for one photoreceptor outer segment (36, 37). In rhesus monkeys, every RPE cell interacts with 20–45 photoreceptor tips (38), and the human macula support the RPE/photoreceptor interaction in a ratio of 1 to 23 (39).
The constant light reception activity by photoreceptors exhausts pigments and other molecules involved in the photo-transduction process. As a consequence, there is a need to replace these molecules. In this context, the reconstruction of the outer segments requires molecular building blocks and energy. In fact, the length of the outer segments remains constant as a consequence of a well-regulated biogenesis of photoreceptor membranes in the inner segments coupled to phagocytosis of the shed tips. During photoreceptor outer segment renewal, proteins turn over and are continually replaced (37). In contrast, docosahexaenoic acid (DHA) and vitamin A from the opsin chromophore are recycled back from the RPE to inner segments through the interphotoreceptor matrix. The retinoid in the rods includes the reisomerization of all-trans retinal back to 11-cis retinal in the RPE. Furthermore, the interdependence between RPE cells and photoreceptors is notable in Usher type 1B syndrome. The lack of myosin VIIa in this progressive disease affects the ability of RPE cells to phagocytize photoreceptor outer segments, leading to retinal degeneration in a mouse model (40).
The RPE-photoreceptor outer segments are potentially highly susceptible to oxidative stress because of the high oxygen consumption of the retina, active flux of PUFAs (e.g., omega-3 and also omega-6), and exposure to light (41, 42). Recently it was shown that phagocytosis (24–48 h) of oxidized photoreceptor outer segments containing high oxidative products induces the downregulation of complement factor H in RPE cells, similar to the effect of pro-inflammatory cytokines tumor necrosis factor-α (TNFα) and interleukin (IL)-6 (43). The RPE complement regulatory system, in this manner, may be suppressed by pro-inflammatory conditions as well as phagocytosis of oxidized photoreceptor outer segments. Surprisingly, the process enhances refractoriness to oxidative stress-induced apoptosis in RPE cells (Fig. 2A) (44). The protective effect of photoreceptor outer segments is specific, because the phagocytosis of polystyrene microspheres by RPE cells does not lead to a protective response against oxidative stress. Furthermore, polystyrene microspheres failed to induce DHA release and activate synthesis of neuroprotectin D1 (NPD1); this will be discussed in the following section. Interestingly, photoreceptor outer segment-mediated RPE cell protection against oxidative stress, with concurrent activation of NPD1 synthesis, was shown in ARPE-19 cells (44), a spontaneously immortalized human cell line (45), as well as in low passage primary human RPE cells prepared from National Disease Research Interchange-supplied eyes (unpublished observations).
Fig. 2.

Photoreceptor outer segment phagocytosis elicits protection in RPE cells subjected to oxidative stress. A: Quantitative analysis of Hoechst stained ARPE-19 cells indicates that photoreceptor outer segment phagocytosis significantly decreases the amount of apoptosis observed during oxidative stress. Phagocytosis of polystyrene microspheres during oxidative stress did not alter the amount of apoptosis observed during oxidative stress alone. Results represent averages ± SEM of repeats of two independent experiments. B: NPD1 changes as a function of time after photoreceptor outer segment phagocytosis or microspheres: effect of oxidative stress. NPD1 has been quantified in cells as well as in incubation media. Data represents average ± SEM of two independent studies. Statistical analysis is Student's t-test. NS, not statistically significant.
DHA release and NPD1 formation
RPE cells respond to oxidative stress by activating synthesis of NPD1 from DHA (46). The name NPD1 was suggested based upon its neuroprotective bioactivity in oxidative stressed RPE cells and the brain (46, 47) and its potent ability to inactivate pro-apoptotic and pro-inflammatory signaling. D1 refers to its being the first identified stereoselective mediator derived from DHA. NPD1 can be formed from free (unesterified) DHA released from membrane phospholipids by a phospholipase A2 (PLA2) upon stimulation (Fig. 3A). DHA belongs to the essential omega-3 essential fatty acid family (derived from linolenic acid, 18:3, n-3). Photoreceptor cells are highly enriched in DHA, and they tenaciously retain DHA even during very prolonged periods of omega-3 fatty acid deprivation (41, 48, 49).
Fig. 3.

A: NPD1 biosynthesis. Representation of the oxygenation of DHA to form NPD1. PLA2 releases DHA from the second carbon position of the phospholipids upon stimulation. 15-Lipoxygenase-1 catalyzes the synthesis of 17S-H(p)DHA, which is converted to a 16(17)- epoxide and then is enzymatically converted to NPD1. B: Comparison of NPD1/PD1 biosynthesis with that of 10S,17S-diHDHA isomer (see detailed discussion in the text).
The amount of unesterified DHA simultaneously measured in RPE cells and in incubation media by MS/MS was found to be increased as a function of time during exposure to oxidative stress in RPE cells. Specifically, the free intracellular DHA pool size showed a moderate increase after 6 h when cells were subjected only to photoreceptor outer segment phagocytosis (44). Oxidative stress, however, strongly enhanced free DHA accumulation in a time-dependent fashion, peaking at 16 h (44). Interestingly, although the overall increase reached 10-fold, photoreceptor outer segment phagocytosis kept the DHA pool size at a constant 2.4-fold increased level. This implies that NPD1 synthesis does not result from the simple enhancement of the overall availability of free DHA upon phagocytosis. There is a general correlation between increases in free DHA pool size and in NPD1 synthesis. Photoreceptor outer segment phagocytosis stimulates NPD1 synthesis at 3–6 h in cells and accumulation in media after 16 h, while free DHA increases earlier and keeps accumulating up to 16 h. These enhancements in DHA and NPD1 pool size are much larger when photoreceptor outer segment phagocytosis takes place on RPE cells exposed to oxidative stress. Interestingly, microsphere phagocytosis does not cause enhanced changes in DHA and NPD1 (Fig. 2B). As such, a very specific free DHA pool may be the precursor for NPD1.
Therefore, the supply of DHA and the induction of NPD1 synthesis during photoreceptor outer segment phagocytosis represents a homeostatic regulatory event for RPE cell protection in conditions of oxidative stress challenge and, as a consequence, the fostering of photoreceptor cell integrity (44). In this context, not only is photoreceptor outer segment phagocytosis in RPE cells essential for photoreceptor cell function, but the survival of the RPE promoted by this process correlates with NPD1 synthesis.
On the structure of NPD1, biosynthesis, and stereochemical assignment
The results discussed in this review provide the biological basis for the important actions of NPD1 derived from DHA. In this section, we shall consider the results obtained from several independent lines of investigation required to address the structure of the potent bioactive NPD1. As with other bioactive mediators, such as the eicosanoids (50), it is important to establish the stereochemistry of the compound and/or mediator, because many structurally related products can be less active, inactive, or even in some cases display opposing biologic actions as a result of subtle changes in stereochemistry that are recognized in biologic systems. To confirm the proposed basic structure and establish the complete stereochemistry, these studies on the 10,17S-docosatriene termed NPD1 included results from biosynthesis studies, matching of materials prepared by total organic synthesis with defined stereochemistry, and the actions of these and related compounds in biological systems (42, 44, 46, 47, 51–55). We also considered the chronology in which these findings appear in the literature with the goal of providing a clear and rigorous account of the evidence that supports the structure and bioactions of NPD1/protectin D1 (PD1) for the readership of The Journal of Lipid Research. As interested JLR readers will surmise, investigations along these lines were essential to establish the complete structure of the potent NPD1/PD1 and related endogenous products biosynthesized from DHA in vivo/in situ because of the small amounts of NPD1 attainable from biological systems at the time, which precluded direct stereochemical analyses of the products identified in RPE cells. Thus, in this section, we focus on the evidence for NPD1/PD1 structural elucidation. JLR readers interested in the structural elucidation of the resolvins and their complete stereochemical assignments are directed to other recent reviews (56, 57).
In 1984, the first evidence was obtained for the conversion of DHA to mono-, di-, and tri- DHA-derived products, named docosanoids, in the retina (an integral part of the central nervous system) (58). Use of available inhibitors of the time suggested a role for lipoxygenase in the biosynthesis of these compounds. An initial step in docosanoid synthesis was envisioned to be the release of DHA from membrane phospholipids by PLA/sA2, early demonstrated to be rapidly activated by ischemia or seizures (41). The structure of 10,17-docosatriene was first disclosed while reporting on the characterization of the novel bioactive resolvins that were identified using a systems approach with resolving inflammatory exudates and LC-MS-MS-based lipidomics (51). These new compounds (resolvins and docosatrienes) were biosynthesized from omega-3 essential fatty acids during the resolution phase of acute inflammatory reactions in vivo that promote resolution of inflammation in vivo [see Fig. 8 in reference (51) and related text]. Since the DHA-derived compounds we identified in resolving inflammatory exudates, additional evidence was obtained for their biosynthesis from murine brain and vascular endothelial cells for the new bioactive compounds (47). These investigations focused on aspirin and its impact in the biosynthesis of 17R-hydroxy-containing resolvins and related structures. The initial results indicated that DHA-derived products reduced cytokine IL-1β production by human glioma cells stimulated with TNFα. In parallel, studies with human cells were carried out to reconstruct the potential biosynthetic routes involved in the biosynthesis of these mediators. In this context, hypoxic endothelial cells exposed to inflammatory stimuli in vitro converted DHA and eicosapentaenoic acid to intermediates that were taken up by human leukocytes and further converted to bioactive products that showed potent activities relevant to the control of inflammation (51, 59, 60).
Of interest to the present review, in these investigations without aspirin treatment, 17S-HDHA and corresponding 17S-hydroxy-containing di- and trihydroxy products were reported in murine exudates and isolated human cells (51). The formation of some of these compounds was modeled in vitro and formed by sequential lipoxygenation reactions. These products were investigated with 15-lipoxygenase and included the double dioxygenation products, namely 7S,17S-diHDHA and 10,17S-diHDHA, which were identified along with trihydroxy-containing products formed via epoxide-containing intermediates from DHA (51). The well-established lipoxygenase reaction mechanism suggested that new products 7S,17S-diHDHA and 10,17S-diHDHA, which could easily be made in vitro, each contained two diene conjugated double bond systems both in a trans,cis geometry. For example, the well-known 5S,12S-diHETE formed from arachidonate via double dioxygenation is an isomer of the potent chemoattractant LTB4 (50). This isomer 5S12S-diHETE separates in SP-HPLC from LTB4 and are very similar to each other, but 5S12S-diHETE shows little chemotactic activity compared with LTB4 (61).
DHA is also precursor to a novel family of endogenous docosatrienes formed in blood, leukocytes, brain, and glial cells (46, 47, 52). The main bioactive member of the docosatrienes from the 17S-hydroxy-containing docosanoids proved to be 10,17S-docosatriene, in addition to the resolvins (47, 52). Also, human polymorphonuclear leukocytes (PMN) convert 10,17S-docosatriene to its omega-22 hydroxy product with DHA as the precursor; this is likely an inactivation route for this compound (47). As with the new bioactive products from omega-3 precursors, the basic structures and proposed stereochemical assignments reported were based on results of biosynthesis studies and given as tentative stereochemical assignments, because matching studies with synthetic reference compounds of known stereochemistry were still underway (see below). At the time, some of the newly identified compounds were matched to reference compounds prepared with plant lipoxygenases, e.g., 17S-HDHA and 7S,17S-diHDHA, which matched to those profiled by LC-MS-MS in exudates and with isolated cells. It was clear that the 10,17S-docosatriene from exudates did not coelute with the major 10,17S-docosatriene produced in vitro with plant enzymes, suggesting it was an isomer; however, this system could be used to prepare related docosatrienes via the LTA4 synthase activity of lipoxygenases to further probe the bioactions of the 10,17S-docosatriene while complete matching and total synthesis were in progress.
Of note, glial cells generate both 17S series resolvins and the 10,17S-docosatriene. Importantly, evidence for a novel omega-22-hydroxy-16,17S-trihydroxydocosatriene was obtained from these cells and human PMN, which suggest that the 10,17S-docosatriene biosynthesis is via a 16(17S)-epoxide-containing intermediate, because the identified vicinal diol could be a product of this epoxide intermediate. Hence, a series of alcohol trapping studies were undertaken to address the potential role of epoxide-containing intermediates in the biosynthesis of the bioactive compounds, in particular the 10,17S-docosatriene. Indeed, evidence for epoxide-containing intermediates in the biosynthesis of docosatrienes and 17S series resolvins was obtained from human PMN (52). In these incubations with human cells, two 16-OCH3 and two 10-OCH3 methoxy-trapping products, likely all-trans in their triene conjugation, were obtained, which implicates production of a 16(17S)-epoxide intermediate that was proposed in the biosynthetic pathway for the bioactive 10,17S-docosatriene (52). Also, 10,17S-docosatriene proved to display potent actions with human glial cells and produced 10,17S-docosatriene, which reduced IL-1β production at 1–50 nM and evoked ligand-operated extracellular acidification with glial cells in a microphysiometer (52). These findings indicated that DHA is precursor to novel potent protective mediators and that 10,17S-docosatriene carries potent anti-inflammatory activity in mice in vivo and with human cells in vitro as well as activated surface receptors present on human glial cells to regulate their function. Thus, the basic structure of the novel 10,17S-docosatriene, later coined NPD1/PD1 (see below) due to its potent actions in vivo and in vitro in cell cultures, was established (47, 51, 52). This docosatriene also displayed potent anti-inflammatory actions, i.e., reducing PMN numbers in vivo and reducing the production of inflammatory cytokines by glial cells in vitro. Moreover, during the resolution phase of peritonitis, unesterified DHA levels increase in resolving exudates, where it appears to promote catabasis or the return to homeostasis following tissue insult via conversion to D-series resolvins and also 10,17S-docosatrienes (62) by shortening the resolution interval of an inflammatory response in vivo (63).
We then found that the DHA-derived 10,17S-docosatriene proved to be generated in vivo during experimental stroke in the ipsilateral cerebral hemisphere following focal ischemia and also demonstrated potent bioactions in this system, where it limits the entry of leukocytes, downregulates cyclooxygenase-2 expression and nuclear factor κB activation, and decreases infarct volume (47). Next, we found that 10,17S-docosatriene is formed in the human retinal pigment epithelial cell line, ARPE-19, and introduced the term NPD1 based on its neuroprotective bioactivity (46, 47).
Taken together, these findings in ARPE-19 cells (46), inflammatory murine exudates, human PMN, glial cells, and the brain (47, 51, 52) underscored the need to establish the complete stereochemistry of endogenous, biologically generated, active 10,17S-docosatriene, namely the chirality of its carbon 10 position alcohol and its triene double geometry, which remained to be established (Fig. 3B). These involved the total organic synthesis and matching of both bioactivity and physical properties of the endogenous compound and those of the synthetic with established stereochemistry. In recognition of its wide scope of formation and uncovered actions, PD1 was introduced and used to denote the structure of this chemical mediator in the immune system. The prefix Neuro before PD1 (NPD1) was proposed to denote its biosynthesis and potent neuroprotective actions (53). It was also apparent that NPD1/PD1 was a member of a larger family of 17-hydroxy-containing docosatrienes, termed protectins.
Biosynthesis and function studies were undertaken with human TH2-skewed peripheral blood mononuclear cells (PBMC) that specifically express 15-lipoxygenase type 1 and convert DHA to the 10,17S-docosatrienes by serving as a 17-lipoxygenase with DHA as a substrate. When produced by these cells, PD1 promotes T cell apoptosis via the formation of lipid raft-encoded signaling complexes and reduces T-cell traffic in vivo. These results were consistent with the physical properties of NPD1. Matching materials prepared by total organic synthesis determined the complete stereochemistry of the PBMC DHA-derived product. NPD1/PD1 generated by human PBMC carried the complete stereochemistry of (10R,17S)-dihydroxydocosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid and was matched to the most potent bioactive product using several dihydroxytriene-containing, DHA-derived products isolated from human PBMC, human PMN, and murine exudates (53, 54).
During the course of these investigations, Butovich et al. (64) reported that NPD1 had the complete structure of 10S,17S-diHDHA. This was based on results obtained with isolated lipoxygenase enzymes incubated with DHA without mammalian cell/tissue biosynthesis or authentic NPD1, as defined earlier in the literature (46, 51, 52). Importantly, neither the bioactivity of the product nor appropriate comparisons with authentic NPD1 was presented to support the conclusions in this report (64) in regards to the complete structure of NPD1. Note that 10S,17S-diHDHA is an isomer of NPD1/PD1 (Fig. 3B).
With the preparation of six stereochemically defined, 10,17-dihydroxy-containing geometric isomers by total organic synthesis that had been initiated earlier, it was possible to match the stereochemistry and biological actions of the endogenously produced materials (54). In addition to PD1 formed from human leukocytes, additional isomers were identified in inflammatory exudates, including Δ15-trans-PD1 (isomer III), 10S,17S-dihydroxy-docosa-4Z,7Z,11E,13Z,15Z,19Z-hexaenoic acid (isomer IV), and the expected double dioxygenation product 10S,17S-dihydroxy-docosa-4Z,7Z,11E,13Z,15E,19Z-hexaenoic acid (isomer I), which are present in inflammatory exudates obtained from mice. 18O-labeling results provided evidence that this isomer I was a double dioxygenation product and that the 10 position of NPD1/PD1 originated from enzymatic conversion. Also, the rank order of activities was established between these isomers, and NPD1/PD1 proved to be most potent (55), with doses as low as 1–10 ng reducing murine peritonitis, followed by Δ15-trans-PD1 > 10S,17S-diHDHA (isomer I). Hence, although the double dioxygenation product 10S,17S-diHDHA was generated in murine exudates, it was far less active than PD1/NPD1 both in vitro and in vivo.
The proposed biosynthetic route for NPD1/PD1 is shown in Fig. 3B from results previously reported (54). Following 17S-HpDHA formation from 15-lipoxygenase action on DHA, an epoxide intermediate is formed that requires enzymatic transformation to obtain the correct double bond geometry, namely cis,trans,trans present in NPD1/PD1. This double bond geometry and chirality of the carbon 10 position and the R configuration were established from the matching of synthetic compounds of defined chirality. Of interest, both 7S,17S-diHDHA (resolvin D5) and 10S,17S-diHDHA are double dioxygenation products, and at this point they appear to be less active than endogenous or synthetic NPD1/PD1. Without endogenous biosynthesis studies or assessment of biological actions, it is surprising that a claim could be made for assessment of the complete stereochemistry of NPD1 based only on nuclear magnetic resonance results obtained for 10S,17S-diHDHA from plant lipoxygenase-prepared material with DHA (64).
With the stereochemistry of NPD1/PD1 established, its identification in human material was sought and found to be present in breath condensates from human asthmatics (65) and in the human brain, both under basal conditions and from patients with AD (66). In addition, PD1 was found to be a major product in bone marrow of female rats fed eicosapentaenoic acid and DHA (67). PD1 was generated in vivo during ischemia-reperfusion of renal tissues, where it has profound actions, namely reversing the deleterious consequences of ischemia-reperfusion in renal tissues (68), in agreement with our previous observations in the brain (47).
With the complete stereochemistry and synthetic compound in hand, it was possible to demonstrate for the first time that PD1 activates resolution programs in vivo and that it shortens the resolution time of experimental inflammation in animal models (63). With the total organic synthesis route of NPD1/PD1 in place, it was possible to radiolabel and purify 3H-NPD1/PD1 made from the synthetic intermediate. With this radiolabel, it was then possible to define for the specific binding sites present with ARPE-19 cells (Kd ∼31 pM/mg cell protein) for 3H-NPD1/PD1 as well as specific binding to human neutrophils that gave a Kd of ∼25 nM (55). Most importantly, critical information on NPD1 biosynthesis with ARPE-19 cells was obtained, namely identification of alcohol-trapping products, indicating the formation of a 16,17S-epoxide-containing intermediate from DHA in the biosynthesis of NPD1 (55). These results, as well as the rank order of potency established for NPD1/ PD1 of defined stereochemical analysis, indicated that the most potent of the isomers prepared was NPD1/PD1. Also in the ARPE-19 cells, NPD1 was more potent than either resolvin D1 or resolvin E1. In other systems, resolvin E1 and resolvin D1 established higher potencies than PD1 (69).
Along with the complete stereochemical identification of NPD1/PD1, recent studies using an unbiased LC-MS-MS profiling approach demonstrated that PD1 is made during the resolution of Lyme disease infections in mice (70). NPD1 inhibits retinal ganglion cell death (71), is renal protective (72), and regulates adiponectin (73). Of interest, the double dioxygenation product 10S,17S-diHDHA isomer of NPD1/PD1 was recently shown to have actions on platelets, reducing platelet aggregation at 0.3 μM, 1 μM, and higher concentrations (74). In peritonitis, this isomer also showed biological activity but was less potent than NPD1/PD1 (54, 74). It is noteworthy that NPD1/PD1 and the resolvins also are produced by trout tissues, including trout brain, from endogenous DHA, suggesting that these structures are highly conserved from fish to humans (75). This shows we still have much to learn regarding the bioactions and functions of NPD1/PD1, the D-series and E-series resolvins, and related products in human physiology and pathophysiology as well as in biological systems such as fish, where the actions of NPD1/PD1 remain to be fully appreciated.
Neurotrophins induce the synthesis and release of NPD1 from human RPE cells
RPE cells also are capable of producing a wide variety of growth factors (76). These trophic factors support surrounding cells by paracrine and autocrine signaling and hence promote the communication and structure of the retina as well as photoreceptor survival (77, 78). It is important to note that neurotrophins enhance the production of NPD1 in RPE cells. In turn, NPD1 is released and serves as a lipid signaling messenger in its surroundings. This observation was made in human RPE cells grown to confluence using a specialized culture (79) that allows the cells to develop a high degree of differentiation, preserving the apical-basolateral polarization (Fig. 4A, C). Neurotrophins [pigment epithelium derived factor (PEDF), BDNF, ciliary neurotrophic factor, FGF, glial-derived neurotrophic factor (GDNF), leukemia inhibitory factor, NT3, or persephin], which have bioactivities that promote neuronal and/or photoreceptor cell survival, are also agonists of NPD1 synthesis (Fig. 4B), favoring the release of this lipid messenger through the cell's apical surface (80). Among all the growth factors tested, PEDF is by far the most potent stimulator of NPD1 synthesis. PEDF, a member of the serine protease inhibitor (serpin) family, was identified in human RPE cells. If PEDF or ciliary neurotrophic factor are added to the incubation media, bathing the basolateral side in increasing concentrations, they evoke a lesser degree of NPD1 release on the apical side. Conversely, if these neurotrophins are added to the apical side, they exert concentration-dependent increases in NPD1 release only on the apical side (Fig. 4B, C) (80).
Fig. 4.
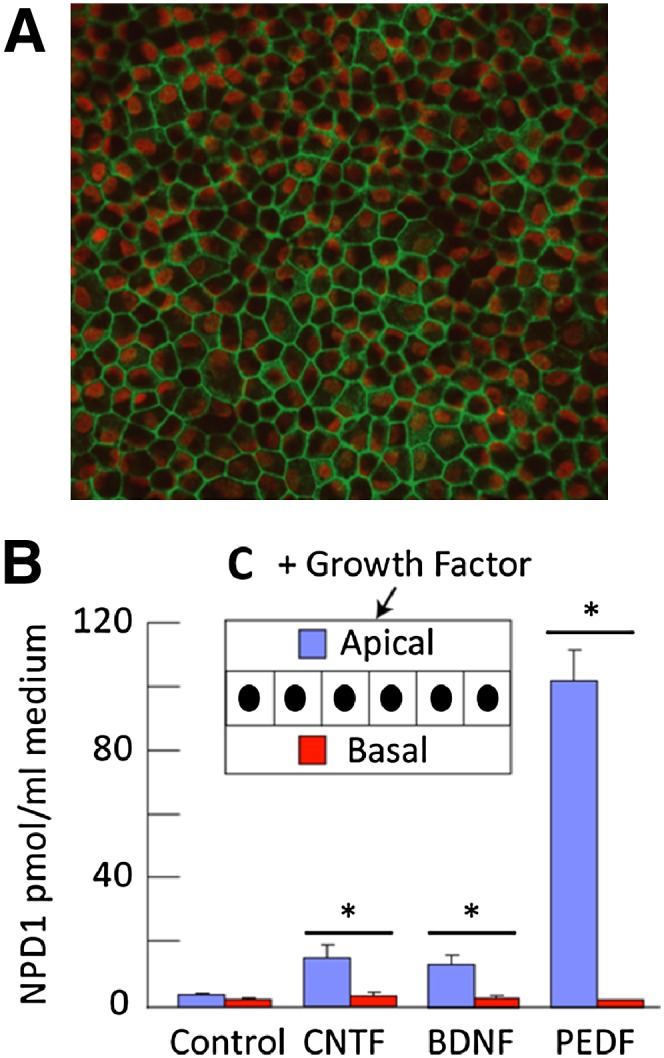
Neurotrophins activate NPD1 synthesis in cultured primary human RPE cells. A: Zonula occludens-1 (ZO-1) antibody immunoreactivity (green) illustrates confluence of the monolayer polyhedric-shape of the cells. B: Differential ability of growth factors to selectively release NPD1 through the apical surface of the cell. Growth factors (20 ng/ml) were added to the apical medium. Apical and basal media were collected separately after 72 h and subjected to lipidomic analysis. Each bar is an average ± SEM of four or five independent wells. Values are averages ± SEM of five independent wells. Statistical analysis was performed using Student's t-test shows *P < 0.05. C: Schematic representation of the monolayer orientation within the insert. [Fig. 4 A, C, modified with permission from reference (40)].
NPD1 biosynthesis is an endogenous response to oxidative stress
Previous studies have shown that the retina forms mono-, di-, and trihydroxy derivatives of DHA and that lipoxygenase inhibitors block this synthesis, indicating an enzymatic process of a lipoxygenase nature (42). Although the stereochemistry and bioactivity of DHA-oxygenated derivatives were not defined at the time of these observations, it was suggested that these lipoxygenase products might be neuroprotective (and therefore the name docosanoids was introduced) (42, 81). Liquid chromatography-photodiode array-electrospray ionization MS/MS-based lipidomic analysis was used to identify oxygenation pathways for the synthesis of the docosanoid NPD1 during brain ischemia-reperfusion (47) and the retinal pigment epithelium (46). Moreover, it was also found that RPE cells have the ability to synthesize NPD1 (46). Photoreceptors and RPE cells, although they contain phospholipids richly endowed with DHA (as docosahexaenoyl- or DHA-elongated fatty acyl-chains), display an undetectable quantity of unesterified (free) DHA [as is the case with unesterified arachidonic acid (AA)] under basal, nonstimulated conditions (82–86). This means that the pool size of unesterified DHA is tightly regulated by production (PLA2), by its removal (e.g., by reacylation), and by peroxidation. Free DHA incorporated into membrane phospholipids first becomes the substrate of docosahexaenoyl-CoA synthesis for its channeling through acyltransferases, which incorporate this fatty acid into phospholipids (87–90). The RPE cell thus modulates the uptake, conservation, and delivery of DHA to photoreceptors (81). In addition, RPE cells utilize a specific DHA-phospholipid pool as a precursor for NPD1 synthesis. Then this stereospecific mediator is synthesized by 15-lipoxygenase-1 (15-LOX-1) (91) (Fig. 3A). In AD brain (short postmortem time), cPLA2α and 15-LOX-1 expression changed in concert with NPD1-decreased content and DHA-enhanced pool size in the CA1 area of the hippocampus (66). In ARPE-19 cells (spontaneously transformed human RPE cells), IL-1β, oxidative stress, or the Ca2+ ionophore A23187 activates the synthesis of NPD1 (46). In turn, NPD1 might act in an autocrine fashion and/or diffuse through the interphotoreceptor matrix to act in a paracrine mode on photoreceptor cells and/or Müller cells (41).
15-LOX-1 deficiency in RPE cells promotes apoptosis and it is rescued selectively by NPD1
The increased availability of DHA is followed by NPD1 synthesis. This is of particular interest in the process of phagocytosis of photoreceptor outer segments, given that the endogenous pool of DHA is augmented upon activation of the RPE cell phagolysosomal system. In this context, it is known that NPD1 production and release is increased (Fig. 2). Recent evidence shows 15-LOX-1 as the enzyme that oxygenates DHA into NPD1 (91). In ARPE-19 cells where 15-LOX-1 protein expression was knocked down by 70% posttranscriptionally, the production of NPD1 was not increased, in contrast with normal cells (Fig. 5). In normal cells, NPD1 production was induced as early as 4 h after oxidative stress was applied. In this way, preexisting pools of 15-LOX-1 are activated upon stress stimulation (Fig. 5B). Thus, phagocytosis triggers the activation of 15-LOX-1 to oxygenate DHA into NPD1.
Fig. 5.

NPD1 synthesis is mediated by 15-lipoxygenase-1. A: Immunocytochemistry showing localization of 15-LOX-1 in ARPE-19 cells. Right column shows normal cells and left column shows 15-LOX-1 silenced cells. The four upper panels depict the localization of 15-LOX1 (green) relative to the nuclei (blue) and Actin (red). The four lower panels display nuclear localization of 15-LOX-2 (red) in relationship with nuclei (blue) and Actin (green). B: Histograms showing the differential production of NPD1 upon different strength of oxidative stress treatment (0, 400, 600, 800 μM H2O2 and 10 ng/ml TNFα). In each cell, as in the medium, the production of NPD1 was almost completely abolished (*P < 0.01). C: Apoptosis percentage measured by Hoechst staining of ARPE-19 cultures of control and silenced cells subjected to oxidative stress and treated with different metabolites of 15-LOX-1 and NPD1 precursor DHA. In silenced cells, apoptosis was augmented by oxidative stress. In normal cells, PEDF/DHA, NPD1, and lipoxin A4 did prevent apoptotic cell death, but in the silenced cells, neither DHA nor lipoxin A4 had any effect. Only NPD1 was able to rescue these cells from apoptosis. Data represents average ± SEM of two independent studies; statistical analysis is Student's t-test (*P < 0.01, **P < 0.001, ***P < 0.0001 and ****P < 0.00001). Modified with permission from reference (91).
15-LOX-1, a nonheme iron-containing dioxygenase, stereospecifically inserts oxygen into AA, dually forming 15(S)-hydroxyeicosatetraenoic acid (HETE) and 12(S)-HETE as well as lipoxin A4, a product of its joint activity with 5-LOX. 15-LOX-1 also has the capability to oxygenate linoleic acid into 13-hydroxyoctadecadienoic acid (92). Human 15-LOX-1 and 12-LOX are highly homologous proteins (65% identity) encoded by different genes, and their mRNAs are similar (70% identity). On the other hand, 15-LOX-2 is a different lipoxygenase that shares only 39% identity with human 15-LOX-1 (93). 15-LOX 2 and human 12-LOX differ from 15-LOX-1 not only in the ratio of 15-HETE and 12-HETE produced from AA but also in their localization (Fig. 5A). This means that they possess different selective product formations, and thus their activities contribute to different lipid mediators as well as display different substrate availability.
Silencing 15-LOX-1 also leads to enhanced susceptibility to apoptosis (Fig. 5C). It is worth noting that only NPD1 could rescue these cells from the exaggerated apoptosis experienced under oxidative stress conditions. This indicates that cells failing to produce this lipid mediator have an increased sensibility to oxidative stress caused by lack of the pro-survival signaling elicited by NPD1.
Photoreceptor outer segment phagocytosis induces RPE cell survival: NPD1-signaling pathway
The novel signaling pathway, resulting from the NPD1 action, may be initiated through the photoreceptor outer segment phagocytosis. A significant body of evidence has been accumulated that supports this idea.
FGF2 promotes bovine RPE cell survival through a sustained adaptive phenomenon that involves both FGF1-mediated activation of extracellular signal-regulated kinase andextracellular signal-regulated kinase 2-dependent Bcl-xL production (94). Bcl-xL may play a key role in integrating and transmitting exogenous FGF2 signals for RPE cell survival. Moreover, a well-organized signaling regulatory mechanism on the apical side of the RPE cell is reflected by the ability of neurotrophins to induce NPD1 synthesis and release (80). The response of human RPE cell monolayers in culture with NPD1 synthesis and release upon addition of certain neurotrophins to the apical side suggests sidedness of receptors for these ligands (80). Persephin is a novel neurotrophin with homology to GDNF (95, 96). Both persephin and GDNF are agonists of NPD1 synthesis and activators of its release from the apical surface of RPE cells (80). The same was true for leukemia inhibitory factor and FGF2 as well as for other neurotrophins (80). The finding that there is polarized (apically) neurotrophin/mediated NPD1 release has relevance for the initiation and progression of retinal degenerations. This is because when RPE cell polarization in the plane of the epithelium is disrupted, dysregulated growth factor secretion and pro-inflammatory signaling arises (2, 97, 98), thereby setting in motion pathological changes that include the proliferative component of macular degeneration: choroidal neovascularization (99–101).
Bcl-2 family proteins regulate apoptotic signaling at the mitochondrial level and at the endoplasmic reticulum. As a consequence, cytochrome c is released from mitochondria and caspase-3, a downstream effector of pro-apoptotic and anti-apoptotic Bcl-2 proteins, is activated (102). Also, oxidative stress-induced activation of caspase-3 in RPE cells is decreased by NPD1 (46). Apoptosis is an outcome of excessive oxidative stress in RPE cells, and NPD1 is effective in counteracting this oxidative stress-induced cell death (46). It is interesting that DHA itself inhibits apoptosis, concomitant with a remarkable, time-dependent formation of NPD1. Significantly, the potency of DHA for cytoprotection is much higher than that of added NPD1 (46), suggesting that NPD1 might exert its action close to the subcellular site of its synthesis. Importantly, these actions of DHA cannot be mimicked by other PUFAs (e.g., 20:4, n-6). Alternatively, it implies that other NPD-like mediators participate in promoting RPE cell survival, even though DHA fails to exert protection in 15-LOX-1-deficient cells, confirming that its pro-survival effect is mediated via NPD1. In RPE cells, cleavage of endogenous substrates by caspase-3 is enhanced by oxidative stress, as indicated by increased accumulation of poly(ADP-ribose) polymerases. NPD1 inhibits caspase-3 activation when added at the onset of oxidative stress (46), likely reflecting a downstream consequence of NPD1 modulation of Bcl-2 proteins.
A consequence of RPE cell damage and apoptosis is impaired photoreceptor cell survival, a dominant factor in AMD (103). The pigment lipofuscin, which increases in the RPE during aging, accumulates further during AMD. The progressively greater onslaught of photooxidative damage to the RPE affects photoreceptor survival. For example, in the juvenile form of macular degeneration known as Stargardt's disease, oxidative stress mediated by the lipofuscin fluorophore N-retinylidene-N-retinylethanolamine produces RPE damage; caspase-3 is part of the damaging cascade, whereas Bcl-2 exerts cellular protection (104). NPD1 downregulates lipofuscin fluorophore N-retinylidene-N-retinylethanolamine-mediated apoptosis induced by oxidative stress, restoring the integrity of the RPE and its relationship with the photoreceptor (80).
Anti-apoptotic and anti-inflammatory bioactivity of NPD1 is mediated in part through modulation of gene expression
Photoreceptor outer segment phagocytosis is a mechanism that initiates NPD1 signaling. This signaling promotes modulation of protein expression and activity involved in counteracting apoptosis at the cell fate decision level. For instance, NPD1 promotes differential changes in expression of Bcl-2 family proteins, upregulating protective Bcl-2 proteins (Bcl-2, Bcl-xL, and Bfl-1/A1) and attenuating expression of proteins that challenge cell survival (e.g., Bax, Bad, Bid, and Bik). Thus, an NPD1-mediated and coordinated regulation of Bcl-2 protein availability for subsequent downstream signaling may be crucial for cell survival (46, 80). NPD1 regulates expression of the genes encoding death repressors and effectors of the Bcl-2 family of proteins, e.g., Bcl-2-associated athanogene domain 3, whose mRNA is upregulated in mouse retinas subjected to light damage (105, unpublished observations). Bcl-2-associated athanogene domain 3 is a co-chaperone protein involved in release of the refolded protein by the chaperone Hsp70 (106–108).
NPD1 activity lessens the effects of stressors such as oxidative stress not only by its production and release upon stimulation but also because its down-stream signaling modulates gene transcription by compensating the activation or inhibition of gene expression. Thus, the consequence of the signaling is to return the system to normal levels. This is the case with NPD1 downregulation of the pro-apoptotic death-associated protein kinase 1, which displays increased expression under oxidative stress treatment (unpublished observations). In RPE cells, NPD1 downregulates the expression of pro-inflammatory genes, such as cycloxygenase 2 (COX-2), which is induced by cytokines such as IL-1β (46, 109). In ischemia reperfusion-injured hippocampus, as well as in neural progenitor cells stimulated by IL-1β, NPD1 also inhibits COX-2 induction (46, 47). In brain ischemia reperfusion, NPD1 decreases infarct size and inhibits PMN infiltration (47). Moreover, our laboratory, through a genome-wide screen in human brain progenitor cells in culture (66), has identified other NPD1-targeted pro-inflammatory genes; they include IL-1β, cytokine exodus protein-1, and TNFα-inducible pro-inflammatory element (B94, TNFAIP2). NPD1 bioactivity acts as a modulatory signal that counteracts pro-inflammatory injury to the RPE, a condition involved in pathoangiogenic signaling in the wet form of AMD and in proliferative vitreoretinopathy of diabetic retinopathy.
Oxidative stress enhances pro-inflammatory gene expression that leads to RPE cell injury. The inducible enzyme COX-2 is the rate-limiting step in prostaglandin synthesis and is involved in oxidative stress as well as cell function. COX-2 expression is regulated in RPE cells by photoreceptor outer-segment phagocytosis and by growth factors (110), and IL-1β activates expression of the proximal human COX-2 promoter. In the latter case, NPD1 potently counteracts the activation of the transcription mediated by IL-1β, displaying an IC50 of <5 nM (46).
CONCLUDING REMARKS
NPD1 is a pro-survival, anti-inflammatory, and homeostatic mediator that, by acting on RPE cells, promotes photoreceptor cell integrity (42). NPD1 synthesis agonists include neurotrophins and oxidative stress, and thus this lipid mediator modulates signaling pathways related with the pro- and anti-apoptotic balance (Fig. 6). As a consequence, NPD1 tilts the equilibrium toward cell survival. Recently, the presence of stereoselective specific binding of NPD1 was shown in ARPE-19 cells and in human leukocytes, suggesting a specific receptor in immune and retinal cells (55).
Fig. 6.

Diagram outlining 15-LOX-1 activity of NPD1 synthesis and bioactivity in RPE cells. Noxious stimuli activate 15-LOX-1, promoting the synthesis of NPD1 from DHA. NPD1 signaling modulates the activity and gene expression of proteins involved in pro- and anti-inflammatory signaling in apoptosis, ultimately fostering photoreceptor cell integrity and overall homeostasis.
A proof of principle of the in vivo bioactivity of NPD1 was obtained in a mouse model of choroidal neovascularization by clinically grading retinal laser-induced lesions, measuring leakage area, and volumetrically quantifying vascular endothelial cell proliferation. NPD1 injected intraperitoneally was found to inhibit choroidal neovascularization, thus suggesting a sustained protection and highlighting the potential applicability of this lipid mediator in preventing or ameliorating endothelial cell growth in pathoangiogenesis (111).
Several questions remain: What are the molecular mechanisms through which NPD1 synthesis is stimulated? What are the early molecular NPD1 interactors/effectors in the signaling pathway? The evolving information on the bioactivity of NPD1 in RPE/photoreceptor interactions suggests that DHA/NPD1 signaling may contribute to preventing and/or halting progression of retinal degenerative diseases.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- AD
- Alzheimer's disease
- AMD
- age-related macular degeneration
- C2
- component 2
- CNTF
- ciliary neurotrophic factor
- COX-2
- cycloogenase-2
- DHA
- docosahexaenoic acid, 22:6,n-3
- GDNF
- glial-derived neurotrophic factor
- HETE
- 15(S)-hydroxyeicosatetraenoic acid
- IL
- interleukin
- NPD1
- neuroprotectin D1
- PBMC
- peripheral blood mononuclear cell
- PD1
- protectin D1
- PEDF
- pigment epithelium derived factor
- PLA2
- phospholipase A2
- PMN
- polymorphonuclear leukocyte
- RP
- retinitis pigmentosa
- RPE
- retinal pigment epithelial
- TNFα
- tumor necrosis factor-α
- VEGF
- vascular endothelial growth factor
This work was supported by National Institutes of Health, National Eye Institute Grant EY-005121, and National Center for Research Resources grant P20 RR-016816, by the Eye, Ear, Nose, and Throat Foundation, New Orleans, LA, and by the Ernest C. and Yvette C. Villere Endowed Chair. C.N.S. acknowledges support from National Institutes of Health Grant GM-38765 for the work reviewed herein. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Papermaster D. S. 2002. The birth and death of photoreceptors: the Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 43: 1300–1309. [PubMed] [Google Scholar]
- 2.Rattner A., Nathans J. 2006. Macular degeneration: recent advances and therapeutic opportunities. Nat. Rev. Neurosci. 7: 860–872. [DOI] [PubMed] [Google Scholar]
- 3.Chang G. Q., Hao Y., Wong F. 1993. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 11: 595–605. [DOI] [PubMed] [Google Scholar]
- 4.Portera-Cailliau C., Sung C. H., Nathans J., Adler R. 1994. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 91: 974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird A. C. 2003. The Bowman lecture. Towards an understanding of age-related macular disease. Eye (Lond.). 17: 457–466. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q., Zuo J., Pierce E. A. 2004. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 24: 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dryja T. P., McGee T. L., Reichel E., Hahn L. B., Cowley G. S., Yandell D. W., Sandberg M. A., Berson E. L. 1990. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 343: 364–366. [DOI] [PubMed] [Google Scholar]
- 8.Mendes H. F., van der Spuy J., Chapple J. P., Cheetham M. E. 2005. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 11: 177–185. [DOI] [PubMed] [Google Scholar]
- 9.de Jong P. T. 2006. Age-related macular degeneration. N. Engl. J. Med. 355: 1474–1485. [DOI] [PubMed] [Google Scholar]
- 10.Sreekumar P. G., Kannan R., Yaung J., Spee C. K., Ryan S. J., Hinton D. R. 2005. Protection from oxidative stress by methionine sulfoxide reductases in RPE cells. Biochem. Biophys. Res. Commun. 334: 245–253. [DOI] [PubMed] [Google Scholar]
- 11.Shen J. K., Dong A., Hackett S. F., Bell W. R., Green W. R., Campochiaro P. A. 2007. Oxidative damage in age-related macular degeneration. Histol. Histopathol. 22: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 12.Dunaief J. L., Dentchew T., Ying G. S., Milam A. H. 2002. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 120: 1435–1442. [DOI] [PubMed] [Google Scholar]
- 13.Feher J., Kovacs I., Artico M., Cavallotti C., Papale A., Balacco Gabrieli C. 2006. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging. 27: 983–993. [DOI] [PubMed] [Google Scholar]
- 14.Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., Hageman J. L., Stockman H. A., Borchardt J. D., Gehrs K. M., et al. 2005. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisphotoreceptor outer segment es individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., Henning A. K., SanGiovanni J. P., Mane S. M., Mayne S. T., et al. 2005. Complement factor H polymorphism in age-related macular degeneration. Science. 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards A. O., Ritter R., III, Abel K. J., Manning A., Panhuysen C., Farrer L. A. 2005. Complement factor H polymorphism and age-related macular degeneration. Science. 308: 421–424. [DOI] [PubMed] [Google Scholar]
- 17.Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., et al. 2005. Complement factor H variant increases the risk of age-related macular degeneration. Science. 308: 419–421. [DOI] [PubMed] [Google Scholar]
- 18.Gold B., Merriam J.E., Zernant J., Hancox L.S., Taiber A.J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G.R., et al. 2006. Variation in factor B (BF) and complement 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 38: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bok D. 2005. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc. Natl. Acad. Sci. USA. 102: 7053–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spencer K. L., Hauser M. A., Olson L. M., Schmidt S., Scott W. K., Gallins P., Agarwal A., Postel E. A., Pericak-Vance M. A., Haines J. L. 2007. Protective effect of complement factor B and complement component 2 variants in age-related macular degeneration. Hum. Mol. Genet. 16: 1986–1992. [DOI] [PubMed] [Google Scholar]
- 21.Pournaras C. J., Rungger-Brändle E., Riva C. E., Hardarson S. H., Stefansson E. 2008. Regulation of retinal blood flow in health and disease. Prog. Retin. Eye Res. 27: 284–330. [DOI] [PubMed] [Google Scholar]
- 22.Strauss O. 2005. The retinal pigment epithelium in visual function. Physiol. Rev. 85: 845–881. [DOI] [PubMed] [Google Scholar]
- 23.Wang J. S., Kefalov V. J. 2009. An alternative pathway mediates the mouse and human cone visual cycle. Curr. Biol. 19: 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navid A., Nicholas S. C., Hamer R. D. 2006. A proposed role for all-trans retinal in regulation of rhodopsin regeneration in human rods. Vision Res. 46: 4449–4463. [DOI] [PubMed] [Google Scholar]
- 25.Saari J. C. 2000. Biochemistry of visual pigment regeneration: the Friedenwald lecture. Invest. Ophthalmol. Vis. Sci. 41: 337–348. [PubMed] [Google Scholar]
- 26.Lamb T. D., Pugh E. N., Jr 2004. Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 23: 307–380. [DOI] [PubMed] [Google Scholar]
- 27.Besch D., Jägle H., Scholl H. P. N., Seeliger M. W., Zrenner E. 2003. Inherited multifocal RPE-diseases: mechanisms for local dysfunction in global retinoid cycle gene defects. Vision Res. 43: 3095–3108. [DOI] [PubMed] [Google Scholar]
- 28.Cideciyan A. V., Aleman T. S., Swider M., Schwartz S. B., Steinberg J. D., Brucker A. J., Maguire A. M., Bennett J., Stone E. M., Jacobson S. G. 2004. Mutations in ABCA4 result in accumulation of lipofucsin before slowing the retinoid cycle: a reappraisal of the human disease sequence. Hum. Mol. Genet. 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 29.Zlokovic B. V. 2005. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 28: 202–208. [DOI] [PubMed] [Google Scholar]
- 30.Zhao S., Overbeek P. A. 2001. Regulation of choroid development by the retinal pigment epithelium. Mol. Vis. 2: 277–282. [PubMed] [Google Scholar]
- 31.Lee S. J., Kim J. H., Kim J. H., Chung M. J., Wen Q., Chung H., Kim K. W., Yu Y. S. 2007. Human apolipoprotein E2 transgenic mice show lipid accumulation in retinal pigment epithelium and altered expression of VEGF and bFGF in the eyes. J. Microbiol. Biotechnol. 17: 1024–1030. [PubMed] [Google Scholar]
- 32.Gao G., Li Y., Zhang D., Gee S., Crosson C., Ma J. 2001. Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett. 489: 270–276. [DOI] [PubMed] [Google Scholar]
- 33.Dawson D. W., Volpert O. V., Gillis P., Crawford S. E., Xu H., Benedict W., Bouck N. P. 1999. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 285: 245–248. [DOI] [PubMed] [Google Scholar]
- 34.Bok D. 1993. The retinal pigment epithelium: a versatile partner in vision. J. Cell Sci. Suppl. 17: 189–195. [DOI] [PubMed] [Google Scholar]
- 35.Kolko M., Wang J., Zhan C., Poulsen K. A., Prause J. U., Nissen M. H., Heegaard S., Bazan N. G. 2007. Identification of intracellular phospholipases A2 in the human eye: involvement in phagocytosis of photoreceptor outer segments. Invest. Ophthalmol. Vis. Sci. 48: 1401–1409. [DOI] [PubMed] [Google Scholar]
- 36.LaVail M. M. 1980. Circadian nature of rod outer segment disc shedding in the rat. Invest. Ophthalmol. Vis. Sci. 19: 407–411. [PubMed] [Google Scholar]
- 37.Young R. W., Droz B. 1968. The renewal of protein in retinal rods and cones. J. Cell Biol. 39: 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young R. W. 1971. The renewal of the rod and cone outer segments in the rhesus monkey. J. Cell Biol. 49: 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao H., Hollyfield J. G. 1992. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 33: 1–17. [PubMed] [Google Scholar]
- 40.Gibbs D., Kitamoto J., Williams D. S. 2003. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl. Acad. Sci. USA. 100: 6481–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bazan N. G. 2006. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 29: 263–271. [DOI] [PubMed] [Google Scholar]
- 42.Bazan N. G. 2007. Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 48: 4866–4881. [DOI] [PubMed] [Google Scholar]
- 43.Chen M., Forrester J. V., Xu H. 2007. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 84: 635–645. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee P. K., Marcheselli V. L., de Rivero Vaccari J. C., Gordon W. C., Jackson F. E., Bazan N. G. 2007. Photoreceptor outer segment phagocytosis selectively attenuates oxidative stress-induced apoptosis with concomitant neuroprotectin D1 synthesis. Proc. Natl. Acad. Sci. USA. 104: 13158–13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunn K. C., Aotaki-Keen A. E., Putkey F. R., Hjelmeland L. M. 1996. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 62: 155–169. [DOI] [PubMed] [Google Scholar]
- 46.Mukherjee P. K., Marcheselli V. L., Serhan C. N., Bazan N. G. 2004. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA. 101: 8491–8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcheselli V. L., Hong S., Lukiw W. J., Tian X. H., Gronert K., Musto A., Hardy M., Gimenez J. M., Chiang N., Serhan C. N., et al. 2003. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278: 43807–43817. [DOI] [PubMed] [Google Scholar]
- 48.SanGiovanni J. P., Chew E. Y. 2005. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog. Retin. Eye Res. 24: 87–138. [DOI] [PubMed] [Google Scholar]
- 49.Marszalek J. R., Lodish H. F. 2005. Docosahexaenoic acid, fatty acid interacting proteins, and neuronal function: breast milk and fish are good for you. Annu. Rev. Cell Dev. Biol. 21: 633–657. [DOI] [PubMed] [Google Scholar]
- 50.Samuelsson B., Dahlen S. E., Lindgren J. A., Rouzer C. A., Serhan C. N. 1987. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 237: 1171–1176. [DOI] [PubMed] [Google Scholar]
- 51.Serhan C. N., Hong S., Gronert K., Colgan S. P., Devchand P. R., Mirick G., Moussignac R-L. 2002. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. J. Exp. Med. 196: 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hong S., Gronert K., Devchand P., Moussignac R-L., Serhan C. N. 2003. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood and glial cells: autacoids in anti-inflammation. J. Biol. Chem. 278: 14677–14687. [DOI] [PubMed] [Google Scholar]
- 53.Ariel A., Li P-L., Wang W., Tang W-X., Fredman G., Hong S., Gotlinger K. H., Serhan C. N. 2005. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 280: 43079–43086. [DOI] [PubMed] [Google Scholar]
- 54.Serhan C. N., Gotlinger K., Hong S., Lu Y., Siegelman J., Baer T., Yang R., Colgan S. P., Petasis N. A. 2006. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J. Immunol. 176: 1848–1859. [DOI] [PubMed] [Google Scholar]
- 55.Marcheselli V. L., Mukherjee P. K., Arita M., Hong S., Antony R., Sheets K., Petasis N., Serhan C. N., Bazan N. G. 2010. Neuroprotectin D1/protectin D1 stereoselective and specific binding with human retinal pigment epithelial cells and neutrophils. Prostaglandins Leukot. Essent. Fatty Acids. 82: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serhan C. N. 2007. Resolution phases of inflammation: novel endogenous anti-inflammatory and pro-resolving lipid mediators and pathways. Annu. Rev. Immunol. 25: 101–137. [DOI] [PubMed] [Google Scholar]
- 57.Serhan C. N., Chiang N., Van Dyke T. E. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bazan N. G., Birkle D. L., Reddy T. S. 1984. Docosahexaenoic acid (22:6, n-3) is metabolized to lipoxygenase reaction products in the retina. Biochem. Biophys. Res. Commun. 125: 741–747. [DOI] [PubMed] [Google Scholar]
- 59.Serhan C. N., Clish C. B., Brannon J., Colgan S. P., Chiang N., Gronert K. 2000. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arita M., Bianchini F., Aliberti J., Sher A., Chiang N., Hong S., Yang R., Petasis N. A., Serhan C. N. 2005. Stereochemical assignment, anti-inflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serhan C. N., Lundberg U., Weissmann G., Samuelsson B. 1984. Formation of leukotrienes and hydroxy acids by human neutrophils and platelets exposed to monosodium urate. Prostaglandins. 27: 563–581. [DOI] [PubMed] [Google Scholar]
- 62.Bannenberg G. L., Chiang N., Ariel A., Arita M., Tjonahen E., Gotlinger K. H., Hong S., Serhan C. N. 2005. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174: 4345–4355. [DOI] [PubMed] [Google Scholar]
- 63.Schwab J. M., Chiang N., Arita M., Serhan C. N. 2007. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 447: 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butovich I. A. 2005. On the structure and synthesis of neuroprotectin D1, a novel anti-inflammatory compound of the docosahexaenoic acid family. J. Lipid Res. 46: 2311–2314. [DOI] [PubMed] [Google Scholar]
- 65.Levy B. D., Kohli P., Gotlinger K., Haworth O., Hong S., Kazani S., Israel E., Haley K. J., Serhan C. N. 2007. Protectin D1 is generated in asthma and dampens airway inflammation and hyper-responsiveness. J. Immunol. 178: 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lukiw W. J., Cui J. G., Marcheselli V. L., Bodker M., Botkjaer A., Gotlinger K., Serhan C. N., Bazan N. G. 2005. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115: 2774–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poulsen R. C., Gotlinger K. H., Serhan C. N., Kruger M. C. 2008. Identification of inflammatory and pro-resolving lipid mediators in bone marrow and their profile alteration with ovariectomy and omega-3 intake. Am. J. Hematol. 83: 437–445. [DOI] [PubMed] [Google Scholar]
- 68.Duffield J. S., Hong S., Vaidya V., Lu Y., Fredman G., Serhan C. N., Bonventre J. V. 2006. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 177: 5902–5911. [DOI] [PubMed] [Google Scholar]
- 69.Sun Y-P., Oh S. F., Uddin J., Yang R., Gotlinger K., Campbell E., Colgan S. P., Petasis N. A., Serhan C. N. 2007. Resolvin D1 and its aspirin-triggered 17R epimer: stereochemical assignments, anti-inflammatory properties and enzymatic inactivation. J. Biol. Chem. 282: 9323–9334. [DOI] [PubMed] [Google Scholar]
- 70.Blaho V. A., Buczynski M. W., Brown C. R., Dennis E. A. 2009. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J. Biol. Chem. 284: 21599–21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin Q., Patil K. A., Gronert K., Sharma S. C. 2008. Neuroprotectin D1 inhibits retinal ganglion cell death following axotomy. Prostaglandins Leukot. Essent. Fatty Acids. 79: 201–207. [DOI] [PubMed] [Google Scholar]
- 72.Hassan I. R., Gronert K. 2009. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J. Immunol. 182: 3223–3232. [DOI] [PubMed] [Google Scholar]
- 73.González-Périz A., Horrillo R., Ferré N., Gronert K., Dong B., Morán-Salvador E., Titos E., Martínez-Clemente M., López-Parra M., Arroyo V., et al. 2009. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 23: 1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen P., Fenet B., Michaud S., Tomczyk N., Véricel E., Lagarde M., Guichardant M. 2009. Full characterization of PDX, a neuroprotectin/protectin D1 isomer, which inhibits blood platelet aggregation. FEBS Lett. 583: 3478–3484. [DOI] [PubMed] [Google Scholar]
- 75.Hong S., Tjonahen E., Morgan E. L., Yu L., Serhan C. N., Rowley A. F. 2005. Rainbow trout (Oncorhynchus mykiss) brain cells biosynthesize novel docosahexaenoic acid-derived resolvins and protectins–mediator lipidomic analysis. Prostaglandins Other Lipid Mediat. 78: 107–116. [DOI] [PubMed] [Google Scholar]
- 76.Tanihara H., Inatani M., Honda Y. 1997. Growth factors and their receptors in the retina and pigment epithelium. Prog. Retin. Eye Res. 16: 271–301. [Google Scholar]
- 77.LaVail M. M., Yasumura D., Matthes M. T., Lau-Villacorta C., Unoki K., Sung C. H., Steinberg R. H. 1998. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest. Ophthalmol. Vis. Sci. 39: 592–602. [PubMed] [Google Scholar]
- 78.Politi L. E., Rotstein N. P., Carri N. G. 2001. Effect of GDNF on neuroblast proliferation and photoreceptor survival: additive protection with docosahexaenoic acid. Invest. Ophthalmol. Vis. Sci. 42: 3008–3015. [PubMed] [Google Scholar]
- 79.Hu J., Bok D. 2001. A cell culture medium that supports the differentiation of human retinal pigment epithelium into functionally polarized monolayers. Mol. Vis. 7: 14–19. [PubMed] [Google Scholar]
- 80.Mukherjee P. K., Marcheselli V. L., Barreiro S., Hu J., Bok D., Bazan N. G. 2007. Neurotrophins enhance retinal pigment epithelial cell survival through neuroprotectin D1 signaling. Proc. Natl. Acad. Sci. USA. 104: 13152–13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bazan N. G., Birkle D. L., Reddy T. S. 1985. Biochemical and nutritional aspects of the metabolism of polyunsaturated fatty acids and phospholipids in experimental models of retinal degeneration. Retinal Degeneration: Experimental and Clinical Studies. LaVail M.M., Hollyfield J.G., Anderson R.E., Alan R. Liss, Inc; New York, NY: 159–187. [Google Scholar]
- 82.Bazan N. G. 2003. Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J. Lipid Res. 44: 2221–2233. [DOI] [PubMed] [Google Scholar]
- 83.Aveldano M. I., Bazan N. G. 1974. Displacement into incubation medium by albumin of highly unsaturated retina free fatty acids arising from membrane lipids. FEBS Lett. 40: 53–56. [DOI] [PubMed] [Google Scholar]
- 84.Aveldano M. I., Bazan N. G. 1975. Differential lipid deacylation during brain ischemia in a homeotherm and a poikilotherm. Content and comphotoreceptor outer segment ition of free fatty acids and triacylglycerols. Brain Res. 100: 99–110. [DOI] [PubMed] [Google Scholar]
- 85.Horrocks L. A., Farooqui A. A. 1994. NMDA receptor-stimulated release of arachidonic acid: mechanisms for the Bazan effect. Cell Signal Transduction, Second Messengers, and Protein Phosphorylation in Health and Disease. Municio A.M., Miras-Portugal M.T., Plenum Press, New York, NY: 113–128. [Google Scholar]
- 86.Sun G. Y., Xu J., Jensen M. D., Simonyi A. 2004. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J. Lipid Res. 45: 205–213. [DOI] [PubMed] [Google Scholar]
- 87.Reddy T. S., Bazan N. G. 1984. Activation of polyunsaturated fatty acids by rat tissues in vitro. Lipids. 19: 987–989. [DOI] [PubMed] [Google Scholar]
- 88.Reddy T. S., Bazan N. G. 1984. Synthesis of arachidonoyl coenzyme A and docosahexaenoyl coenzyme A in retina. Curr. Eye Res. 3: 1225–1232. [DOI] [PubMed] [Google Scholar]
- 89.Reddy T. S., Bazan N. G. 1985. Synthesis of arachidonoyl coenzyme A and docosahexaenoyl coenzyme A in synaptic plasma membranes of cerebrum and microsomes of cerebrum, cerebellum, and brain stem of rat brain. J. Neurosci. Res. 13: 381–390. [DOI] [PubMed] [Google Scholar]
- 90.Reddy T. S., Bazan N. G. 1985. Synthesis of docosahexaenoyl-, arachidonoyl- and palmitoyl-coenzyme A in ocular tissues. Exp. Eye Res. 41: 87–95. [DOI] [PubMed] [Google Scholar]
- 91.Calandria J. M., Marcheselli V. L., Mukherjee P. K., Uddin J., Winkler J. W., Petasis N. A., Bazan N. G. 2009. Selective survival rescue in 15-lipoxygenase-1 deficient retinal pigment epithelial cells by the novel docosahexaenoic acid-derived mediator, neuroprotectin D1. J. Biol. Chem. 284: 17877–17882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kühn H., Thiele B. J., Ostareck-Lederer A., Stender H., Suzuki H., Yoshimoto T., Yamamoto S. 1993. Bacterial expression, purification and partial characterization of recombinant rabbit reticulocyte 15-lipoxygenase. Biochim. Biophys. Acta. 1168: 73–78. [DOI] [PubMed] [Google Scholar]
- 93.Brash A. R., Boeglin W. E., Chang M. S. 1997. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA. 94: 6148–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bryckaert M., Guillonneau X., Hecquet C., Courtois Y., Mascarelli F. 1999. Both FGF1 and bcl-x synthesis are necessary for the reduction of apoptosis in retinal pigmented epithelial cells by FGF2: role of the extracellular signal-regulated kinase 2. Oncogene. 18: 7584–7593. [DOI] [PubMed] [Google Scholar]
- 95.Bilak M. M., Shifrin D. A., Corse A. M., Bilak S. R., Kuncl R. W. 1999. Neuroprotective utility and neurotrophic action of neurturin in photoreceptor outer segment tnatal motor neurons: comparison with GDNF and persephin. Mol. Cell. Neurosci. 13: 326–336. [DOI] [PubMed] [Google Scholar]
- 96.Milbrandt J., de Sauvage F. J., Fahrner T. J., Baloh R. H., Leitner M. L., Tansey M. G., Lampe P. A., Heuckeroth R. O., Kotzbauer P. T., Simburger K. S., et al. 1998. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron. 20: 245–253. [DOI] [PubMed] [Google Scholar]
- 97.Kannan R., Zhang N., Sreekumar P. G., Spee C. K., Rodriguez A., Barron E., Hinton D. R. 2006. Stimulation of apical and basolateral VEGF-A and VEGF-C secretion by oxidative stress in polarized retinal pigment epithelial cells. Mol. Vis. 12: 1649–1659. [PubMed] [Google Scholar]
- 98.Bhutto I. A., McLeod D. S., Hasegawa T., Kim S. Y., Merges C., Tong P., Lutty G. A. 2006. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp. Eye Res. 82: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Frank R. N., Amin R. H., Eliott D., Puklin J. E., Abrams G. W. 1996. Basic fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. Am. J. Ophthalmol. 122: 393–403. [DOI] [PubMed] [Google Scholar]
- 100.Frank R. N. 1997. Growth factors in age-related macular degeneration: pathogenic and therapeutic implications. Ophthalmic Res. 29: 341–353. [DOI] [PubMed] [Google Scholar]
- 101.Frank R. N., Amin R. H., Puklin J. E. 1999. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am. J. Ophthalmol. 127: 694–709. [DOI] [PubMed] [Google Scholar]
- 102.Mattson M. P., Bazan N. G. 2006. Apoptosis and necrosis. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 7th ed Siegel G.J., Albers R.W., Brady S.T., Price D.L., Elsevier Academic Press, Burlington, MA: 603–615. [Google Scholar]
- 103.Hinton D. R., He S., Lopez P. F. 1998. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch. Ophthalmol. 116: 203–209. [DOI] [PubMed] [Google Scholar]
- 104.Sparrow J. R., Cai B. 2001. Blue light-induced apoptosis of A2E containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest. Ophthalmol. Vis. Sci. 42: 1356–1362. [PubMed] [Google Scholar]
- 105.Chen L., Wu W., Dentchev T., Zeng Y., Wang J., Tsui I., Tobias J. W., Bennett J., Baldwin D., Dunaief J. L. 2004. Light damage induced changes in mouse retinal gene expression. Exp. Eye Res. 79: 239–247. [DOI] [PubMed] [Google Scholar]
- 106.Stuart J. K., Myszka D. G., Joss L., Mitchell R. S., McDonald S. M., Xie Z., Takayama S., Reed J. C., Ely K. R. 1998. Characterization of interactions between the anti-apoptotic protein BAG-1 and Hsc70 molecular chaperones. J. Biol. Chem. 273: 22506–22514. [DOI] [PubMed] [Google Scholar]
- 107.Takayama S., Bimston D. N., Matsuzawa S., Freeman B. C., Aime-Sempe C., Xie Z., Morimoto R. I., Reed J. C. 1997. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 16: 4887–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takayama S., Xie Z., Reed J. C. 1999. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 274: 781–786. [DOI] [PubMed] [Google Scholar]
- 109.Belvisi M. G., Saunders M. A., Haddad E. B., Hirst S. J., Yacoub M. H., Barnes P. J., Mitchell J. A. 1997. Induction of cyclo-oxygenase-2 by cytokines in human cultured airway smooth muscle cells: novel inflammatory role of this cell type. Br. J. Pharmacol. 120: 910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ershov A. V., Bazan N. G. 1999. Induction of cyclooxygenase-2 gene expression in retinal pigment epithelium cells by photoreceptor rod outer segment phagocytosis and growth factors. J. Neurosci. Res. 58: 254–261. [PubMed] [Google Scholar]
- 111.Sheets K. G., Zhou Y., Ertel M. E., Knott E. J., Regan C. E., Jr., Elison J. R., Gordon W. C., Gjorstrup P., Bazan N. G. 2010. Neuroprotectin D1 attenuates laser-induced choroidal neovascularization in mouse. Mol. Vis. 16: 320–329. [PMC free article] [PubMed] [Google Scholar]