

Abstract
A deficiency of lysosomal phospholipase A2 (LPLA2) causes macrophage-associated phospholipidosis, suggesting that the enzyme is important in the lipid catabolism. Because LPLA2 is secreted by macrophages, extracellular LPLA2 activity may potentially reflect a change in macrophage activation. In this report, the detection of LPLA2 activity in plasma was established by the measurement of the transacylase activity of LPLA2 under acidic conditions. No transacylase activity of LPLA2 was detected in normal human plasma when the plasma was incubated with liposomes consisting of 1,2-dioleoylphosphatidylcholine/sulfatide/N-acetylsphingosine (NAS) at pH 4.5. However, the transacylase activity in the plasma was detected when liposomes consisting of 1,2-dioleoylphosphatidylglycerol/NAS were used as a substrate. To establish the specificity of the assay, ceramide transacylase activity was detected in the plasma of wild-type mice. By contrast, the plasma obtained from LPLA2-knockout mice had no measurable transacylase activity under the same conditions. The enzymatic activity of recombinant LPLA2 was inhibited by treatment with methylarachidonylfluorophosphonate. The inhibitor also suppressed the transacylase activity observed in both normal human and wild-type mouse plasma, establishing that the transacylase activity observed in plasma is due to LPLA2. Plasma LPLA2 activity may be a useful bioassay marker for the identification of LPLA2-related disorders.
Keywords: lysosomal phospholipase A2-knockout mouse, group XV phospholipase A2, serum, dioleoylphosphatidylglycerol, N-acetylsphingosine, 1-O-acyl-N-acetylsphingosine
A decade ago, we reported the discovery of a novel enzyme present in the lysosomal fraction of MDCK cells and characterized by dual enzyme activities, transacylase, and phospholipase A2 under acidic conditions (1). Later, the enzyme was purified, cloned, sequenced, expressed, and named lysosomal phospholipase A2 (LPLA2) (2, 3). The enzyme purified from calf brain is a water-soluble glycoprotein consisting of a single polypeptide chain with a molecular weight of 45 kDa with Ca2+-independent phospholipase A2 activity at a pH optimum of 4.5 (2). LPLA2 belongs to the αβ-hydrolase super-family of enzymes and has 49% protein sequence identity to lecithin cholesterol acyltransferase, with the latter thought to have arisen as a gene duplication product (3). LPLA2 is now classified as the only member of the group XV phospholipase A2 family (4).
LPLA2 in mouse and rat is highly expressed in alveolar macrophages and may be centrally involved in the catabolism of glycerophospholipids in pulmonary surfactant (5). Additionally, both alveolar and peritoneal macrophages obtained from LPLA2 knockout mice are characterized by the significant accumulation of phospholipids and the formation of numerous cytoplasmic multi-lamellar inclusion bodies, a hallmark of cellular phospholipidosis (6). The phospholipid accumulation found in alveolar macrophages obtained from LPLA2 knockout mice is markedly reduced by the addition of recombinant mouse LPLA2 (7). The recombinant LPLA2 is taken into the cells via mannose receptors and translocated into acidic compartments such as late endosomes and lysosomes (7). In addition, endogenous LPLA2 in the alveolar macrophage is secreted following stimulation with zymosan, indicating that LPLA2 is a secreted enzyme as well as a lysosomal enzyme. Recently, we have demonstrated that negatively charged lipids mediate LPLA2 membrane binding through an electrostatic interaction and amplify apparent LPLA2 activity (8). These observations indicate that cationic amphiphilic drug-induced phospholipidosis in some cases could result from the inhibition of the interaction of LPLA2 with phospholipid-containing membranes by cationic amphiphilic drugs.
Because LPLA2 is a secreted protein, the LPLA2 activity in extracellular fluids may be altered in disorders associated with LPLA2 deficiency and provides valuable information. For example, older LPLA2-knockout mice phenotypically resemble human autoimmune disease (unpublished observations). These observations raise the possibility that a loss or deficiency in LPLA2 activity may be associated with important clinical disorders. Because LPLA2-related disorders have yet to be established, an assay method for the detection LPLA2 activity in plasma is potentially useful.
LPLA2 shows the transacylase activity when the enzyme is incubated with glycerophospholipid liposomes containing the short-chain ceramide N-acetylsphingosine (NAS) under acidic conditions (1–3). In the transacylase reaction, the hydroxyl group at the C1 position of NAS is esterified with an acyl group from a glycerophospholipid donor by LPLA2. The reaction product, 1-O-acyl-NAS, is easily isolated by TLC and detected by charring. To the best of our knowledge, the ceramide transacylation found in cell extracts is LPLA2 specific (3, 8, 9). Therefore, to establish a conventional assay method to detect LPLA2 activity in extracellular fluids, we applied and developed the transacylation-based assay method to measure LPLA2 activities in plasma and serum.
MATERIALS AND METHODS
Reagents
1,2-Dioleoylphosphatidylcholine (DOPC), 1,1′, 2,2′-tetraoleoyl cardiolipin, 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DOPG), 1,2-dioleoyl-sn-glycero-3-phosphate, 1,2-dioleoyl-sn-glycero-3-phosphoinositol, 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), 1,2-dioleoyl-sn-glycero-3-phosphoethanol (DOPEt) and 1,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (1,2-di-(9Z-octadecenyl)-PC) were purchased from Avanti (Alabaster, AL). para-Nitro-phenylbutyrate (p-NPB), methylarachidonylfluorophosphonate (MAFP), and ceramide (type-III) were obtained from Sigma (St. Louis, MO). Recombinant mouse LPLA2 was from Proteos (Kalamazoo, MI). NAS was from Matreya (Pleasant Gap, PA). Bicinchoninic acid protein assay reagent was from Pierce Chemical (Rockford, IL). High performance TLC (HPTLC) silica gel plates, 10 × 20 cm, were from Merck (Darmstadt, Germany). Sulfatide was prepared in our laboratory. Human plasma and sera were obtained from healthy volunteers under institutional review board approval through the University of Michigan.
Transacylase activity of LPLA2
The preparation of liposomes was performed as follows. Glycerophospholipid, NAS, and/or another lipid were mixed in a glass tube and dried down under a stream of nitrogen gas. The dried lipid mixture was dispersed into 50 mM sodium citrate (pH 4.5) by a probe-type sonicator for 8 min in an ice-water bath.
For the regular assay, the reaction mixture consisted of 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, 38 µM NAS incorporated into phospholipid liposomes (containing 12.7 µM sulfatide and 127 µM DOPC), and recombinant mouse LPLA2, human or mouse plasma, or human serum in a total volume of 500 µl. The reaction was initiated by adding the enzyme source, incubated for 5–90 min at 37°C, and terminated by adding 3 ml of chloroform/methanol (2:1, v/v) plus 0.3 ml of 0.9% (w/v) NaCl. The mixture was centrifuged at 800 g for 5 min at room temperature. The resultant lower organic layer was transferred into another glass tube and dried down under a stream of nitrogen gas. The dried lipid was dissolved in 40 µl of chloroform/methanol (2:1), applied on an HPTLC plate, and developed in a solvent system consisting of chloroform/acetic acid (90:10, v/v). The plate was dried and soaked in 8% (w/v) CuSO4, 5H2O, 6.8% (v/v) H3PO4, and 32% (v/v) methanol. The uniformly wet plate was briefly dried by a hair dryer and charred for 15 min in a 150°C oven. The plate was scanned and the content of the product (1-O-acyl-NAS) was estimated by NIH-ImageJ 1.37v. The known concentration of ceramide (type-III) was used to obtain a standard curve in this assay. For determining the substrate specificity of LPLA2, liposomes consisting of DOPC, sulfatide, and NAS used in regular assay were replaced by liposomes consisting of 1,2-di-(9Z-octadecenyl)-PC, DOPC or anionic glycerophospholipid, and NAS (54:22:24, molar ratio). For all other assays, liposomes consisting of DOPG and NAS (3:1, molar ratio) were used in the assay system.
Binding assay
Recombinant mouse LPLA2 was incubated with liposomes consisting of DOPC and sulfatide (10:1, molar ratio) or liposomes consisting of DOPG in a total volume of 500 µl for 30 min on ice. The reaction mixture contained 48 mM Na-citrate (pH 4.5), 130 µM phospholipid, and 8 µg/ml LPLA2. Following the incubation, the reaction mixture was centrifuged for 1 h at 150,000 g at 4°C. The resultant pellet was rinsed with 500 µl of cold 50 mM Na-citrate (pH 4.5) briefly, dissolved with 35 µl of SDS-PAGE sample loading buffer, and run on 12% separating gel by the method of Laemmli (10). After electrophoresis, the proteins in the gel were stained with Coomassie Brilliant Blue R-250.
RESULTS
Effect of plasma or serum on LPLA2 activity
The transacylase activity of LPLA2 was first examined in normal human plasma and serum. No acylated-NAS formation was detected when liposomes consisting of DOPC, sulfatide, and NAS (DOPC/sulfatide/NAS-liposomes) were incubated with 1.3% of plasma or serum under acidic conditions (Fig. 1A). In addition, the transacylase activity of recombinant mouse LPLA2 was completely suppressed in the presence of 1.3% of plasma or serum in the same assay system (Fig. 1A). Many hydrolases including LPLA2 show esterase activity on a small artificial molecule, p-NPB (8, 11). As shown in Fig. 1B, the esterase activity of the recombinant LPLA2 toward p-NPB was not inhibited in the presence of 1.3% serum in the reaction mixture, indicating that the catalytic site of LPLA2 remains active in the presence of serum. Therefore, altering the liposome composition was considered as means for the detection of the transacylase activity of LPLA2 in serum and plasma.
Fig. 1.

Effect of plasma or serum on LPLA2 activity. A: Transacylase activity of LPLA2. The reaction mixture contained 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, liposomes (130 µM phospholipid), 1.3% human serum, 1.3% human plasma, or 14.5 ng/ml of recombinant mouse LPLA2 in the presence or absence of 1.3% human serum or 1.3% human plasma in 500 µl of total volume. Liposomes consisting of DOPC/sulfatide/NAS (3:0.3:1, molar ratio) were incubated with the enzyme for 2 h or 15 min at 37°C as shown in A. The reaction products were extracted and separated by an HPTLC plate using a solvent system consisting of chloroform/acetic acid (9:1, v/v). The reaction product, 1-O-acyl-NAS, is produced by LPLA2. B: Esterase activity of LPLA2. The reaction mixture contained 200 µM p-nitro-phenylbutyrate, 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, and 14.5 ng/ml of recombinant mouse LPLA2 in the presence or absence of 1.3% serum in 500 µl of total volume. The reaction was initiated by adding the recombinant LPLA2 and kept for 10, 20, 30, and 40 min at 37°C. One hundred microliters of the reaction mixture was taken at each time point and mixed with 100 µl of cold 0.2 M NaHCO3. The absorbance of the mixture at 400 nm was measured immediately.
Substrate specificity of LPLA2
Anionic glycerophospholipids not only enhance LPLA2 activity but also serve as a better substrate of LPLA2 than zwitterionic glycerophospholipids (8), suggesting that the anionic phospholipids have higher affinity for LPLA2 than zwitterionic phospholipids. Thus, we considered the possibility that anionic phospholipids would be better substrates and might improve the usual assay system containing DOPC/sulfatide/NAS-liposomes. The substrate specificity of LPLA2 for anionic phospholipids was therefore investigated (Fig. 2A).
Fig. 2.

Substrate specificity of LPLA2. A. The reaction mixture contained 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, liposomes (130 µM phospholipid) and recombinant mouse LPLA2 in 500 µl of total volume. Before starting the reaction, liposomes consisting of 1,2-di-(9Z-octadecenyl)-PC, DOPC or anionic glycerophospholipid and NAS (54:22:24, molar ratio) were preincubated for 5 min at 37°C. The reaction was initiated by the addition of recombinant LPLA2 and kept for 1.5 to 10 min at 37°C. In the reaction, 4.83 ng/ml and 58 ng/ml of recombinant mouse LPLA2 were used for DOPC and anionic phospholipid liposomes, respectively. The reaction products were extracted and separated with an HPTLC plate using a solvent system consisting of chloroform/acetic acid (9:1, v/v). The reaction product, 1-O-oleoyl-NAS, was quantified by scanning the plate. Error bars indicate S.D. (n = 3). PC, PI, CL, PA, PS, PG and PEt indicate 1,2-dioleoyl phosphatidylcholine, 1,2-dioleoyl phosphatidylinositol, 1,1′, 2,2′-tetraoleoyl cardiolipin, 1,2-dioleoyl phosphatidic acid, 1,2-dioleoyl phosphatidylserine, 1,2-dioleoyl phosphatidylglycerol and 1,2-dioleoyl phosphatidylethanol, respectively. B: Different concentrations of liposomes consisting of DOPC/sulfatide/NAS (3:0.3:1, molar ratio) or DOPG/NAS (3:1, molar ratio) were preincubated for 5 min at 37°C and then incubated with the recombinant LPLA2. The reaction was performed as described in A.
Anionic phospholipids do not always form liposomes in a lipid bilayer structure at an acidic pH. Thus, to compare the specificity of each anionic phospholipid as a substrate of LPLA2, the phospholipids were incorporated into liposomes consisting of 1,2-di-(9Z-octadecenyl)-PC, and NAS. 1,2-Di-(9Z-octadecenyl)-PC is a di-alkenyl-glycerophospholipid and is not a substrate for LPLA2. The various substrates assayed, including PC and anionic phospholipids, contained the same acyl chains (dioeoyl groups), which exhibit very similar thermal behavior, including phase transition temperature. All anionic phospholipids assayed were better substrates than DOPC (Fig. 2A). In particular, LPLA2 showed much higher activity against DOPS, DOPG, and DOPEt than other anionic phospholipids (Fig. 2A). Unlike other anionic phospholipids, DOPG itself can form anionic vesicles even under acidic conditions, implying that the anionic liposomes consisting of DOPG and NAS (DOPG/NAS-liposomes) are suitable for measuring LPLA2 activity.
LPLA2-transacylase activity toward DOPG/NAS-liposomes was compared with that against DOPC/sulfatide/NAS-liposomes (Fig. 2B). The values of Km and Vmax of recombinant mouse LPLA2 for DOPC were estimated to be 171 µM and 5.55 µmol/min/mg protein, respectively. Because the activity of the recombinant LPLA2 against DOPG/NAS-liposomes was much higher than against the DOPC/sulfatide/NAS-liposomes, we could not correctly determine the initial velocity of the transacylase activity of LPLA2 at lower concentrations of DOPG. The enzyme may potentially be saturated with substrate at 10 µM of DOPG when LPLA2 was incubated with DOPG/NAS-liposomes, (Fig. 2B). Thus, based on these considerations, the values of Km and Vmax of LPLA2 for DOPG were roughly estimated to be <10 µM and 22 µmol /min/mg protein, respectively (Fig. 2).
The binding of LPLA2 to the lipid membrane is one of the rate-limiting steps in lipid-water interfacial reaction of the enzyme. The binding affinity of LPLA2 to DOPG-liposomes was compared with that to DOPC/sulfatide-liposomes using a very high-speed centrifugation (150,000 g). The binding affinity of LPLA2 to DOPG-liposomes was higher than that to DOPC/sulfatide-liposomes (Fig. 5C, lanes 1, 3, and 5).
Fig. 5.

Effect of MAFP on LPLA2. A: Recombinant mouse LPLA2 was treated with 0, 10, or 100 µM MAFP for 1 h on ice. LPLA2 was incubated with DOPG/NAS (3:1, molar ratio) liposomes. The reaction mixture contained 130 µM phospholipid, 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, and 14.5 ng/ml of the enzyme in 500 µl of total volume and was incubated for 1.5–4.5 min at 37°C. The reaction products were extracted, separated, and quantified as described in Fig. 2. Error bars indicate SD (n = 3). B: Esterase activity of MAFP-treated LPLA2 was performed as described in Fig. 1. The reaction was initiated by adding vehicle or the enzyme and kept for 5, 15, 25, and 35 min at 37°C. C: Recombinant mouse LPLA2 (4 µg) pretreated with or without 100 µM MAFP as described in A was incubated with DOPC/ sulfatide-liposomes or DOPG-liposomes for 30 min on ice and centrifuged at 150,000 g for 1 h at 4°C. The resultant pellet was applied on SDS-PAGE as described in “Materials and Methods.” D and E: Human plasma (D) and mouse WT and LPLA2-knockout plasma (E) were pretreated with or without 100 µM MAFP for 1 h on ice. The treated plasma samples (2% for human and 4% for mice) were incubated with DOPG/NAS liposomes for 90 min at 37°C. The reaction products were analyzed by TLC as described in Fig. 1.
Transacylase activity in plasma
We used DOPG/NAS-liposomes for the detection of LPLA2 activity in human plasma. In distinction from studies with DOPC/sulfatide/NAS-liposomes, a reaction product with the same mobility as 1-O-acyl-NAS on TLC appeared. This reaction product increased linearly over 90 min when DOPG/NAS-liposomes were incubated with 1.6% of human plasma under acidic conditions (Fig. 3A, B). The reaction product was converted to NAS and methyl acyl ester by alkaline methanolysis (data not shown), demonstrating that the reaction product was 1-O-acyl-NAS. In addition, the formation of acylated NAS increased as a function of plasma concentration (Fig. 3C, D) despite the observation that DOPG/NAS-liposomes generated aggregates in the presence of plasma or serum under acidic conditions. In general, the transacylase activity was decreased in the presence of >4% plasma. In addition, the repeated freeze-thawing of plasma and serum resulted in a decrease of LPLA2 activity and an increase in the background of the TLC (data not shown).
Fig. 3.

Transacylase activity in human plasma. A: The reaction mixture contained 48 mM sodium citrate (pH 4.5), liposomes (130 µM phospholipid), and 1.6% of human plasma in 500 µl of total volume. The liposomes consisted of DOPG/NAS (3:1, molar ratio). The reaction was initiated by adding 8 µl of human plasma and kept for 30, 60, and 90 min at 37°C. The reaction product was determined and quantified as described in Fig. 2 and plotted against incubation time (B). C: Different concentrations of plasma were incubated with DOPG/NAS liposomes for 90 min at 37°C in 500 µl of 48 mM Na-citrate (pH 4.5). D: The 1-O-acyl-NAS formed after 90 min incubation was plotted against the plasma concentration in the reaction mixture.
No transacylase activity in LPLA2-knockout mice plasma
To confirm whether the transacylase activity found in plasma is due to LPLA2, plasma samples from wild-type and knockout LPLA2 mice were examined. There was no formation of 1-O-acyl-NAS when DOPG/NAS-liposomes were incubated with 4% LPLA2-knockout mouse plasma (Fig. 4). On the contrary, 1-O-acyl-NAS was formed when the same liposomes were incubated with 4% of wild-type mouse plasma (Fig. 4).
Fig. 4.
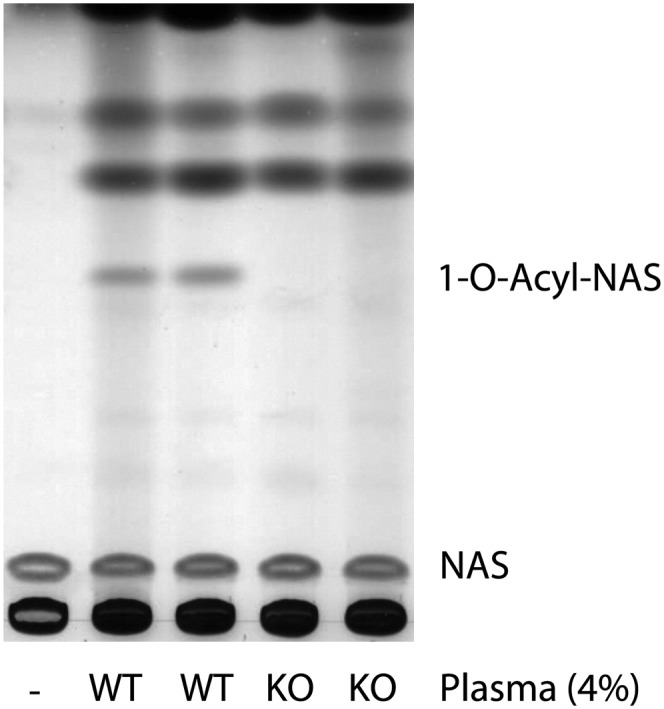
The absence of transacylase activity in LPLA2-knockout mouse plasma. The reaction mixture contained 48 mM sodium citrate (pH 4.5), 10 µg/ml BSA, liposomes (130 µM phospholipid), 4% mouse plasma, or 4% LPLA2-knockout mouse plasma in 500 µl of total volume. Liposomes consisting of DOPG/NAS (3:1, molar ratio) were incubated with the mouse plasma for 90 min at 37°C. The reaction products were extracted and separated by an HPTLC plate using a solvent system consisting of chloroform/acetic acid (9:1, v/v).
MAFP inhibits LPLA2 activity
LPLA2 contains a catalytic triad structure that consists of three amino residues, serine, aspartic acid, and histidine, and is essential for the enzymatic activity (9). Such triad structures are found in many hydrolases such as proteases, lipases, phospholipases, and esterases. A group of inhibitors has been described that acts through the modification of the catalytic serine residue. These include diisopropyl fluorophosphates, phenylmethyl sulfonyl fluoride (PMSF), arachidonyl trifluoromethyl ketone (AACOCF3) and MAFP. The inhibition of LPLA2 activity was observed using MAFP (Fig. 5A, B). Previously, we observed that both PMSF and AACOCF3 failed to inhibit the catalytic reaction of LPLA2 (2). Both the transacylase and esterase activities of LPLA2 were almost completely abolished by pretreatment of LPLA2 with 100 µM of MAFP (Fig. 5A, B). Of note, the binding of LPLA2 with liposomes was not affected by the same treatment of LPLA2 with MAFP (Fig. 5C). When human and mouse plasma were pretreated with 100 µM MAFP, there was a complete loss of the transacylase activity (Fig. 5D, E).
DISCUSSION
LPLA2 catabolizes endogenous and exogenous glycerophospholipids in acidic cellular compartments, including late endosomes and lysosomes. We recently reported that LPLA2 is not only a lysosomal enzyme but also a secreted enzyme (7). This finding suggests that LPLA2 might normally circulate extracellularly.
Plasma and serum contain lipids, including triglycerides, phospholipids, free and esterified cholesterols, and fatty acids. These lipids are present in plasma in association with lipoproteins and serum albumin. Endogenous lipase and phospholipase A activities are also present in plasma. Therefore, in establishing this assay, it was important to insure that any fatty acid released in the presence of plasma or serum originates from LPLA2 activity. LPLA2 cleaves an acyl group from glycerophospholipids and forms an acyl-enzyme intermediate at the catalytic serine residue (3, 8). In the following step, the enzyme displays both phospholipase A activity and transacylase activity when the intermediate reacts with water and primary alcohols, respectively (3, 7, 8, 12, 13). Based on our previous studies, the transacylase reaction of LPLA2 is a highly specific reaction under acidic conditions (3, 6, 8). Therefore, to confirm LPLA2 activity, we have previously used liposomes consisting of zwitterionic phospholipid, anionic lipid, and NAS. Phospholipid and NAS serve as an acyl donor and an acyl acceptor, respectively. To enhance the reaction, an anionic lipid such as sulfatide is usually incorporated into liposomes. 1-O-Acyl-NAS is produced by the transacylase activity. We therefore employed this method in LPLA2 assay in plasma.
In the preliminary study, we failed to detect LPLA2 activity in both human plasma and serum when DOPC/sulfatide/NAS-liposomes were used in the assay system (Fig. 1A). Moreover, recombinant mouse LPLA2 activity was completely blocked in the presence of 1.3% of plasma or serum in the same assay system (Fig. 1A), suggesting that the failure to detect LPLA2 activity might be the result of the presence of inhibitory factors. Interestingly, the same concentration of serum did not inhibit the esterase activity of the recombinant LPLA2 when a small water-soluble molecule, pNPB, was used as a substrate (Fig. 1B). Because the catalytic triad of LPLA2 is essential for the LPLA2 activity (9), the catalytic site of LPLA2 must remain active in the presence of serum. In this assay system, pNPB was dispersed into the reaction mixture as a monomer, indicating that LPLA2 can interact with substrates dispersed as a monomer but not aggregated substrates such as DOPC/sulfatide/NAS-liposomes in the presence of serum. Therefore, human plasma and serum probably contain factors that block LPLA2 activity against zwitterionic phospholipids, but not against water-soluble substrates. The blocking of the transacylase activity by such a factor could result from the interference of the interaction of LPLA2 with the phospholipids and render them more resistant to LPLA2 activity. For example, anionic liposomes are known to be associated with serum proteins such as serum albumin and β2-glycoprotein I (14).
We previously reported that anionic lipids greatly affect phospholipid hydrolysis by LPLA2 via an electrostatic interaction of LPLA2 with the negatively charged lipid membrane surface (8), suggesting that anionic phospholipids improve the deficiency of DOPC/sulfatide/NAS liposomes in the assay. Anionic phospholipids not only worked to enhance the reaction but also as a preferred substrate compared to DOPC in the LPLA2 reaction (Fig. 2A). In particular, DOPS, DOPG, and DOPEt were more preferential substrates (Fig. 2A). Shinozaki and Waite (15) partially purified a phosphatidylglycerol-selective LPLA2 (PG-LPLA2) from RAW 264.7 macrophage-like cells. They suggested the physiological role of PG-LPLA2 in the biosynthesis of bis(monoacylglycero)phosphates. Interestingly, physicochemical properties and substrate and inhibitor specificities of PG-LPLA2 are very similar to those of LPLA2, raising the possibility that PG-LPLA2 is the same enzyme as LPLA2.
Unlike DOPS and DOPEt, DOPG itself can form stable anionic liposomes even under acidic conditions. It was therefore possible to replace DOPC and sulfatide with DOPG in anionic liposomes containing NAS. This replacement rendered the liposomes even more anionic. The use of DOPG/NAS-liposomes resulted in an increase in the apparent affinity and catalytic rate of LPLA2 for the substrate (Fig. 2B). In fact, the Km and Vmax values of LPLA2 for DOPG were at least less 17-fold lower and 4-fold higher, respectively, than those for DOPC. Both human LPLA2 and mouse LPLA2 are positively charged at pH 4.5. DOPG-liposomes are more negatively charged vesicles than DOPC/sulfatide liposomes at pH 4.5. As expected, the sedimentation study revealed that LPLA2 has higher affinity for DOPG-liposomes than DOPC-liposomes (Fig. 5C). Our prior study showed that negatively charged lipids mediate LPLA2-lipid membrane binding through an electrostatic interaction and amplify apparent LPLA2 activity (8). Thus, the observed differences in LPLA2 activity between DOPG/NAS-liposomes and DOPC/sulfatide/NAS liposomes can be explained by the differences in binding affinities between liposomes and LPLA2 as well as between substrate and LPLA2. These observations led to the realization that the use of DOPG/NAS-liposomes yields a more sensitive method for the measurement of LPLA2 activity.
We observed the quantitative formation of 1-O-acyl-NAS by human plasma when DOPG/NAS-liposomes were used in the LPLA2 assay under acidic conditions (Fig. 3). Although the formation of acyl products was linear up to 4% of plasma in the assay system (Fig. 3C, D), the use of 1–2% of plasma concentrations in the assay always provided satisfactory results. During the reaction, plasma and serum induced formation of insoluble materials under acidic conditions. The transacylase activity appeared to be dependent on the amount of insoluble material formed by the addition of plasma or serum, and the activity was diminished in the presence of >4% of plasma or serum.
The DOPG/NAS-liposome assay also showed that the identical transacylase activity is found in the plasma obtained from wild-type mice but not LPLA2-knockout mice (Fig. 4). Therefore, the transacylase activity detected in plasma using DOPG/NAS-liposomes under acidic conditions is truly due to LPLA2, indicating that LPLA2 circulates in both human and mouse plasma. Thus, DOPG/NAS-liposomes provide a useful tool for detecting LPLA2 activity in plasma, serum, and perhaps other body fluids.
Finally, the present study demonstrated that the catalytic activity of the recombinant LPLA2 on phospholipid and p-NPB is almost completely inhibited by pretreatment of the enzyme with 100 µM MAFP (Fig. 5A, B). However, the interaction of LPLA2 with liposomes was unaffected by the same treatment (Fig. 5C). Unlike PMSF and AACOCF3, MAFP can covalently and specifically modify the nucleophilic hydroxyl group of the serine residue of the catalytic triad of LPLA2. Thus, the catalytic serine residue of LPLA2 is essential for the enzyme activity but not for the binding ability to anionic lipid bilayers at lipid-water interfaces. In addition, in light of the persistently observed esterase activity of LPLA2 on p-NPB, the binding of LPLA2 to the lipid membrane may be distinguished from the catalytic reaction of LPLA2. Both human and mouse plasma pretreated with 100 µM MAFP failed to produce 1-O-acyl-NAS. These results offer further demonstration that the acidic transacylase and phospholipase A2 activities in plasma detected in this assay system is of LPLA2 origin.
In summary, we have shown that the anionic glycerophospholipid, DOPG, is a favored substrate for LPLA2 and can be utilized to provide sufficient sensitivity for the detection of LPLA2 activity in plasma and serum. The preference of LPLA2 for DOPG may be due to the high affinities of LPLA2 for DOPG-containing anionic liposomes and due to the utilization of DOPG as substrate under acidic conditions. Both DOPG and NAS are commercially available. The reaction product, 1-O-acyl-NAS, is easily separated from substrates and other materials in plasma and serum by TLC. Thus, the assay method using DOPG/NAS-liposomes is convenient as well as specific for detecting LPLA2 activity in plasma and serum. Establishing a link between LPLA2 deficiency and susceptibility to drug-induced phospholipidosis or autoimmunity will require the use of facile LPLA2 assays such as described in this report.
Footnotes
Abbreviations:
- 1,2-di-(9Z-octadecenyl)-PC
- 1,2-di-(9Z-octadecenyl)-phosphatidylcholine
- AACOCF3
- arachidonyl trifluoromethyl ketone
- DOPC
- 1,2-dioleoylphosphatidylcholine
- DOPEt
- 1,2-dioleoyl-sn-glycero-3-phosphoethanol
- DOPG
- 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phosphoserine
- HPTLC
- high performance TLC
- LPLA2
- lysosomal phospholipase A2
- MAFP
- methylarachidonylfluorophosphonate
- NAS
- N-acetylsphingosine
- PG-LPLA2
- phosphatidylglycerol-selective lysosomal phospholipase A2
- PMSF
- phenylmethyl sulfonyl fluoride
- p-NPB
- para-nitro-phenylbutyrate
This work was supported by a Merit Review Award from the Department of Veterans Affairs and National Institutes of Health Grant 5RO1AR056991. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies.
REFERENCES
- 1.Abe A., Shayman J. A., Radin N. S. 1996. A novel enzyme that catalyzes the esterification of N-acetylsphingosine. Metabolism of C2-ceramides. J. Biol. Chem. 271: 14383–14389. [DOI] [PubMed] [Google Scholar]
- 2.Abe A., Shayman J. A. 1998. Purification and characterization of 1-O-acylceramide synthase, a novel phospholipase A2 with transacylase activity. J. Biol. Chem. 273: 8467–8474. [DOI] [PubMed] [Google Scholar]
- 3.Hiraoka M., Abe A., Shayman J. A. 2002. Cloning and characterization of a lysosomal phospholipase A2, 1-O-acylceramide synthase. J. Biol. Chem. 277: 10090–10099. [DOI] [PubMed] [Google Scholar]
- 4.Schaloske R. H., Dennis E. A. 2006. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 1761: 1246–1259. [DOI] [PubMed] [Google Scholar]
- 5.Abe A., Hiraoka M., Wild S., Wilcoxen S. E., Paine R., III, Shayman J. A. 2004. Lysosomal phospholipase A2 is selectively expressed in alveolar macrophages. J. Biol. Chem. 279: 42605–42611. [DOI] [PubMed] [Google Scholar]
- 6.Hiraoka M., Abe A., Lu Y., Yang K., Han X., Gross R. W., Shayman J. A. 2006. Lysosomal phospholipase A2 and phospholipidosis. Mol. Cell. Biol. 26: 6139–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe A., Kelly R., Kollmeyer J., Hiraoka M., Lu Y., Shayman J. A. 2008. The secretion and uptake of lysosomal phospholipase A2 by alveolar macrophages. J. Immunol. 181: 7873–7881. [DOI] [PubMed] [Google Scholar]
- 8.Abe A., Shayman J. A. 2009. The role of negatively charged lipids in lysosomal phospholipase A2 function. J. Lipid Res. 50: 2027–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiraoka M., Abe A., Shayman J. A. 2005. Structure and function of lysosomal phospholipase A2: identification of the catalytic triad and the role of cysteine residues. J. Lipid Res. 46: 2441–2447. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli U. K., Favre M. 1973. Maturation of the head of bacteriophage T4. I. DNA packaging events. J. Mol. Biol. 80: 575–599. [DOI] [PubMed] [Google Scholar]
- 11.Taniyama Y., Shibata S., Kita S., Horikoshi K., Fuse H., Shirafuji H., Sumino Y., Fujino M. 1999. Cloning and expression of a novel lysophospholipase which structurally resembles lecithin cholesterol acyltransferase. Biochem. Biophys. Res. Commun. 257: 50–56. [DOI] [PubMed] [Google Scholar]
- 12.Abe A., Hiraoka M., Shayman J. A. 2006. Positional specificity of lysosomal phospholipase A2. J. Lipid Res. 47: 2268–2279. [DOI] [PubMed] [Google Scholar]
- 13.Abe A., Hiraoka M., Shayman J. A. 2007. The acylation of lipophilic alcohols by lysosomal phospholipase A2. J. Lipid Res. 48: 2255–2263. [DOI] [PubMed] [Google Scholar]
- 14.Chonn A., Semple S. C., Cullis P. R. 1995. Beta 2 glycoprotein I is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J. Biol. Chem. 270: 25845–25849. [DOI] [PubMed] [Google Scholar]
- 15.Shinozaki K., Waite M. 1999. A novel phosphatidylglycerol selective phospholipase A2 from macrophages. Biochemistry. 38: 1669–1675. [DOI] [PubMed] [Google Scholar]