

Abstract
Integrated Raman and angular-scattering microscopy (IRAM) is a multimodal platform capable of noninvasively probing both the chemistry and morphology of a single cell without prior labeling. Using this system, we are able to detect activation-dependent changes in the Raman and elastic-scattering signals from CD8+ T cells stimulated with either Staphylococcal enterotoxin B (SEB) or phorbol myristate acetate (PMA). In both cases, results obtained from the IRAM instrument correlate well with results obtained from traditional fluorescence-based flow cytometry for paired samples. SEB-mediated activation was distinguished from resting state in CD8+ T cells by an increase in the number and mean size of small (∼500-nm) elastic scatterers as well as a decrease in Raman bands, indicating changes in nuclear content. PMA-mediated activation induced a different profile in CD8+ T cells from SEB, showing a similar increase in small elastic scatterers but a different Raman change, with elevation of cellular protein and lipid bands. These results suggest the potential of this multimodal, label-free optical technique for studying processes in single cells.
Keywords: Raman spectroscopy, elastic scattering, multimodal microscopy
Introduction
T-cell activation is one of the essential events in adaptive immunity and an area of extensive research.1, 2, 3 Activation involves a series of morphological and biochemical changes both inside the cell and on the cell surface. Currently, the most commonly used technique to track these changes is fluorescence-activated cell sorting (FACS) analysis, which relies on labeling cells with fluorochrome-conjugated antibodies specific for molecules of interest such as CD69 (an early activation marker on T cells) to assess their activation status. Although FACS analysis is a very powerful tool to monitor T-cell activation, recently there have been advances in new technologies that are capable of identifying activated T cells and yet require no labeling. Label-free study of activated cells is advantageous from several standpoints. Not only does it simplify the experimental protocol by minimizing sample preparation, but it also preserves cell integrity and function. This enables cells to be followed longitudinally to examine, for example, heterogeneity of response to a stimulus. Two such optical techniques that can provide insight into microscopic changes in cellular status are Raman spectroscopy and angle-resolved elastic scattering. Raman spectroscopy, a technique that measures vibrational modes of molecules, can reveal the details of chemical content in individual cells.4, 5, 6 Recent studies have shown that T-cell activation state can be determined from an individual cell’s Raman spectrum7, 8 by examining changes in Raman peaks associated with DNA. Elastic scattering, which can be used to determine cell size as well as detailed subcellular morphology, has been previously used to distinguish between cancerous and noncancerous cells and tissues9, 10, 11 and to identify organelle damage and ablation effects during photodynamic therapy,12, 13 among other applications. Recently, it has also been applied at the single-cell level14, 15 to distinguish alterations in mitochondrial morphology and to detect organelle reorganization during apoptosis. Although either of these techniques alone can provide some insights into a cell’s status at a subcellular level, an instrument that characterizes a cell’s contents both chemically and morphologically can provide a more complete picture of a wide range of biological processes. Our group has recently reported on the development of a technique, integrated Raman and angular-scattering microscopy (IRAM), that is capable of simultaneously measuring a Raman spectrum and an angle-resolved elastic scattering pattern from a single cell.16, 17, 18 Previously, we reported that clear differences exist between the IRAM measurements of granulocytes and lymphocytes,17 which are two major immune cell populations with distinct phenotypes and functions characterized by a priori known chemical and morphological differences.
In this study, we turned our attention to CD8+ T cells, a subset of T lymphocytes expressing the CD8 (cluster of differentiation 8) protein on their cell surface. CD8+ T cells are also known as cytotoxic T cells, as their primary function is to kill host cells with aberrant antigen expressions, such as tumor cells and cells infected with viruses. Thus, CD8+ T cells are critical in host defense against many viral infections and tumors. On appropriate signaling through T-cell antigen∕peptide receptors, CD8+ T cells undergo a series of biochemical and morphological changes, collectively referred to as “activation,” that eventually leads to the T cell acquiring effector functions, such as proliferation, cytokine production, and cytotoxicity. Here, we present the results of early CD8+ T cell activation events stimulated by one of two different agents and monitored with the IRAM instrument.
Materials and Methods
Preparation of Unlabeled, Pure CD8+ Cell Populations
Peripheral blood mononuclear cells (PBMCs) from human whole blood samples obtained using informed consent from healthy adult donors under an Institutional Review Board (IRB)-approved protocol and collected in CPT tubes with heparin (BD, Franklin Lakes, New Jersey) were isolated following the manufacturer’s protocol. CD8+ T cells were isolated from the PBMCs using a negative selection process where CD8+ cells remained unlabeled. Briefly, PBMCs were first incubated with the biotinylated antibody cocktail from the CD8+ isolation kit (130-091-154, Miltenyi Biotec, Auburn, California) to label non-CD8+ T cells, washed once with 1% bovine serum albumin (BSA)∕Hank’s balanced salt solution (HBSS), followed by secondary staining with streptavidin-phycoerythrin (PE). Cells were then washed twice and resuspended in 1% BSA∕HBSS at a final concentration of 4×107 cells∕mL. The CD8+ T cells were isolated by fluorescent activated cell sorting the PE-negative population using a FACSAria cell sorter (BD Biosciences, San Jose, California).
Protocol for Activation of CD8+ T Cells with Staphylococcal Enterotoxin B and PMA
To activate CD8+ T cells for IRAM, the sorted CD8+ T cells were stimulated with either staphylococcal enterotoxin B (SEB) or phorbol myristate acetate (PMA), or were left untreated as a negative control. For the stimulated samples, CD8+ T cells were incubated with either 1 μg∕mL of SEB or 10 ng∕mL of PMA diluted in RPMI-1640 (Roswell Park Memorial Institute-1640) medium supplemented with 8% fetal bovine serum (FBS) at a concentration of 2.5×106 cells∕mL for 18 h at 37 °C and 5% CO2. The control samples were incubated with the RPMI-1640∕8% FBS medium alone under the same conditions. The cultured cells were then collected and washed three times with 1% BSA∕HBSS to remove the SEB, resuspended in 1% BSA∕HBSS at a concentration of 5×105 cells∕mL, and analyzed either on the IRAM system or by flow cytometry on a BD LSR II flow cytometer following immunofluorescent labeling. The viability of cells after overnight SEB stimulation was greater than 95% as measured by trypan blue exclusion.
Flow Cytometry of CD8+ Cells
To assess the ability of the IRAM instrument to measure T-cell activation status, samples were analyzed by flow cytometry in parallel with IRAM measurements. Cells were stained with an antibody cocktail composed of CD8-PE-Texas Red (Invitrogen, Carlsbad, California), CD69-PE (BD Biosciences, San Jose, California), and CD137-APC (allophycocyanin) (BD Biosciences, San Jose, California). A vital dye 7-AAD (7-Amino-Actinomycin D) was also included in the antibody cocktail to determine cell viability. For each staining, 5×105 cells were incubated with 10 μL of antibody cocktail at 4 °C for 20 min, washed twice with 1% BSA∕HBSS buffer, and then resuspended in 200 μL of fixation buffer [10% formaldehyde in phosphate-buffered saline (PBS)]. The unstained and single stained compensation controls were set up by staining antibody-capturing compensation beads (Bangs Laboratories, Fishers, Indiana) with 1% BSA∕HBSS buffer alone or the optimal dilution of antibody-fluorochrome conjugate. For each sample, at least 100,000 cells total were analyzed.
IRAM Measurements and Analysis
Raman scattering spectra and scattergrams of elastic scattered intensity versus angle were recorded using the IRAM instrument described in Ref. 18. Briefly, a 500-μL volume of either stimulated or control cells resuspended in HBSS with 1% BSA were placed in the instrument’s sample chamber (Attofluor, Invitrogen, Carlsbad, California). The sample chamber was then placed on the sample stage of a Nikon inverted microscope (TE-2000, Nikon Instruments, Tokyo, Japan) into which 60 mW of 785-nm laser light was coupled either through the microscope objective from below (epi-illumination mode) or through a specialized condenser assembly from above (transillumination mode). In the epi-illumination mode, used to perform the Raman scattering measurements, the light was focused to a diffraction-limited spot size of approximately 2 μm. In transillumination mode, used to perform the elastic scattering measurements, the light was focused to a diffraction-limited spot of approximately 7 μm. Cells were first allowed to settle to the bottom of the chamber, and then were individually brought into the center of the microscope’s field of view by manual translation of the sample stage. The measurements were performed identically as described in Smith and Berger.17 Each elastic scattering measurement, made in transillumination mode, had a total exposure time of 10 s. Each cell was measured 10 times (for a total of 100 s of data acquisition), with the cell being moved beneath the laser focus between each measurement to reduce speckle arising from interference between the scattering of individual particles within the cell. Raman measurements were made in the epi-illumination geometry, with each cell being measured for 200 s. We note that at the fluences used in this study, the cells remained viable (as measured by trypan blue dye exclusion), and no time-correlated changes in cell spectra during the course of the measurement were observed (data not shown).
Data analysis proceeded identically to that described in Smith and Berger.17 Measured scattering patterns were first divided into four pie-slice-shaped bins, representing an averaging over the azimuthal angle ϕ to obtain four curves of scattered intensity versus polar angle θ. These four curves were then simultaneously compared to the predictions of generalized Lorenz-Mie theory19 computed by incoherently summing the contributions of two log-normal populations of small scatterers, with the means, standard deviations, and relative amplitude of the two populations being varied in a downhill-simplex search to obtain the best fit to the experimental measurement. Raman spectra were background corrected, smoothed, and normalized to the area under the 1450-cm−1 peak to account for variations in overall density, as the 1450-cm−1 peak roughly reports on total organic content within a sample.
Results
Flow Cytometry Results
Results of the flow cytometry analysis for the SEB-stimulated CD8+ T cells are shown in Fig. 1. Activated cells are defined as those cells that express either CD69 or CD137, both activation-specific surface markers that may occur at different times during activation. The left-hand plot in Fig. 1 shows the flow results for the control sample, with the presence of CD69 (marked by fluorochrome PE) indicated along the x axis and CD137 (marked by fluorochrome APC) along the y axis. As is customary, a trained user drew the bounding lines on the control sample, defining the region within which cells are classified as resting, and outside of which cells are classified as activated. These bounding lines were then kept fixed when examining the stimulated sample. By these definitions, 99.9% of the cells in the control sample were in a resting state. In contrast, the right-hand plot shows the results for the SEB-stimulated sample, where 26.38% of the cells were considered activated by SEB, with 20.93% of the cells being CD69+, 23.65% of the cells being CD137+, and 18.20% of the cells double positive for CD69 and CD137.
Figure 1.

Flow cytometric results for control (a) and SEB-stimulated (b) CD8+ cells. Stimulation was defined as expression of either CD69 (x axis) or CD137 (y axis). Numbers in each quadrant indicate the percentage of total cells falling into that area. The control sample shows 99.9% of the measured cells as unactivated, while the stimulated sample shows 26.38% are activated.
Results of the flow cytometry analysis for the PMA-stimulated CD8+ T cells in a separate experiment are shown in Fig. 2. The definitions of activated and nonactivated cells were identical to that for the SEB flow analysis described above. The left-hand plot in Figure 2 shows the flow results for the control sample. In this case, 97.5% of the cells in the control sample were in a resting state, whereas 97.66% of the cells in the stimulated cells were considered activated by PMA, with 96.3% of the cells being CD69+, 42.98% of the cells being CD137+, and 41.60% of the cells double positive for CD69 and CD137.
Figure 2.

Flow cytometric results for control (a) and PMA-stimulated (b) CD8+ cells. Stimulation was defined as expression of either CD69 (x axis) or CD137 (y axis). Numbers in each quadrant indicate the percentage of total cells falling into that area. The control sample shows 97.5% of the measured cells as unactivated, while the stimulated sample shows 97.66% are activated.
We emphasize here that the terms “stimulated” and “activated” are not interchangeable. A cell is said to have been stimulated if it was exposed to an antigen such as SEB or a pharmacological agent such as PMA. However, depending on the stimulus used in the experiments, there could have been a substantial number of the stimulated T cells that did not exhibit activation phenotypes. Additionally, there is a basal level of T cell activation within each human subject at any given time, reflecting the fact that the human body is constantly exposed to various environmental antigens. Consequently, there were a few cells within the unstimulated control samples showing activation phenotypes.
IRAM Measurements of SEB- and PMA-Stimulated CD8+ Cells
Bright Field Imaging
A bright field image was captured for each cell measured with the IRAM microscope. The images for the SEB and PMA experiments are shown in Figs. 34, respectively. One can see morphological differences, such as swelling and the growth of protrusions in several of the activated cells. However, it is important to note that several of the stimulated cells for both experiments are nonetheless indistinguishable from the control cell population. This hints at the variability of responses of cells to a stimulus.
Figure 3.

Bright field images of 10 control and 10 SEB-stimulated cells measured by the IRAM microscope. Scale bar (shown on control cell 1) indicates 5 μm.
Figure 4.

Bright field images of 10 control and 10 PMA-stimulated cells measured by the IRAM microscope. Scale bar (shown on control cell 1) indicates 5 μm.
Elastic Scattering
Elastic scattering measurements from SEB-stimulated and control lymphocytes are summarized in Fig. 5. Figure 5a shows the mean elastic scattergram for the 10 control CD8+ T cells, while Fig. 5b shows the mean scattergram for the 10 stimulated CD8+ T cells. Figures 5c, 5d show the data corresponding to the first azimuthal bin for control and SEB-stimulated cells, respectively (black dots). Also shown in Figs. 5c, 5d are the best fits of theory to each experiment (solid curves, red online) with the contribution from the larger log-normal scatterer populations shown with a dashed line (green online), and the contribution from the smaller scatterer population shown with a dash-dotted line (magenta online). There is an increased contribution from smaller scatterers in the stimulated population. Figures 5e, 5f show the population extractions for control and stimulated cells. The thick curves (blue online) are the extractions performed on the mean data for each cell type, while the solid and dashed curves (magenta and green online) are the 2 log-normal scatterer populations extracted from modeling of the scattering patterns of individual cells.
Figure 5.
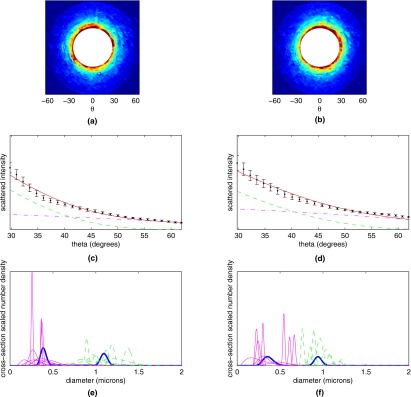
Averaged scattergrams from (a) 10 control CD8+ T cells and (b) 10 CD8+ T cells stimulated with SEB. Color scale runs from blue (low) to red (high). (c) and (d) Averaged experimental data from (c) 10 control CD8+ T cells and (d) 10 SEB-stimulated CD8+ T cells (black dots) from the first of the four annular bins, along with theoretical fits to the experimental data (solid curve, red online). Also plotted are the contributions from the population of smaller scatterers (dash-dotted curve, magenta online) and population of larger scatterers (dashed curve, green online). (e) and (f) Extracted populations for (e) control and (f) SEB-stimulated CD8+ T cells. Extractions for individual cells are shown in magenta and green online (for the small and large scatterer population, respectively), while population extractions for the cell-averaged signals are shown with a thick blue line. (Color online only.)
For each cell type a population around 300 nm and a population around 1000 nm was extracted, consistent with the results presented for lymphocytes in Ref. 17. However, it is evident from Figs. 5e, 5f that several of the stimulated cells have a shift of the small-diameter population to a larger mean size. This shift among several of the individual cells is not reflected in the mean population extraction, highlighting the power of single-cell analysis for heterogeneous populations.
Elastic scattering measurements from PMA-stimulated and control lymphocytes are summarized in Fig. 6. As in Fig. 5, Fig. 6a shows the mean elastic scattergram for the 10 control CD8+ T cells, while Fig. 6b shows the mean scattergram for the 10 stimulated CD8+ T cells. Figures 6c, 6d show the data corresponding to the first azimuthal bin for control and PMA-stimulated cells, respectively (black dots). Also shown in Figures 6c, 6d are the best fits of theory to each experiment (solid curves, red online) with the contribution from the larger log-normal populations shown with a dashed line (green online), while the contributions from the smaller populations are shown with a dash-dotted line (magenta online). In this case, the PMA-stimulated cells show a slight decrease in total scattering and similar contributions from the two scattering populations when compared to the control cells. Figures 6e, 6f show the population extractions for control and stimulated cells. The thick blue curves are the extractions performed on the mean data for each cell type, while the magenta and green curves are the extractions performed on the scattering patterns of individual cells, showing the heterogeneity of scattering response to PMA-stimulation.
Figure 6.
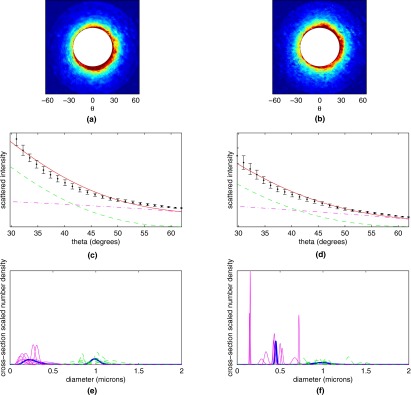
Averaged scattergrams from (a) 10 control CD8+ T cells and (b) 10 CD8+ T cells stimulated with PMA. Color scale runs from blue (low) to red (high). (c) and (d) Averaged experimental data from (c) 10 control CD8+ T cells and (d) 10 PMA-stimulated CD8+ T cells (black dots) from first of the four annular bins, along with theoretical fits to the experimental data (solid curve, red online). Also plotted are the contributions from the population of smaller scatterers (dash-dotted curve, magenta online) and population of larger scatterers (dashed curve, green online). (e) and (f) Extracted populations for (e) control and (f) PMA-stimulated CD8+ T cells. Extractions for individual cells are shown in magenta and green (for the small and large scatterer population, respectively), while population extractions for the cell-averaged signals are shown with a thick blue line. (Color online only.)
The extracted size information for the control and PMA-stimulated cells was similar to the previous experiment with SEB. For the control group, two populations of approximately equal cross-section weighted amplitude centered at 300 and 1000 nm were found to be the best fit to the data. For the PMA-stimulated lymphocytes, as in the SEB-stimulated lymphocytes, we found that although the larger population continued to be centered around 1000 nm, the small population tended to be shifted to larger mean diameters.
Raman Scattering of Single Lymphocytes
Raman scattering measurements from SEB-stimulated and control CD8+ lymphocytes are summarized in Fig. 7. Using the peaks marked in Figure 7, the stimulated cells were classified as belonging to two groups. One, containing cells whose spectra (shown in red online in Fig. 7) were indistinguishable from the mean control spectrum (shown as the thick curve in blue online in Fig. 7), was denoted as stimulated but spectrally unresponsive. The other group, containing cells whose spectra (bottom four curves, green online, in Fig. 7) showed clear differences compared to the mean control spectrum, was denoted as stimulated and responsive. Several areas of spectral difference between the responsive and unresponsive cells are immediately noticeable by visual inspection of the spectra. Gray rectangles mark those differences that were found to be statistically significant at the 95% confidence level. The chemical assignments for those peaks, taken from Uzunbajakava et al.,20 are summarized in Table 1.
Figure 7.

Plot of the mean Raman spectrum for 10 control CD8+ T cells (thick blue online). Also shown are color-coded spectra of individual cells from the SEB-stimulated sample. Cells were labeled as unresponsive, or nonactivated (six curves below thick blue curve, red online), and responsive, or activated (bottom four curves, green online), based on the heights of the peaks marked by the gray rectangles. These marked bands represent areas of statistically significant difference between the magenta spectra and the blue mean control spectrum. Band assignments for the marked peaks are given in Table 1, showing the differences are primarily due to a decrease in DNA signal. (Color online only.)
Table 1.
Raman peak differences between SEB- and PMA-stimulated and control CD8+ lymphocytes.
| Stimulation Type | Wavenumber Value (cm−1) | Peak Assignment20 |
|---|---|---|
| SEB | 788 | T, O–P–O symmetric stretch |
| 1095 | PO2 stretching | |
| 1375 | T, A, G | |
| 1487 | A, G | |
| 1510 | A | |
| PMA | 1049 | C–O stretching |
| 1656 | ν(C=C)cis | |
| 1659 | Amide I α-helix |
Raman scattering measurements from PMA-stimulated and control CD8+ lymphocytes are summarized in Fig. 8. The upper spectrum (blue online) represents the mean Raman spectrum of 10 control CD8+ cells, while the bottom spectrum (red online) is the mean Raman spectrum for ten PMA-stimulated CD8+ cells. Shaded areas indicate the standard deviation of the spectra. Although the differences between stimulated and control cells are more subtle than in the SEB stimulation experiment, two areas of spectral difference, marked by asterisks, were found to be statistically significant at the 95% confidence level. The chemical assignments for those peaks, taken from Uzunbajakava et al.,20 are summarized in Table 1.
Figure 8.

Mean Raman spectra for control (top, blue online) and PMA-stimulated (bottom, red online) CD8+ T cells. Shading indicates ±1 standard deviation. Asterisks indicate bands whose areas are statistically significantly different between the two cell types. Band assignments for the marked peaks are given in Table 1, showing the differences are primarily due to an increase in protein and lipid signal. (Color online only.)
Elastic and Raman Scattering Indices
To explore the power of the IRAM instrument to objectively classify cells by activation state, we compress the elastic scattering and Raman scattering results into an elastic index and a Raman index. Together these two indices construct a 2-D space analogous to the 2-D space of the fluorescence flow measurements shown in Figs. 12. Extending the analogy, each index is constructed to increase when a cell is measurably different from the control cells. The elastic index has two terms. The first is computed by taking the ratio of the area under the small log-normal population to the area under the large log-normal population in the two-population fit. Physically, a change in this ratio means that the fraction of light being scattered by small organelles, such as granules, has changed relative to the control population. The index also includes a term that responds to fractional increases in the smaller population’s mean diameter, relative to the control cells. This is shown as
| (1) |
where X is the amount of scattering by the small population relative to the large population, and is given by
| (2) |
where σ(r), computed by generalized Lorenz-Mie theory, is the scattering cross section (for our range of collection angles) for each radius, ρ(r) is a log-normal distribution (small-diameter population indicated by subscript sm, large-diameter population indicated by subscript lg), and is the mean value of X for all control cells. In the second term of Eq. 1, μsm is the cross-section weighted mean diameter for the small-diameter distribution, and is the mean diameter of the small-diameter population across the control samples.
The Raman index is constructed by first selecting bands that are statistically significantly different (as chosen by a one-sided t test) between the stimulated and control populations. Several bands corresponding to DNA, protein, and lipid content within the cell, all possible areas of chemical difference between activated and unactivated cells, are considered. The area of each band is computed for each cell and compared to the mean area under the band for all control cells. For each peak, if the cell has the same spectrum as the mean control cell spectrum, the value of that ratio is unity. The normalized areas under each of the peaks are added together as
| (3) |
where N is the total number of statistically significant bands being considered, Aj is the area of the jth band, is the area of the jth band for the mean control spectrum, and Φ(j) is 1 if the control mean is greater than the stimulated mean for a given peak, and −1 if smaller. This exponent is used so that all changes in the stimulated mean increase the index above zero. As is evident from Eqs. 3, 1, the mean values of the two indices for the control samples should lie at zero. As already noted, both the Raman and elastic scattering indices are constructed such that, analogously to the flow plots, the unactivated cells should cluster toward the lower left corner of the plot, and increases in either index correspond to cellular activation. Note that, as in the FACS analysis, the control data points will be nonidentical and appear as a cloud due to biological variability.
Figure 9 depicts the Raman and elastic scattering results for the SEB stimulation experiment compressed onto a 2-D space with equal weight given to each measurement modality. The Raman index is plotted along the x axis, while the elastic index is on the y axis. Each cell is shown as a single point in this space, with the control cells being labeled with circles and the stimulated cells being labeled as squares. Echoing the flow cytometry results shown in Fig. 1, the control cells cluster toward the lower left-hand corner of the plot, while the stimulated cells are spread from the lower left-hand corner up toward the upper right-hand corner of the plot.
Figure 9.
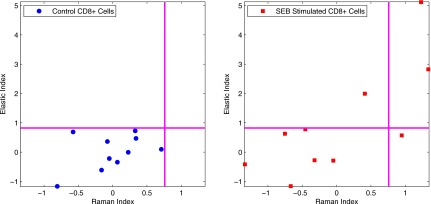
Scatter plots of Raman index versus elastic index for SEB-stimulated cells. Left-hand plot shows control cells, while stimulated cells are shown on the right-hand panel.
Figure 10 depicts the results from the PMA stimulation experiment compressed onto the Raman and elastic scattering index space. In this plot, the control cells once again cluster at the lower left-hand corner of the plot, while the stimulated cells cluster towards the right-hand side of the plot, extending from top to bottom, echoing the flow results shown in Fig. 2.
Figure 10.
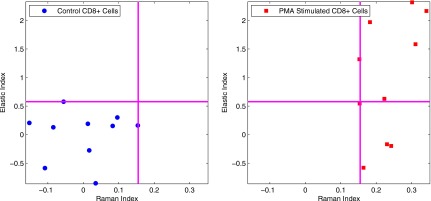
Scatter plot of Raman index versus elastic index for PMA-stimulated cells. Left hand-plot shows control cells, while stimulated cells are shown on the right-hand panel.
Discussion
The chief aim of this work was a proof-of-principle demonstration that the IRAM platform provides sensitivity to changes that occur during immune cell activation. Using the current system, only 10 cells per cohort could be studied in the time that the cells remained viable; a faster, automated instrument is currently under construction as part of an effort to increase these numbers. Even this current study, however, exhibited several trends for which interpretations can be offered.
With both SEB and PMA stimulation, subtle but clear differences are apparent in the Raman spectra, shown in Figs. 78. In the case of SEB stimulation, significant decreases were seen in several bands, summarized in Table 1, indicating a decrease in DNA signal compared to total protein content. Both Mannie et al. and Chan et al., who both observed the decrease in DNA signal upon cell activation, have speculated on the reason for this decrease.7, 8 As described by Branco et al., on T-cell activation there is a decondensation of DNA related to active transcription to produce new cytokines, cell surface markers, and cytotoxic proteins.2 This decondensation is associated with nuclear swelling, leading to an overall lower density of DNA within the nucleus. Chan et al. speculate that as such changes in density occur while the microscope’s (subnuclear) sampling volume stays constant, the total DNA signal measured is decreased. In other words, since the Raman spectrum for a particular vibration is proportional to the number of molecules excited, if the number of molecules within a fixed sampling volume decreases, the Raman spectrum will report that change in density.8
By contrast, with PMA stimulation we do not see the same magnitude of change in the DNA bands as we do for cells undergoing SEB-induced activation. In PMA-induced activation we see a decrease in the C–O stretching vibration, and an increase in the amide I and C=C bands. At present we hypothesize the increase in amide I and C=C vibrations is due to an increase in total protein and lipid content of the cell as it assembles new proteins and packages them into new cytolytic granule populations. However, we currently do not have a clear understanding of the decrease in the C–O stretching vibration, except to speculate that since the assembly of amides involves the replacement of a hydroxyl group with an amino group, a rapid construction of such proteins may decrease the total number of C–O bonds in the cell. As further support for this hypothesis, we note that Puppels et al. found that lymphocytes contain high concentrations of antioxidant carotenoids,21 with CD8+ lymphocytes having carotenoids collocated with the Golgi body. Here they might be recruited to scavenge the large numbers of highly reactive hydroxyl radicals that would be created during rapid protein assembly occurring at the Golgi body and nearby endoplasmic reticulum. We note that this increase in protein and lipid construction occurring in the cytoplasm is also likely taking place within the SEB-activated cells despite the fact that we observe no evidence of this in the SEB-activated cells’ Raman spectra. This could be because the decondensed nucleus pushes the cytoplasm farther away from the focal region of the Raman-sampling beam.
Elastic scattering changes, by contrast, appear to be similar between the two activation experiments, as presented in Figs. 56. SEB activation resulted in both an increase in the relative contribution of scattering by small scatterers to the scattering pattern, presumably due to construction of new cytolytic granules or new podosomes (organelles that facilitate cell adhesion and motility), and a shift of the small population to larger mean diameters. Some cells in the SEB-stimulated cohort exhibited scattering very similar to the scattering of the control population, consistent with the expectation that many of the SEB-stimulated cells would be unresponsive to the antigen. PMA activation had a heterogenous response with some cells exhibiting the same changes seen in the SEB-stimulated experiment, while some of the PMA-stimulated cells exhibited no change in their scattering when compared to control cells, despite the fact that nearly all of the cells were defined as activated by the paired flow cytometry analysis.
One possible explanation for the difference between CD8+ lymphocyte responses to the two agents is that PMA and SEB stimulations evolve following different temporal kinetics. This is especially plausible given that the two exploit fundamentally different mechanisms of cell activation, with the PMA directly stimulating the PKC (protein kinase C) signaling pathway, while SEB stimulation requires several intermediate steps between cell recognition of SEB and PKC activation. Therefore, since both experiments had the same 18 hour stimulation time, it is possible that cells stimulated with PMA were measured by the IRAM microscope at a different stage in their activation process.
When the Raman and elastic scattering results are compressed into the index space, as shown in Figs. 910, clear separations can be seen between the stimulated and control cells. For the SEB case, we see that some of the cells in the stimulated sample register as unactivated, as they land within the box defined by the control cells. This matches what we expect based both on previous SEB stimulation studies as well as our own flow cytometry results, where only a fraction of the exposed cells are shown as responsive to SEB stimulation. We further note that 4 out of 10 of the stimulated cells are marked as activated, which is consistent with the flow cytometry result of 26.38% of the stimulated cells activating, considering our small number of IRAM-measured cells. Among the activated cells we see a broad distribution of Raman and elastic scattering responses, suggesting the heterogeneity in terms of activated phenotypes among different cells. This heterogeneity also loosely correlates with the heterogeneity seen in the flow cytometry data, as some cells express only CD69 or CD137, while the majority of activated cells express both. This heterogeneity of response is also evidenced in the Raman versus elastic index plot for PMA stimulation, as seen in Fig. 10. In this case, all of the stimulated cells cluster differently from the control cells, mirroring the flow results that reported a 96.3% activation rate among the stimulated cells. However, we again see a large spread in the elastic scattering response of the stimulated cells. Some cells show a strong Raman and elastic scattering response, while others show a strong Raman response but a weak elastic response.
The typical criteria for “activation” used in flow cytometric studies of immune cell activation usually measure surface protein expression, whereas IRAM measurements report on a cell’s internal contents. To explore the various hypotheses proposed in this study, it will be necessary to utilize other methodologies such as fluorescence microscopy and scanning electron microscopy to examine internal properties that are more closely linked to the type of data acquired by IRAM, such as numbers of granules per cell, intracellular cytokine and granzyme production, and distribution of chromatin within the nucleus.
Such parallel experiments will also help validate the robustness of our fitting techniques. We note that there appears to be a consistent discrepancy between the collected data and our best fit theory curve, indicating that there is likely something in the experimental measurement that the model is not accounting for. However, attempts to fit the data with three log-normal distributions or including a single large scatterer representing the nucleus have not resulted in improved fits. This is consistent with the experimental observations of Wilson and Foster22 as well as with simulations performed by our group. We speculate that this residual is due to either unscattered laser light that interferes with the scattered light (a particular concern when the scattered intensity is low as it is for lymphocytes) or to insufficient averaging of speckle.
Conclusion
We showed evidence of stimulus-dependent changes in the elastic and Raman scattering signatures of single activated immune cells relative to their resting counterparts. These changes suggest chromatin decondensation, increases in protein and lipid content, and assembly of new cytotoxic organelles. Emerging questions about the heterogeneity and kinetics of activations between single cells could be answered by the IRAM instrument, as it is, in principle, capable of measuring a cell longitudinally over several hours or days. Tracking a multitude of cells over time as they traverse the 2-D Raman and elastic scattering index space could provide detailed insights into the biochemical and morphological nature of internal, activation-dependent cell processes, and is a direction of inquiry our group is actively pursuing.
Acknowledgments
This work was supported in part by National Science Foundation (NSF) Grant CBET-0754698 and by National Institutes of Health (NIH) National Institute of Alergy and Infectious Deseases (NIAID) Grant No. R24A1054953 (Rochester Human Immunology Center).
References
- Auphan-Anezin N., Verdeil G., and Schmitt-Verhulst A.-M., “Distinct thresholds for CD8 T cell activation lead to functional heterogeneity: CD8 T cell priming can occur independently of cell division,” J. Immunol. 170(5), 2442–2448 (2003). [DOI] [PubMed] [Google Scholar]
- Branco M. R., Branco T., Ramirez F., and Pombo A., “Changes in chromosome organization during PHA-activation of resting human lymphocytes measured by cryo-FISH,” Chromosome Res. 16(3), 413–426 (2008). 10.1007/s10577-008-1230-x [DOI] [PubMed] [Google Scholar]
- Wölfl M., Kuball J., Ho W. Y., Nguyen H., Manley T. J., Bleakley M., and Greenberg P. D., “Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities,” Blood 110(1), 201–210 (2007). 10.1182/blood-2006-11-056168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft C., Knetschke T., Siegner A., Funk R. H. W., and Salzer R., “Mapping of single cells by near infrared Raman microspectroscopy,” Vib. Spectrosc. 32(5), 75–83 (2003). 10.1016/S0924-2031(03)00049-3 [DOI] [Google Scholar]
- Xie C., Chen D., and Li Y.-Q., “Raman sorting and identification of single living micro-organisms with optical tweezers,” Opt. Lett. 30(14), 1800–1802 (2005). 10.1364/OL.30.001800 [DOI] [PubMed] [Google Scholar]
- Chan J. W., Taylor D. S., Zwerdling T., Lane S. M., Ihara K., and Huser T., “Micro-Raman spectroscopy detects individual neoplastic and normal hematopoietic cells,” Biophys. J. 90(2), 648–656 (2006). 10.1529/biophysj.105.066761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannie M. D., McConnell T. J., Xie C., and Li Y.-Q., “Activation-dependent phases of T cells distinguished by use of optical tweezers and near infrared Raman spectroscopy,” J. Immunol. Methods 297(1), 53–60 (2005). 10.1016/j.jim.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Chan J. W., Taylor D. S., Lane S. M., Zwerdling T., Tuscano J., and Huser T., “Nondestructive identification of individual leukemia cells by laser trapping Raman spectroscopy,” Anal. Chem. 80(6), 2180–2187 (2008). 10.1021/ac7022348 [DOI] [PubMed] [Google Scholar]
- Mourant J. R., Hielscher A. H., Eick A. A., Johnson T. M., and Freyer J. P., “Evidence of intrinsic differences in the light scattering properties of tumorigenic and nontumorigenic cells,” Cancer Cytopathol. 84(6), 366–374 (1998). [PubMed] [Google Scholar]
- Backman V., Gurjar R., Badizadegan K., Itzkan I., Dasari R. R., Perelman L. T., and Feld M. S., “Polarized light scattering spectroscopy for quantitative measurement of epithelial cellular structures in situ,” IEEE J. Sel. Top. Quantum Electron. 5(4), 1019–1026 (1999). 10.1109/2944.796325 [DOI] [Google Scholar]
- Wax A., Pyhtila J. W., Graf R. N., Nines R., Boone C. W., Dasari R. R., Feld M. S., Steele V. E., and Stoner G. D., “Prospective grading of neoplastic change in rat esophagus epithelium using angle-resolved low-coherence interferometry,” J. Biomed. Opt. 10(25), 051604 (2005). 10.1117/1.2102767 [DOI] [PubMed] [Google Scholar]
- Wilson J. D., Bigelow C. E., Calkins D. J., and Foster T. H., “Light scattering from intact cells reports oxidative-stress-induced mitochondrial swelling,” Biophys. J. 88, 2929–2938 (2005). 10.1529/biophysj.104.054528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. D., Giesselman B. R., Mitra S., and Foster T. H., “Lysosome-damage-induced scattering changes coincide with release of cytochrome c,” Opt. Lett. 32(17), 2517–2519 (2007). 10.1364/OL.32.002517 [DOI] [PubMed] [Google Scholar]
- Boustany N. N., Kuo S. C., and Thakor N. V., “Optical scatter imaging: subcellular morphometry in situ with Fourier filtering,” Opt. Lett. 26(14), 1063–1065 (2001). 10.1364/OL.26.001063 [DOI] [PubMed] [Google Scholar]
- Fang H., Qiu L., Vitkin E., Zaman M. M., Andersson C., Salahuddin S., Kimerer L. M., Cipolloni P. B., Modell M. D., Turner B. S., Keates S. E., Bigio I., Itzkan I., Freedman S. D., Bansil R., Hanlon E. B., and Perelman L. T., “Confocal light absorption and scattering spectroscopic (CLASS) microscopy,” Appl. Opt. 46(10), 1760–1769 (2007). 10.1364/AO.46.001760 [DOI] [PubMed] [Google Scholar]
- Smith Z. J. and Berger A. J., “Integrated Raman- and angular-scattering microscopy,” Opt. Lett. 33(7), 714–716 (2008). 10.1364/OL.33.000714 [DOI] [PubMed] [Google Scholar]
- Smith Z. J. and Berger A. J., “Validation of an integrated Raman- and angular-scattering microscopy system on heterogeneous bead mixtures and single human immune cells,” Appl. Opt. 48(10), D109–D120 (2009). 10.1364/AO.48.00D109 [DOI] [PubMed] [Google Scholar]
- Smith Z. J. and Berger A. J., “Construction of an integrated Raman- and angular-scattering microscope,” Rev. Sci. Instrum. 80, 044302 (2009). 10.1063/1.3124797 [DOI] [PubMed] [Google Scholar]
- Gouesbet G., Grehan G., and Maheu B., “Scattering of a Gaussian beam by a Mie scatter center using a Bromwich formalism,” J. Opt. (Paris) 16(2), 83–93 (1985). [Google Scholar]
- Uzunbajakava N., Lenferink A., Kraan Y., Willekens B., Vrensen G., Greve J., and Otto C., “Nonresonant Raman imaging of protein distribution in single human cells,” Biopolymers 72(1), 1–9 (2003). 10.1002/bip.10246 [DOI] [PubMed] [Google Scholar]
- Puppels G. J., Garritsen H. S. P., Kummer J. A., and Greve J., “Carotenoids located in human lymphocyte subpopulations and natural killer cells by Raman microspectroscopy,” Cytometry A 14(3), 251–256 (1993). 10.1002/cyto.990140303 [DOI] [PubMed] [Google Scholar]
- Wilson J. D. and Foster T. H., “Mie theory interpretations of light scattering from intact cells,” Opt. Lett. 30(18), 2442–2444 (2005). 10.1364/OL.30.002442 [DOI] [PubMed] [Google Scholar]