

Abstract
Progranulin (also known as granulin/epithelin precursor, GEP) is composed of seven granulin/epithelin repeats (granulins) and functions both as a full-length protein and as individual granulins. It is a secretory protein but a substantial amount of GEP is found inside cells, some in complexes with positive transcription elongation factor b (P-TEFb). GEP and certain granulins interact with the cyclin T1 subunit of P-TEFb, and with its HIV-1 Tat co-factor, leading to repression of transcription from the HIV promoter. We show that GEP lacking the signal peptide (GEPspm) remains inside cells and, like wild-type GEP, interacts with cyclin T1 and Tat. GEPspm represses transcription from the HIV-1 promoter at the RNA level. Granulins that bind cyclin T1 are phosphorylated by P-TEFb in vivo and in vitro on serine residues. GEPspm and those granulins that interact with cyclin T1 also inhibit transcription from cellular cad and c-myc promoters, which are highly dependent on P-TEFb, but not from the PCNA promoter. In addition, GEPspm and granulins repress transcriptional activation by VP16 or c-Myc, proteins that bind and recruit P-TEFb to responsive promoters. These data suggest that intracellular GEP is a promoter-specific transcriptional repressor that modulates the function of cellular and viral transcription factors.
Keywords: GEP, Progranulin, P-TEFb, HIV-1, c-Myc, cad promoter, cyclin T1, CDK9, transcription elongation, c-myc promoter
Introduction
The product of the GRN gene is a pluripotent protein involved in development, cell growth and proliferation, host defense, wound repair and transcription (He and Bateman, 2003; Hoque et al., 2003). The full-length protein product of the GRN gene is variously known as granulin/epithelin precursor (GEP), progranulin, PC cell-derived growth factor (PCDGF), proepithelin, and acrogranin. GRN mRNA is prominent in several human cell lines including epithelial cells and many hematopoietic cells (Ong and Bateman, 2003). High levels of GEP expression are found in human cancers including breast cancer (Serrero, 2003), invasive ovarian cancers (Jones et al., 2003) and glioblastomas (Liau et al., 2000) and its expression is directly correlated with tumorigenicity in breast cancer (Lu and Serrero, 2000). In the last three years, GEP has become a focus of intense attention in the field of neurodegenerative dementia since reduced levels of this protein are genetically and biochemically connected with FTLD-U (frontotemporal lobar degeneration with ubiquitin-positive inclusions) (Bateman and Bennett, 2009; Cruts and Van Broeckhoven, 2008). In FTLD-U, most GRN mutations lead to the disappearance of mRNA from the defective allele with concomitant depletion of the protein (Baker et al., 2006). The molecular mechanism whereby this haploinsufficiency leads to nerve degeneration remains to be discovered, but microarray analysis of FTLD-U brains indicated that the expression of hundreds of genes is affected (Chen-Plotkin et al., 2008).
GEP contains seven and a half repeats of a highly conserved cysteine-rich motif in the order P-G-F-B-A-C-D-E, where A-G are granulin repeats and P is a half repeat containing a putative secretion signal sequence (Fig. 1A). GEP is processed to its granulin repeats by the enzyme elastase which is regulated by the secretory leukocyte protease inhibitor (SLPI) that forms a complex with GEP (Zhu et al., 2002). Single repeats, called granulins or epithelins, are individually biologically active although their functions are little understood (Ong and Bateman, 2003). GEP is secreted as a 88 kDa glycoprotein (GP88) or a 68 kDa form depending on the producing cells (Xu et al., 1998; Zhou et al., 1993). We discovered that a fraction of GEP is retained inside cells and interacts with the cyclin T1 subunit of the general positive transcription elongation factor b (P-TEFb). Intriguingly, GEP also interacts with the HIV-1 transactivator Tat (transactivator of transcription) (Hoque et al., 2005; Hoque et al., 2003; Shoham et al., 2003; Trinh et al., 1999). During infection, Tat cooperates with the HIV TAR (transactivation response element) to recruit P-TEFb to viral transcription complexes via direct and specific binding to cyclin T1, resulting in the formation of Tat/TAR/P-TEFb ternary complexes. This interaction allows the generation of full-length rather than prematurely terminated HIV-1 RNA chains (Wei et al., 1998; Zhu et al., 1997). Hence, the HIV-1 promoter is highly dependent on P-TEFb and Tat. P-TEFb is involved in numerous cellular functions including cell growth, cellular proliferation, apoptosis, cellular differentiation, and response to stress (Bres et al., 2008; De Falco and Giordano, 2002; Kohoutek et al., 2009). It is required for transcription elongation from most cellular genes by RNA polymerase II (Pol II), but promoters have differing P-TEFb requirements: while some are highly dependent on P-TEFb, others display relatively little dependence (Haaland et al., 2005; Lam et al., 2001).
Fig. 1. GEP lacking the signal peptide inhibits expression from the HIV-1 promoter.
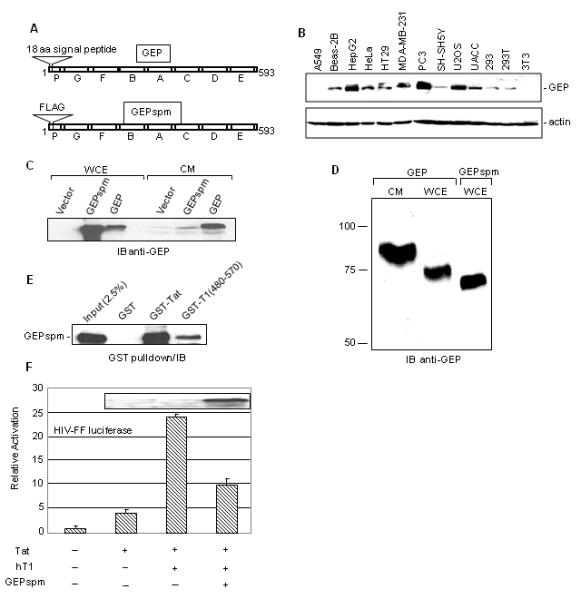
A. Schematic representation of GEP and FLAG-GEPspm (signal peptide minus). B. Detection of endogenous GEP and actin in whole cell extract (WCE) prepared from the depicted epithelial cell lines and from 3T3 cells. C. FLAG-GEPspm is retained inside the cells. 293T cells (5×105) were transfected with 2 μg of GEP or FLAG-GEPspm or empty vector using lipofectamine 2000 (Invitrogen, CA) and 24 hrs post-transfection WCE and culture medium (CM) were analyzed by immunoblotting with anti-GEP antibody. D. Mobility of GEP from CM or WCE in denaturing gel. CM or WCE from transfected cells was resolved on 7% SDS gel and GEP was detected by immunoblotting (IB) with anti-GEP antibody. E. FLAG-GEPspm forms complexes with Tat and cyclin T1. WCE of 293T cells expressing FLAG-GEPspm was analyzed in GST pull-down assays. Complexes bound to beads containing GST, GST-Tat or GST-T1 (aa 480-570) were separated in denaturing gels and immunoblotted with anti-GEP antibody. F. Inhibition of Tat transactivation by GEPspm. 3T3 cells (2×105) were co-transfected with 0.1 μg HIV-FF luciferase, 0.25 μg hT1 and 0.5 μg of FLAG-GEPspm expression vector or an equimolar amount of empty vector. Luciferase expression was analyzed 24 hr post transfection. The activation of HIV-FF luciferase was normalized to Renilla luciferase expressed from the RSV promoter. Results are the average of two experiments in duplicates ±SD. Extracts from the transfected cells were immunoblotted with anti-GEP antibody (inset).
P-TEFb contains, in addition to cyclin T1 (or cyclin T2 or K), the cyclin-dependent kinase CDK9. CDK9 catalyzes phosphorylation at specific serines in the carboxy-terminal domain (CTD) of the large subunit of Pol II, allowing its transition into a productive elongation mode (Peterlin and Price, 2006; Ramanathan et al., 1999). P-TEFb is targeted by numerous cellular and viral transcription regulators that activate or inhibit its function (Bres et al., 2008; Zhou and Yik, 2006). As in the HIV paradigm, it does not bind directly to DNA but is recruited to genes by cellular DNA-bound activators such as NF-κB and steroid hormone receptors, and by Brd4, a general chromatin-bound remodeling protein that recruits P-TEFb to actively transcribed chromatin. The interactions of P-TEFb with c-Myc have been studied in some detail (Gargano et al., 2007; Kanazawa et al., 2003). The Myc activation domain (in its N terminus) is sufficient for binding to cyclin T1, and expression of this domain elevates P-TEFb kinase activity resulting in increased CTD phosphorylation (Cowling and Cole, 2007). Like the HSV transcription activator VP16 and the cellular activators CIITA and Rel A, c-Myc binds to the cyclin domain of cyclin T1, recruits P-TEFb and activates transcription elongation (Kurosu and Peterlin, 2004).
The activity of P-TEFb is also subjected to negative regulation. A substantial fraction of the cell’s P-TEFb is found in inactive ribonucleoprotein particles containing the small nuclear RNA, 7SK, and one of two HEXIM proteins (Brigati et al., 2003; Michels and Bensaude, 2008; Michels et al., 2003; Nguyen et al., 2001; Ping and Rana, 1999; Zhou and Yik, 2006; Zhu et al., 1997). HEXIM and 7SK RNA bind reversibly to P-TEFb, inhibiting its transcriptional function by reducing its kinase activity and by preventing the recruitment of P-TEFb to the pre-initiation complex. P-TEFb activity or recruitment is also inhibited, specifically and in a cell-dependent manner, by a number of cellular proteins including HIC and I-mfa, PIE-1, Runx-1, TAF7, Pcg and the glucocorticoid receptor (Gegonne et al., 2008; Hanyu-Nakamura et al., 2008; Hoque et al., 2003; Jiang et al., 2005; Wang et al., 2007; Young et al., 2003; Zhang et al., 2003).
In its interactions with both cyclin T1 and Tat, GEP targets sites that are critical for transcription from the HIV-1 promoter. GEP binds directly to the histidine-rich (His-rich) domain of cyclin T1 required for its interaction with Pol II (Hoque et al., 2003), and to the activation domain of Tat required for its interactions with TAR and P-TEFb (Hoque et al., 2005). It also binds to the Tat/P-TEFb complex, thereby inhibiting transcription from the HIV-1 promoter (Hoque et al., 2005). Some granulin repeats, such as GrnDE and GrnE, bind to Tat but do not bind directly to cyclin T1 although they do interact with the Tat/P-TEFb complex (Hoque et al., 2005). The C-terminal part of GEP, GrnCDE, also serves as a specific substrate for CDK9 in vitro.
The dual nature of GEP, as an intracellular as well as a secreted protein, may explain its ability to regulate a large number of cellular functions via signal transduction pathways from outside the cell and via transcriptional modulation from within. In this study we investigated the function of intracellular GEP and granulins on P-TEFb dependent transcription. GEP lacking its secretory signal peptide is retained inside cells and inhibits transcription from the HIV-1 promoter, showing that active secretion of GEP is not necessary for its inhibitory function. GEP fractionates with complexes containing active P-TEFb, and its repression activity correlates with its ability to bind to the Pol II binding site of cyclin T1. Granulins inhibit expression from two cellular promoters that, like the HIV promoter, are highly dependent on P-TEFb. Furthermore, granulins are able to modulate the transcriptional activation function of c-Myc and VP16.
Materials and Methods
Cell culture
293, 293T, NIH 3T3, Beas-2B, A549, Hep G2, HT-29, PC-3, MDA-MB-231, SH-SY5Y, U2OS, UACC and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 8% fetal bovine serum (Sigma-Aldrich).
Reagents
Mouse anti-FLAG monoclonal antibody (M2) was from Sigma-Aldrich. Rabbit anti-HA, rabbit anti-CDK9, goat anti-human and mouse cyclin T1, goat anti-GEP and secondary antibody linked to HRP were from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Rabbit anti-GEP antibody was from Dr. G. Serrero (Lu and Serrero, 2000). Synthesis and purification of CTD4 was described earlier (Hoque et al., 2003).
Plasmids
The plasmids expressing CDK9, cyclin T1 and truncated versions of cyclin T1, GEP (pcDNA3-PCDGF), HIV-FF luciferase (pLTR-luciferase), Renilla luciferase (Ren) (pRSV-luciferase), Gal4-human cyclin T1 and pcDNA3.HA-Tat were described earlier (Hoque et al., 2003). Vectors expressing FLAG-tagged granulin repeats, GrnCDE, GrnPGFBA, GrnCD, GrnDE, GrnD, and GrnE, were described previously (Hoque et al., 2005). The pFLAG-GEPspm expression vector was generated by cloning PCR-amplified GEP cDNA without its signal peptide sequence into the pFLAG-CMV2 vector. Plasmids pGL3-G5-ML1-FF luciferase and PCNA-FF luciferase were described earlier (Reichman et al., 2002; Reichman et al., 2003). cad3G4luc (cad-FF luciferase) and Gal4–myc (aa 1-262) were obtained from Dr. P.J. Farnham (Eberhardy and Farnham, 2001). Gal4-VP16 was obtained from Dr. C.-G. Lee (New Jersey Medical School).
Preparation of whole cell extract (WCE)
Cells were grown to confluency in 100-mm tissue culture dishes or in suspension to a density of 1 × 106 cells/ml, and washed twice with 10 ml of phosphate-buffered saline (minus calcium and magnesium). Cells were then lysed as described earlier (Hoque et al., 2005).
Co-immunoprecipitation
Mouse 3T3, human 293T or HeLa cells (5 × 106 cells) were transfected with 10 μg of DNA using Jet-PEI (Polyplus) according to the manufacturer’s instructions. Cells were harvested at 24 h post-transfection and lysed in 1 ml of immunoprecipitation (IP) lysis buffer (50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 1%Triton X-100, 1 mM EDTA) containing 1X protease inhibitor cocktail (Sigma). Extracts were cleared, and supernatants were collected after 15-min centrifugation at 4 °C (Eppendorf, 13,000 rpm). Antibody (2 μg) was bound to protein G-Sepharose by incubation at 4 °C for 2 h with rocking. The conjugates were mixed with cell extract (500 μg of total protein) and incubated 3 h at 4 °C with rocking. Unbound proteins were removed by extensive washing with lysis buffer containing 0.03% SDS. Protein complexes bound to the conjugated antibody were separated by denaturing polyacrylamide gel electrophoresis and subjected to immunoblotting as described previously (Ramanathan et al., 1999). The same protocol was used to immunoprecipitate and detect endogenous proteins.
RNase protection assay (RPA)
3T3 cells (2 × 106 cells) were seeded in 10-cm-diameter plates and transfected 20 h later by using lipofectamine 2000 (Invitrogen). Cells were harvested at 24 h post-transfection. Total RNA was prepared by using Trizol (Invitrogen) according to the manufacturer’s instructions. Radioactively labeled firefly and Renilla antisense RNA probes were generated as described previously (Young et al., 2003). The RNase protection assay was performed with 20 μg of RNA, using the RPAIII kit from Ambion (Austin, Texas.) according to the manufacturer’s instructions.
Transfection and luciferase assays
3T3 cells (2 × 105 cells) were transfected using Lipofectamine™ 2000 according to the manufacturer’s instructions. After 24 h the cells were harvested, washed with phosphate-buffered saline, and lysed in 0.1 ml of 1x Promega lysis buffer and assayed for luciferase activity. Luciferase assays were performed with the Promega Dual Luciferase Reporter System according to the manufacturer’s instructions.
Phospho-amino acid analysis
Phosphoamino acid analysis was performed essentially as described by Ausbel et al. 1991. (Current protocols in molecular biology, 2nd ed., vol. 1). Briefly, the phosphorylated protein were transferred from gels to PVDF membranes, hydrolyzed in 6 M HCl, dried in vacuum and resuspended in 2 to 5 μl of water. In some experiments, the membranes were treated with alkali before acid hydrolysis. A portion of the digest containing 1,000 to 5,000 CPM was spotted on a cellulose thin-layer chromatography plate, and 1 μl of a solution containing about 1 μg of each unlabeled phosphoamino acid was spotted as a reference. The amino acids were resolved by two-dimensional electrophoresis (HTLE 7000; CBS Scientific). Phospho-amino acid standards were visualized by ninhydrin staining, and 32P-labeled amino acids were detected by autoradiography.
Sedimentation analysis
Glycerol gradients (5 - 45%) were prepared in buffer A (10 mM HEPES, 15 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% PMSF, 1mM DTT and 40 U/ml RNasin) (Michels et al., 2003). Cell extracts prepared in buffer A were layered on top of the glycerol gradient and centrifuged for 16 h at 40,000 rpm at 4°C in a SW41 rotor. Fractions collected from the top were analyzed by immunoblotting or immunoprecipitatin followed by RNA extraction and RT-PCR (Young et al., 2007).
In vitro binding assays
GST-tagged granulins bound in equimolar amounts to glutathione beads were incubated for 3 h with 500 μg of 293T cell WCE. After extensive washing, bound proteins were separated in a denaturing gel and detected by immunoblotting (Hoque et al., 2005).
Kinase assays
GST-granulin pull-down kinase assays, IP-kinase assays, and purification of GST and GST-granulin substrates were described previously (Hoque et al., 2003).
Results
Granulin secretion is not required for its transcriptional effect
Expression driven by the HIV-1 promoter was inhibited by expression of full-length GEP and certain granulin repeats in human and mouse cells (Hoque et al., 2005; Hoque et al., 2003). Even though it is a secreted growth factor, endogenous GEP co-immunoprecipitated with P-TEFb from extracts of Daudi cells (Hoque et al., 2003), indicating that at least a fraction of the protein is intracellular. The presence of GEP has also been reported in extracts of PC cells (Zhang and Serrero, 1998). To determine whether endogenous GEP is generally found intracellularly, we tested extracts of a number of cell lines. Cells were thoroughly washed to remove traces of medium. GEP was detected at varying levels in nearly all epithelial cells examined (Fig. 1B). Exceptionally, GEP was not detected in A549 human epithelial cells although this cell line contains GEP mRNA (Daniel et al., 2000), suggesting that GEP expression is controlled post-transcriptionally in these cells or that they secrete the protein especially efficiently. Little GEP was detected in U937 cells (data not shown), but it is inducible by mitogens (data not shown, (Ong et al., 2006)). GEP was also undetectable in 3T3 cells, in keeping with previous studies and with the failure to detect its mRNA in these cells (Bhandari et al., 1992; Zhang and Serrero, 1998).
Since GEP is commonly found inside cells, we asked whether secretion is necessary for its intracellular function. To address this question, we constructed a vector encoding a GEP signal-peptide-minus variant (GEPspm) that lack the protein’s putative 18-amino acid long signal peptide (Fig. 1A). The whole cell extract, as well as the culture media, from human 293T cells transfected with GEP or GEPspm vectors were analyzed for GEP by immunoblotting (Fig. 1C). Confirming the functionality of the putative signal peptide, GEPspm was overwhelmingly retained in the cells whereas substantial amounts of GEP were detected in both fractions. Endogenous GEP expression in 293T cell extract is very low (Fig. 1B). Accordingly, we observed a very faint band of endogenous GEP in the culture medium of vector-transfected cells (Fig. 1C). High-resolution gels resolved three forms of ectopically expressed GEP (Fig. 1D). The secreted form of GEP, which is presumably glycosylated (Zhou et al., 1993) migrated more slowly than cell-associated GEP, and GEPspm migrated faster than cell-associated GEP as expected. Like full-length GEP (Hoque et al., 2003), GEPspm bound to Tat and to the His-rich domain of cyclin T1 in GST pulldown experiments (Fig. 1E). To determine its ability to inhibit Tat-activated gene expression from the HIV-1 promoter, we conducted co-transfection assays in 3T3 cells. The levels of GEP expressed were comparable to those seen in cells such as HeLa and did not affect the growth of the 3T3 cells (data not shown). Human cyclin T1 is needed for Tat transactivation in mouse cells (Wei et al., 1998). As with GEP (Hoque et al., 2003), GEPspm inhibited the human cyclin T1-dependent expression of luciferase from the HIV-1 promoter by ~2.5 fold (Fig. 1F). Neither GEP nor GEPspm had any discernible toxic effect on the cells. Thus, active secretion of GEP is not necessary for its repressive function in this system.
GEP is present in complexes that co-sediment with active P-TEFb
GEPspm and some granulin repeats repress the production of firefly luciferase driven by the HIV-1 promoter. Accumulated evidence argues that GEP inhibits gene expression via P-TEFb, possibly by driving it into inactive complexes containing HEXIM and 7SK RNA. To test this idea, we first verified that the effect is at the RNA level. The N-terminal part of GEP, GrnPGFBA (which lacks the signal peptide), is a potent inhibitor of HIV-driven luciferase expression, essentially eliminating the activation elicited by human cyclin T1 in mouse 3T3 cells (Fig. 2A). When total RNA was examined by RNase protection assay, GrnPGFBA largely neutralized the effect of human cyclin T1 on the accumulation of luciferase RNA driven by the HIV-1 promoter (Fig. 2B).
Fig. 2. Granulins inhibit expression from the HIV-1 promoter at the RNA level but do not associate with the inactive P-TEFb complexes.
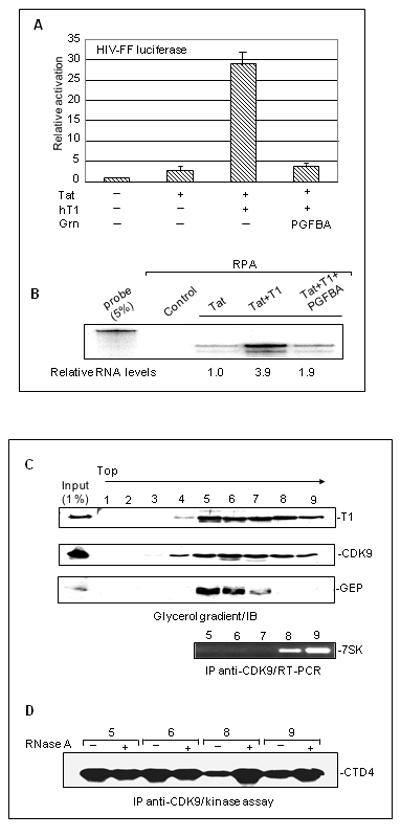
A. Inhibition of Tat transcativation by GrnPGFBA. 3T3 cells (2×105) were co-transfected with 0.5 μg HIV-FF luciferase, 1 μg RSV-renilla, 1 μg hT1, 2.5 μg FLAG-GrnPGFBA or an equimolar amount of empty vectors as indicated. Luciferase expression was quantified as in Fig. 1F. B. The levels of FF luciferase RNA are reduced in the presence of FLAG-GrnPGFBA. RNA extracted from 3T3 cells (2×106) was subjected to RNA protection assays (RPA). Autoradigrams display RPA fragments corresponding to FF-luciferase and Renilla luciferase. Probe: undigested probe was run as a control. C. GEP is not present in fractions containing 7SK RNA. Daudi cell extracts containing GEP were fractionated in a 5%-45% glycerol gradient. The fractions were immunoblotted with anti-cyclin T1 (upper panel) anti-CDK9 (middle panel) and anti-GEP (lower panel) antibody. Aliquot (200 μl) of gradient fractions 5-9 were immunoprecipitated with anti-CDK9 antibody and 7SK RNA was detected by RT-PCR. D. Kinase-active P-TEFb complexes are present in fractions that contain GEP. 200 μl from gradient fractions were immunoprecipitated with anti-CDK9 antibody and subjected to RNase A treatment as shown, followed by kinase assays using CTD4 as substrate.
Since the inhibitory effect of granulin is exerted at the RNA level, we considered the possibility that granulins inhibit P-TEFb by binding to the repressed P-TEFb/HEXIM/7SK complexes and stabilizing them. Endogenous GEP co-immunoprecipitates with P-TEFb from extracts of Daudi cells, a cell line that expresses relatively large amounts of endogenous GEP (Hoque et al., 2003). Therefore we fractionated Daudi cell extract in glycerol gradients and tested fractions for the presence of cyclin T1, CDK9, 7SK and GEP (Fig. 2C). The CDK9 and cyclin T1 components of P-TEFb were present in a broad region of the gradient, from fractions 4-9 (~130 to 600 kDa; Fig. 2C, top panels). P-TEFb immunoprecipitated from gradient fractions, was tested in kinase assays using CTD4 as substrate (Fig. 2D). As expected, active kinase was present in fractions 5 and 6. Fractions 8 and 9, which contained 7SK (Fig. 2C, bottom panel), exhibited reduced kinase activity that was greatly increased by treatment with RNase A (Fig. 2D). GEP was present in the slower-sedimenting part of the region (fractions 5 and 6, with a lesser amount in fraction 7; Fig. 2C) containing active P-TEFb. Thus GEP and 7SK displayed reciprocal, not overlapping, distributions. We conclude that its inhibitory activity is not associated with the larger 7SK inhibitory complex.
Granulin phosphorylation and binding to P-TEFb
The ability of granulins to interact with cyclin T1 and/or Tat correlates with their ability to inhibit gene expression, and previous work suggested a connection between the inhibition of gene expression and granulin phosphorylation (Hoque et al., 2005; Hoque et al., 2003). To substantiate this connection, we first determined whether GrnCDE is phosphorylated in vivo. Human 293T cells transfected with FLAG-GrnCDE vector, or empty FLAG vector as control, were labeled with radioactive inorganic phosphate. FLAG-GrnCDE was readily detected by immunoblotting, both in WCE and in anti-FLAG immunoprecipitates (Fig. 3A, left panel). The same band was strongly phosphorylated as detected by autoradiography (right panel). Analysis of 32P-labeled FLAG-GrnCDE revealed serine as the sole phospho-amino acid in GrnCDE (Fig. 3B). CDK9 is a serine/threonine kinase that is autophosphorylated on both serine and threonine residues but it exclusively targets serine residues in the Pol II CTD (Ramanathan et al., 1999). Thus GrnCDE is phosphorylated in cells with the same specificity as P-TEFb substrates.
Fig. 3. GrnCDE is phosphorylated on serine residues inside cells.
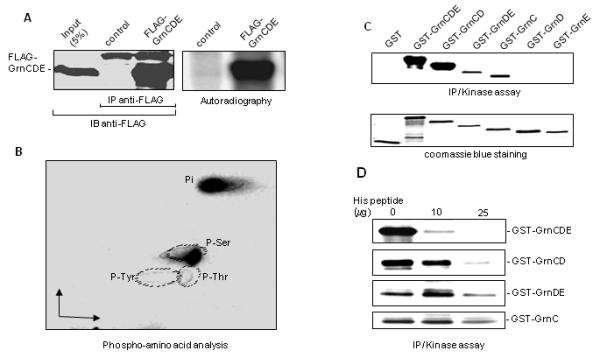
A. GrnCDE is phosphorylated inside cells. The 32P labeled FLAG-GrnCDE was immunoprecipitated from WCE with anti-FLAG antibody and immunoblotted with the same antibody (left panel). An autoradigram of the blotted FLAG-GrnCDE is shown in the right panel. B. Only serine residues in GrnCDE are targeted for phosphorylation. 32P-labeled FLAG-GrnCDE was subjected to Phospho-amino acid analysis and the labeled amino acids were detected by autoradiography. C. Some GST-granulins are substrates of P-TEFb. P-TEFb was immunoprecipitated with anti-CDK9 antibody and P-TEFb complexes were tested for their ability to phosphorylate various GST-granulins as indicated. Coomassie blue staining of GST and GST-granulins is shown in the lower panel. D. His peptide inhibits the phosphorylation of granulins that bind to cyclin T1. IP-kinase assay was performed as in C except that increasing amounts of his peptide were added as shown.
We next tested the correlation between the binding of granulins to P-TEFb and their ability to serve as substrates for this kinase. In the yeast two-hybrid system (Hoque et al., 2005) and in pulldown assays from WCE (data not shown), GrnCDE and GrnCD bound tightly to cyclin T1 while GrnDE and the GrnC, GrnD and GrnE single repeats were poor binding partners. When assayed as P-TEFb substrates in vitro using IP-kinase reactions, GrnCDE and GrnCD were strongly phosphorylated whereas GrnD and GrnE were not labeled to a detectable extent (Fig. 3C). Although they did not form complexes with cyclin T1 that were detectable by immunoblotting or yeast two-hybrid assays, GrnDE and GrnC were labeled weakly in the IP-kinase reactions. Similar results were obtained using the GST-pulldown kinase assay (Hoque et al., 2003), another highly sensitive technique for detecting granulin binding to and phosphorylation by CDK9 (data not shown). These data suggested that GrnDE and GrnC are able to interact transiently with P-TEFb, although the interactions are not sufficiently stable to register in IP-western or yeast two-hybrid assays (Hoque et al., 2005). To examine the specificity of the granulin/P-TEFb interactions, we evaluated their sensitivity to competition by a peptide corresponding to the His-rich domain of cyclin T1. The synthetic His peptide, amino acids 511-530 of cyclin T1, efficiently blocks the binding of GrnCDE to P-TEFb as well as the phosphorylation of GrnCDE by P-TEFb (Hoque et al., 2003). In the IP-kinase assay, phosphorylation of GrnCDE and GrnCD was greatly reduced by the His peptide whereas phosphorylation of GrnDE and GrnC was little affected (Fig. 3D). Evidently the transient interaction of P-TEFb with GrnDE and GrnC, observed only with the highly sensitive kinase assay, is not mediated via the cyclin T1 His-rich domain. These results support the conclusion that granulins that inhibit HIV-directed gene expression are effective P-TEFb substrates and bind tightly to the Pol II binding site of cyclin T1. Ectopically expressed GEP or granulins did not prevent P-TEFb from phosphorylating serine 2 in the Pol II CTD, however. No decrease in the phosphorylation levels of these serines was detected using specific antibodies (data not shown), suggesting that GEP and granulins do not exert a global inhibitory effect on P-TEFb function.
Granulin inhibits transcription from P-TEFb-dependent promoters
Like the HIV-1 viral promoter, a number of cellular promoters are known to be highly dependent on P-TEFb. One such promoter is that of the cad gene encoding carbamoyl-phosphate synthase/aspartate carbamoyltransferase/dihydroorotase, a trifunctional enzyme that catalyzes the first three rate-limiting steps of pyrimidine synthesis. This gene is a well-studied target of c-Myc which binds directly to cyclin T1 (Eberhardy and Farnham, 2001; Eberhardy and Farnham, 2002). Transcription from the cad promoter is normally dependent on the recruitment of P-TEFb to the promoter via c-Myc, but the need for c-Myc is bypassed when the Gal4-human cyclin T1 fusion protein is tethered to a modified cad promoter (Eberhardy and Farnham, 2001; Eberhardy and Farnham, 2002). We examined the effect of granulin on the cad promoter using the cad3G4luc reporter construct which contains the minimal cad promoter, furnished with three Gal4 DNA binding elements in place of the E box, driving firefly luciferase (Eberhardy and Farnham, 2001). When transfected into 3T3 cells together with Gal4-cyclin T1, the expression of firefly luciferase was increased ~8 fold. Co-transfection of human CDK9 expression vector further increased the expression of firefly luciferase to ~11 fold. GEPspm inhibited this activation by ~3-4 fold (Fig. 4). By contrast, GrnDE, which does not stably bind cyclin T1 (Hoque et al., 2005), had no effect on expression from the cad promoter (data not shown).
Fig. 4. GEP inhibits expression from the cad promoter.
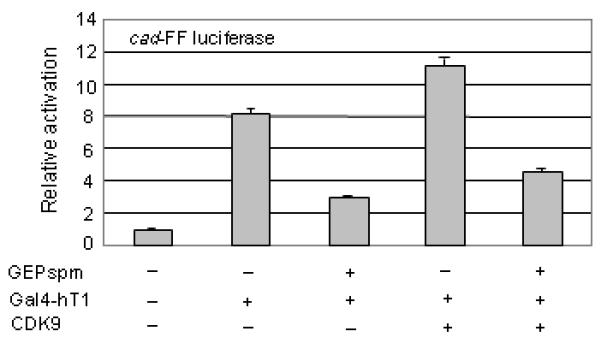
mmmmGEPspm inhibits the cyclin T1/ CDK9 dependent expression from the cad promoter. NIH 3T3 cells (2×105) were co-transfected with 0.2 μg cad-FF luciferase, 0.25 μg Gal4-hT1, 0.25 μg CDK9, 0.5 μg GEPspm expression vectors or equimolar amounts of control vectors as indicated. Luciferase expression was quantified as in Fig. 1F.
These experiments were conducted in 3T3 cells expressing human cyclin T1. To examine the effect of granulin on transcription in the absence of ectopic cyclin T1, we conducted experiments with two additional promoter constructs that are highly dependent on P-TEFb. The human c-myc promoter was identified as potentially P-TEFb dependent in a survey of genes that are down-regulated by flavopiridol, a potent inhibitor of P-TEFb (Haaland et al., 2005; Lam et al., 2001). As shown in Figure 5A, GrnPGFBA and GrnCDE inhibited expression from the c-myc promoter by 2.5-3 fold, whereas GrnD which does not interact with cyclin T1 was not inhibitory. The second promoter is an adenovirus major late promoter (MLP) modified to contain five upstream Gal4 binding sites. The Gal4 sites serve to recruit the Gal4-VP16 fusion protein. Transcriptional activation of the G4-Ad.ML-luciferase reporter plasmid by VP16 is dependent on P-TEFb because the VP16 activation domain is necessary for binding to the cyclin domain of cyclin T1 (Kurosu and Peterlin, 2004). In this Gal4 tethering system, GrnPGFBA or GrnCDE also reduced Gal4-VP16 activated expression by 3-4 fold (Fig. 5B). On the other hand, expression driven by the PCNA promoter was not significantly affected by GEP or other granulins (Fig. 5C). The PCNA promoter is not highly dependent on P-TEFb (Haaland et al., 2005; Lam et al., 2001), exemplifying the promoter specificity and P-TEFb dependence of the response to granulins. Interpretation of these experiments assumes that mouse cyclin T1 binds human granulins with the same specificity as human cyclin T1. This seemed likely because the His-rich domain that determines granulin binding is identical in mouse and human cyclins T1 (Kwak et al., 1999), and was borne out by the co-immunoprecipitation of mouse cyclin T1 with human FLAG-tagged GrnCDE but not GrnDE (Fig. 5D). We therefore conclude that granulins bind P-TEFb and inhibit transcription from viral and cellular promoters that are highly dependent on recruitment of P-TEFb.
Fig. 5. Granulins repress expression from the c-myc promoter and the activation of transcription by VP16.
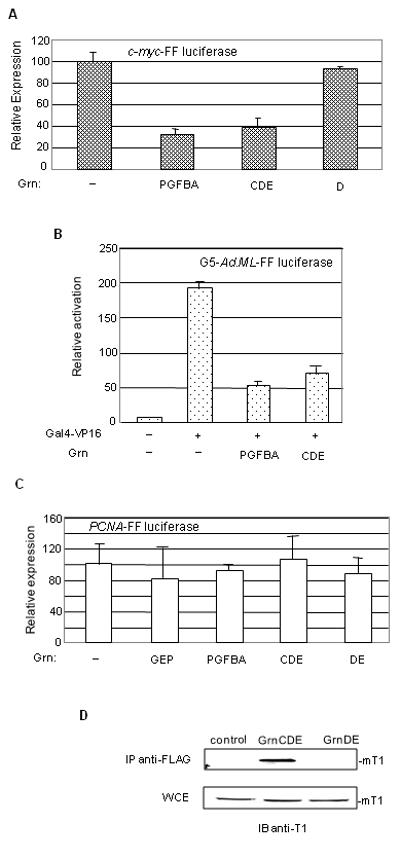
A. 3T3 cells were co-transfected with 0.2 μg c-myc-FF luciferase, 0.5 μg of Grn-PFGBA or equimolar amounts of the indicated granulin expression vectors. Empty vector was used as control. Expression of FF luciferase was normalized to protein level. Results are the average of two experiments in duplicates ±SD. B. 3T3 cells were co-transfected with 0.2 μg of the reporter vector G5-Ad.ML-FF luciferase, 0.1 μg of Gal4-VP16 and the granulin expressing vectors as indicated and as in panel A. Empty vector was used as control. Luciferase expression was quantified as in Fig. 1F. C. As in A, except that 0.2 μg of the reporter vector PCNA-FF luciferase was used. D. Human granulin interact with mouse cyclin T1. 3T3 cells (2×106) were transfected with 5 μg of FLAG-GrnCDE or equimolar amounts of FLAG-GrnDE or empty vector (control) as indicated. Anti-FLAG antibody was used to immunoprecipitate complexes containing FLAG-granulins from WCE. The immunoprecipitated complexes were immunoblotted with anti-mouse cyclin T1 antibody (upper panel). The same extracts were immunoblotted with anti-mouse cyclin T1 (lower panel).
GEP inhibits c-Myc activation and is present in c-Myc complexes
Activation of the cad promoter by c-Myc is formally analogous to activation of the HIV-1 promoter by Tat. In both cases, the activator recruits P-TEFb by binding to the N-terminal region of cyclin T1 (Eberhardy and Farnham, 2002). We have shown that granulins can inhibit P-TEFb mediated transcription from the HIV-1 promoter by binding directly to Tat, instead of cyclin T1 (Hoque et al., 2005). Therefore, we speculated that GEP might bind c-Myc and inhibit Myc-activated transcription. Anti-Myc antibody co-immunoprecipitated FLAG-tagged GEPspm and both the N- and C-terminal parts of GEP (PGFBA and CDE) from transfected HeLa cells (Fig. 6A). The reciprocal experiment, immunoprecipitation with anti-FLAG antibody followed by immunoblotting with anti-Myc antibody, was uninformative for technical reasons (i.e., that the anti-Myc antibodies are weak in immunoblots and c-Myc migrates close to the mouse IgG heavy chain which reacts strongly with the secondary antibody). However, anti-Myc antibody immunoprecipitated endogenous GEP (Fig. 6B). GEP also co-immunoprecipitated with the human Myc-interacting zinc-finger protein 1, Miz-1 (Fig. 6B), as reported previously for rat (Sui and Wilson, 2000). These data are consistent with functional interplay between GEP and the c-Myc regulatory network. Finally, GEPspm caused a ~ 2.5 fold inhibition of expression from the c-Myc activated cad promoter (Fig. 6C), indicating that GEP may exert control over c-Myc dependent gene expression similar to that mediated by HIV Tat.
Fig. 6. GEP is present in c-Myc and Miz-1 containing complexes and inhibits activation by c-Myc.
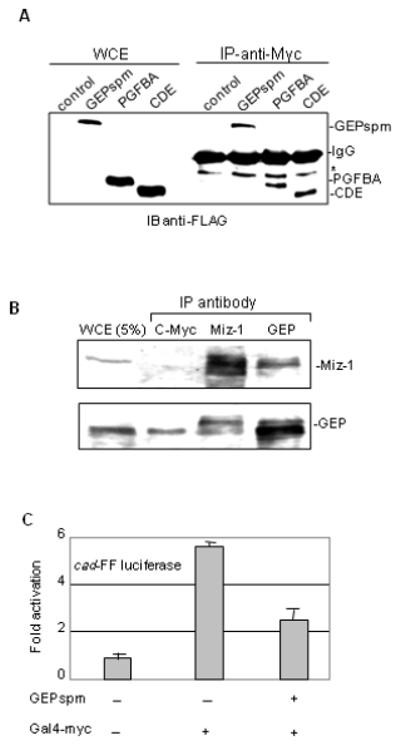
A. HeLa cells (2×106) were transfected with 5 μg of FLAG-GEPspm or equimolar amounts of FLAG-GrnPGFBA or FLAG-GrnCDE or empty vector. Antibodies against c-Myc were used to immunoprecipitate complexes containing granulins from WCE. The expression of the specific granulin was monitored by immunoblotting with anti-FLAG antibody. B. Complexes from HeLa WCE were immunoprecipitated with antibodies against GEP, Miz-1 and c-Myc and separated on denaturing gels. Antibodies against GEP or Miz-1 were used to detect the proteins by immunoblotting. C. 3T3 cells (2×105) were co-transfected with 0.2 μg cad-FF luciferase, 0.25 μg Gal4-Myc and 0.5 μg GEPspm expression vectors as indicated. Empty vector was used as control and samples were processed as in Fig. 1F.
Discussion
GEP is an enigmatic protein, functioning as a potent full-length multi-repeat entity as well as the precursor for granulins/epithelins which display diverse biological functions. To date, it has been investigated chiefly as a secreted growth factor that stimulates the proliferation of cells in tissue culture. We previously reported that GEP interacts inside cells with human cyclin T1, the major cyclin component of the transcription elongation factor P-TEFb, and inhibits transcription from the HIV-1 promoter. The specificity of GEP binding to human cyclin T1 is conferred by its His-rich domain containing a sequence of five histidines. This sequence is conserved in mouse cyclin T1, which also interacts with granulin (Fig. 5), but not in human cyclin T2 which does not bind granulin (Hoque et al., 2003). Remarkably, GEP also binds specifically to HIV-1 Tat (Hoque et al., 2005; Hoque et al., 2003; Shoham et al., 2003; Trinh et al., 1999), and Tat itself binds to cyclin T1 but not cyclin T2 (Napolitano et al., 1999; Ramanathan et al., 1999). Thus granulin binds to both viral and cellular components of P-TEFb complexes that are responsible for productive transcription from the HIV promoter. These two cyclins, which are both regulators of CDK9, are encoded by unique genes that are non-redundant in mouse, indicating unique functions (Kohoutek et al., 2009). Therefore we predict that promoters that preferentially utilize P-TEFb complexes containing cyclin T2 will not be repressed by granulin. These findings point to an intracellular role for GEP in the selective regulation of both viral and cellular genes.
The present study demonstrates that the intracellular function of GEP is independent of its active secretion from the cell. GEP lacking the signal peptide (GEPspm) is retained in the cell, binds to the His-rich domain of cyclin T1 and to Tat, and inhibits transcription from the HIV-1 promoter. Consistent with an intracellular function, GEP co-sediments in glycerol gradients with active P-TEFb in fractions containing complexes of ~200 kDa. Furthermore, the C-terminal region of GEP, GrnCDE, is phosphorylated in vivo by P-TEFb on serine residues, suggesting that GEP itself is a phospho-protein (Fig. 3). The binding of granulin repeats to the cyclin T1 His-rich domain is required for their efficient phosphorylation (Fig. 3). Phosphorylation by CDK9 regulates the function of a number of cellular proteins, the best studied of which is the Pol II CTD, where serine phosphorylation drives processive transcription elongation (Peterlin and Price, 2006). Granulin appears to be a preferred substrate for P-TEFb, and we proposed that it inhibits transcriptional elongation by blocking CTD phosphorylation (Hoque et al., 2003). As with GEP, the CTD binds the conserved His-rich domain of cyclin T1. This domain also binds to the I-mfa domain of the two I-mfa family proteins, HIC and I-mfa, that are also phosphorylated by CDK9 in vitro (Wang et al., 2007). Additional substrates of CDK9 are the elongation factors NELF and DSIF. Phosphorylation of the hSPT5 subunit of the elongation factor DSIF has a profound effect on transcription elongation, switching DSIF from a negative factor to an activator of transcription elongation (Fujinaga et al., 2004; Yamada et al., 2006). We speculate that GEP intracellular function is regulated by its phosphorylation.
The levels of both cyclin T1 and GEP are greatly increased after stimulation of U937 cells with phorbol ester, treatment which causes their differentiation to macrophages (Ong et al., 2006; Reza et al., 2003) and unpublished data. These observations are consistent with the conclusion that granulin is a cyclin T1-dependent gene in an activated monocytic cell line (Yu et al., 2008). Recent studies reported the expression of progranulin in tissue macrophages (Kojima et al., 2009), and we have found GEP to be highly expressed in HIV-infected microglia in brains from HIVE patients relative to controls (Sharer LR, Hoque M and Pe’ery T, in preparation). Since macrophages are early targets of HIV-1 infection, it is possible that HIV-1 Tat has evolved to bind GEP and thereby prevent its interactions with P-TEFb. This strategy is analogous to that exemplified by the competition of Tat and HEXIM1 for binding to P-TEFB (Barboric et al., 2007; Sedore et al., 2007).
Our study raises three general models for its intracellular function (Fig. 7). First, GEP and specific granulins bind P-TEFb and inhibit transcription from the HIV-1 promoter (scheme i) as reported previously (Hoque et al., 2003). Data shown here demonstrate that this repression is not restricted to the HIV promoter. Transcription from the cellular promoters cad and c-myc that are highly dependent on P-TEFb is also repressed by GEP and granulins. The cad promoter is dependent on c-Myc for recruiting cyclin T1/P-TEFb but this dependence can be bypassed by direct recruitment of cyclin T1 via Gal4 binding sites (Eberhardy and Farnham, 2002; Gargano et al., 2007). In this system granulins that bind P-TEFb prevent gene expression from the cad promoter (Fig. 4). The high sensitivity of the c-Myc promoter to inhibition by GEP is consistent with the finding that c-Myc and the HIV promoters share common specific elements that are regulated by P-TEFb (Haaland et al., 2005; Montanuy et al., 2008). c-Myc functions as a master regulator of cell growth and inhibitor of cell cycle arrest. Consequently regulation of c-Myc protein levels has wide reaching effects on many cellular processes including differentiation, growth and cell proliferation (Eilers and Eisenman, 2008). The ability of granulins to inhibit expression from the c-Myc promoter suggests that they are linked to some of these cellular processes. Moreover the involvement of c-Myc in many different cancers makes it a target for drug inhibition (Soucek et al., 2008). In this context the inhibition by granulins of expression from the c-Myc promoter should be explored.
Fig. 7. Models of granulin interactions with the cyclin T1/P-TEFb complex and activators.
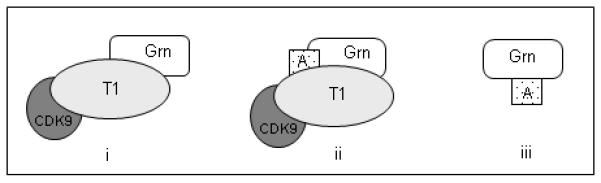
GEP and some granulin repeats bind directly to (i) the cyclin T1 component of the P-TEFb complex, or (iii) HIV-1 Tat and possibly other viral and cellular activators (A) such as VP16 and c-Myc. The activators may themselves be present in multi-component complexes. As with Tat, the activators may bridge between granulin and P-TEFb (ii).
Second, some granulins that inhibit transcription from the HIV-1 promoter bind Tat without binding directly to P-TEFb (Hoque et al., 2005; Hoque et al., 2003). Similarly, granulins can bind to cellular activators such as c-Myc and Miz-1 (Fig. 7. scheme iii). GEP is present in high molecular weight complexes in glycerol gradient fractions (Fig. 2) and we confirmed the interaction between endogenous GEP and Miz-1. We also discovered the existence of GEP in complex with c-Myc in HeLa cells (Fig. 6). GEP has been identified as an interacting partner of type III hexokinase in rat. In addition to GEP, four further proteins bind to hexokinase type III (insulin-like growth factor binding protein-4, Miz-1, leptin, and lipocalin-type prostaglandin D synthase) and appear to be present together in a single complex (Sui and Wilson, 2000). Miz-1 is a DNA binding transcriptional activator that binds to core promoters and regulates the transcription of many genes (Ziegelbauer et al., 2004). It was originally isolated as a c-Myc interacting protein and is targeted by c-Myc for repression of transcription from a number of genes, notably the cell-cycle inhibitors p15Ink4b and p21Cip1. Miz-1 interacts with c-Myc to form inhibitory complexes on core promoters (Wanzel et al., 2003). Hence it is possible that GEP regulates cellular function by sequestering Miz-1 and c-Myc.
Third, as exemplified by HIV, GEP can bind to both P-TEFb and the transcription activator, Tat (Fig. 7, scheme ii). Tat is able to replace c-Myc in recruiting P-TEFb to the cad promoter (Eberhardy and Farnham, 2002). It will be interesting to test whether GEP and c-Myc, which co-immunoprecipitate (Fig. 6), can bind each other directly and form tripartite complexes with P-TEFb. VP16-activated transcription is also susceptible to inhibition by granulins, raising the possibility of direct interactions of VP16 with GEP. We speculate that promoters that are highly sensitive to inhibition by GEP are defined by two features: they are highly dependent on P-TEFb and are regulated by activators that themselves bind GEP.
Acknowledgments
We thank Drs. Tara Young, Chee-Gun Lee, Ginette Serrero, Peggy Farnham and David Price for reagents. We are grateful to Anita Antes and Y. Ramanathan for technical assistance. This work was supported by grants from from The New Jersey Commission on Cancer Research (NJCCR) and from the NIH to Tsafi Pe’ery.
Contract grant sponsor and number: New Jersey Commission on Cancer Research - 03-1134-CCR-EO NIH - AI 060403
References
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Peterlin B Matija, Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35(6):2003–2012. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31(11):1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- Bhandari V, Palfree RG, Bateman A. Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine-rich granulin domains. Proc Natl Acad Sci U S A. 1992;89(5):1715–1719. doi: 10.1073/pnas.89.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol. 2008;20(3):334–340. doi: 10.1016/j.ceb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigati C, Giacca M, Noonan DM, Albini A. HIV Tat, its TARgets and the control of viral gene expression. FEMS Microbiol Lett. 2003;220(1):57–65. doi: 10.1016/S0378-1097(03)00067-3. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, Van Deerlin VM, Trojanowski JQ, Lee VM. Variations in the Progranulin Gene Affect Global Gene Expression in Frontotemporal Lobar Degeneration. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Cole MD. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol Cell Biol. 2007;27(6):2059–2073. doi: 10.1128/MCB.01828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24(4):186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- De Falco G, Giordano A. CDK9: from basal transcription to cancer and AIDS. Cancer Biol Ther. 2002;1(4):342–347. [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem. 2001;276:48562–48571. doi: 10.1074/jbc.M109014200. [DOI] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem. 2002;277(42):40156–40162. doi: 10.1074/jbc.M207441200. [DOI] [PubMed] [Google Scholar]
- Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22(20):2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24(2):787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargano B, Amente S, Majello B, Lania L. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle. 2007;6(16):2031–2037. doi: 10.4161/cc.6.16.4554. [DOI] [PubMed] [Google Scholar]
- Gegonne A, Weissman JD, Lu H, Zhou M, Dasgupta A, Ribble R, Brady JN, Singer DS. TFIID component TAF7 functionally interacts with both TFIIH and P-TEFb. Proc Natl Acad Sci U S A. 2008;105(14):5367–5372. doi: 10.1073/pnas.0801637105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaland RE, Herrmann CH, Rice AP. siRNA depletion of 7SK snRNA induces apoptosis but does not affect expression of the HIV-1 LTR or P-TEFb-dependent cellular genes. J Cell Physiol. 2005;205(3):463–470. doi: 10.1002/jcp.20528. [DOI] [PubMed] [Google Scholar]
- Hanyu-Nakamura K, Sonobe-Nojima H, Tanigawa A, Lasko P, Nakamura A. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature. 2008;451(7179):730–733. doi: 10.1038/nature06498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81(10):600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- Hoque M, Tian B, Mathews MB, Pe’ery T. Granulin and granulin repeats interact with the Tat:P-TEFb complexand inhibit tat transactivation. J Biol Chem. 2005 doi: 10.1074/jbc.M409575200. [DOI] [PubMed] [Google Scholar]
- Hoque M, Young TM, Lee CG, Serrero G, Mathews MB, Pe’ery T. The Growth Factor Granulin Interacts with Cyclin T1 and Modulates P-TEFb-Defendent Transcription. Mol Cell Biol. 2003;23:1688–1702. doi: 10.1128/MCB.23.5.1688-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhang F, Kurosu T, Peterlin BM. Runx1 binds positive transcription elongation factor b and represses transcriptional elongation by RNA polymerase II: possible mechanism of CD4 silencing. Mol Cell Biol. 2005;25(24):10675–10683. doi: 10.1128/MCB.25.24.10675-10683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Michener CM, Blanchette JO, Kuznetsov VA, Raffeld M, Serrero G, Emmert-Buck MR, Petricoin EF, Krizman DB, Liotta LA, Kohn EC. The granulin-epithelin precursor/PC-cell-derived growth factor is a growth factor for epithelial ovarian cancer. Clin Cancer Res. 2003;9(1):44–51. [PubMed] [Google Scholar]
- Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene. 2003;22(36):5707–5711. doi: 10.1038/sj.onc.1206800. [DOI] [PubMed] [Google Scholar]
- Kohoutek J, Li Q, Blazek D, Luo Z, Jiang H, Peterlin BM. Cyclin T2 is essential for mouse embryogenesis. Mol Cell Biol. 2009;29(12):3280–3285. doi: 10.1128/MCB.00172-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Ono K, Inoue K, Takagi Y, Kikuta K, Nishimura M, Yoshida Y, Nakashima Y, Matsumae H, Furukawa Y, Mikuni N, Nobuyoshi M, Kimura T, Kita T, Tanaka M. Progranulin expression in advanced human atherosclerotic plaque. Atherosclerosis. 2009;206(1):102–108. doi: 10.1016/j.atherosclerosis.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Kurosu T, Peterlin BM. VP16 and ubiquitin; binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr Biol. 2004;14(12):1112–1116. doi: 10.1016/j.cub.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Kwak YT, Ivanov D, Guo J, Nee E, Gaynor RB. Role of the human and murine cyclin T proteins in regulating HIV-1 tat-activation. J Mol Biol. 1999;288(1):57–69. doi: 10.1006/jmbi.1999.2664. [DOI] [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, Wahl LM, Harris ED, Mikovits JA, Monks AP, Hollingshead MG, Sausville EA, Staudt LM. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2(10):0041.0041–0011. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau LM, Lallone RL, Seitz RS, Buznikov A, Gregg JP, Kornblum HI, Nelson SF, Bronstein JM. Identification of a human glioma-associated growth factor gene, granulin, using differential immuno-absorption. Cancer Res. 2000;60:1353–1360. [PubMed] [Google Scholar]
- Lu R, Serrero G. Inhibition of PC cell-derived growth factor (PCDGF, epithelin/granulin precursor) expression by antisense PCDGF cDNA transfection inhibits tumorigenicity of the human breast carcinoma cell line MDA-MB-468. Proc Natl Acad Sci USA. 2000;97:3993–3998. doi: 10.1073/pnas.97.8.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels AA, Bensaude O. RNA-driven cyclin-dependent kinase regulation: when CDK9/cyclin T subunits of P-TEFb meet their ribonucleoprotein partners. Biotechnol J. 2008;3(8):1022–1032. doi: 10.1002/biot.200800104. [DOI] [PubMed] [Google Scholar]
- Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23(14):4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanuy I, Torremocha R, Hernandez-Munain C, Sune C. Promoter influences transcription elongation: TATA-box element mediates the assembly of processive transcription complexes responsive to cyclin-dependent kinase 9. J Biol Chem. 2008;283(12):7368–7378. doi: 10.1074/jbc.M706243200. [DOI] [PubMed] [Google Scholar]
- Napolitano G, Licciardo P, Gallo P, Majello B, Giordano A, Lania L. The CDK9-associated cyclins T1 and T2 exert opposite effects on HIV-1 Tat activity. AIDS. 1999;13(12):1453–1459. doi: 10.1097/00002030-199908200-00003. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- Ong CH, Bateman A. Progranulin (Granulin-epithelin precursor, PC-cell derived growth factor, Acrogranin) in proliferation and tumorigenesis. Histol Histopathol. 2003;18(4):1275–1288. doi: 10.14670/HH-18.1275. [DOI] [PubMed] [Google Scholar]
- Ong CH, He Z, Kriazhev L, Shan X, Palfree RG, Bateman A. Regulation of progranulin expression in myeloid cells. Am J Physiol Regul Integr Comp Physiol. 2006;291(6):R1602–1612. doi: 10.1152/ajpregu.00616.2005. [DOI] [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23(3):297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Ping YH, Rana TM. Tat-associated kinase (P-TEFb): a component of transcription preinitiation and elongation complexes. J Biol Chem. 1999;274:7399–7404. doi: 10.1074/jbc.274.11.7399. [DOI] [PubMed] [Google Scholar]
- Ramanathan Y, Reza SM, Young TM, Mathews MB, Pe’ery T. Human and rodent transcription elongation factor P-TEFb: interactions with human immunodeficiency virus type 1 tat and carboxy-terminal domain substrate. J Virol. 1999;73:5448–5458. doi: 10.1128/jvi.73.7.5448-5458.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman TW, Muniz LC, Mathews MB. The RNA binding protein nuclear factor 90 functions as both a positive and negative regulator of gene expression in mammalian cells. Mol Cell Biol. 2002;22:343–356. doi: 10.1128/MCB.22.1.343-356.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman TW, Parrott AM, Fierro-Monti I, Caron DJ, Kao PN, Lee CG, Li H, Mathews MB. Selective regulation of gene expression by nuclear factor 110, a member of the NF90 family of double-stranded RNA-binding proteins. J Mol Biol. 2003;332:85–98. doi: 10.1016/s0022-2836(03)00885-4. [DOI] [PubMed] [Google Scholar]
- Reza SM, Rosetti M, Mathews MB, Pe’ery T. Differential activation of Tat variants in mitogen-stimulated cells: implications for HIV-1 postintegration latency. Virology. 2003;310(1):141–156. doi: 10.1016/s0042-6822(03)00106-5. [DOI] [PubMed] [Google Scholar]
- Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35(13):4347–4358. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrero G. Autocrine growth factor revisited: PC-cell-derived growth factor (progranulin), a critical player in breast cancer tumorigenesis. Biochem Biophys Res Commun. 2003;308(3):409–413. doi: 10.1016/s0006-291x(03)01452-9. [DOI] [PubMed] [Google Scholar]
- Shoham N, Cohen L, Gazit A, Yaniv A. The Tat protein of the caprine arthritis encephalitis virus interacts with the Notch2 EGF-like repeats and the epithelin/granulin precursor. Intervirology. 2003;46(4):239–244. doi: 10.1159/000072434. [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui D, Wilson JE. Interaction of insulin-like growth factor binding protein-4, Miz-1, leptin, lipocalin-type prostaglandin D synthase, and granulin precursor with the N-terminal half of type III hexokinase. Arch Biochem Biophys. 2000;382:262–274. doi: 10.1006/abbi.2000.2019. [DOI] [PubMed] [Google Scholar]
- Trinh DP, Brown KM, Jeang KT. Epithelin/granulin growth factors: extracellular cofactors for HIV-1 and HIV-2 Tat proteins. Biochem Biophys Res Commun. 1999;256:299–306. doi: 10.1006/bbrc.1999.0317. [DOI] [PubMed] [Google Scholar]
- Wang Q, Young TM, Mathews MB, Pe’ery T. Developmental Regulators Containing the I-mfa Domain Interact with T cyclins and Tat and Modulate Transcription. J Mol Biol. 2007;367(3):630–646. doi: 10.1016/j.jmb.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanzel M, Herold S, Eilers M. Transcriptional repression by Myc. Trends Cell Biol. 2003;13(3):146–150. doi: 10.1016/s0962-8924(03)00003-5. [DOI] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Xu SQ, Tang D, Chamberlain S, Pronk G, Masiarz FR, Kaur S, Prisco M, Zanocco-Marani T, Baserga R. The granulin/epithelin precursor abrogates the requirement for the insulin-like growth factor 1 receptor for growth in vitro. J Biol Chem. 1998;273:20078–20083. doi: 10.1074/jbc.273.32.20078. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21(2):227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Young TM, Tsai M, Tian B, Mathews MB, Pe’ery T. Cellular mRNA activates transcription elongation by displacing 7SK RNA. PLoS One. 2007;2(10):e1010. doi: 10.1371/journal.pone.0001010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TM, Wang Q, Pe’ery T, Mathews MB. The Human I-mfa Domain-Containing Protein, HIC, Interacts with Cyclin T1 and Modulates P-TEFb-Dependent Transcription. Mol Cell Biol. 2003;23(18):6373–6384. doi: 10.1128/MCB.23.18.6373-6384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Ramakrishnan R, Wang Y, Chiang K, Sung TL, Rice AP. Cyclin T1-dependent genes in activated CD4 T and macrophage cell lines appear enriched in HIV-1 co-factors. PLoS One. 2008;3(9):e3146. doi: 10.1371/journal.pone.0003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Barboric M, Blackwell TK, Peterlin BM. A model of repression: CTD analogs and PIE-1 inhibit transcriptional elongation by P-TEFb. Genes Dev. 2003;17(6):748–758. doi: 10.1101/gad.1068203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Serrero G. Inhibition of tumorigenicity of the teratoma PC cell line by transfection with antisense cDNA for PC cell-derived growth factor (PCDGF, epithelin/granulin precursor) Proc Natl Acad Sci U S A. 1998;95:14202–14207. doi: 10.1073/pnas.95.24.14202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gao G, Crabb JW, Serrero G. Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J Biol Chem. 1993;268(15):10863–10869. [PubMed] [Google Scholar]
- Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006;70(3):646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111(6):867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelbauer J, Wei J, Tjian R. Myc-interacting protein 1 target gene profile: a link to microtubules, extracellular signal-regulated kinase, and cell growth. Proc Natl Acad Sci U S A. 2004;101(2):458–463. doi: 10.1073/pnas.0307562100. [DOI] [PMC free article] [PubMed] [Google Scholar]