

Abstract
Activating mutations in CDK4 and inactivation of its key kinase inhibitor, p16INK4A, have been implicated in the genesis and progression of human cancer. Previous work has demonstrated that CDK4 expression is required for Neu-induced but not Wnt-induced breast tumorigenesis in mice. However, the role that CDK4 plays in ras-mediated breast tumor development is not well defined. To gain an understanding of the role of Cdk4 in ras-induced breast tumorigenesis, MMTV-v-Ha-ras transgenic mice were bred with Cdk4(+/neo) and Cdk4(R24C/R24C) mice to generate Cdk4(neo/neo):MMTV-v-Ha-ras, Cdk4(+/+):MMTV-v-Ha-ras, and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. The studies presented here demonstrate that Cdk4 expression is essential for Ras-mediated breast tumorigenesis. Surprisingly, the results also show that coexpression of mutant ras and Cdk4R24C genes in breast epithelial cells leads to an activation of senescent pathways that delay tumorigenesis. Analysis of the phosphorylated form of H2AX, a marker for DNA damage, indicated its increased presence in the tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. These observations indicate that the increased apoptosis and senescence seen in breast tumors of these mice might be due to increased DNA damage response in cells expressing activated forms of ras and Cdk4(R24C).
Keywords: Cdk4, Ras, breast cancer, senescence, apoptosis
Introduction
In the mammalian system, mitogenic growth factors bind to their cognate receptors and initiate a cascade of events that culminate in the expression and assembly of different kinase holoenzymes composed of a regulatory subunit, called cyclin, and a catalytic subunit, named cyclin-dependent kinase (CDK). These protein complexes are formed at specific stages of the cell cycle, and their activities trigger progression through the G1, S, and M phases of the cell cycle.1-5 Of these, CDK4 and CDK6 are thought to cooperate with CDK2 in inactivating members of the retinoblastoma family of proteins, thereby activating an E2F-dependent gene expression program required for passage through the restriction point in mid-to-late G1.
A key response to many mitogens in a variety of cell types is the activation of CDK4 or CDK6 by members of the cyclin D family (D1, D2, and D3). D-type cyclins are typically expressed at low levels in quiescent cells, and their expression is stimulated by mitogens and growth factors. Consequently, if mitogens are removed, the levels of D-type cyclins drop immediately, regardless of the stage of the cell cycle. Thus, D-type cyclins are considered to be important integrators of mitogenic signaling. Cyclin D1 is an unstable molecule, and its availability is controlled by a balance between the RAS/RAF/MAPK pathway (which promotes cyclin D1 synthesis) and RAS/PI3/AKT pathway (which ensures cyclin D1 stability).6-8
Deregulation of cyclins and CDKs has been shown to play an etiological role in many different cancers. Thus, Cdk4 has been shown to be amplified in 16% of sporadic breast tumors, and this amplification correlates with high Cdk4 protein levels.9 Furthermore, R24C mutation in Cdk4, which confers resistance to p16INK4a, has been associated with familial melanoma.10,11 It has also been demonstrated that the mice that are homozygous for this mutation are susceptible to tumors in different tissues, including those of the mammary gland.12,13 We, along with others, have recently shown that Cdk4 is critical for ErbB2 (ErbB2/HER2)–induced breast tumorigenesis14-16 but not for that induced by Wnt-1.14 Oncogenic signaling by ErbB/HER signaling primarily involves the Ras pathway. Approximately 25% of human tumors exhibit mutations in the ras oncogene, and mutant ras potently stimulates neoplasia and tumor progression in cooperation with other oncoproteins. However, it has also been shown that strong signaling by Ras results in the activation of pathways that lead to senescence.17,18 In this regard, it is significant to note that CDK4 cooperates with Ras in the tumorigenesis of many different tissues. In primary epidermal cells, coexpression of wild-type Cdk4 along with Ras generates invasive neoplasia.19 In addition, Yu et al.16 have shown that Ras requires cyclin D1 for breast tumor induction in mice. However, although it is now well established that both cyclin D1 and CDK4 are required for ErbB2-induced breast tumorigenesis in mice in a manner that requires CDK4 activity,14-16 others have suggested that the oncogenic activity of cyclin D1 is independent of its CDK activity but dependent on the ability of cyclin D1 to modulate transcription.20 Therefore, it is important to examine whether Cdk4 is also required for Ras-induced mammary tumors and whether mutations in the Cdk4 gene cooperate with mutant ras in tumorigenesis. To address this question, we have investigated the requirement of Cdk4 in Ras-mediated breast tumorigenesis using transgenic and gene knockout mouse model systems and demonstrate that Cdk4 expression is essential for Ras-mediated breast tumorigenesis. Surprisingly, our results also show that coexpression of mutant ras and Cdk4 R24C genes in breast epithelial cells leads to an unexpected activation of senescent pathways that delay tumorigenesis. The results of this study illustrate the importance of Cdk4 in Ras-mediated breast tumorigenesis and expand our knowledge of different roles played by Ras in breast tumor cell growth, senescence, and apoptosis.
Results
Importance of Cdk4 in v-Ha-ras–Induced Mammary Tumorigenesis
To gain an understanding of the role of Cdk4 in ras-induced breast tumorigenesis, Cdk4(+/neo) mice were bred with MMTV-v-Ha-ras transgenic mice to generate Cdk4(neo/neo):MMTV-v-Ha-ras mice and Cdk4(+/+):MMTV-v-Ha-ras mice. Since Cdk4(neo/neo) mice are infertile, we used Cdk4(+/neo) mice for these matings, and this approach also yielded mice with an identical genetic background. The histopathological sections of the mammary glands derived from virgin adult mice (approximately 14 weeks) from these crosses showed that Cdk4(+/+):MMTV-v-Ha-ras mice exhibit abnormal morphology of the mammary epithelium as evidenced by the appearance of multiple hyperplastic and dysplastic changes that resulted in the loss of ductal architecture (Fig. 1D). Similar examination of the histopathological sections of mammary tissue derived from Cdk4(neo/neo):MMTV-v-Ha-ras mice showed a well-defined ductal architecture with very little or complete absence of any hyperplastic and dysplastic changes (Fig. 1C), which was very similar to that seen in Cdk4(+/+) and Cdk4(neo/neo) mice (Figs. 1 A and B). These results suggest that Cdk4 expression is essential for the appearance of MMTV-v-Ha-ras–induced proliferative disturbances. This is very similar to what was observed in Cdk4(neo/neo):MMTV-ErbB2 mice,14 which also show a requirement for Cdk4 expression for ErbB2-induced tumorigenesis.
Figure 1.
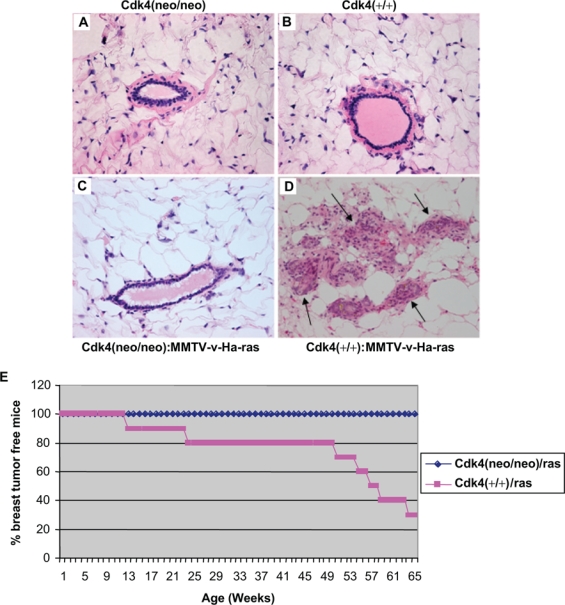
Loss of Cdk4 abrogates Ras-induced hyperplastic and dysplastic changes in the epithelial tissue of mammary glands and blocks breast tumor formation. Formalin-fixed paraffin-embedded mammary gland sections of Cdk4(neo/neo) (A), Cdk4(+/+) (B), Cdk4(neo/neo):MMTV-v-Ha-ras (C), and Cdk4(+/+):MMTV-v-Ha-ras (D) mice were deparaffinized and stained with H&E stain (40x). Arrows in (D) indicate the loss of ductal architecture. (E) H-Ras transgenic mice on Cdk4(neo/neo) and Cdk4(+/+) backgrounds were examined for breast tumor formation for a period of 65 weeks, and the number of mice that developed breast tumors was plotted against age. For these studies, 9 Cdk4(neo/neo):MMTV-v-Ha-ras mice and 10 Cdk4(+/+):MMTV-v-Ha-ras mice were used.
Loss of Expression of Cdk4 Influences the Incidence of Mammary Carcinomas in MMTV-v-Ha-ras Transgenic Mice
It has been previously reported that MMTV-v-Ha-ras–induced breast carcinomas require the expression of cyclin D1.21 To determine whether Cdk4 plays a similar role in the development of breast carcinomas induced by v-Ha-ras, we observed the two groups of transgenic mice described above for the appearance of breast tumors. The results of this study presented in Figure 1E show that approximately 70% of the Cdk4(+/+):MMTV-v-Ha-ras mice develop mammary tumors between 12 to 64 weeks of age. In contrast, none of the Cdk4(neo/neo):MMTV-v-Ha-ras mice develop any signs of mammary tumors and remain tumor-free beyond 65 weeks. These observations suggest that the development of breast tumors in MMTV-v-Ha-ras transgenic mice requires normal expression of Cdk4, which is in accordance with the requirement of Cdk4 for Ras-dependent skin tumor development.22
Upregulation of the Canonical Ras Pathway in MMTV-ras–Induced Tumors
To ascertain that the Ras pathway is activated in the breast tissues of Cdk4(+/+):MMTV-ras mice, we examined the steady-state levels of Ras as well as the steady-state levels and phosphorylation status of downstream effector molecules such as Raf and MEK kinases by Western blotting and immunohistochemical methods (Figs. 2 A and B). As expected, Ras expression was observed in the normal tissues as well as tumors of the mammary glands of cdk4(+/+):MMTV-v-Ha-ras mice (Fig. 2B). Consistent with the expression of Ras, we could observe the activation of the Raf-MEK-ERK pathway, as evidenced by the detection of phospho-Raf and phospho-MEK1/2 (Fig. 2A). While we could readily detect steady-state levels of these proteins in both normal and tumor tissues, Raf and MEK1/2 were expressed in greater abundance in tumor tissues, which paralleled Ras protein expression (Fig. 2A). This elevation in the levels of Ras pathway proteins might be due to enrichment of tumors with epithelial cells, while the normal glands are known to contain a heterogeneous population of cells where epithelial cells constitute a small percentage of total cell population (Fig. 2B).
Figure 2.
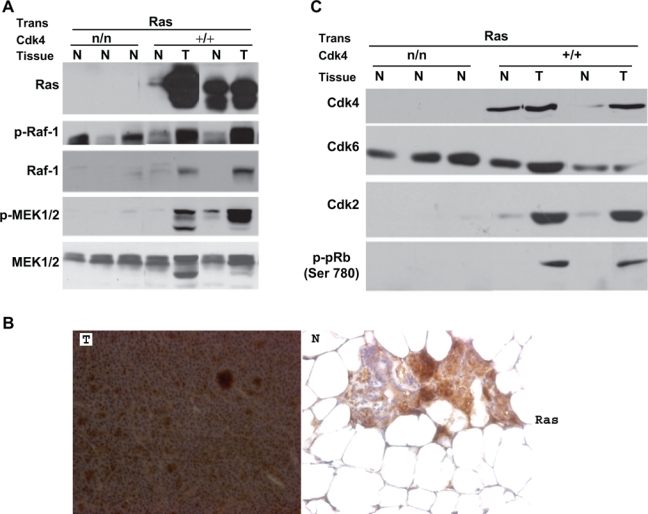
Activation of MAPK pathway in the mammary tumors of H-Ras transgenic mice. (A) Protein extracted from normal (N) and mammary tumor tissues (T) of Cdk4(neo/neo):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice was analyzed for Ras, Phospho-Raf1 (P-Raf1), Raf-1, Phospho-MEK1/2 (P-MEK1/2), and MEK1/2 by Western blot analysis. (B) Formalin-fixed paraffin-embedded normal and tumor sections of the mammary glands of Cdk4(+/+):MMTV-v-Ha-ras mice were subjected to immunohistochemical analysis using antibody against Ras (40x). (C) Presence of increased levels of cell cycle proteins in Ras-induced breast tumors. Protein extracted from normal (N) and mammary tumor tissues (T) of Cdk4(neo/neo):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice was analyzed for Cdk4, Cdk6, Cdk2, and P-pRb (Ser780).
Note: Trans. = Transgene; n/n = neo/neo.
Analysis of Cell Cycle Proteins
To determine the changes in the expression patterns of proteins associated with cell cycle progression, we analyzed the steady-state levels of Cdk4, Cdk6, Cdk2, and phosphorylation status of retinoblastoma protein (Rb) in tissues derived from Cdk4(neo/neo):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice (Fig. 2C). As can be expected, results from these studies showed the absence of Cdk4 in the mammary tissues of Cdk4(neo/neo):MMTV-v-Ha-ras mice (Fig. 2C), while abundant levels of this protein were seen in both normal and tumor tissues of the mammary glands of Cdk4(+/+):MMTV-v-Ha-ras mice. Interestingly, we also observed an increase in the levels of Cdk2 protein and Cdk4-specific phosphorylation of pRb at the Ser780 residue in the tumor tissues of Cdk4(+/+):MMTV-v-Ha-ras mice (Fig. 2C). Such an elevation in the expression of Cdk6 or Cdk2 was not seen in the mammary tissues of Cdk4(neo/neo):MMTV-v-Ha-ras mice, suggesting that loss of Cdk4 was not compensated through the expression of Cdk6 or Cdk2. The presence of increased levels of cell cycle proteins in the tumor tissues might be due to the presence of an increased number of epithelial cells as the tumors arise from the epithelial cell compartment.
Effect of Cdk4R24C Mutation on Ras-Induced Breast Tumorigenesis
Mutation of residue 24 from an Arg to Cys in Cdk4 occurs in familial cases of melanomas and confers resistance of the protein to inhibition by INK4 family members.10,11 Mouse modeling experiments have shown that mice harboring a knock-in cdk4R24C mutation develop tumors at an accelerated rate.12,13 Furthermore, these studies showed that Cdk4(R24C/R24C) mice exhibit a higher susceptibility to carcinogen-induced tumorigenesis.23,24 Since some of the carcinogens such as DMBA used in these studies are known to mediate their oncogenic function by targeting ras gene, it was of interest to examine whether Cdk4R24C mutation promotes Ras-induced tumorigenesis in breast tissue. To perform these studies, MMTV-v-Ha-ras transgenic mice were bred with either Cdk4(R24C/R24C) mice or Cdk4(+/+) mice to generate Cdk4(R24C/R24C):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice and examined for the onset and progression of mammary tumors. Histopathological analysis of breast tissues derived from 10- to 14-week-old mice showed that Cdk4(R24C/R24C):MMTV-v-Ha-ras mice exhibit abnormal morphology in the mammary epithelium similar to that seen with Cdk4(+/+):MMTV-v-Ha-ras mice (Fig. 3A). However, when these mice were compared with Cdk4(+/+):MMTV-v-Ha-ras mice for the appearance of breast tumors, they were found to develop breast tumors with a longer latency period. Thus, the results of this study, presented in Figure 3B, show that 100% of the Cdk4(+/+):MMTV-v-Ha-ras mice developed breast tumors in about 33 weeks, while the Cdk4(R24C/R24C):MMTV-v-Ha-ras mice developed breast tumors in about 65 weeks, suggesting a lag in the tumor development of these mice (Fig. 3B). This result suggests that R24C mutation not only does not accelerate breast tumor formation but instead impedes the development of Ras-induced breast tumors. To rule out the possibility that this retardation of tumor development is not due to inadequate activation of the Ras pathway in these compound mice, we examined the steady-state levels of Ras as well as the steady-state levels and phosphorylation status of downstream effector molecules such as Raf and MEK kinases by Western blotting and immunohistochemical methods. The results of these studies, presented in Figures 3C and D, show that tumors that develop in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice express robust levels of Ras and activation of the Ras-Raf-MAPK pathway, as judged by the presence of phospho-Raf and phospho-MEK1/2, but the elevation in the levels of these proteins might be partially due to enrichment of tumors with epithelial cells.
Figure 3.
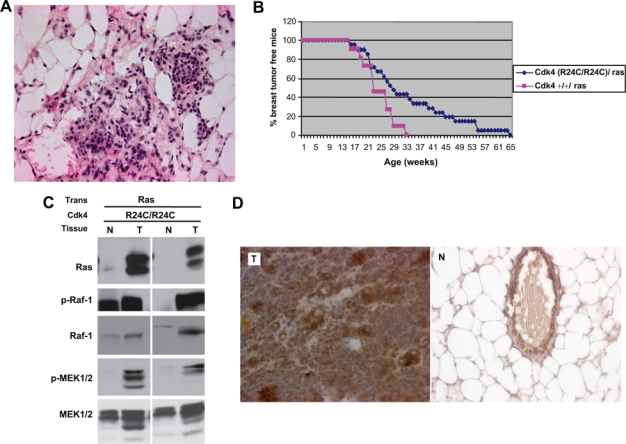
R24C mutation of Cdk4 does not accelerate Ras-induced breast tumorigenesis. (A) Formalin-fixed paraffin-embedded mammary gland sections of Cdk4 (R24C/R24C):MMTV-v-Ha-ras mice were deparaffinized and stained with H&E stain (40×). (B) Cdk4(R24C/R24C):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice were examined for Ras-induced breast tumor incidence over a period of 65 weeks. For these studies, 21 Cdk4(R24C/R24C):MMTV-v-Ha-ras mice and 11 Cdk4(+/+):MMTV-v-Ha-ras mice were used. (C) Protein extracted from normal (N) and mammary tumor tissues (T) of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice was analyzed for Ras, Phospho-Raf1 (P-Raf1), Raf-1, Phospho-MEK1/2 (P-MEK1/2), and MEK1/2. (D) Formalin-fixed paraffin-embedded normal and tumor sections of the mammary glands of Cdk4 (R24C/R24C):MMTV-v-Ha-ras mice were subjected to immunohistochemical analysis using antibody against Ras (40x).
An analysis of the steady-state levels of Cdk4, Cdk6, and Cdk2 proteins in tumor tissues derived from Cdk4(R24C/R24C):MMTV-v-Ha-ras revealed robust expression of CDK4, CDK6, and CDK2 and cyclin D1 with concomitant elevation Rb phosphorylation at Ser780, the phosphorylation site of CDK4 (Fig. 4). These results suggest that both Ras and CDK4 (R24C) proteins are expressed at high levels in these tumor tissues, and the observed lag in tumor growth cannot be attributed to inadequate expression of these proteins.
Figure 4.

Increased expression of cell cycle proteins in breast tumors derived from Cdk4 (R24C/R24C):MMTV-v-Ha-ras mice. Protein extracted from normal (N) and mammary tumor tissues (T) of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice was analyzed for Cdk4, Cdk6, Cdk2, cyclin D1, and P-pRb (Ser780) by Western blot analysis.
The Delayed Tumorigenesis in Cdk4(R24C/R24C):MMTV-v-Ha-ras Is Not Due to Reduced Proliferation
To gain an understanding of the molecular mechanisms associated with the delayed tumorigenesis seen in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice, we performed immunohistochemical analysis of the formalin-fixed breast tumor sections for the proliferation-specific marker Ki67. In this study, tumor tissue sections derived from cdk4(+/+):MMTV-v-Ha-ras mice were used for comparison. The results from these studies showed the presence of a number of cells positive for this proliferation marker in the tumor tissues of both Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. Further quantitation of the Ki67 positive areas using the Bioquant program indicated no significant difference in the proliferation index of these tumors, suggesting similar levels of cell proliferation in both types of tumor tissues (Fig. 5A, bottom panel).
Figure 5.
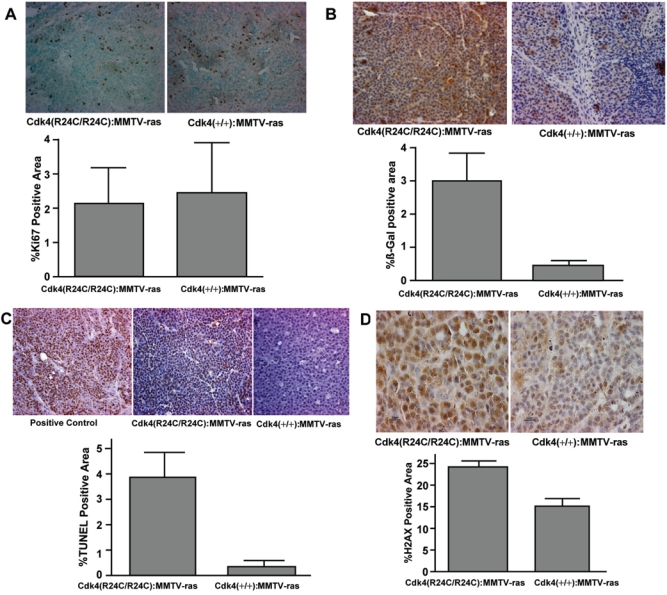
Analysis of tumor sections of Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice for proliferation, senescence, apoptosis, and DNA damage markers. (A) Analysis of proliferation marker Ki67 in the tumor sections. (A, top) Formalin-fixed paraffin-embedded normal and tumor sections of the mammary glands of Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice were subjected to immunohistochemical analysis using antibody against proliferation marker Ki67 (40x). (A, bottom) Ki67-positive area (dark brown–stained nuclei) of the tumor sections was measured using Bioquant software and analyzed using an unpaired t test provided in Prism software. (B) Increased senescence in the mammary tumor tissues of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice when compared to Cdk4(+/+):MMTV-v-Ha-ras mice. (B, top) Formalin-fixed paraffin-embedded normal and tumor sections of the mammary glands of Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice were subjected to immunohistochemical analysis using antibody against β-galactosidase protein (40x). (B, bottom) β-galactosidase positive area (brown-stained area ) of the tumor sections was measured using Bioquant software and analyzed using an unpaired t test provided in Prism software. (C) Mammary tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice exhibit enhanced apoptosis when compared to Cdk4(+/+):MMTV-v-Ha-ras mice. (C, top) Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) analysis of formalin-fixed paraffin-embedded normal and tumor sections of the mammary glands of Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice (40x). (C, bottom) TUNEL-positive area of the tumor sections was measured using Bioquant software and analyzed using an unpaired t test provided in Prism software. (D) Increased levels of γ-H2AX in the tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. (D, top) Immunohistochemical analysis of formalin-fixed paraffin-embedded sections for γ-H2AX (100x). (D, bottom) γ-H2AX-positive area of the tumor sections was measured using Bioquant software and analyzed using an unpaired t test provided in Prism software.
Increased Senescence in the Tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras Mice
Overexpression of mutant Ras has been shown to induce a permanent growth arrest in cultured primary cells in the absence of cooperating oncogenic mutations.17,25 Studies have also indicated that high levels of mutant Ras induce irreversible cellular senescence.26 To determine whether induction of senescence plays a role in the delayed tumor progression seen in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice, we analyzed tumor tissues derived from Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice for the expression of β-galactosidase, a marker for senescence, using an immunohistochemical technique. The results of this study (presented in Fig. 5B) show an elevated expression of β-galactosidase marker in the breast tumor sections of cdk4(R24C/R24C):MMTV-v-Ha-ras mice when compared to their wild-type counterparts. Computer-assisted quantitation of the immunohistochemical staining using the Bioquant program further confirmed a significant increase in the levels of β-galactosidase-positive areas in the tumor sections of cdk4(R24C/R24C):MMTV-v-Ha-ras mice when compared to their wild-type counterparts (Fig. 5B, bottom panel). These results suggest that coexpression of mutant Ras and Cdk4R24C proteins in breast epithelial tissue results in an increased activation of senescence pathways that could explain the lag seen in the growth of breast tumors in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice.
Tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras Mice Exhibit Enhanced Apoptosis Compared to Their Wild-Type Counterparts
To determine whether coexpression of activated Ras and Cdk4 proteins results in increased apoptotic death of breast epithelial cells, we used terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining to identify apoptotic cells in tumor tissue sections derived from Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. Results from these studies, presented in Figure 5C, show a significant increase in TUNEL-positive cells in the tumors of Cdk4 (R24C/R24C):MMTV-v-Ha-ras mice when compared to their wild-type counterparts.
Increased Ser139-Specific Phosphorylation of H2AX (γ-H2AX) in the Tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras Mice When Compared to Their Wild-Type Counterparts
Ras oncoprotein has previously been shown to induce senescence through the activation of p38MAPK-MKK3/6 pathway27 or via the activation of DNA damage checkpoint response (DDR)28-30 and apoptosis through the JNK pathway,31 which in turn leads to apoptosis or cell cycle arrest. Our analysis of the p38MAPK-MKK3/6 pathway did not show an increase in the activity of p38MAPK or JNK in tumor tissues derived from Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice, suggesting that activation of p38 MAPK is not responsible for the observed differences in senescence (data not shown).
To determine whether DNA damage checkpoint response (DDR) is the major cause for the enhanced senescence and apoptosis seen in the breast tumor tissues of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice, we examined tissue sections for the formation of subnuclear foci containing γ-H2AX, one of the markers that is commonly used for detecting DNA double-strand breaks.32,33 Our immunohistochemical analysis of tumor tissue sections revealed a significant increase in the appearance of subnuclear foci containing γ-H2AX protein in mouse tissues derived from Cdk4(R24C/R24C):MMTV-v-Ha-ras mice compared to their wild-type counterparts (Fig. 5D). These results suggest that the enhanced senescence and apoptosis seen in breast tumor tissues of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice could be due to oncogenic stress-induced DNA damage checkpoint response.
Discussion
It is now well established that members of the ras gene family are mutated in approximately 30% of human tumors, and mutant Ras proteins promote growth and tumorigenesis of mammalian cells.5 Ectopic expression of mutant ras gene under the control of a mouse mammary tumor virus promoter (MMTV:LTR) was found to result in the development of mammary tumors in transgenic mouse models with a latency of 12 to 24 weeks.34 This mouse model system has been used extensively to examine the effects of various oncogenes on breast tumor development and dissect the signaling pathways that lead to the transformation of breast epithelial cells. The ras oncogenes, acting through the Ras-Raf-MAPK pathway, are known to transcriptionally activate the expression of cyclin D1,6,7 which in turn forms complexes with Cdk4 and/or Cdk6 and mediates cell cycle progression by phosphorylating members of the Retinoblastoma protein family. Our results, presented in this article, show that Cdk4 is essential for oncogenic Ras-induced breast tumorigenesis. Previously, cyclin D1 was similarly shown to be required for Ras-induced breast tumorigenesis.21 Consistent with these results, we detected increased phosphorylation of the retinoblastoma protein in Ras; cdk4 +/+ tumors. Since it is unlikely that the functions of cyclin D1 and CDK4 would be independently required for Ras-induced tumorigenesis, these results suggest that cyclin D1 and Cdk4 form a kinase complex required for Ras-mediated breast tumorigenesis. Our results also show that Cdk6 and Cdk2, which are expressed at normal levels in most tissues derived from Cdk4(neo/neo) mice, are unable to compensate for the loss of Cdk4 in Ras-induced breast tumorigenesis. This observation is in accordance with the previous studies where loss of Cdk4 leads to similar abrogation of ErbB2-induced breast tumorigenesis.14-16 It is interesting to note that the survival rate of Cdk4(+/+):MMTV-v-Ha-ras mice used in Figures 1E and 3B was considerably different. The differences in the survival rate of transgenic mice used for the two studies appear to be due to differences in their genetic backgrounds. The mice represented in Figure 1E had a mixed background of Cdk4(+/neo) mice (ICR background) and MMTV-v-Ha-ras mice (FVB/N background), whereas the mice represented in Figure 3B had a mixed background of Cdk4(+/R24C) mice (ICR and BALB/c-J) and MMTV-v-Ha-ras mice (FVB/N background).
Paradoxically, it was also observed that overexpression of oncogenic Ras in normal primary cells induces premature senescence, an irreversible growth arrest that is morphologically indistinguishable from replicative senescence observed with aging primary cells.17,18 This phenomenon appears to be mediated by the p53 and Ink4a-Arf, which accumulate in cells undergoing oncogene-induced senescence. Recently, Sarkisian et al.35 have examined the effects of low and high doses of Ras expression on mammary tumorigenesis and observed that high-level expression of oncogenic Ras triggers an irreversible senescent growth arrest of mammary epithelial cells in vivo. These investigators also observed that chronic low-level Ras expression results in tumor formation but only after spontaneous upregulation of activated Ras and evasion of senescence checkpoints. Our findings presented in this report show that coexpression of oncogenic Ras and Cdk4R24C proteins results in a delay in the development of breast tumors. This does not appear to be due to an impairment in the expression of other Cdks as we did not observe major differences in the steady-state levels of Cdk4 and Cdk2, proteins in the tumors of Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice. As can be expected, we observed a 2- to 3-fold increase in the levels of P-pRb (Ser 780) in the tumors derived from Cdk4R24C mice. This increase of P-pRb (Ser 780) could be attributed to the constitutive activation of Cdk4R24C protein, which is refractory to inhibition mediated by the Ink4 family of proteins.13,24 Our studies also show that tumors that develop in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice express robust levels of Ras and activation of the Ras-Raf-MAPK pathway, as judged by the presence of phospho-Raf and phospho-MEK1/2. Hence, the observed lag in the development of breast tumors in these mice cannot be attributed to an inefficient activation of the Ras-Raf-MAPK pathway.
To gain an understanding of the molecular mechanisms associated with the delayed tumorigenesis seen in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice, we performed immunohistochemical analysis of breast tumor sections for the proliferation-specific marker Ki67 and did not observe any significant difference between the proliferation indices of breast tumors arising in Cdk4(R24C/R24C):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice, suggesting that reduced cell proliferation is not the cause for the observed lag in R24C mice.
To test the possibility that coexpression of a mutant ras allele along with a mutant form of Cdk4, such as Cdk4R24C, might mimic the effects of Ras overexpression and lead to premature senescence, we examined the tumor tissues derived from Cdk4(+/+):MMTV-v-Ha-ras and Cdk4(R24C/R24C):MMTV-v-Ha-ras mice for the expression of markers for senescence (β-galactosidase) and apoptosis (TUNEL). The results of this study revealed a significant elevation in the expression of β-galactosidase marker in the breast tumor sections of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice when compared to their wild-type counterparts. We also observed a significant increase in TUNEL-positive cells in the tumors of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice when compared to their wild-type counterparts. These results suggest that coexpression of mutant Ras and Cdk4R24C proteins in breast epithelial tissue results in the increased activation of senescence pathways that also results in the appearance of apoptotic cells, which could explain the lag seen in the growth of breast tumors that develop in Cdk4(R24C/R24C):MMTV-v-Ha-ras mice.
Recent studies also demonstrate that oncogene-induced senescence is associated with DNA replication stress, including prematurely terminated DNA replication forks and DNA double-strand breaks.29 This appears to result in a robust DNA damage checkpoint response (DDR). It is now known that one of the markers that is commonly associated with DNA double-strand breaks is the formation of subnuclear foci containing γ-H2AX.28,32,33 We therefore used the γ-H2AX foci formation as an indicator of DNA damage and compared the tissue sections derived from Cdk4(R24C/R24C):MMTV-v-Ha-ras and Cdk4(+/+):MMTV-v-Ha-ras mice for the appearance of this marker. These studies revealed a significant increase in the appearance of subnuclear foci containing γ-H2AX protein in tumor tissues derived from Cdk4(R24C/R24C):MMTV-v-Ha-ras mice compared to their wild-type counterparts, suggesting that the enhanced senescence and apoptosis seen in breast tumor tissues of Cdk4(R24C/R24C):MMTV-v-Ha-ras mice could be due to oncogenic stress-induced DNA damage checkpoint response, which results in an impairment of tumor development. This delay appears to occur only in Ras-mediated breast tumor development as we did not observe such a lag in Cdk4(R24C/R24C):MMTV-erbB2 transgenic mice (data not shown). Thus, expression of the Cdk4(R24C/R24C) oncogene results in distinct outcomes depending on the cooperating oncogene, despite the possibility that Ras operates downstream of Erb B2 in the same pathway, illustrating the complexity of the signaling downstream of these oncogenes. Also, it is important to comment on the unexpected finding that an oncogenic form of Cdk4 (Cdk4(R24C/R24C)), which is thought to be resistant to inactivation by a potent inducer of senescence, p16, is a partner to a senescent phenotype in the context of oncogenic Ras expression. It is conceivable that Cdk4(R24C/R24C) enhances the potency of Ras mitogenic functions to a point that triggers DNA damage and the oncogenic checkpoint more efficiently than Ras alone. In this regard, our results are consistent with a previous study describing the induction of invasive melanomas in CDK4(R24C/R24C) mice using a high dose of DMBA (5 mg/mouse) followed by repeated treatment with TPA.23 Interestingly, they found that this carcinogenic protocol, which frequently results in the mutagenic activation of the Ras family of genes in wild-type mice, failed to activate Ras genes in mice that carry the germline Cdk4 R24C mutation. This again suggests that similar senescent pathways that we observe in mammary epithelial cells might be operative in melanocytes, which preclude concomitant activation of Ras and Cdk4 genes.
In summary, our studies demonstrate that the proliferative and tumorigenic response of mammary epithelial cells to Ras activation in vivo is critically dependent on Cdk4 expression and activity. Our results also show that while lack of Cdk4 expression leads to complete abrogation of Ras-induced mammary tumorigenesis, oncogenic mutations in Cdk4 in conjunction with oncogenic Ras expression activate cellular senescence pathways that hinder growth of mammary tumors.
Materials and Methods
Mice
MMTV-v-Ha-ras transgenic mice were obtained from Charles River Laboratories. Generation of cdk4+/- (ICR background) and cdk4 (+/R24C) (ICR and BALB/c-J mixed background) has been described earlier.24
Antibodies
Antibodies directed against MEK1/2 (#9122), p-MEK1/2 (#9121), MKK3 (#9232), pMKK3/6 (#9231), MKK4/SEK1(#9152), pMKK4/SEK1(#9156), p38MAPK(#9212), p-p38 MAPK (#9211), p-Raf1 (#9427), and p-pRb (#9307) were obtained from Cell Signaling Technology. Antibodies directed against cyclin D1 (A12), Cdk2 (M2), Cdk4 (c-20), H-ras (c-20), and Raf1 (c-20) were obtained from Santa Cruz Biotechnologies. Anti-Ras (clone RAS10, cat. #05-516) and γ-H2AX antibody (05-636) were obtained from Upstate Cell Signaling Solutions. Anti-Cdk6 (AB3) (MS-451-p1) was obtained from Neomarkers. Anti-β-galactosidase (ab616) was obtained from Abcam. Anti-Ki 67 (M7249 (TEC-3)) was obtained from DakoCytomation.
Generation of cdk4(neo/neo):MMTV and cdk4(R24C/R24C):MMTV-transgenic mice
To generate compound mice that express v-HA-ras oncogenes in a cdk4 null background, cdk4(neo/+) mice were crossed to MMTV-v-Ha-ras transgenic mice to generate cdk4(neo/+):MMTV-v-Ha-ras mice. Later, these cdk4(neo/+):MMTV-v-Ha-ras transgenic mice were intracrossed to generate cdk4(neo/neo):MMTV-v-Ha-ras transgenic mice. To generate cdk4 (R24C/R24C)/MMTV-v-Ha-ras mice, cdk4 (+/R24C) or cdk4 (R24C/R24C) mice were mated with MMTV-v-Ha-ras transgenic mice. All female mice were maintained as virgins throughout the observation period for all the studies. Only female mice were used for the studies. Mice exhibiting palpable tumors were euthanized and dissected for the collection of either normal or mammary tumor tissues.
Histopathological analysis of mammary glands
The fourth inguinal mammary glands were dissected, formalin fixed, and processed for paraffin embedding and H&E staining. For histopathological analyses, mammary gland tissue from 14- to 17-week Cdk4+/+, Cdk4neo/neo Cdk4(+/+):MMTV-ras, and Cdk4(R24C/R24C):MMTV-ras mice was dissected free of surrounding fat and connective tissue and perfusion fixed in 10% buffered formalin. When formalin-fixed paraffin-embedded sections were used, they were first deparaffinized by immersing the slides in xylene for 5 min (×3) and rehydrated in gradients of ethanol (100%, 95%, 90%, 75%, and 50%) and water. The sections were then antigen retrieved by boiling in 10 mM sodium citrate buffer (pH 6) for 2 min. After cooling down for 30 min, the sections were washed in PBST (phosphate-buffered saline with 0.1% Triton X) for 5 min (×2), incubated in 1.5% H2O2/methanol for 15 min, washed in PBST for 5 min (×2), blocked with 5% serum/PBS for 1 h at room temperature, and probed with 1° antibody overnight at 4°C in a humidified chamber. The sections were then washed with PBST for 5 min (×5), probed with 2° antibody for 1 h at room temperature, and washed again for 5 min (×5) with PBST. The slides were then incubated with ABC reagent (cat. #PK-4000, VECTASTAIN ABC Kit, Vector Laboratories) for 30 min, washed in PBST for 5 min (×2), and treated with DAB (cat. #SK-4100, Vector Laboratories). As soon as the required color intensity developed, the reaction was stopped by immersing the slides in deionized H2O. The sections were counterstained with hematoxylin or methyl green (cat. #H-3402, Vector Labs), dehydrated in gradients of ethanol and xylene, and mounted with permount. Images were acquired on a Nikon microscope with digital imaging capabilities.
Protein analysis
Mammary glands or tumors were homogenized in TNE lysis buffer as described previously.14 The samples were incubated on ice for 30 min, and lysates were cleared by centrifugation (15,000 rpm, 15 min, 4°C). Then, 50 to 100 μg of protein was resolved by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) and transferred to nitrocellulose membranes. The membrane was probed with desired primary antibody overnight at 4°C, washed with TBST (Tris buffer with 0.05% Tween-20) for 5 min (×3), and probed with a suitable secondary antibody for 1 h. After washing with TBST for 5 min (×3), the membrane was incubated with chemiluminescence reagent or chemiluminescence reagent plus for 1 min and exposed to X-ray film.
TUNEL assay
The TUNEL assay was performed using the DeadEnd™ Colorimetric TUNEL System (cat. #G7130, Promega) according to the manufacturer's protocol. Sections were deparaffinized by immersing the slides in xylene for 5 min (×2) at room temperature. Then the slides were washed in 100% ethanol for 5 min followed by 3-min washes in a gradient of ethanol (100%, 95%, 85%, 70%, and 50%). The slides were incubated in 0.85% of NaCl for 5 min at room temperature and washed in PBS for 5 min. Sections were fixed by incubating the slides in 4% methanol-free formaldehyde in PBS for 15 min at room temperature and washed in PBS for 5 min (×2). The slides were placed on a flat surface and treated with 5 μg/mL proteinase K solution at room temperature, washed in PBS for 5 min, refixed in 4% methanol-free formaldehyde in PBS for 5 min at room temperature, and washed in PBS for 5 min (×2). The sections were then equilibrated with 100 μL of equilibration buffer for 10 min at room temperature and treated with rTdT reaction mix. Cover slips were applied to the sections and incubated at 37°C for 60 min inside a humidified chamber to allow the end-labeling reaction to occur. The reaction was stopped by immersing the slides in 2× SSC for 15 min at room temperature. The slides were then washed in PBS for 5 min (×3), blocked with 0.3% H2O2 for 5 min, and washed in PBS for 5 min (×2). The sections were incubated with 100 μL of streptavidin horseradish peroxidase (HRP) solution for 30 min, washed in PBS for 5 min (×2), and incubated with DAB solution until a light brown background appeared. The slides were rinsed in deionized water, counterstained with hematoxylin, dehydrated in gradients of ethanol, and mounted with permount.
A positive control was prepared by treating the sections with DNase I. Formaldehyde fixed sections were treated with proteinase K and incubated with DNase I buffer for 5 min. The DNase I buffer was tapped off, and 10 unit/mL DNase I was added to the sections. After a 10-min incubation at room temperature, excess liquid was removed, and the slides were 4 times rinsed with deionized water, followed by a wash with PBS for 5 min. Sections were then incubated with equilibration buffer for 10 min and continued as described above.
Quantitation of IHC and TUNEL-positive stain
Quantification was performed using a bioquantification software system (Bioquant TCW 98, Nashville, TN), as described by Al-Shatti et al.36 Pixel areas were measured at the selected magnification (100× for γ-H2AX and 40× for all others) using the videocount area option of the Bioquant software system. Videocount area is defined as the number of pixels in a field that meets a user-defined color threshold of staining multiplied by the area of a pixel at the selected magnification.36 A background threshold was set, and the brown HRP-DAB immunostaining above the background levels was measured. The threshold values for each protein were stored in the computer program for consistent auto-measurement of the immunostained slides. Percent area fractions of immunoreaction product for each protein were calculated by dividing the videocount area containing pixels at or above the defined background threshold by the videocount area of the total number of pixels in the chosen field (ROI) and multiplying by 100. Three to 7 individual areas for each mouse with a minimum of 3 mice per genotype were analyzed.
Statistical analysis
Analysis was performed using Prism software. An unpaired t test or F test was used to analyze the statistical significance of the data.
Acknowledgments
We gratefully acknowledge the help of Dr. Mary Barbe in performing statistical analyses.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by NIH grants PO1 CA95569 and RO1 AG22022 to EP Reddy. HKDL Reddy was supported by a graduate student fellowship from USAMRMC Breast Cancer Research Program, W81XWH-05-1-0262.
References
- 1. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004;18:2699-711 [DOI] [PubMed] [Google Scholar]
- 2. Graña X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995;11:211-9 [PubMed] [Google Scholar]
- 3. Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle 2003;1:103-10 [PubMed] [Google Scholar]
- 4. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997;13:261-91 [DOI] [PubMed] [Google Scholar]
- 5. Malumbres A, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Cancer Rev 2001;1:222-35 [DOI] [PubMed] [Google Scholar]
- 6. Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995;270:23589-97 [DOI] [PubMed] [Google Scholar]
- 7. Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996;271:20608-16 [DOI] [PubMed] [Google Scholar]
- 8. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998;12:3499-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. An H-X, Beckmann MW, Reifenberger G, Bender HG, Niederacher D. Gene amplification and overexpression of Cdk4 in sporadic breast carcinomas if associated with high tumor cell proliferation. Am J Pathol 1999;154:113-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995;269:1281-4 [DOI] [PubMed] [Google Scholar]
- 11. Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet 1996;12:97-9 [DOI] [PubMed] [Google Scholar]
- 12. Sotillo R, Dubus P, Martín J, de la Cueva E, Ortega S, Malumbres M, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J 2001;20:6637-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999;22:44-52 [DOI] [PubMed] [Google Scholar]
- 14. Reddy HK, Mettus RV, Rane SG, Graña X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res 2005;65:10174-8 [DOI] [PubMed] [Google Scholar]
- 15. Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 2006;9:13-22 [DOI] [PubMed] [Google Scholar]
- 16. Yu Q, Sicinska E, Geng Y, Ahnström M, Zagozdzon A, Kong Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006;9:23-32 [DOI] [PubMed] [Google Scholar]
- 17. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88:593-602 [DOI] [PubMed] [Google Scholar]
- 18. Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol 1997;17:5588-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med 2002;8:1105-14 [DOI] [PubMed] [Google Scholar]
- 20. Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, et al. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 2003;114:323-34 [DOI] [PubMed] [Google Scholar]
- 21. Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001;411:1017-21 [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez-Puebla ML, Miliani de Marval PL, LaCava M, Moons DS, Kiyokawa H, Conti CJ. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol 2002;161:405-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sotillo R, García JF, Ortega S, Martin J, Dubus P, Barbacid M, Malumbres M. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci USA 2001;98:13312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol 2002;22:644-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997;91:649-59 [DOI] [PubMed] [Google Scholar]
- 26. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature 2005;436:642. [DOI] [PubMed] [Google Scholar]
- 27. Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/ 6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol 2002; 22:3389-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006;444:638-42 [DOI] [PubMed] [Google Scholar]
- 29. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006;444:633-7 [DOI] [PubMed] [Google Scholar]
- 30. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005;434:907-13 [DOI] [PubMed] [Google Scholar]
- 31. Kennedy NJ, Sluss HK, Jones SN, Bar-Sagi D, Flavell RA, Davis RJ. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes Dev 2003;17:629-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998;273:5858-68 [DOI] [PubMed] [Google Scholar]
- 33. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000;10:886-95 [DOI] [PubMed] [Google Scholar]
- 34. Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 1987;49:465-75 [DOI] [PubMed] [Google Scholar]
- 35. Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 2007;9:493-505 [DOI] [PubMed] [Google Scholar]
- 36. Al-Shatti T, Barr AE, Safadi FF, Amin M, Barbe MF. Increase in inflammatory cytokines in median nerves in a rat model of repetitive motion injury. J Neuroimmunol 2005;167:13-22 [DOI] [PMC free article] [PubMed] [Google Scholar]