

Abstract
Prospective identification of which individuals with diabetes mellitus (DM) are at greatest risk of developing cardiovascular (CVD) complications would have considerable public health importance by allowing the allocation of limited resources to be focused on those individuals who would most benefit from aggressive intervention. Over the past 20 years genetic disease association studies have demonstrated that polymorphisms at specific genetic loci may identify those individuals at greatest risk of developing CVD in the setting of DM. This article reviews the evidence accumulated to date on four polymorphic loci with the aim of explaining how these polymorphisms modify the risk of CVD in DM by modifying the functional activity of a specific gene. Utilization of the knowledge of these genetic differences among individuals in targeting drug therapy (pharmacogenomics) is also discussed.
Keywords: Diabetes, CVD, polymorphism, genetic variance, genotype, phenotype
1. Introduction
Due to increasing prevalence rates in the past decade, Diabetes Mellitus (DM) has become a major public health issue. Both macrovascular and microvascular complications are common long term sequalae of the disease. Cardiovascular disease is the single most major cause of death among DM patients, accounting for approximately 65% of mortality in DM[1]. End stage renal disease is also a major complication of DM with nearly a third of all individuals with DM eventually requiring treatment with dialysis. Overall, DM accounts for 35% of hospitalizations due to CVD, and is the leading cause of blindness in the western world. Identification of those individuals with DM who are at greatest risk for the development of CVD would have considerable public health importance as it would allow for a more efficient allocation of resources in order to alleviate the burden of disease[2]. Beginning over 20 years ago hundreds of polymorphisms in genes that are involved in the pathogenesis of CVD and DM have been examined for their ability to predict which individuals with DM will develop CVD. These polymorphisms may modify the activity of proteins, making them more or less active, or alter their expression and stability, thus modulating their ability to retain normal vascular physiology and metabolism. Figure 1 provides a functional categorization of these polymorphic genes. Table 1 provides a comprehensive list of the SNPs and polymorphisms that have been assessed to date in the search for loci which may predict CVD in DM. However, only a handful of these polymorphisms have been shown reproducibly to predict diabetic CVD across various populations and ethnic groups. These polymorphisms will be reviewed in this paper.
Figure 1.
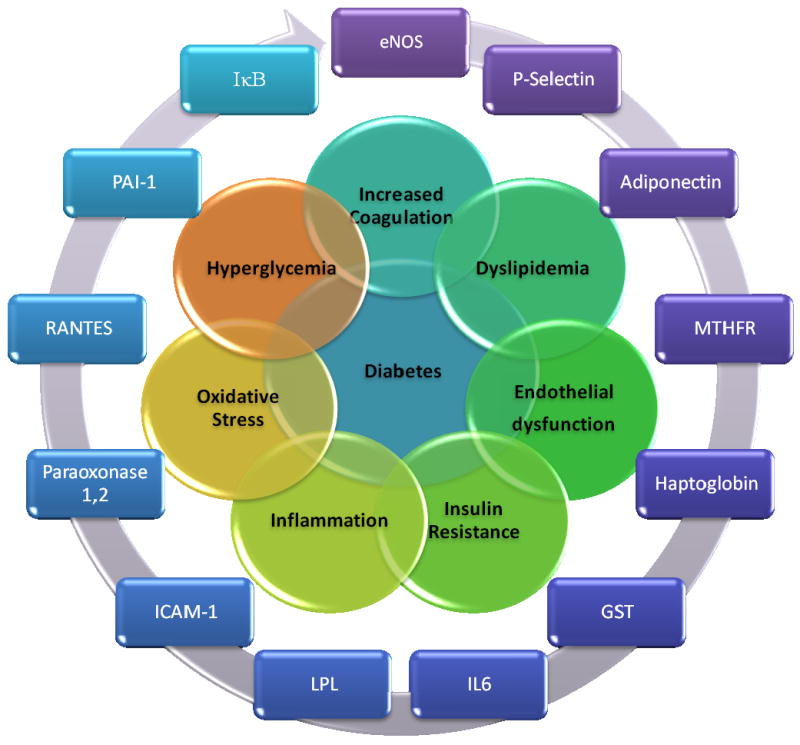
Candidate genes for diabetic CVD. eNOS: endothelial nitric oxide synthase, MTHFR: methylenetetrahydrofolate reductase, GST: glutathione S transferase, IL6: interleukin 6, LPL: lipoprotein lipase, PON: paraoxonase, RANTES: regulated on activation, normal T cell expressed and secreted, PAI-1: plasminogen activator inhibitor 1, IκB: inhibitor of NFκB.
Table 1. SNPs and genetic variations implicated in the risk of cardiovascular disease, in the settings of Diabetes Mellitus.
| Gene | Protein | Polymorphism | References |
|---|---|---|---|
| 16p3 | - | [3] | |
| 9p21 | - | rs2383206 | [4] |
| ACE | Angiotensin Converting Enzyme | Insertion/Deletion | [5, 6] |
| ADIPOQ/ACDC | Adiponectin | −11391G>A | [7] |
| −11377C>G | [7-9] | ||
| -11365C>G | [10] | ||
| -4041 A>C | [9] | ||
| -4034A>C | [10] | ||
| -3964A>G | [10] | ||
| 45T>G | [7, 9-13] | ||
| 276G>T | [7, 9-13] | ||
| A349G | [11] | ||
| +2019 delA | [9] | ||
| ADIPOR1 | Adiponectin Receptor 1 | rs2232853 | [14] |
| rs12733285 | [14] | ||
| rs1342387 | [14] | ||
| rs7539542 | [14] | ||
| rs10920531 | [14] | ||
| rs4950894 | [14] | ||
| AGT | Angiotensinogen | M235T | [6, 15] |
| AGT1R | Angiotensin-II Receptor 1 | T573C | [15] |
| A1166C | [15] | ||
| AT1R | Angiotensin Receptor 1 | rs5186 | [6] |
| aP2 | Fatty-acid binding protein | T-78C | [16] |
| APOA4 | Apolipoprotein A-IV | Gln360His | [17] |
| Thr347Ser | [17] | ||
| APOE | Apolipoprotein E | ε2/ε3/ε4 | [18] |
| CD40 | CD-40 | rs1535045 | [19] |
| rs3765459 | [19] | ||
| CD40L | CD-40 Ligand | rs3092948 | [19] |
| rs3092929 | [19] | ||
| rs3092923 | [19] | ||
| rs3092920 | [19] | ||
| CETP | Cholesteryl ester transfer protein | rs289714 | [20] |
| I405V | [20] | ||
| R451Q | [20] | ||
| COX2 | Cyclooxygenase-2 | rs689466 | [21] |
| rs20417 | [21] | ||
| rs2745557 | [21] | ||
| rs5277 | [21] | ||
| rs20432 | [21] | ||
| rs2066826 | [21] | ||
| rs5275 | [21] | ||
| rs10911902 | [21] | ||
| CX3CR1 | Fractalkine receptor | T280M | [22] |
| ENPP1 | Ectoenzyme nucleotide pyrophosphate phosphodiesterase 1 | K121Q | [23] |
| EPHX2 | Epoxide hydrolase 2 | rs7003694 | [24] |
| rs7837347 | [24] | ||
| R287Q | [24] | ||
| rs721619 | [24] | ||
| rs747276 | [24] | ||
| HL | Hepatic Lipase | C-480T | [20] |
| HSP70-2 | Heat Shock Protein 70-2 | A1267G (A/B) | [25] |
| GCH1 | GTP cyclohydrolase 1 | C59038T | [26] |
| GSTT1 | Glutathione S transferase theta-1 | 1/0 | [27, 28] |
| GSTM1 | Glutathione S transferase mu-1 | 1/0 | [27, 28] |
| GSTP1 | Glutathione S transferase pi-1 | Ile105Val | [28] |
| IL6 | Interleukin 6 | -174G/C | [29] |
| LPL | Lipoprotein Lipase | rs285 | [20] |
| rs320 | [20] | ||
| MT1A | Metallothioneins | A647C | [30] |
| A1245G | [30] | ||
| MCP-1 | Monocyte Chemoattractant Protein 1 | A-2518G | [22] |
| NFKBIA | Inhibitor of NFκB | A/G in the 3′-UTR | [31] |
| NFKB1 | Nuclear factor κ B | CA repeat | [31] |
| NQO1 | Nicotinamide adenine dinucleotide phosphate: Quinone oxidoreductase 1 | C609T (rs1800566) | [32] |
| PAI-1 | Plasminogen Activator Inhibitor 1 | −675 4G/5G insertion/deletion | [33] |
| PON2 | Paraoxonase 2 | S311C | [20] |
| PPARA | Peroxisome proliferator activated receptor α | L162V | [34, 35] |
| C2528G | [35] | ||
| P-Selectin | P-Selectin | Tyr715Pro | [36] |
| Asn562Asp | [36] | ||
| Ser290Asn | [36] | ||
| PTPN1 | Protein Tyrosine Phosphatase 1B | rs2904268 | [37] |
| rs803742 | [37] | ||
| rs1967439 | [37] | ||
| rs718630 | [37] | ||
| rs4811078 | [37] | ||
| rs2206656 | [37] | ||
| rs932420 | [37] | ||
| rs3787335 | [37] | ||
| rs2426158 | [37] | ||
| rs2904269 | [37] | ||
| rs941798 | [37] | ||
| rs1570179 | [37] | ||
| rs3787345 | [37] | ||
| rs1885177 | [37] | ||
| rs754118 | [37] | ||
| rs3215684 | [37] | ||
| rs968701 | [37] | ||
| rs2282147 | [37] | ||
| rs718049 | [37] | ||
| rs718050 | [37] | ||
| rs3787348 | [37] | ||
| 1484insG | [37] | ||
| rs914458 | [37] | ||
| RANTES | regulated on activation, normal T cell expressed and secreted | A(−403)G | [22] |
| C-28G | [22] | ||
| In1.1C/T | [22] | ||
| VEGF | Vascular Endothelial Growth Factor | Insertion / Deletion | [38] |
| G405C | [38] | ||
| T(-1498)C | [39] | ||
| G(-1190)A | [39] | ||
| C(-634)G | [39] | ||
| C(-7)T | [39] | ||
2. Paraoxonase
2.1 The Paraoxonase Family
Located at locus 7q21.3, the cluster of the Paraoxonase (PON) gene family is comprised of three members: PON1, PON2 and PON3. The three members are highly homogenous, presenting with 70% similarity at the nucleotide level and 60% similarity of the amino acid sequence[40]. However, their expression patterns are varied. While PON1 and PON3 are expressed mainly in the liver and associate with high density lipoprotein (HDL) in the circulation[41, 42], PON2 is expressed in a variety of tissues and is found on the endoplasmic reticulum (ER) and the nuclear membrane[43]. While only PON1 exhibits the ability to hydrolyze organophosphates, the three enzymes share the ability to metabolize different lactones[44], some of which are the product of phospholipid peroxidation[45]. It has been suggested that this enzymatic activity has a pivotal role in preserving the integrity of cell membranes, protecting it from a wide variety of both endogenous and exogenous toxins. Supporting this hypothesis is the localization of PON1 to the same HDL subfraction as clusterin, which is also suspected to have a role in cell membrane protection[46]. Recently, it has been suggested that the PON enzymes have a role in innate immunity, being able to hydrolyze the quorum sensing signal molecule N-acyl-homoserine-lactone[47].
2.2 Antiatherogenic properties of Paraoxonase
HDL is known to attenuate the development of atherosclerosis by a variety of mechanisms, including removal of excess cholesterol from cells of the vessel wall (reverse cholesterol transport) and limiting low density lipoprotein (LDL) oxidation. These antiatherogenic activities are catalyzed by the many proteins associated with the HDL particle. PON1 and PON3, which are associated with HDL in the plasma, take part in the antioxidant activity of HDL. PON1 was shown to diminish LDL oxidation[48] and prevent the pro-inflammatory response elicited by oxidized LDL (OxLDL), the latter probably resulting from the metabolism of lipid peroxides[49]. Moreover, PON1 was shown to be critical for preventing the oxidation of HDL, allowing it to maintain its function[50]. Similar results were obtained in a study of PON1 knockout mice, where the ability of HDL isolated from these mice to limit lipid peroxidation and LDL induced inflammation was decreased[51]. PON1 knockout mice were also more susceptible to the development of atherosclerosis in dietary or genetic models[51, 52]. Demonstrating a therapeutic potential for PON1 elevation, mice overexpressing PON1 were more resistant to atherosclerosis compared to wild-type mice[53].
2.3 Paraoxonase genotype and relation to CVD
Genetic polymorphisms have been discovered in all the three members of the Paraoxonase gene family. In PON1, which has gained the most attention concerning its relationship with CVD, several polymorphisms have been identified, both in coding regions, affecting the amino acid sequence, and in the promoter region. Of the polymorphisms of the promoter, the C(-107)T polymorphism has been most widely studied. Due to differences in the affinity of the transcription factor Sp1[54], this polymorphism has a dramatic effect on gene transcription, the -107C allele having significantly increased transcriptional activity compared to the -107T allele. This variation is reflected in higher plasma PON1 concentrations and activity in carriers of the -107C allele[54, 55]. However, although significantly decreasing enzyme concentration and activity, this polymorphism has not consistently been shown to be associated with CVD[56-59]. Interestingly, a recent trial has shown that although the promoter polymorphisms do not affect the risk for CVD, they do influence the extent of the disease as measured by the number of coronary vessels undergoing stenosis[58].
Of the polymorphisms in the coding region, the Glu192Arg polymorphism has gained the most interest [60]. This polymorphism results in a decrease in serum Paraoxonase activity and concentration[61], possibly due to decreased affinity of the Arg192 polymorphism to the HDL, which leads to decreased protein stability and activity[62]. Of the many polymorphisms of the PON enzymes, the Glu192Arg polymorphism alone was found to be associated with CVD in a large meta-analysis, although the external validity of this association was questioned due to the lack of a significant association between this polymorphism and CVD in large trials and due to possible publication bias[63].
Following the many failures to associate specific polymorphisms of the PON1 gene with vascular disease, it has been suggested that the relationship between PON1 phenotype expressed by serum concentration and activity, rather than genotype, and CVD should be explored. Indeed, it has been found that PON1 activity is a predictor of CVD, regardless of PON1 genotype[64, 65]. A recent large trial in which both PON1 activity and genotype were tested has shown that not only do both PON1 phenotype and Glu192Arg genotype determine the risk for CVD, but also that the PON1 genotype is a predictor of its activity and concentration[61].
2.4 Paraoxonase and Diabetes Mellitus
The relationship between PON1 and DM appears to be bidirectional with DM significantly decreasing PON1 concentration[66] and activity[66-70], and in turn, PON1 genotype modulating the risk for type 2 DM[68, 71]. The importance of Paraoxonase activity in the prevention of DM was also demonstrated in an in vivo model, where increased PON1 expression in mice has impeded the development of streptozotocin induced DM[72].
Similar to what is seen in non-DM individuals, PON1 concentration and activity were found to be negatively associated with CVD[73-76]. In the settings of Type 2 DM only a single study has focused on the C(-107)T polymorphism, indicating an increased risk for CVD in carriers of the TT allele compared to carriers of the CT or CC alleles[77]. This genotype was also associated with decreased PON1 concentration and activity[67], and increased plasma OxLDL/Apolipoprotein B (ApoB) ratio among DM individuals[78]. The Glu192Arg polymorphism was extensively studied in the settings of DM. In these settings, HDL from 192Arg allele homozygotes was demonstrated to be less efficient in the metabolism of oxidized phospholipids[69], although these results were contested by others showing either no difference in plasma oxidized LDL[79], or decreased oxidation in the aforementioned genotype[80]. In most epidemiological studies, the 192Arg allele was shown to be significantly correlated with CVD[81-84], although several studies have presented contradicting results[74]. Several studies have highlighted the importance of interaction between the PON1 Glu192Arg polymorphism and DM, arguing that this polymorphism most significantly increases risk for CVD in the presence of DM[85, 86].
3. Methyltetrahydrofolate Reductase & Homocysteine Metabolism
3.1 Metabolic pathways of Homocysteine
Homocysteine (Hcy) is positioned at the crossroads of several metabolic pathways. Hcy is synthesized from methionine in a 3-steps reaction, which includes activation of methionine by ATP, loss of a methyl group and enzymatic hydrolysis. Remethylation of Hcy to methionine is catalyzed by betaine-homocysteine methyltransferase (BHMT) in the liver or by Methionine Synthase (MS) in most bodily tissues, the latter depending on methyltetrahydrofolate as a methyl donor and vitamin B12 as a cofactor. Synthesis of methyltetrahydrofolate is catalyzed by the enzyme methylenetetrahydrofolate reductase (MTHFR), in the presence of the vitamin B2. In a state of methionine excess, Hcy is irreversibly transsulfurated by CBS and vitamin B6 to cystathionine, which can be converted to cysteine[87]. A third elimination pathway of Hcy is the pathological synthesis of the Hcy-Thiolactone. This reaction is carried out by methionyl-tRNA synthetase (MetRS) when Hcy is mistakenly recognized as methionine[88].
3.2 Atherogenic effects of Homocysteine
Homocysteine and its metabolites were shown to have an atherogenic potential, affecting cell survival, proliferation and apoptosis, thrombosis and lipid metabolism and peroxidation.
Hcy-thiolactone is capable of forming amide bonds with lysine residues, thus creating n-homocysteinylated proteins, altering their activity and solubility.
N-homocysteinylation of the HDL-associated enzyme PON1 that hydrolyses Hcy-thiolactone and oxidized phospholipids renders it inactive thus decreasing the anti-atherogenic activity of HDL[88]. Hyperhomocysteinemia has also been associated with decreased expression of HDL-associated proteins such as LCAT and Apo-A1, and accelerated HDL catabolism, resulting in overall decreased HDL levels and activity[89]. Although LDL oxidation by Hcy was not proven to take place in vivo[90], modification of ApoB100 by Hcy-thiolactone leads to its aggregation, making it cytotoxic to endothelial cells[88].
Because of the strong relationship between oxidative stress and CVD, Hcy oxidative potential has been extensively studied. A central role in Hcy-mediated oxidative stress has been attributed to its reaction with copper to produce hydrogen peroxide (H2O2) and other reactive oxygen species (ROS). Although Hcy promotes the synthesis of gluthathione and Nitrous Oxide (NO), the former scavenging H2O2 and the latter scavenging Hcy itself, prolonged exposure to high levels of Hcy leads to saturation of these reactions, thus allowing excess Hcy to produce free H2O2 and ROS[91].
The prothrombotic effects of Hcy and its metabolites are a result of increased procoagulatory properties of the endothelium and decreased fibrinolysis. Treatment of endothelial cells with Hcy increased the synthesis and activation of procoagulatory molecules such as tissue factor and Factor V, and decreased the synthesis and activation of anticoagulatory molecules such as heparan sulfate, Protein C, NO and prostacyclins[91]. N-Homocysteinylation of fibrinogen renders it more resistant to fibrinolysis[88], a process that is attenuated further by decreased tissue plasminogen activator (TPA) activation by the endothelium[91].
Disruption of normal endothelial function by Hcy and its metabolites is not restricted to its role in coagulation and thrombolysis. Treatment with Hcy or Hcy thiolactone resulted in increased apoptosis[92] and decreased proliferation[93-95] of cultured endothelial cells. Impaired endothelial dependent vasodilation, most likely resulting from both endothelial injury, increased oxidative stress and decreased NO bioavailability, is another manifestation of hyperhomocysteinemia[90, 91].
Increased proliferation of vascular smooth muscle cells (VSMCs) has been shown to occur following exposure to Hcy[96, 97]. Thickening of the vessel wall is also the result of the increased collagen production exerted by Hcy[91].
3.3 Hyperhomocysteinemia and Cardiovascular Disease
Hyperhomocysteinemia was first implicated in the pathogenesis of vascular disease in 1969, following an observation that children carrying inherited deficiencies in the enzyme Cystathionine β-Synthase (CBS) presenting with homocysteinuria commonly suffer from vascular diseases[98]. This observation was the cornerstone for extensive research regarding the relationship between Hcy and vascular diseases. Over the years, this relationship has been thoroughly studied, with considerable evidence pointing towards a significant association between cardiovascular disease and elevated Hcy level which is independent of other known cardiovascular risk factors[99-103]. Consequently, Hcy-reducing treatments, generally including folate, vitamin B6 and vitamin B12, were tested for their ability to reduce the risk for CVD among individuals with hyperhomocysteinemia. Although supplementation decreases homocysteine levels, in most studies this has not been accompanied by a reduction in the risk of CVD[102, 104-106]. Moreover, the results of two studies suggested a potentially harmful effect of vitamin supplementation[107, 108]. An exception to these findings is the effect of folate and vitamin supplementation on stroke, where a significant risk reduction was noted[109]. It has been suggested that Hcy is only a marker of other pathologic phenomena related to CVD. However, other explanations for the discrepancy between the results of the observational studies and the clinical trials have been offered, amongst which are the duration of Hcy lowering treatments and the confounding effect of folate fortification of grains[110]. Another possibility is that Hcy reduction may only be helpful in early stages of CVD.
Similarly to what is seen in the general population, hyperhomocysteinemia is associated with increased CVD among DM patients as well[111-115]. Although it is unclear whether hyperhomocysteinemia is one of the manifestations of DM[116-118], in-group studies have found several correlates to Hcy levels, the most prominent being renal function[115, 118].
3.4 The MTHFR 677 CT Polymorphism
Following the discovery of the relationship between CBS deficiency, increased Hcy and CVD, other polymorphisms and mutations that may interfere with Hcy metabolism have been examined. The most documented polymorphism is that of the enzyme MTHFR, where a substitution of C to T occurs at position 677. This substitution results in a missense polymorphism, producing a thermolabile enzyme with decreased reactivity. The 677T allele has been shown to have a frequency of 30%-40% with 10% of all individuals homozygous for the T allele. TT homozygotes have been shown to have marked hyperhomocysteinemia in states of folate deficiency[91]. Although initial studies demonstrated a strong relationship between the 677CT polymorphism and CVD[119], these results have not been replicated[120] leading to the notion that this polymorphism may only be a minor risk factor for CVD[121, 122]. The relationship between the 677 CT polymorphism and CVD in DM is complicated as well. While significant evidence exist linking the 677 CT polymorphism to diabetic retinopathy[123-125] and nephropathy[126-128], evidence is disputed regarding its role in stroke and peripheral and coronary artery disease[129-136].
4. Endothelial Nitric Oxide Synthase and Nitric Oxide metabolism
4.1 Role of NO and eNOS in cardiovascular physiology
NO has been identified as an important factor in maintaining normal cardiovascular function and preserving the integrity of the vascular bed. It inhibits thrombosis and coagulation not only by maintaining anti-coagulatory and anti-thrombogenic properties of the endothelium[137], but also by inhibiting platelet activation and aggregation and thereby reducing platelet derived growth factor (PDGF) induced proliferation of vascular smooth muscle cells in the vessel wall[138]. Acting directly on VSMCs, NO is a potent vasodilator[138] and a regulator of cell proliferation[139]. NO protects the endothelium and the underlying intima from inflammatory processes, inhibiting the expression of chemoattractants such as MCP-1 and of adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1)[138]. NO is also capable of acting as an anti-oxidant, inhibiting pro-oxidative reactions catalyzed by H2O2[140].
In vivo, NO is synthesized by the Nitric Oxide Synthase (NOS) family of enzymes. The endothelial Nitric Oxide Synthase (eNOS) is synthesized from the gene NOS3, which is located at chromosomal locus 7q35-36[141]. Despite its name, eNOS expression is not restricted to endothelial cells and can be found in other cell types such as erythrocytes, leukocytes and mast cells[137]. It acts as a membrane bound homodimer[142] catalyzing the synthesis of NO from L-arginine, a reaction that demands the presence of several cofactors such as heme, tetrahydrobiopterin and nicotinamide-adenine-dinucleotide phosphate (NADPH)[139]. eNOS is a constitutively expressed enzyme that is subjected to regulation by a variety of factors such as calcium and calmodulin, phosphorylation, protein-protein interaction and fatty-acid modification. Altogether, these factors determine NO production and release by the endothelium[143].
4.2 Polymorphisms of the NOS3 gene and their relation to CVD
As a result of the importance of NO and eNOS in vascular physiology, polymorphisms were examined in the gene NOS3. In the coding region, the Glu298Asp (G894T) polymorphism has been most widely studied. Several studies reported differences in NO synthesis and reduced NO availability in carriers of the 298Asp variant, probably due to protein cleavage[144, 145], but these results were contradicted by others[146, 147]. Characteristics of enzymatic activity, such as KM, Vmax and Ki for various inhibitors did not differ between the 298Asp and 298Glu proteins[147, 148]. In several independent studies, the 298Asp variant has been associated with poor endothelial and vasomotor function, carriers of the allele presenting with decreased flow or stimuli dependent vasodilation[149, 150], increased coronary vascular resistance[150] and increased vasoconstriction in response to stimuli[151]. However, these results were not observed in all trials, some finding no relation between the 298Asp allele and endothelial function[152-154]. This polymorphism has also been identified as an important determinant of collateral vessels development[155, 156].
In the promoter region, the T(-786)C polymorphism was identified as a predictor of eNOS expression, the -786C allele associated with decreased mRNA levels, which translate to decreased NO production[157] and decreased endothelial function[153, 158, 159]. The variability in mRNA expression most likely results from differential binding of an inhibitory transcription factor to the -786C allele[158], perhaps the replication protein A1[157].
A third polymorphism that has gained much attention is a 27 base pair tandem repeat in intron 4, where one allele, denoted a, presents with 4 tandem repeats and the second allele, denoted b, presents with 5 tandem repeats. Sparse evidence exists regarding the effect of this polymorphism on eNOS expression and activity. While the b allele has been linked to protein expression[160] and increased plasma NO concentrations in some[161] but not all trials[162-164], the a allele was associated with increased specific activity[160]. Lacking a mechanism that links it with protein activity or expression, it has been suggested that this intron polymorphism is found in linkage disequilibrium with other functional polymorphisms. This theory was supported by several studies that found linkage disequilibrium between this polymorphism and the T-786C polymorphism[160, 162].
The effect of the aforementioned polymorphisms on CVD has been evaluated in a recent meta-analysis. When all studies were taken into account, all three polymorphisms were found to be mild risk factors for CHD. However, when only the larger studies were examined, this association was either lost or was no longer significant[165]. In this meta-analysis, no significant association was found between these polymorphisms and other CVD outcomes such as stroke and carotid stenosis.
4.3 NOS3 polymorphisms and Diabetes Mellitus
Endothelial dysfunction and decreased NO bioavailability is a prominent feature of T2DM, and may even appear before the onset of the disease[166]. Decreased NO release by endothelial cells may be the result of several mechanisms, including a deficiency in L-arginine, deficiencies in the various co-factors and increased NO scavenging by ROS. Aberrant insulin signaling through the PI3K/Akt pathway in endothelial cells prevents eNOS phosphorylation on Serine 1177, thus decreasing its activity[167]. Similar to the observations in non-DM patients, the evidence regarding the impact of NOS3 polymorphisms on NO production and endothelial function is inconclusive[168-170]. A limited body of data exists regarding the relationship between NOS3 polymorphisms and cardiovascular outcomes in the setting of DM. A recent review that studied the relationship between the different NOS3 polymorphisms and diabetic nephropathy was able to show a significant effect of the Glu298Asp across different populations. A specific interaction between the Glu298Asp and the 4a/b polymorphisms and severe diabetic nephropathy was shown in East Asian populations[171]. A recent meta-analysis also investigated the relationship between NOS3 polymorphism and diabetic retinopathy, failing to find a significant association between the two[172]. The relationship between the Glu298Asp polymorphism and other cardiovascular diseases in the setting of DM was identified by several[131, 173] but not all[174, 175] studies. However, these were mostly retrospective studies, with only one prospective study demonstrating such a relationship in patients with Type 1 DM[176].
5. Haptoglobin and hemoglobin-mediated oxidative stress
5.1 Haptoglobin Metabolism
Haptoglobin (Hp) is an acute-phase plasma born glycoprotein produced mainly by hepatocytes, most widely known for its ability to strongly bind free hemoglobin (Hb) following its release from erythrocytes[177]. The concentrations of Hp in the plasma are high, ranging from 0.3mg/ml to 3mg/ml, producing an Hp:Hb molar ratio of 400:1. This allows effective scavenging of free Hb, even in the scenario of hemolysis when its levels are sharply increased[178]. In fact, Hp has a major role in iron preservation during hemolysis, as it prevents Hb filtration in the glomeruli[179] and renal damage[180]. The Hp-Hb complex is transported to the liver and other tissues to be degraded by Hp-Hb scavenger receptors, such as the CD163 receptor present on macrophages and liver Kupfer cells[181, 182]. Another important aspect of Hb scavenging by Hp is the reduction in oxidative stress. Extracorpuscular Hb can initiate a free radical reaction by releasing heme iron, which acts as a potent Fenton reagent. This reaction results in the production of ROS that cause oxidative damage to their surroundings[183]. Hp binding to Hb prevents this cascade by shielding the heme iron from its aqueous surrounding[184, 185]. Moreover, Hp maintains Hb integrity by preventing oxidation of the globin by heme iron. This allows effective clearance of Hb by the CD163 receptor[186].
5.2 The Hp Polymorphism
The Hp gene has been localized to chromosome 16q22. Two Hp alleles exist in man: Hp1, with an allele frequency of 0.4 and Hp2, with an allele frequency of 0.6 in most western populations. The alleles are found in a Hardy-Weinberg equilibrium, the frequency of the Hp 1-1, Hp 2-1 and Hp 2-2 genotypes being 16%, 48% and 36% respectively[177]. The Hp2 allele, whose development most likely occurred in early human evolution, has evolved from the Hp1 allele via a duplication of exons 3 and 4 present in the Hp 1 allele. Exon 3 contains a cysteine residue that can form a disulfide bridge between Hp monomers. Therefore, its duplication in the Hp 2 allele makes the Hp2 protein monomer bivalent, while the Hp1 protein monomer is monovalent. This has significant implications for the stoichiometry and structure of Hp found in serum. Being monovalent, the Hp1 monomer can only bind to one other Hp molecule, forming linear dimers in the Hp 1-1 genotype. The Hp2 monomer, conversely, binds two other Hp molecules and forms cyclic polymers in individuals with the Hp 2-2 genotype. In the Hp 2-1 genotype, heteromeric linear polymers are formed, with Hp1 proteins bracketing a chain of linear Hp2 proteins. Hp1-1 dimers may also be found in the Hp2-1 genotype[187].
5.3 Hp genotype and diabetic CVD -a specific gene-disease interaction
As opposed to the other CVD-related genes discussed above, the Hp polymorphism appears to have a unique interaction with DM. In the setting of DM, it has been shown by several groups that the Hp 2-2 polymorphism confers a 2-5 fold increased risk for CVD compared to the Hp 1-1 and Hp 2-1 genotype[188-192]. This was also seen in in vivo studies, where Hp 2-2 DM mice were more prone to develop retinopathy[193], nephropathy[194] and atherosclerosis[195, 196]. Interestingly in the absence of DM, the Hp 1-1 genotype may be associated with increased CVD[197, 198].
5.4 Hp polymorphism and Oxidative Stress – a mechanism for the gene-disease interaction
The underlying mechanism for the specific interaction between the Hp 2-2 genotype and DM appears to be the result of its interaction with Hb. In DM individuals, extravascular and intravascular hemolysis occur in higher rates compared to non-DM individuals, thus increasing the amount of Hp-Hb molecules in plasma and tissues. As already mentioned, Hp is considered an antioxidant due to its ability to scavenge Hb and prevent the initiation of radical chain reactions. However, this antioxidant activity varies greatly between the different Hp genotypes[184]. While the affinity for Hb is similar for Hp 1-1 and Hp 2-2[199], the ability to seclude the heme-iron from its aqueous surrounding is greatly decreased in the latter. This disparity is further magnified by oxidation and glycation of Hb, both common in DM[200]. Under hyperglycemic conditions, Hp 2-Hb, as compared to Hp 1-Hb evoked increased oxidative stress in cultured CD163-transfected CHO cells[200]. In vivo, the ability of Hp 1-1 to better shield Hb is translated to a decrease in redox active heme iron in Hp 1-1 DM compared to Hp 2-2 DM mice and humans, both in blood and tissues[200, 201]. The increase in plasma oxidative stress in Hp 2-2 individuals is also indicated by the decrease in antioxidants, such as vitamin C[187] in the serum of Hp 2-2 individuals. Furthermore, increased oxidative stress and hyperglycemia lead to decreased expression of CD163 on macrophages by inducing its shedding and decreasing transcription[202, 203]. Finally, clearance rate of the Hp-Hb-2-2 complex by the CD163 receptor is decreased compared to Hp-Hb-1-1, prolonging its presence in tissues[199] and perhaps decreasing the expression of CD163 itself[204].
Another novel aspect of Hp-Hb-2-2 mediated oxidative stress is related to the association of Hp with HDL. It was shown that Hp, either free or Hb-bound, is a member of the HDL proteome[205]. Since the Hp 2-2 polymers contain more Hp monomers than the Hp 1-1 dimers, Hp is more abundant in the HDL of Hp 2-2 individuals[205]. In Hp 2-2 DM individuals, as a result of the impaired clearance of Hp-Hb, there is an increased binding of Hp-Hb to HDL which along with the decreased antioxidant properties of Hp 2-2, expose HDL particles of Hp 2-2 DM individuals to increased oxidative stress, expressed by an increase in HDL associated lipid peroxides. This oxidative modification of HDL in Hp 2-2 DM individuals results in a decrease in HDL related functions such as reverse cholesterol efflux and cholesterol esterification[195, 205] and paradoxically may result in the transformation of the HDL particle in Hp 2-2 DM individuals into a proatherogenic species. This pathway may form the basis for the pharmacogenetic relationship between Vitamin E and the Hp 2-2 genotype whereby vitamin E can markedly reduce CVD in Hp 2-2 DM individuals, which was recently reviewed[206, 207].
6. Conclusion and Future Perspectives
With the advent of genome wide association studies, hundreds of genetic polymorphisms with a possible impact on diabetic CVD are being invesigated. However, most of these polymorphisms have failed to show any significant effect when tested across various populations. This has been due to the nature of the polymorphisms being tested, which are usually SNPs (single nucleotide polymorphisms) that have no established effect on protein activity or expression. Such polymorphisms are likely in linkage disequilibrium with other genetic markers which directly alter disease progression and therefore these SNP-disease associations may not be preserved in all populations and may be subject to population stratification. Polymorphisms in the Hp or PON genes discussed here, do not suffer from this setback. Having a direct effect on the pathophysiology of the disease, the Hp and PON polymorphisms have been shown to be risk factors for CVD in diverse populations.
Genetic testing certainly portends to be an important component of personalized medicine, but only when if it can be accompanied by a treatment plan that would match the genetic profile of the patient. While such treatment plans may include more aggressive treatment to at-risk individuals, they may also include more frequent screening and closer monitoring, as is customarily done for carriers of genetic mutations associated with increased risk for cancer. In addition, as illustrated by the pharmacogenomic interaction between the Hp genotype and vitamin E on CVD risk [206,207], these genetic markers might be useful in the identification of which individuals may benefit from specific drug treatments.
Acknowledgments
This work was supported by grants from the United States-Israel Binational Science Foundation, Israel Science Foundation, Juvenile Diabetes Research Foundation, the Kennedy Leigh Charitable Trust and RO1KD085226 from the NIH to APL
Footnotes
Financial Disclosure Information: Dr Levy has served in the past as a consultant for Synvista Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Economic costs of diabetes in the U.S. In 2007. Diabetes Care. 2008 Mar;31(3):596–615. doi: 10.2337/dc08-9017. [DOI] [PubMed] [Google Scholar]
- 2.Levy AP. Application of pharmacogenomics in the prevention of diabetic cardiovascular disease: mechanistic basis and clinical evidence for utilization of the haptoglobin genotype in determining benefit from antioxidant therapy. Pharmacol Ther. 2006 Nov;112(2):501–512. doi: 10.1016/j.pharmthera.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Bowden DW, Lehtinen AB, Ziegler JT, et al. Genetic epidemiology of subclinical cardiovascular disease in the diabetes heart study. Ann Hum Genet. 2008 Sep;72(Pt 5):598–610. doi: 10.1111/j.1469-1809.2008.00446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doria A, Wojcik J, Xu R, et al. Interaction between poor glycemic control and 9p21 locus on risk of coronary artery disease in type 2 diabetes. Jama. 2008 Nov 26;300(20):2389–2397. doi: 10.1001/jama.2008.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grammer TB, Renner W, von Karger S, Boehm BO, Winkelmann BR, Maerz W. The angiotensin-I converting enzyme I/D polymorphism is not associated with type 2 diabetes in individuals undergoing coronary angiography. (The Ludwigshafen Risk and Cardiovascular Health Study) Mol Genet Metab. 2006 Aug;88(4):378–383. doi: 10.1016/j.ymgme.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Burdon KP, Langefeld CD, Wagenknecht LE, et al. Association analysis of genes in the renin-angiotensin system with subclinical cardiovascular disease in families with Type 2 diabetes mellitus: the Diabetes Heart Study. Diabet Med. 2006 Mar;23(3):228–234. doi: 10.1111/j.1464-5491.2005.01777.x. [DOI] [PubMed] [Google Scholar]
- 7.Gable DR, Matin J, Whittall R, et al. Common adiponectin gene variants show different effects on risk of cardiovascular disease and type 2 diabetes in European subjects. Ann Hum Genet. 2007 Jul;71(Pt 4):453–466. doi: 10.1111/j.1469-1809.2006.00340.x. [DOI] [PubMed] [Google Scholar]
- 8.Prior SL, Gable DR, Cooper JA, et al. Association between the adiponectin promoter rs266729 gene variant and oxidative stress in patients with diabetes mellitus. Eur Heart J. 2009 May;30(10):1263–1269. doi: 10.1093/eurheartj/ehp090. [DOI] [PubMed] [Google Scholar]
- 9.Lacquemant C, Froguel P, Lobbens S, Izzo P, Dina C, Ruiz J. The adiponectin gene SNP+45 is associated with coronary artery disease in Type 2 (non-insulin-dependent) diabetes mellitus. Diabet Med. 2004 Jul;21(7):776–781. doi: 10.1111/j.1464-5491.2004.01224.x. [DOI] [PubMed] [Google Scholar]
- 10.Qi L, Doria A, Manson JE, et al. Adiponectin genetic variability, plasma adiponectin, and cardiovascular risk in patients with type 2 diabetes. Diabetes. 2006 May;55(5):1512–1516. doi: 10.2337/db05-1520. [DOI] [PubMed] [Google Scholar]
- 11.Vendramini MF, Pereira AC, Ferreira SR, Kasamatsu TS, Moises RS. Association of genetic variants in the adiponectin encoding gene (ADIPOQ) with type 2 diabetes in Japanese Brazilians. J Diabetes Complications. 2009 Mar 5; doi: 10.1016/j.jdiacomp.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Kim SH, Kang ES, Hur KY, et al. Adiponectin gene polymorphism 45T>G is associated with carotid artery plaques in patients with type 2 diabetes mellitus. Metabolism. 2008 Feb;57(2):274–279. doi: 10.1016/j.metabol.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Bacci S, Menzaghi C, Ercolino T, et al. The +276 G/T single nucleotide polymorphism of the adiponectin gene is associated with coronary artery disease in type 2 diabetic patients. Diabetes Care. 2004 Aug;27(8):2015–2020. doi: 10.2337/diacare.27.8.2015. [DOI] [PubMed] [Google Scholar]
- 14.Soccio T, Zhang YY, Bacci S, et al. Common haplotypes at the adiponectin receptor 1 (ADIPOR1) locus are associated with increased risk of coronary artery disease in type 2 diabetes. Diabetes. 2006 Oct;55(10):2763–2770. doi: 10.2337/db06-0613. [DOI] [PubMed] [Google Scholar]
- 15.Lin J, Hu FB, Qi L, Curhan GC. Genetic polymorphisms of angiotensin-2 type 1 receptor and angiotensinogen and risk of renal dysfunction and coronary heart disease in type 2 diabetes mellitus. BMC Nephrol. 2009;10:9. doi: 10.1186/1471-2369-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuncman G, Erbay E, Hom X, et al. A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc Natl Acad Sci U S A. 2006 May 2;103(18):6970–6975. doi: 10.1073/pnas.0602178103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kretowski A, Hokanson JE, McFann K, et al. The apolipoprotein A-IV Gln360His polymorphism predicts progression of coronary artery calcification in patients with type 1 diabetes. Diabetologia. 2006 Aug;49(8):1946–1954. doi: 10.1007/s00125-006-0317-1. [DOI] [PubMed] [Google Scholar]
- 18.Winkler K, Hoffmann MM, Krane V, Marz W, Drechsler C, Wanner C. Apolipoprotein E genotype predicts cardiovascular endpoints in dialysis patients with type 2 diabetes mellitus. Atherosclerosis. 2009 Jul 8; doi: 10.1016/j.atherosclerosis.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 19.Burdon KP, Langefeld CD, Beck SR, et al. Variants of the CD40 gene but not of the CD40L gene are associated with coronary artery calcification in the Diabetes Heart Study (DHS) Am Heart J. 2006 Mar;151(3):706–711. doi: 10.1016/j.ahj.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Burdon KP, Langefeld CD, Beck SR, et al. Association of genes of lipid metabolism with measures of subclinical cardiovascular disease in the Diabetes Heart Study. J Med Genet. 2005 Sep;42(9):720–724. doi: 10.1136/jmg.2004.029850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudock ME, Liu Y, Ziegler JT, et al. Association of polymorphisms in cyclooxygenase (COX)-2 with coronary and carotid calcium in the Diabetes Heart Study. Atherosclerosis. 2009 Apr;203(2):459–465. doi: 10.1016/j.atherosclerosis.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boger CA, Fischereder M, Deinzer M, et al. RANTES gene polymorphisms predict all-cause and cardiac mortality in type 2 diabetes mellitus hemodialysis patients. Atherosclerosis. 2005 Nov;183(1):121–129. doi: 10.1016/j.atherosclerosis.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Chen MP, Chung FM, Chang DM, et al. ENPP1 K121Q polymorphism is not related to type 2 diabetes mellitus, features of metabolic syndrome, and diabetic cardiovascular complications in a Chinese population. Rev Diabet Stud. 2006 Spring;3(1):21–30. doi: 10.1900/RDS.2006.3.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burdon KP, Lehtinen AB, Langefeld CD, et al. Genetic analysis of the soluble epoxide hydrolase gene, EPHX2, in subclinical cardiovascular disease in the Diabetes Heart Study. Diab Vasc Dis Res. 2008 Jun;5(2):128–134. doi: 10.3132/dvdr.2008.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giacconi R, Caruso C, Lio D, et al. 1267 HSP70-2 polymorphism as a risk factor for carotid plaque rupture and cerebral ischaemia in old type 2 diabetes-atherosclerotic patients. Mech Ageing Dev. 2005 Aug;126(8):866–873. doi: 10.1016/j.mad.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Liao YF, Zeng TS, Chen LL, et al. Association of a functional polymorphism (C59038T) in GTP cyclohydrolase 1 gene and Type 2 diabetic macrovascular disease in the Chinese population. J Diabetes Complications. 2009 Jun 8; doi: 10.1016/j.jdiacomp.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Hayek T, Stephens JW, Hubbart CS, et al. A common variant in the glutathione S transferase gene is associated with elevated markers of inflammation and lipid peroxidation in subjects with diabetes mellitus. Atherosclerosis. 2006 Feb;184(2):404–412. doi: 10.1016/j.atherosclerosis.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Doney AS, Lee S, Leese GP, Morris AD, Palmer CN. Increased cardiovascular morbidity and mortality in type 2 diabetes is associated with the glutathione S transferase theta-null genotype: a Go-DARTS study. Circulation. 2005 Jun 7;111(22):2927–2934. doi: 10.1161/CIRCULATIONAHA.104.509224. [DOI] [PubMed] [Google Scholar]
- 29.Danielsson P, Truedsson L, Eriksson KF, Norgren L. Inflammatory markers and IL-6 polymorphism in peripheral arterial disease with and without diabetes mellitus. Vasc Med. 2005 Aug;10(3):191–198. doi: 10.1191/1358863x05vm617oa. [DOI] [PubMed] [Google Scholar]
- 30.Giacconi R, Bonfigli AR, Testa R, et al. +647 A/C and +1245 MT1A polymorphisms in the susceptibility of diabetes mellitus and cardiovascular complications. Mol Genet Metab. 2008 May;94(1):98–104. doi: 10.1016/j.ymgme.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Romzova M, Hohenadel D, Kolostova K, et al. NFkappaB and its inhibitor IkappaB in relation to type 2 diabetes and its microvascular and atherosclerotic complications. Hum Immunol. 2006 Sep;67(9):706–713. doi: 10.1016/j.humimm.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Han SJ, Kang ES, Kim HJ, et al. The C609T variant of NQO1 is associated with carotid artery plaques in patients with type 2 diabetes. Mol Genet Metab. 2009 May;97(1):85–90. doi: 10.1016/j.ymgme.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Saely CH, Muendlein A, Vonbank A, et al. Type 2 diabetes significantly modulates the cardiovascular risk conferred by the PAI-1 -675 4G/5G polymorphism in angiographied coronary patients. Clin Chim Acta. 2008 Oct;396(1-2):18–22. doi: 10.1016/j.cca.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Tai ES, Collins D, Robins SJ, et al. The L162V polymorphism at the peroxisome proliferator activated receptor alpha locus modulates the risk of cardiovascular events associated with insulin resistance and diabetes mellitus: the Veterans Affairs HDL Intervention Trial (VA-HIT) Atherosclerosis. 2006 Jul;187(1):153–160. doi: 10.1016/j.atherosclerosis.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 35.Doney AS, Fischer B, Lee SP, Morris AD, Leese G, Palmer CN. Association of common variation in the PPARA gene with incident myocardial infarction in individuals with type 2 diabetes: a Go-DARTS study. Nucl Recept. 2005 Nov 25;3:4. doi: 10.1186/1478-1336-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zalewski G, Ciccarone E, Di Castelnuovo A, et al. P-selectin gene genotypes or haplotypes and cardiovascular complications in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2006 Sep;16(6):418–425. doi: 10.1016/j.numecd.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Burdon KP, Bento JL, Langefeld CD, et al. Association of protein tyrosine phosphatase-N1 polymorphisms with coronary calcified plaque in the Diabetes Heart Study. Diabetes. 2006 Mar;55(3):651–658. doi: 10.2337/diabetes.55.03.06.db05-0058. [DOI] [PubMed] [Google Scholar]
- 38.Buraczynska M, Ksiazek P, Baranowicz-Gaszczyk I, Jozwiak L. Association of the VEGF gene polymorphism with diabetic retinopathy in type 2 diabetes patients. Nephrol Dial Transplant. 2007 Mar;22(3):827–832. doi: 10.1093/ndt/gfl641. [DOI] [PubMed] [Google Scholar]
- 39.Suganthalakshmi B, Anand R, Kim R, et al. Association of VEGF and eNOS gene polymorphisms in type 2 diabetic retinopathy. Mol Vis. 2006;12:336–341. [PubMed] [Google Scholar]
- 40.Primo-Parmo SL, Sorenson RC, Teiber J, La Du BN. The human serum paraoxonase/arylesterase gene (PON1) is one member of a multigene family. Genomics. 1996 May 1;33(3):498–507. doi: 10.1006/geno.1996.0225. [DOI] [PubMed] [Google Scholar]
- 41.Blatter MC, James RW, Messmer S, Barja F, Pometta D. Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem. 1993 Feb 1;211(3):871–879. doi: 10.1111/j.1432-1033.1993.tb17620.x. [DOI] [PubMed] [Google Scholar]
- 42.Reddy ST, Wadleigh DJ, Grijalva V, et al. Human paraoxonase-3 is an HDL-associated enzyme with biological activity similar to paraoxonase-1 protein but is not regulated by oxidized lipids. Arterioscler Thromb Vasc Biol. 2001 Apr;21(4):542–547. doi: 10.1161/01.atv.21.4.542. [DOI] [PubMed] [Google Scholar]
- 43.Ng CJ, Wadleigh DJ, Gangopadhyay A, et al. Paraoxonase-2 is a ubiquitously expressed protein with antioxidant properties and is capable of preventing cell-mediated oxidative modification of low density lipoprotein. J Biol Chem. 2001 Nov 30;276(48):44444–44449. doi: 10.1074/jbc.M105660200. [DOI] [PubMed] [Google Scholar]
- 44.Draganov DI, Teiber JF, Speelman A, Osawa Y, Sunahara R, La Du BN. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J Lipid Res. 2005 Jun;46(6):1239–1247. doi: 10.1194/jlr.M400511-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Shih DM, Lusis AJ. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr Opin Lipidol. 2009 Aug;20(4):288–292. doi: 10.1097/MOL.0b013e32832ca1ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durrington PN, Mackness B, Mackness MI. Paraoxonase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2001 Apr;21(4):473–480. doi: 10.1161/01.atv.21.4.473. [DOI] [PubMed] [Google Scholar]
- 47.Yang F, Wang LH, Wang J, Dong YH, Hu JY, Zhang LH. Quorum quenching enzyme activity is widely conserved in the sera of mammalian species. FEBS Lett. 2005 Jul 4;579(17):3713–3717. doi: 10.1016/j.febslet.2005.05.060. [DOI] [PubMed] [Google Scholar]
- 48.Mackness MI, Arrol S, Durrington PN. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991 Jul 29;286(1-2):152–154. doi: 10.1016/0014-5793(91)80962-3. [DOI] [PubMed] [Google Scholar]
- 49.Watson AD, Berliner JA, Hama SY, et al. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995 Dec;96(6):2882–2891. doi: 10.1172/JCI118359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest. 1998 Apr 15;101(8):1581–1590. doi: 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shih DM, Gu L, Xia YR, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. 1998 Jul 16;394(6690):284–287. doi: 10.1038/28406. [DOI] [PubMed] [Google Scholar]
- 52.Shih DM, Xia YR, Wang XP, et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J Biol Chem. 2000 Jun 9;275(23):17527–17535. doi: 10.1074/jbc.M910376199. [DOI] [PubMed] [Google Scholar]
- 53.Tward A, Xia YR, Wang XP, et al. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation. 2002 Jul 23;106(4):484–490. doi: 10.1161/01.cir.0000023623.87083.4f. [DOI] [PubMed] [Google Scholar]
- 54.Deakin S, Leviev I, Brulhart-Meynet MC, James RW. Paraoxonase-1 promoter haplotypes and serum paraoxonase: a predominant role for polymorphic position -107, implicating the Sp1 transcription factor. Biochem J. 2003 Jun 1;372(Pt 2):643–649. doi: 10.1042/BJ20021670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leviev I, James RW. Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxonase activities and concentrations. Arterioscler Thromb Vasc Biol. 2000 Feb;20(2):516–521. doi: 10.1161/01.atv.20.2.516. [DOI] [PubMed] [Google Scholar]
- 56.Leviev I, Righetti A, James RW. Paraoxonase promoter polymorphism T(-107)C and relative paraoxonase deficiency as determinants of risk of coronary artery disease. J Mol Med. 2001 Aug;79(8):457–463. doi: 10.1007/s001090100240. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Fan Z, Huang J, et al. Extensive association analysis between polymorphisms of PON gene cluster with coronary heart disease in Chinese Han population. Arterioscler Thromb Vasc Biol. 2003 Feb 1;23(2):328–334. doi: 10.1161/01.atv.0000051702.38086.c1. [DOI] [PubMed] [Google Scholar]
- 58.Najafi M, Gohari LH, Firoozrai M. Paraoxonase 1 gene promoter polymorphisms are associated with the extent of stenosis in coronary arteries. Thromb Res. 2009;123(3):503–510. doi: 10.1016/j.thromres.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 59.Yamada Y, Izawa H, Ichihara S, et al. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N Engl J Med. 2002 Dec 12;347(24):1916–1923. doi: 10.1056/NEJMoa021445. [DOI] [PubMed] [Google Scholar]
- 60.Adkins S, Gan KN, Mody M, La Du BN. Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes. Am J Hum Genet. 1993 Mar;52(3):598–608. [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. Jama. 2008 Mar 19;299(11):1265–1276. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaidukov L, Rosenblat M, Aviram M, Tawfik DS. The 192R/Q polymorphs of serum paraoxonase PON1 differ in HDL binding, lipolactonase stimulation, and cholesterol efflux. J Lipid Res. 2006 Nov;47(11):2492–2502. doi: 10.1194/jlr.M600297-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Wheeler JG, Keavney BD, Watkins H, Collins R, Danesh J. Four paraoxonase gene polymorphisms in 11212 cases of coronary heart disease and 12786 controls: meta-analysis of 43 studies. Lancet. 2004 Feb 28;363(9410):689–695. doi: 10.1016/S0140-6736(04)15642-0. [DOI] [PubMed] [Google Scholar]
- 64.Jarvik GP, Hatsukami TS, Carlson C, et al. Paraoxonase activity, but not haplotype utilizing the linkage disequilibrium structure, predicts vascular disease. Arterioscler Thromb Vasc Biol. 2003 Aug 1;23(8):1465–1471. doi: 10.1161/01.ATV.0000081635.96290.D3. [DOI] [PubMed] [Google Scholar]
- 65.Mackness B, Davies GK, Turkie W, et al. Paraoxonase status in coronary heart disease: are activity and concentration more important than genotype? Arterioscler Thromb Vasc Biol. 2001 Sep;21(9):1451–1457. doi: 10.1161/hq0901.094247. [DOI] [PubMed] [Google Scholar]
- 66.Mackness B, Mackness MI, Arrol S, et al. Serum paraoxonase (PON1) 55 and 192 polymorphism and paraoxonase activity and concentration in non-insulin dependent diabetes mellitus. Atherosclerosis. 1998 Aug;139(2):341–349. doi: 10.1016/s0021-9150(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 67.Inoue M, Suehiro T, Nakamura T, Ikeda Y, Kumon Y, Hashimoto K. Serum arylesterase/diazoxonase activity and genetic polymorphisms in patients with type 2 diabetes. Metabolism. 2000 Nov;49(11):1400–1405. doi: 10.1053/meta.2000.17724. [DOI] [PubMed] [Google Scholar]
- 68.Flekac M, Skrha J, Zidkova K, Lacinova Z, Hilgertova J. Paraoxonase 1 gene polymorphisms and enzyme activities in diabetes mellitus. Physiol Res. 2008;57(5):717–726. doi: 10.33549/physiolres.931285. [DOI] [PubMed] [Google Scholar]
- 69.Mastorikou M, Mackness M, Mackness B. Defective metabolism of oxidized phospholipid by HDL from people with type 2 diabetes. Diabetes. 2006 Nov;55(11):3099–3103. doi: 10.2337/db06-0723. [DOI] [PubMed] [Google Scholar]
- 70.Mackness B, Durrington PN, Boulton AJ, Hine D, Mackness MI. Serum paraoxonase activity in patients with type 1 diabetes compared to healthy controls. Eur J Clin Invest. 2002 Apr;32(4):259–264. doi: 10.1046/j.1365-2362.2002.00977.x. [DOI] [PubMed] [Google Scholar]
- 71.van den Berg SW, Jansen EH, Kruijshoop M, et al. Paraoxonase 1 phenotype distribution and activity differs in subjects with newly diagnosed Type 2 diabetes (the CODAM Study) Diabet Med. 2008 Feb;25(2):186–193. doi: 10.1111/j.1464-5491.2007.02328.x. [DOI] [PubMed] [Google Scholar]
- 72.Rozenberg O, Shiner M, Aviram M, Hayek T. Paraoxonase 1 (PON1) attenuates diabetes development in mice through its antioxidative properties. Free Radic Biol Med. 2008 Jun 1;44(11):1951–1959. doi: 10.1016/j.freeradbiomed.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 73.Ikeda Y, Inoue M, Suehiro T, Arii K, Kumon Y, Hashimoto K. Low human paraoxonase predicts cardiovascular events in Japanese patients with type 2 diabetes. Acta Diabetol. 2009 Sep;46(3):239–242. doi: 10.1007/s00592-008-0066-3. [DOI] [PubMed] [Google Scholar]
- 74.Mackness B, Durrington PN, Abuashia B, Boulton AJ, Mackness MI. Low paraoxonase activity in type II diabetes mellitus complicated by retinopathy. Clin Sci (Lond) 2000 Mar;98(3):355–363. [PubMed] [Google Scholar]
- 75.Kosaka T, Yamaguchi M, Motomura T, Mizuno K. Investigation of the relationship between atherosclerosis and paraoxonase or homocysteine thiolactonase activity in patients with type 2 diabetes mellitus using a commercially available assay. Clin Chim Acta. 2005 Sep;359(1-2):156–162. doi: 10.1016/j.cccn.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 76.Ikeda Y, Suehiro T, Inoue M, et al. Serum paraoxonase activity and its relationship to diabetic complications in patients with non-insulin-dependent diabetes mellitus. Metabolism. 1998 May;47(5):598–602. doi: 10.1016/s0026-0495(98)90246-3. [DOI] [PubMed] [Google Scholar]
- 77.James RW, Leviev I, Ruiz J, Passa P, Froguel P, Garin MC. Promoter polymorphism T(-107)C of the paraoxonase PON1 gene is a risk factor for coronary heart disease in type 2 diabetic patients. Diabetes. 2000 Aug;49(8):1390–1393. doi: 10.2337/diabetes.49.8.1390. [DOI] [PubMed] [Google Scholar]
- 78.Tsuzura S, Ikeda Y, Suehiro T, et al. Correlation of plasma oxidized low-density lipoprotein levels to vascular complications and human serum paraoxonase in patients with type 2 diabetes. Metabolism. 2004 Mar;53(3):297–302. doi: 10.1016/j.metabol.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 79.Sampson MJ, Braschi S, Willis G, Astley SB. Paraoxonase-1 (PON-1) genotype and activity and in vivo oxidized plasma low-density lipoprotein in Type II diabetes. Clin Sci (Lond) 2005 Aug;109(2):189–197. doi: 10.1042/CS20050089. [DOI] [PubMed] [Google Scholar]
- 80.Agachan B, Yilmaz H, Ergen HA, Karaali ZE, Isbir T. Paraoxonase (PON1) 55 and 192 polymorphism and its effects to oxidant-antioxidant system in turkish patients with type 2 diabetes mellitus. Physiol Res. 2005;54(3):287–293. [PubMed] [Google Scholar]
- 81.Pfohl M, Koch M, Enderle MD, et al. Paraoxonase 192 Gln/Arg gene polymorphism, coronary artery disease, and myocardial infarction in type 2 diabetes. Diabetes. 1999 Mar;48(3):623–627. doi: 10.2337/diabetes.48.3.623. [DOI] [PubMed] [Google Scholar]
- 82.Murata M, Maruyama T, Suzuki Y, Saruta T, Ikeda Y. Paraoxonase 1 Gln/Arg polymorphism is associated with the risk of microangiopathy in Type 2 diabetes mellitus. Diabet Med. 2004 Aug;21(8):837–844. doi: 10.1111/j.1464-5491.2004.01252.x. [DOI] [PubMed] [Google Scholar]
- 83.Odawara M, Tachi Y, Yamashita K. Paraoxonase polymorphism (Gln192-Arg) is associated with coronary heart disease in Japanese noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1997 Jul;82(7):2257–2260. doi: 10.1210/jcem.82.7.4096. [DOI] [PubMed] [Google Scholar]
- 84.Ruiz J, Blanche H, James RW, et al. Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes. Lancet. 1995 Sep 30;346(8979):869–872. doi: 10.1016/s0140-6736(95)92709-3. [DOI] [PubMed] [Google Scholar]
- 85.Aubo C, Senti M, Marrugat J, et al. Risk of myocardial infarction associated with Gln/Arg 192 polymorphism in the human paraoxonase gene and diabetes mellitus. The REGICOR Investigators. Eur Heart J. 2000 Jan;21(1):33–38. doi: 10.1053/euhj.1999.1660. [DOI] [PubMed] [Google Scholar]
- 86.Li J, Wang X, Huo Y, et al. PON1 polymorphism, diabetes mellitus, obesity, and risk of myocardial infarction: Modifying effect of diabetes mellitus and obesity on the association between PON1 polymorphism and myocardial infarction. Genet Med. 2005 Jan;7(1):58–63. doi: 10.1097/01.gim.0000151152.78092.ca. [DOI] [PubMed] [Google Scholar]
- 87.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990 May;1(5):228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 88.Jakubowski H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J Physiol Pharmacol. 2008 Dec;59 9:155–167. [PubMed] [Google Scholar]
- 89.Obeid R, Herrmann W. Homocysteine and lipids: S-adenosyl methionine as a key intermediate. FEBS Lett. 2009 Apr 17;583(8):1215–1225. doi: 10.1016/j.febslet.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 90.Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 91.Nygard O, Vollset SE, Refsum H, Brattstrom L, Ueland PM. Total homocysteine and cardiovascular disease. J Intern Med. 1999 Nov;246(5):425–454. doi: 10.1046/j.1365-2796.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- 92.Kerkeni M, Tnani M, Chuniaud L, Miled A, Maaroufi K, Trivin F. Comparative study on in vitro effects of homocysteine thiolactone and homocysteine on HUVEC cells: evidence for a stronger proapoptotic and proinflammative homocysteine thiolactone. Mol Cell Biochem. 2006 Oct;291(1-2):119–126. doi: 10.1007/s11010-006-9204-9. [DOI] [PubMed] [Google Scholar]
- 93.Chang PY, Lu SC, Lee CM, et al. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ Res. 2008 Apr 25;102(8):933–941. doi: 10.1161/CIRCRESAHA.108.171082. [DOI] [PubMed] [Google Scholar]
- 94.Jamaluddin MD, Chen I, Yang F, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood. 2007 Nov 15;110(10):3648–3655. doi: 10.1182/blood-2007-06-096701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang H, Yoshizumi M, Lai K, et al. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem. 1997 Oct 3;272(40):25380–25385. doi: 10.1074/jbc.272.40.25380. [DOI] [PubMed] [Google Scholar]
- 96.Tsai JC, Wang H, Perrella MA, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J Clin Invest. 1996 Jan 1;97(1):146–153. doi: 10.1172/JCI118383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsai JC, Perrella MA, Yoshizumi M, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci U S A. 1994 Jul 5;91(14):6369–6373. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969 Jul;56(1):111–128. [PMC free article] [PubMed] [Google Scholar]
- 99.Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2009 Sep;38(3):316–322. doi: 10.1016/j.ejvs.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 100.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008 Nov;83(11):1203–1212. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 101.Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. Jama. 2002 Oct 23-30;288(16):2015–2022. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- 102.Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol. 2009 Sep 15;104(6):745–749. doi: 10.1016/j.amjcard.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 103.Van Guelpen B, Hultdin J, Johansson I, et al. Plasma folate and total homocysteine levels are associated with the risk of myocardial infarction, independently of each other and of renal function. J Intern Med. 2009 Aug;266(2):182–195. doi: 10.1111/j.1365-2796.2009.02077.x. [DOI] [PubMed] [Google Scholar]
- 104.Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. Jama. 2008 May 7;299(17):2027–2036. doi: 10.1001/jama.299.17.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. Jama. 2006 Dec 13;296(22):2720–2726. doi: 10.1001/jama.296.22.2720. [DOI] [PubMed] [Google Scholar]
- 106.Ebbing M, Bleie O, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. Jama. 2008 Aug 20;300(7):795–804. doi: 10.1001/jama.300.7.795. [DOI] [PubMed] [Google Scholar]
- 107.Bonaa KH, Njolstad I, Ueland PM, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med. 2006 Apr 13;354(15):1578–1588. doi: 10.1056/NEJMoa055227. [DOI] [PubMed] [Google Scholar]
- 108.Lonn E, Yusuf S, Arnold MJ, et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006 Apr 13;354(15):1567–1577. doi: 10.1056/NEJMoa060900. [DOI] [PubMed] [Google Scholar]
- 109.Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet. 2007 Jun 2;369(9576):1876–1882. doi: 10.1016/S0140-6736(07)60854-X. [DOI] [PubMed] [Google Scholar]
- 110.Carlsson CM. Lowering homocysteine for stroke prevention. Lancet. 2007 Jun 2;369(9576):1841–1842. doi: 10.1016/S0140-6736(07)60830-7. [DOI] [PubMed] [Google Scholar]
- 111.Kark JD, Selhub J, Bostom A, Adler B, Rosenberg IH. Plasma homocysteine and all-cause mortality in diabetes. Lancet. 1999 Jun 5;353(9168):1936–1937. doi: 10.1016/s0140-6736(98)05381-1. [DOI] [PubMed] [Google Scholar]
- 112.Hoogeveen EK, Kostense PJ, Beks PJ, et al. Hyperhomocysteinemia is associated with an increased risk of cardiovascular disease, especially in non-insulin-dependent diabetes mellitus: a population-based study. Arterioscler Thromb Vasc Biol. 1998 Jan;18(1):133–138. doi: 10.1161/01.atv.18.1.133. [DOI] [PubMed] [Google Scholar]
- 113.Brazionis L, Rowley K, Sr, Itsiopoulos C, Harper CA, O'Dea K. Homocysteine and diabetic retinopathy. Diabetes Care. 2008 Jan;31(1):50–56. doi: 10.2337/dc07-0632. [DOI] [PubMed] [Google Scholar]
- 114.Becker A, Kostense PJ, Bos G, et al. Hyperhomocysteinaemia is associated with coronary events in type 2 diabetes. J Intern Med. 2003 Mar;253(3):293–300. doi: 10.1046/j.1365-2796.2003.01113.x. [DOI] [PubMed] [Google Scholar]
- 115.Buysschaert M, Dramais AS, Wallemacq PE, Hermans MP. Hyperhomocysteinemia in type 2 diabetes: relationship to macroangiopathy, nephropathy, and insulin resistance. Diabetes Care. 2000 Dec;23(12):1816–1822. doi: 10.2337/diacare.23.12.1816. [DOI] [PubMed] [Google Scholar]
- 116.Mazza A, Bossone E, Mazza F, Distante A. Reduced serum homocysteine levels in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2005 Apr;15(2):118–124. doi: 10.1016/j.numecd.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 117.Ndrepepa G, Kastrati A, Braun S, et al. Circulating homocysteine levels in patients with type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2008 Jan;18(1):66–73. doi: 10.1016/j.numecd.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 118.Emoto M, Kanda H, Shoji T, et al. Impact of insulin resistance and nephropathy on homocysteine in type 2 diabetes. Diabetes Care. 2001 Mar;24(3):533–538. doi: 10.2337/diacare.24.3.533. [DOI] [PubMed] [Google Scholar]
- 119.Kluijtmans LA, van den Heuvel LP, Boers GH, et al. Molecular genetic analysis in mild hyperhomocysteinemia: a common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am J Hum Genet. 1996 Jan;58(1):35–41. [PMC free article] [PubMed] [Google Scholar]
- 120.Deloughery TG, Evans A, Sadeghi A, et al. Common mutation in methylenetetrahydrofolate reductase. Correlation with homocysteine metabolism and late-onset vascular disease. Circulation. 1996 Dec 15;94(12):3074–3078. doi: 10.1161/01.cir.94.12.3074. [DOI] [PubMed] [Google Scholar]
- 121.Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate? Bmj. 2005 Nov 5;331(7524):1053. doi: 10.1136/bmj.38611.658947.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Khandanpour N, Willis G, Meyer FJ, et al. Peripheral arterial disease and methylenetetrahydrofolate reductase (MTHFR) C677T mutations: A case-control study and meta-analysis. J Vasc Surg. 2009 Mar;49(3):711–718. doi: 10.1016/j.jvs.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 123.Sun J, Xu Y, Zhu Y, et al. The relationship between MTHFR gene polymorphisms, plasma homocysteine levels and diabetic retinopathy in type 2 diabetes mellitus. Chin Med J (Engl) 2003 Jan;116(1):145–147. [PubMed] [Google Scholar]
- 124.Maeda M, Yamamoto I, Fukuda M, et al. MTHFR gene polymorphism is susceptible to diabetic retinopathy but not to diabetic nephropathy in Japanese type 2 diabetic patients. J Diabetes Complications. 2008 Mar-Apr;22(2):119–125. doi: 10.1016/j.jdiacomp.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 125.Maeda M, Yamamoto I, Fukuda M, et al. MTHFR gene polymorphism as a risk factor for diabetic retinopathy in type 2 diabetic patients without serum creatinine elevation. Diabetes Care. 2003 Feb;26(2):547–548. doi: 10.2337/diacare.26.2.547-a. [DOI] [PubMed] [Google Scholar]
- 126.Moczulski D, Fojcik H, Zukowska-Szczechowska E, Szydlowska I, Grzeszczak W. Effects of the C677T and A1298C polymorphisms of the MTHFR gene on the genetic predisposition for diabetic nephropathy. Nephrol Dial Transplant. 2003 Aug;18(8):1535–1540. doi: 10.1093/ndt/gfg211. [DOI] [PubMed] [Google Scholar]
- 127.Ukinc K, Ersoz HO, Karahan C, et al. Methyltetrahydrofolate reductase C677T gene mutation and hyperhomocysteinemia as a novel risk factor for diabetic nephropathy. Endocrine. 2009 Oct;36(2):255–261. doi: 10.1007/s12020-009-9218-7. [DOI] [PubMed] [Google Scholar]
- 128.Sun J, Xu Y, Zhu Y, Lu H. Genetic polymorphism of methylenetetrahydrofolate reductase as a risk factor for diabetic nephropathy in Chinese type 2 diabetic patients. Diabetes Res Clin Pract. 2004 Jun;64(3):185–190. doi: 10.1016/j.diabres.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 129.Mazza A, Motti C, Nulli A, et al. Lack of association between carotid intima-media thickness and methylenetetrahydrofolate reductase gene polymorphism or serum homocysteine in non-insulin-dependent diabetes mellitus. Metabolism. 2000 Jun;49(6):718–723. doi: 10.1053/meta.2000.6254. [DOI] [PubMed] [Google Scholar]
- 130.Hermans MP, Gala JL, Buysschaert M. The MTHFR CT polymorphism confers a high risk for stroke in both homozygous and heterozygous T allele carriers with Type 2 diabetes. Diabet Med. 2006 May;23(5):529–536. doi: 10.1111/j.1464-5491.2006.01841.x. [DOI] [PubMed] [Google Scholar]
- 131.Szabo GV, Kunstar A, Acsady G. Methylentetrahydrofolate Reductase and Nitric Oxide Synthase Polymorphism in Patients with Atherosclerosis and Diabetes. Pathol Oncol Res. 2009 Mar 29; doi: 10.1007/s12253-009-9163-z. [DOI] [PubMed] [Google Scholar]
- 132.Mazza A, Motti C, Nulli A, et al. Serum homocysteine, MTHFR gene polymorphism, and carotid intimal-medial thickness in NIDDM subjects. J Thromb Thrombolysis. 1999 Oct;8(3):207–212. doi: 10.1023/a:1008962220476. [DOI] [PubMed] [Google Scholar]
- 133.Kaye JM, Stanton KG, McCann VJ, et al. Homocysteine, folate, methylene tetrahydrofolate reductase genotype and vascular morbidity in diabetic subjects. Clin Sci (Lond) 2002 Jun;102(6):631–637. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 134.Pollex RL, Mamakeesick M, Zinman B, Harris SB, Hanley AJ, Hegele RA. Methylenetetrahydrofolate reductase polymorphism 677C>T is associated with peripheral arterial disease in type 2 diabetes. Cardiovasc Diabetol. 2005;4:17. doi: 10.1186/1475-2840-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sun J, Xu Y, Xue J, Zhu Y, Lu H. Methylenetetrahydrofolate reductase polymorphism associated with susceptibility to coronary heart disease in Chinese type 2 diabetic patients. Mol Cell Endocrinol. 2005 Jan 14;229(1-2):95–101. doi: 10.1016/j.mce.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 136.Russo GT, Di Benedetto A, Magazzu D, et al. Mild hyperhomocysteinemia, C677T polymorphism on methylenetetrahydrofolate reductase gene and the risk of macroangiopathy in type 2 diabetes: a prospective study. Acta Diabetol. 2009 Nov 25; doi: 10.1007/s00592-009-0169-5. [DOI] [PubMed] [Google Scholar]
- 137.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007 Jul 15;75(2):247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000 Feb;190(3):244–254. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 139.Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch Pharm Res. 2009 Aug;32(8):1103–1108. doi: 10.1007/s12272-009-1801-1. [DOI] [PubMed] [Google Scholar]
- 140.Kagan VE, Laskin JD. Direct and indirect antioxidant effects of nitric oxide: radically unsettled issues. Antioxid Redox Signal. 2001 Apr;3(2):173–175. doi: 10.1089/152308601300185142. [DOI] [PubMed] [Google Scholar]
- 141.Marsden PA, Heng HH, Scherer SW, et al. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem. 1993 Aug 15;268(23):17478–17488. [PubMed] [Google Scholar]
- 142.Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999 Aug 15;43(3):521–531. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 143.Napoli C, Ignarro LJ. Polymorphisms in endothelial nitric oxide synthase and carotid artery atherosclerosis. J Clin Pathol. 2007 Apr;60(4):341–344. doi: 10.1136/jcp.2006.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tesauro M, Thompson WC, Rogliani P, Qi L, Chaudhary PP, Moss J. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs. glutamate at position 298. Proc Natl Acad Sci U S A. 2000 Mar 14;97(6):2832–2835. doi: 10.1073/pnas.97.6.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Persu A, Stoenoiu MS, Messiaen T, et al. Modifier effect of ENOS in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2002 Feb 1;11(3):229–241. doi: 10.1093/hmg/11.3.229. [DOI] [PubMed] [Google Scholar]
- 146.Fairchild TA, Fulton D, Fontana JT, Gratton JP, McCabe TJ, Sessa WC. Acidic hydrolysis as a mechanism for the cleavage of the Glu(298)-->Asp variant of human endothelial nitric-oxide synthase. J Biol Chem. 2001 Jul 13;276(28):26674–26679. doi: 10.1074/jbc.M103647200. [DOI] [PubMed] [Google Scholar]
- 147.McDonald DM, Alp NJ, Channon KM. Functional comparison of the endothelial nitric oxide synthase Glu298Asp polymorphic variants in human endothelial cells. Pharmacogenetics. 2004 Dec;14(12):831–839. doi: 10.1097/00008571-200412000-00006. [DOI] [PubMed] [Google Scholar]
- 148.Golser R, Gorren AC, Mayer B, Schmidt K. Functional characterization of Glu298Asp mutant human endothelial nitric oxide synthase purified from a yeast expression system. Nitric Oxide. 2003 Feb;8(1):7–14. doi: 10.1016/s1089-8603(02)00131-3. [DOI] [PubMed] [Google Scholar]
- 149.Paradossi U, Ciofini E, Clerico A, Botto N, Biagini A, Colombo MG. Endothelial function and carotid intima-media thickness in young healthy subjects among endothelial nitric oxide synthase Glu298-->Asp and T-786-->C polymorphisms. Stroke. 2004 Jun;35(6):1305–1309. doi: 10.1161/01.STR.0000126482.86708.37. [DOI] [PubMed] [Google Scholar]
- 150.Naber CK, Baumgart D, Altmann C, Siffert W, Erbel R, Heusch G. eNOS 894T allele and coronary blood flow at rest and during adenosine-induced hyperemia. Am J Physiol Heart Circ Physiol. 2001 Nov;281(5):H1908–1912. doi: 10.1152/ajpheart.2001.281.5.H1908. [DOI] [PubMed] [Google Scholar]
- 151.Philip I, Plantefeve G, Vuillaumier-Barrot S, et al. G894T polymorphism in the endothelial nitric oxide synthase gene is associated with an enhanced vascular responsiveness to phenylephrine. Circulation. 1999 Jun 22;99(24):3096–3098. doi: 10.1161/01.cir.99.24.3096. [DOI] [PubMed] [Google Scholar]
- 152.Li R, Lyn D, Lapu-Bula R, et al. Relation of endothelial nitric oxide synthase gene to plasma nitric oxide level, endothelial function, and blood pressure in African Americans. Am J Hypertens. 2004 Jul;17(7):560–567. doi: 10.1016/j.amjhyper.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 153.Rossi GP, Taddei S, Virdis A, et al. The T-786C and Glu298Asp polymorphisms of the endothelial nitric oxide gene affect the forearm blood flow responses of Caucasian hypertensive patients. J Am Coll Cardiol. 2003 Mar 19;41(6):938–945. doi: 10.1016/s0735-1097(02)03011-5. [DOI] [PubMed] [Google Scholar]
- 154.Schmoelzer I, Renner W, Paulweber B, et al. Lack of association of the Glu298Asp polymorphism of endothelial nitric oxide synthase with manifest coronary artery disease, carotid atherosclerosis and forearm vascular reactivity in two Austrian populations. Eur J Clin Invest. 2003 Mar;33(3):191–198. doi: 10.1046/j.1365-2362.2003.01108.x. [DOI] [PubMed] [Google Scholar]
- 155.Lamblin N, Cuilleret FJ, Helbecque N, et al. A common variant of endothelial nitric oxide synthase (Glu298Asp) is associated with collateral development in patients with chronic coronary occlusions. BMC Cardiovasc Disord. 2005;5:27. doi: 10.1186/1471-2261-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gulec S, Karabulut H, Ozdemir AO, et al. Glu298Asp polymorphism of the eNOS gene is associated with coronary collateral development. Atherosclerosis. 2008 Jun;198(2):354–359. doi: 10.1016/j.atherosclerosis.2007.09.037. [DOI] [PubMed] [Google Scholar]
- 157.Miyamoto Y, Saito Y, Nakayama M, et al. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a -786T-->C mutation associated with coronary spastic angina. Hum Mol Genet. 2000 Nov 1;9(18):2629–2637. doi: 10.1093/hmg/9.18.2629. [DOI] [PubMed] [Google Scholar]
- 158.Cattaruzza M, Guzik TJ, Slodowski W, et al. Shear stress insensitivity of endothelial nitric oxide synthase expression as a genetic risk factor for coronary heart disease. Circ Res. 2004 Oct 15;95(8):841–847. doi: 10.1161/01.RES.0000145359.47708.2f. [DOI] [PubMed] [Google Scholar]
- 159.Asif AR, Oellerich M, Armstrong VW, Hecker M, Cattaruzza M. T-786C polymorphism of the NOS-3 gene and the endothelial cell response to fluid shear stress-a proteome analysis. J Proteome Res. 2009 Jun;8(6):3161–3168. doi: 10.1021/pr800998k. [DOI] [PubMed] [Google Scholar]
- 160.Wang XL, Sim AS, Wang MX, Murrell GA, Trudinger B, Wang J. Genotype dependent and cigarette specific effects on endothelial nitric oxide synthase gene expression and enzyme activity. FEBS Lett. 2000 Apr 7;471(1):45–50. doi: 10.1016/s0014-5793(00)01356-9. [DOI] [PubMed] [Google Scholar]
- 161.Tsukada T, Yokoyama K, Arai T, et al. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochem Biophys Res Commun. 1998 Apr 7;245(1):190–193. doi: 10.1006/bbrc.1998.8267. [DOI] [PubMed] [Google Scholar]
- 162.Jeerooburkhan N, Jones LC, Bujac S, et al. Genetic and environmental determinants of plasma nitrogen oxides and risk of ischemic heart disease. Hypertension. 2001 Nov;38(5):1054–1061. doi: 10.1161/hy1101.092967. [DOI] [PubMed] [Google Scholar]
- 163.Yoon Y, Song J, Hong SH, Kim JQ. Plasma nitric oxide concentrations and nitric oxide synthase gene polymorphisms in coronary artery disease. Clin Chem. 2000 Oct;46(10):1626–1630. [PubMed] [Google Scholar]
- 164.Salimi S, Firoozrai M, Nourmohammadi I, et al. Lack of evidence for contribution of intron4a/b polymorphism of endothelial nitric oxide synthase (NOS3) gene to plasma nitric oxide levels. Acta Cardiol. 2008 Apr;63(2):229–234. doi: 10.2143/AC.63.2.2029533. [DOI] [PubMed] [Google Scholar]
- 165.Casas JP, Cavalleri GL, Bautista LE, Smeeth L, Humphries SE, Hingorani AD. Endothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: a HuGE review. Am J Epidemiol. 2006 Nov 15;164(10):921–935. doi: 10.1093/aje/kwj302. [DOI] [PubMed] [Google Scholar]
- 166.Vallance P, Chan N. Endothelial function and nitric oxide: clinical relevance. Heart. 2001 Mar;85(3):342–350. doi: 10.1136/heart.85.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009 Aug;20(6):295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Guang-da X, Qiong-shu W, Wen J. A T-786C polymorphism in 5′-flanking region of the endothelial nitric oxide synthase gene and endothelium-dependent arterial dilation in Type 2 diabetes. Diabet Med. 2005 Dec;22(12):1663–1669. doi: 10.1111/j.1464-5491.2005.01713.x. [DOI] [PubMed] [Google Scholar]
- 169.Rittig K, Holder K, Stock J, et al. Endothelial NO-synthase intron 4 polymorphism is associated with disturbed in vivo nitric oxide production in individuals prone to type 2 diabetes. Horm Metab Res. 2008 Jan;40(1):13–17. doi: 10.1055/s-2007-1004527. [DOI] [PubMed] [Google Scholar]
- 170.Sandrim VC, de Syllos RW, Lisboa HR, Tres GS, Tanus-Santos JE. Influence of eNOS haplotypes on the plasma nitric oxide products concentrations in hypertensive and type 2 diabetes mellitus patients. Nitric Oxide. 2007 May;16(3):348–355. doi: 10.1016/j.niox.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 171.Zintzaras E, Papathanasiou AA, Stefanidis I. Endothelial nitric oxide synthase gene polymorphisms and diabetic nephropathy: a HuGE review and meta-analysis. Genet Med. 2009 Oct;11(10):695–706. doi: 10.1097/GIM.0b013e3181b2046b. [DOI] [PubMed] [Google Scholar]
- 172.Abhary S, Hewitt AW, Burdon KP, Craig JE. A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes. 2009 Sep;58(9):2137–2147. doi: 10.2337/db09-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Odeberg J, Larsson CA, Rastam L, Lindblad U. The Asp298 allele of endothelial nitric oxide synthase is a risk factor for myocardial infarction among patients with type 2 diabetes mellitus. BMC Cardiovasc Disord. 2008;8:36. doi: 10.1186/1471-2261-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang C, Lopez-Ridaura R, Hunter DJ, Rifai N, Hu FB. Common variants of the endothelial nitric oxide synthase gene and the risk of coronary heart disease among U.S. diabetic men. Diabetes. 2006 Jul;55(7):2140–2147. doi: 10.2337/db05-1535. [DOI] [PubMed] [Google Scholar]
- 175.Cai H, Wang X, Colagiuri S, Wilcken DE. A common Glu298-->Asp (894G-->T) mutation at exon 7 of the endothelial nitric oxide synthase gene and vascular complications in type 2 diabetes. Diabetes Care. 1998 Dec;21(12):2195–2196. doi: 10.2337/diacare.21.12.2195. [DOI] [PubMed] [Google Scholar]
- 176.Mollsten A, Lajer M, Jorsal A, Tarnow L. The endothelial nitric oxide synthase gene and risk of diabetic nephropathy and development of cardiovascular disease in type 1 diabetes. Mol Genet Metab. 2009 May;97(1):80–84. doi: 10.1016/j.ymgme.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 177.Bowman BH, Kurosky A. Haptoglobin: the evolutionary product of duplication, unequal crossing over, and point mutation. Adv Hum Genet. 1982;12:189–261. 453–184. doi: 10.1007/978-1-4615-8315-8_3. [DOI] [PubMed] [Google Scholar]
- 178.Garby L, Noyes WD. Studies on hemoglobin metabolism. I. The kinetic properties of the plasma hemoglobin pool in normal man. J Clin Invest. 1959 Sep;38:1479–1483. doi: 10.1172/JCI103925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fagoonee S, Gburek J, Hirsch E, et al. Plasma protein haptoglobin modulates renal iron loading. Am J Pathol. 2005 Apr;166(4):973–983. doi: 10.1016/S0002-9440(10)62319-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lim SK, Kim H, Lim SK, et al. Increased susceptibility in Hp knockout mice during acute hemolysis. Blood. 1998 Sep 15;92(6):1870–1877. [PubMed] [Google Scholar]
- 181.Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001 Jan 11;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 182.Graversen JH, Madsen M, Moestrup SK. CD163: a signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int J Biochem Cell Biol. 2002 Apr;34(4):309–314. doi: 10.1016/s1357-2725(01)00144-3. [DOI] [PubMed] [Google Scholar]
- 183.Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic fenton reagent. J Biol Chem. 1984 Dec 10;259(23):14354–14356. [PubMed] [Google Scholar]
- 184.Melamed-Frank M, Lache O, Enav BI, et al. Structure-function analysis of the antioxidant properties of haptoglobin. Blood. 2001 Dec 15;98(13):3693–3698. doi: 10.1182/blood.v98.13.3693. [DOI] [PubMed] [Google Scholar]
- 185.Miller YI, Altamentova SM, Shaklai N. Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry. 1997 Oct 7;36(40):12189–12198. doi: 10.1021/bi970258a. [DOI] [PubMed] [Google Scholar]
- 186.Buehler PW, Abraham B, Vallelian F, et al. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. 2009 Mar 12;113(11):2578–2586. doi: 10.1182/blood-2008-08-174466. [DOI] [PubMed] [Google Scholar]
- 187.Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin polymorphism in humans. Clin Chem. 1996 Oct;42(10):1589–1600. [PubMed] [Google Scholar]
- 188.Levy AP, Hochberg I, Jablonski K, et al. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J Am Coll Cardiol. 2002 Dec 4;40(11):1984–1990. doi: 10.1016/s0735-1097(02)02534-2. [DOI] [PubMed] [Google Scholar]
- 189.Suleiman M, Aronson D, Asleh R, et al. Haptoglobin polymorphism predicts 30-day mortality and heart failure in patients with diabetes and acute myocardial infarction. Diabetes. 2005 Sep;54(9):2802–2806. doi: 10.2337/diabetes.54.9.2802. [DOI] [PubMed] [Google Scholar]
- 190.Roguin A, Koch W, Kastrati A, Aronson D, Schomig A, Levy AP. Haptoglobin genotype is predictive of major adverse cardiac events in the 1-year period after percutaneous transluminal coronary angioplasty in individuals with diabetes. Diabetes Care. 2003 Sep;26(9):2628–2631. doi: 10.2337/diacare.26.9.2628. [DOI] [PubMed] [Google Scholar]
- 191.Costacou T, Ferrell RE, Orchard TJ. Haptoglobin genotype: a determinant of cardiovascular complication risk in type 1 diabetes. Diabetes. 2008 Jun;57(6):1702–1706. doi: 10.2337/db08-0095. [DOI] [PubMed] [Google Scholar]
- 192.Milman U, Blum S, Shapira C, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008 Feb;28(2):341–347. doi: 10.1161/ATVBAHA.107.153965. [DOI] [PubMed] [Google Scholar]
- 193.Miller-Lotan R, Miller B, Nakhoul F, Aronson D, Asaf R, Levy AP. Retinal capillary basement membrane thickness in diabetic mice genetically modified at the haptoglobin locus. Diabetes Metab Res Rev. 2007 Feb;23(2):152–156. doi: 10.1002/dmrr.654. [DOI] [PubMed] [Google Scholar]
- 194.Miller-Lotan R, Herskowitz Y, Kalet-Litman S, et al. Increased renal hypertrophy in diabetic mice genetically modified at the haptoglobin locus. Diabetes Metab Res Rev. 2005 Jul-Aug;21(4):332–337. doi: 10.1002/dmrr.556. [DOI] [PubMed] [Google Scholar]
- 195.Asleh R, Miller-Lotan R, Aviram M, et al. Haptoglobin genotype is a regulator of reverse cholesterol transport in diabetes in vitro and in vivo. Circ Res. 2006 Dec 8;99(12):1419–1425. doi: 10.1161/01.RES.0000251741.65179.56. [DOI] [PubMed] [Google Scholar]
- 196.Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2007 Jan;27(1):134–140. doi: 10.1161/01.ATV.0000251020.24399.a2. [DOI] [PubMed] [Google Scholar]
- 197.De Bacquer D, De Backer G, Langlois M, Delanghe J, Kesteloot H, Kornitzer M. Haptoglobin polymorphism as a risk factor for coronary heart disease mortality. Atherosclerosis. 2001 Jul;157(1):161–166. doi: 10.1016/s0021-9150(00)00690-0. [DOI] [PubMed] [Google Scholar]
- 198.Holme I, Aastveit AH, Hammar N, Jungner I, Walldius G. Haptoglobin and risk of myocardial infarction, stroke, and congestive heart failure in 342,125 men and women in the Apolipoprotein MOrtality RISk study (AMORIS) Ann Med. 2009 Aug 4;:1–11. doi: 10.1080/07853890903089453. [DOI] [PubMed] [Google Scholar]
- 199.Asleh R, Marsh S, Shilkrut M, et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res. 2003 Jun 13;92(11):1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1. [DOI] [PubMed] [Google Scholar]
- 200.Asleh R, Guetta J, Kalet-Litman S, Miller-Lotan R, Levy AP. Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ Res. 2005 Mar 4;96(4):435–441. doi: 10.1161/01.RES.0000156653.05853.b9. [DOI] [PubMed] [Google Scholar]
- 201.Kalet-Litman S, Moreno PR, Levy AP. The haptoglobin 2-2 genotype is associated with increased redox active hemoglobin derived iron in the atherosclerotic plaque. Atherosclerosis. doi: 10.1016/j.atherosclerosis.2009.06.021. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 202.Levy AP, Purushothaman KR, Levy NS, et al. Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2-2 genotype: implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ Res. 2007 Jul 6;101(1):106–110. doi: 10.1161/CIRCRESAHA.107.149435. [DOI] [PubMed] [Google Scholar]
- 203.Timmermann M, Hogger P. Oxidative stress and 8-iso-prostaglandin F(2alpha) induce ectodomain shedding of CD163 and release of tumor necrosis factor-alpha from human monocytes. Free Radic Biol Med. 2005 Jul 1;39(1):98–107. doi: 10.1016/j.freeradbiomed.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 204.Boyle JJ, Harrington HA, Piper E, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. 2009 Mar;174(3):1097–1108. doi: 10.2353/ajpath.2009.080431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Asleh R, Blum S, Kalet-Litman S, et al. Correction of HDL dysfunction in individuals with diabetes and the haptoglobin 2-2 genotype. Diabetes. 2008 Oct;57(10):2794–2800. doi: 10.2337/db08-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Farbstein D, Levy AP. Pharmacogenomics and the prevention of vascular complications in diabetes mellitus. Therapy. 2009;6(4):531–538. [Google Scholar]
- 207.Levy AP, Asleh R, Blum S, et al. Haptoglobin: Basic and Clinical Aspects. Antioxid Redox Signal. 2009 Dec 5; doi: 10.1089/ars.2009.2793. [DOI] [PubMed] [Google Scholar]