

Abstract
The death domain (DD) superfamily comprising the death domain (DD) subfamily, the death effector domain (DED) subfamily, the caspase recruitment domain (CARD) subfamily and the pyrin domains (PYD) subfamily is one of the largest domain superfamilies. By mediating homotypic interactions within each domain subfamily, these proteins play important roles in the assembly and activation of apoptotic and inflammatory complexes. In this article, we review the molecular complexes that are assembled by these proteins, the structural and biochemical features of these domains and the molecular interactions mediated by them. By analyzing the potential molecular basis for the function of these domains, we hope to provide a comprehensive understanding on the function, structure, interaction and evolution of this important family of domains.
Keywords: Death domain (DD), death effector domain (DED), tandem DED, caspase recruitment domain (CARD), pyrin domain (PYD), crystal structure, NMR structure
INTRODUCTION: APOPTOSIS AND INFLAMMATION
Apoptosis is an orderly cellular suicide program (1, 2) that is critical for the development and homeostasis of a multi-cellular organism. Failure to control apoptosis can lead to serious diseases that threaten the existence of the organism (3, 4). Biologically, apoptosis is highly integrated with inflammation and host defense against intracellular pathogens such as viruses. Because viruses depend on host cells to replicate, the suicide apoptotic program is often elicited in virally infected cells to remove the viruses and to limit viral spread. On the opposing side, viruses often carry genes that induce the host inflammatory processes, which often suppress the cellular apoptotic program.
Apoptosis proceeds through characteristic morphological changes (1, 2) that are dependent on caspase activities. Caspases are cysteine proteases that cleave specifically after aspartic acid residues (5, 6). Because caspases are the executioners of apoptosis, they are the key players in apoptotic cell death. In contrast, inflammatory processes often rely on the activation of protein kinases. One important pro-inflammatory kinase is the IκB kinase (IKK) (7–10), which phosphorylates IκB, leading to its degradation (11). This liberates the NF-κB transcriptional factors, resulting in their nuclear translocation and activation. NF-κBs subsequently induce the transcription of genes for immune and inflammatory responses and for suppression of apoptosis (12–14).
On the molecular level, some protein families are involved in both apoptotic and inflammatory signaling. For example, while caspases are generally apoptotic initiators and effectors, a subclass of caspases are explicitly involved in pro-inflammatory responses. This includes caspase-1, which cleaves pro-IL-1β to IL-1β, leading to NF-κB activation and elicitation of pro-inflammatory responses (15). Proteins in the death domain (DD) superfamily are also shared in apoptotic and inflammatory processes. As shown in the next section, these proteins are often involved in both caspase activation and NF-κB activation. Sometimes, the same molecular complex is regulated to signal either cell death via caspase activation or inflammation via kinase activation.
Because both apoptosis and inflammation are associated with many human diseases, studies in these areas have ultimate biological importance. The past decade has seen an explosion of biochemical and structural studies of proteins involved in apoptotic and inflammatory signaling. These structures and their interactions with partner proteins have revealed the underlying molecular basis for the assembly of important signaling complexes and for the regulation of apoptosis and inflammation. This review will focus on the biology, structure and function of the DD superfamily. For other apoptotic and inflammatory signaling modules, please see related recent reviews (16–18).
THE DD SUPERFAMILY IN THE ASSEMBLY OF APOPTOTIC AND INFLAMMATORY COMPLEXES
The DD superfamily is one of the largest and most studied domain superfamilies (19). It is currently comprised of four subfamilies, the death domain (DD) subfamily, the death effector domain (DED) subfamily, the caspase recruitment domain (CARD) subfamily and the pyrin domain (PYD) subfamily (16). These proteins are evolutionarily conserved in many multicellular organisms such as mammals, Drosophila and C. elegans. Based on a genome analysis, there are 32 DDs, 7 DEDs, 28 CARDs and 19 PYDs in the human genome (16, 17). Perhaps as an evidence of integration with host apoptotic and inflammatory processes, some viruses have acquired DD superfamily sequences. For example, herpesviruses have DED-containing sequences that inhibit cell death and poxviruses have both DED-containing and PYD-containing sequences that interfere with host apoptotic and inflammatory responses to viral infection (20–23). In some cases, the DD superfamily domain is the only motif present in these proteins. In most cases, however, the DD superfamily domain is combined with domains of other subfamily or domains outside the DD superfamily (Figure 1).
Figure 1.

Domain organizations of selective proteins containing the DD superfamily domains.
The unifying feature of the DD superfamily is the six-helical bundle structural fold as first revealed by NMR structures of Fas DD (24), FADD DED (25), RAIDD CARD (26) and NALP1 PYD (27) (Figure 2). Although all members of the DD superfamily have this conserved structural fold, individual subfamilies also exhibit distinct structural and sequence characteristics not shared with other subfamilies.
Figure 2.

Ribbon diagrams for each subfamily of the DD superfamily, Fas DD (a), FADD DED (b), RAIDD CARD (c) and NALP1 PYD (d).
A central paradigm common to both caspase activation and NF-κB activation is the assembly of oligomeric signaling complexes in response to internal or external stimuli. In a simplified view, these molecular complexes activate their effectors via “proximity induced auto-activation” such as dimerization, proteolytic processing and trans-phosphorylation. The DD superfamily plays a critical role in this assembly by participating in both self-association and other protein-protein interactions. Some examples of these important signaling complexes are presented below (Table 1). Interestingly and perhaps surprisingly, interactions within the superfamily are almost all “homotypic” in that only domains within the same subfamily interact with each other.
Table 1.
Selective signaling complexes involving the DD superfamily
| Signaling Complexes | Domains | Protein components |
|---|---|---|
| DISC | DD, DED | Fas, FADD, caspase-8 or -10 |
| complex I | DD | TNFR1, TRADD, RIP, TRAF |
| complex II | DD, DED | TRADD, RIP, FADD, caspase-8 or -10 |
| Apoptosome | CARD | Apaf-1, cytochrome c, ATP/dATP, caspase-9 |
| PIDDosome | DD, CARD | PIDD, RAIDD, caspase-2 |
| TLR/IL-1 R complex | DD | MyD88, TIRAP, IRAKs, TRAF6 |
| Nod pathway | CARD | Nod1 or Nod2, RIP2, caspase-1 |
| RIG pathway | CARD | RIG-I or MDA5, MAVS |
| TCR/BCR signaling | CARD | CARMA1, Bcl-10, MALT1 |
| Inflammasome | PYD | NALP1, ASC, caspase-1, caspase-5 |
The death receptor signaling pathways: the death inducing signaling complex (DISC), complex I and complex II
Death receptors form a subfamily of the TNF receptor superfamily. They mediate the extrinsic cell death pathway. Members of the TNF superfamily of ligands are mostly trimeric and can be either membrane attached or soluble. They activate the TNF receptor superfamily by ligand-induced receptor trimerization (28) and higher order oligomerization (29). The intracellular regions of death receptors contain DDs (30–32), upon which caspase activating and NF-κB activating signaling complexes are assembled. Self association of the DDs in these receptors was shown to be required for the signal transduction (33). Three different types of complexes may be assembled, one by the death receptor Fas and related receptors, and the other two by the death receptor TNFR1 and related receptors.
For Fas, upon ligand activation, its DD recruits the FADD adapter protein via a homotypic interaction with the C-terminal DD of FADD (29, 34). FADD also contains an N-terminal DED that interacts homotypically with the tandem DED in the pro-domain of caspase-8 or -10 (35, 36). These interactions form the ternary DISC containing Fas, FADD and caspase-8 or -10 (32). Recruitment of pro-caspases into the DISC initiates caspase proteolytic auto-processing (37–39). This liberates active caspase-8 or -10 into the cytoplasm to cleave and activate effector caspases such as caspase-3 and caspase-7, leading to a cascade of events in apoptotic cell death (5).
For TNFR1, upon ligand stimulation, its DD recruits the multi-functional TRADD adapter protein via the DD interaction. TRADD in turn recruits RIP via the DD interaction at its C-terminal DD and TRAFs via its N-terminal domain, forming the membrane bound complex I for IKK and NF-κB activation. In a second step, TRADD dissociates from TNFR1 and associates with FADD and caspase-8 to form a cytoplasmic complex II for caspase activation (40). As in the DISC, both DD: DD and DED: DED interactions are important for the assembly of complex II. The regulated assembly of complex I and complex II may underlie the ability of TNF to induce either cell death or cell survival under different cellular contexts.
Caspase activation by both the DISC and complex II is inhibited by FLIPs, a family of cellular and viral tandem DED-containing proteins that interact with FADD (41). Cellular FLIPs (c-FLIPs), comprising the long and short isoforms, c-FLIP-L and c-FLIP-S, are tightly regulated in expression in T cells and may be involved in controlling both T-cell activation and death (41–49). v-FLIPs appear to have evolved to inhibit apoptosis of virally infected host cells and are present in the poxvirus Molluscum contagiosum virus (MCV) as proteins MC159 and MC160 (20–22, 50, 51) and in γ-herpesviruses (20–22, 41, 52).
Deregulation of death receptor signaling is related to many human diseases. Most notably, defective Fas signaling manifested as defective function of the DISC underlies the human genetic disease Autoimmune Lymphoproliferative Syndrome (ALPS) (53, 54). When lymphocytes from patients with ALPS are cultured in vitro, they are resistant to apoptosis as compared to cells from healthy controls. Most patients with ALPS have mutations in the Fas gene and more than 70 mutations have been mapped to its intracellular DD (55–58).
The intrinsic apoptosis pathway: the apoptosome
The intrinsic pathway of apoptotic cell death is induced in a mitochondria-dependent manner in response to intracellular insults. A CARD-containing protein Apaf-1 forms the central platform of a molecular complex known as the apoptosome for caspase activation in this pathway (59, 60). Apaf-1 is comprised of an N-terminal CARD, a central nucleotide binding oligomerization domain (NOD) and a C-terminal (Trp-Asp) WD repeat domain. Upon mitochondrial leakage, cytochrome c, which normally resides at the intermembrane space of the mitochondria, is released to the cytosol. The interaction of cytochrome c with the WD repeat domain of Apaf-1 presumably opens up the Apaf-1 structure, leading to an ATP or dATP-dependent oligomerization of Apaf-1 to form the apoptosome. The apoptosome then recruits caspase-9 via the CARD domain interaction between Apaf-1 and caspase-9.
In Drosophila, the related apoptosome is formed by the Apaf-1 ortholog Dark and the caspase-9 ortholog Dronc (61). In C. elegans, the Apaf-1 ortholog is CED-4, which does not contain an analogous C-terminal WD repeat domain for cytochrome c interaction. Instead, CED-4 is constitutively active but is kept in check by an interaction with CED-9, a Bcl-2 ortholog, which prevents CED-4 oligomerization. Egl-1 is induced to antagonize CED-9 upon apoptosis. CED-4 then oligomerizes to form the worm apoptosome and interacts and activates CED-3, the caspase ortholog, via a CARD interaction (61).
PIDD-mediated responses to DNA damage: caspase-2 activation by the PIDDosome and RIP-mediated NF-κB activation
In response to genotoxic stress, the DD-containing and p53-inducible protein PIDD mediates both caspase-2 and NF-κB activation. Caspase-2 is an evolutionarily conserved initiator caspase with a CARD pro-domain (62). The caspase-2 activation pathway involves the formation of a ternary complex dubbed as the PIDDosome (63, 64), which comprises proteins PIDD (65), RAIDD (66) and caspase-2 and is assembled via the DD interaction between PIDD and RAIDD and the CARD interaction between RAIDD and caspase-2. Accumulating data have shown that caspase-2 acts upstream of the mitochondria to initiate apoptosis (67). Upon genotoxic stress, PIDD also recruits the DD-containing kinase RIP, which in turn interacts with the IKK complex for NF-κB activation (68). Therefore it appears that PIDD acts as a molecular switch to control life and death after DNA damage.
The NF-κB activation pathway of the IL-1 receptor and Toll-like receptors
In the mammalian IL-1 receptor and Toll-like receptor (TLR) pathway, the intracellular TIR domain of these receptors recruits TIR-containing adapters (69). While the IL-1 receptor and most Toll-like receptors recruit MyD88, TLR2 and TLR4 also recruit another TIR-containing protein TIRAP (69). MyD88 contains both a DD and a TIR domain and it in turn recruits IRAKs (which include IRAK1, IRAK2, IRAKM and IRAK4) via a DD interaction. IRAKs contain a DD and a Ser/Thr kinase domain. IRAKs subsequently recruit TRAF6 for NF-κB activation (70). In Drosophila, a heterotrimeric complex of MyD88, Tube and Pelle mediates the orthologous Toll pathway (71), which is important for both immunity against fungal infection (72) and for dorsoventral patterning (73). While Pelle is the functional ortholog of IRAK, Tube is an adapter protein. MyD88, Tube and Pelle all contain a DD and assemble via DD interactions.
Antigen receptor-induced NF-κB activation pathway
NF-κB activation by T-cell and B-cell receptors (TCR/BCR) is initiated by receptor associated tyrosine kinases of the Src and Syk family and a protein kinase C family member, PKC-θ or PKC-β, respectively (74). Upon TCR or BCR activation, the PKC isoforms translocate to membrane bound large molecular complexes in lipid rafts. Although the exact connection to PKC-θ and PKC-β still remains to be resolved, a three-molecule complex containing CARMA1, Bcl-10 and MALT1 appears to act downstream to activate IKK. CARMA1 contains an N-terminal CARD, followed by a coiled-coil domain, a PDZ domain, an SH3 domain, and a C-terminal guanylate kinase-like (GUK) domain (75, 76). The PDZ, SH3 and GUK domains in CARMA classify it into membrane-associated guanylate kinase (MAGUK) family, which plays important roles in regulating the interface between membrane components and cytoskeletal proteins. CARMA1 interacts with Bcl-10, a protein with an N-terminal CARD and a C-terminal Ser/Thr rich region, via the CARD interaction (77). MALT1 is a DD-containing paracaspase, which contains an N-terminal DD, two central immunoglobulin-like (Ig) domains and a C-terminal caspase-like domain for which catalytic activity has not yet been shown (78). The C-terminal Ser/Thr rich region of Bcl-10 and the Ig domains of MALT1 are responsible for the recruitment of MALT1 by Bcl-10.
How the CARMA1, Bcl-10 and MALT1 complex activates IKK is still unclear. One possible model is that this complex further recruits the heterodimeric complex of caspase-8 and c-FLIP-L, which assists caspase-8 activation and in turn is cleaved by activated caspase-8 to an intermediate p43 form (79). The p43 form of c-FLIP-L has been shown to efficiently activate NF-κB via the TRAF pathway and both c-FLIP and caspase-8 are required for the survival and proliferation of T cells following TCR stimulation (79). It has also been shown that MALT1 contains potential TRAF6-binding sites and activates IKK via the TRA6-TAK1 pathway (80).
Intracellular bacterial sensors: the Nod pathway
CARD-containing proteins known as Nods participate in the sensing of intracellular pathogens and the elicitation of innate immunity against these pathogens. For example, Nod1 contains an N-terminal CARD, a central NOD and a C-terminal leucine-rich repeat (LRR) domain (81). This domain architecture is similar to the Apaf-1 protein involved in apoptosome formation and caspase-9 activation. Upon interaction with ligands on intracellular bacteria via its LRR, Nod1 presumably oligomerizes through its NOD. The N-terminal CARD of Nod1 interacts with RIP2, which contains an N-terminal Ser/Thr kinase domain and a C-terminal CARD (82). RIP2 in turn recruits the IKK complex for NF-κB activation (83). In addition to the IKK complex, RIP2 can also interact with caspase-1 via its CARD domain, resulting in the cleavage and activation of pro-IL-1β (84).
In a presumably similar pathway mediated by Nod2, intracellular bacterial LPS are recognized and NF-κB activation is induced, leading to innate immunity and bacterial clearance (85, 86). Nod2 is crucial for immunity against bacteria in the gastrointestinal tract, as a loss of function mutation in Nod2 underlies a susceptibility to Crohn’s disease, a chronic inflammatory disorder of the gut (86).
Several negative regulators of caspase-1 activation have been identified. In particular, both ICEBERG and COP are CARD-containing proteins. They interact with either caspase-1 or RIP2 CARDs and inhibit caspase-1 activation by RIP2 (87, 88). This inhibition may provide a negative feedback loop to terminate inflammatory responses.
Intracellular viral infection sensing and signaling for antiviral immunity: the RIG-like helicase pathway
Like Nods, RIG-like helicases form another family of intracellular pathogen pattern sensors whose primary target is virally derived dsRNA (89). Proteins in this family, such as RIG-I and MDA5, contain tandem CARDs at the N-terminal region and a helicase domain with the DExD/H motif at the C-terminal region. Presumably, binding of dsRNA to the helicase domain of RIG-I or MDA5 would cause a conformational change that exposes the tandem CARDs for recruiting downstream adapter proteins. One such adapter was recently identified independently by several groups and was therefore named independently MAVS, Cardif, IPS-1 and VISA (90–93). MAVS is critical for mediating NF-κB activation and the activation of IRF3/7 to produce antiviral type I IFN (94, 95). It contains an N-terminal CARD that interacts with RIG-I or MDA5 through a homotypic CARD interaction. The C-terminal region of MAVS possesses a mitochondrial targeting sequence that anchors MAVS to the outer membrane of mitochondria, which is critical for its function (90). Although RIG-I and MDA5 share the same domain structure, they appear to detect different viruses (96, 97). The third member of the RIG-like helicase family, LGP2, is devoid of the CARD and prevents activation of antiviral responses, most likely through competitive binding to dsRNA (95, 98).
The caspase-1 activation pathway: the inflammasome
The PYD-containing proteins NALP1 and ASC participate in the formation of the inflammasome for caspase-1 activation (99). NALP1 contains an N-terminal PYD, a central NOD presumably involved in oligomerization, an LRR region, and a C-terminal CARD (100, 101). ASC is an adapter protein containing an N-terminal PYD and a C-terminal CARD (102). NALP1 interacts with ASC via the PYD and with caspase-5 via the CARD. These proteins, together with caspase-1, form the ~700 kD inflammasome for caspase-1 activation (99). Activated caspase-1 cleaves and activates pro-IL-1β, leading to NF-κB activation and elicitation of innate immunity.
STRUCTURAL AND BIOCHEMICAL STUDIES OF THE DD SUBFAMILY
DD structures
Currently, structures of six isolated DDs are available, which include the NMR structures of Fas DD, FADD DD, TNFR1 DD and the p75 NGFR DD, and the crystal structures of IRAK4 DD and RAIDD DD (24, 103–108). Because these domains are involved in protein-protein interactions and have a tendency to aggregate, many of these structures were determined under non-physiological conditions such as extreme pH and/or with “de-aggregating” mutations. While all these DDs exhibit the six-helical bundle fold, variations exist in the length and direction of the helices (Figure 3a). Because of the low sequence homology among DDs, the surface features of these DDs are also entirely different, which may be responsible for their specificity in protein-protein interactions.
Figure 3.

Structural features of DDs and DD: DD interactions. (a) A stereo diagram of the superimposed known DD structures. Green: RAIDD DD; Cyan: Fas DD; Red: FADD DD; Yellow: TNFR1 DD; Gray: p75 DD; Magenta: IRAK4 DD; Blue: Pelle DD; Orange: Tube DD. (b) Pelle DD: Tube DD complex. Pelle is shown in purple with segments for Tube interaction in magenta. Tube is shown in green with segments for Pelle interaction in red. (c) Hypothetical models for a three-fold symmetric Fas DD: FADD DD complex. In one model, FADD DD interacts with one Fas DD only. In the other model, FADD DD interacts with two adjacent Fas DD molecules.
DD: DD interaction
Despite the immense biological importance of DDs, only one structure of a DD: DD complex is available, which is the crystal structure of the monomeric Pelle DD: Tube DD complex (109) (Figure 3b). Regardless of the details of the interaction, the biggest surprise is perhaps the asymmetry of the interaction, considering what might have been expected for homotypic interactions. The first interface involves the H4 helix and the following loop of Pelle and the H1–H2 corner, H6 and the preceding loop in Tube. Most strikingly, the C-terminal tail of Tube wraps around a groove formed by the H4–H5 and H2–H3 hairpins of Pelle to form the second interface and contributes significantly to the interaction (109). While many charged residues at the first interface (such as E50 of Tube and R35 of Pelle) are involved in the interaction, three large hydrophobic residues (I169, L171, and L173) on the C-terminal of Tube dominate the second interface.
Mutational studies based on the crystal structure of Tube showed that the MyD88 DD: Tube DD interaction uses a different surface of the Tube DD (71, 110). Therefore, Tube acts as the bridge between MyD88, which interacts directly with Toll, and Pelle, which signals downstream for the nuclear translocation and activation of the NF-κB ortholog Dorsal, the key event in dorsoventral patterning.
The structural basis of DD: DD interactions involved in death signaling, such as those in the Fas DD: FADD DD complex, the TRADD DD: FADD DD complex and the PIDD DD: RAIDD DD complex, remains largely unresolved. Although no solid experimental evidence is available, the Fas DD: FADD DD complex is likely to be trimeric, most likely with Fas possessing the self-oligomerization surface (Figure 3c). Therefore, the complex might comprise at least two interfaces, a self-oligomerization surface and a Fas DD: FADD DD interaction surface. This conjecture of multiple interaction surfaces in these complexes is supported by mutational data, which showed wide spreads of residues important for binding and/or function on the surfaces of Fas (111), FADD (112), TRADD (113) and TNF-R1 (114). Alternatively, it has been proposed that the Fas DD: FADD DD complex may be constructed from the two types of interfaces observed in the Pelle DD: Tube DD complex and in the Apaf-1 CARD: caspase-9 CARD complex (below) (115). This arrangement, however, does not generate three-fold symmetry.
Controversial data exist on the proposed interaction between Fas DD and FADD DD. While in vitro reconstitution showed that Fas DD interacts strongly with FADD DD (116), an independent experiment showed that GST-Fas DD failed to pulldown FADD DD (117). In addition, while the same in vitro reconstitution showed that Fas DD interacts only weakly with full-length FADD in the absence of the third component of the DISC (116), the pulldown of full-length FADD by GST-Fas DD appeared to be efficient (117).
STRUCTURAL AND BIOCHEMICAL STUDIES OF THE DED SUBFAMILY
DED structures
Currently, four DED structures are available, which include the NMR structures of FADD DED (25) and PEA-15 DED (118) and the DED1 and DED2 structures from the crystal structure of the tandem DED of MC159 (116) (Figure 4a). The NMR structure of FADD DED is also known in the context of its full-length structure comprising both a DED and a DD (119). While FADD is a component of the DISC, PEA-15 appears to participate in MAP kinase activation through a non-homotypic interaction with the kinase ERK (118). MC159 is a v-FLIP from a poxvirus that inhibits caspase activation at the DISC (116). As predicted and consistent with their assignment to the DED subfamily, the structures of FADD DED, PEA-15 DED and MC159 DED2 show the conserved six-helical bundle fold of the DD superfamily and are more similar to each other than to other members of the DD superfamily (Figure 4b).
Figure 4.

Structural features of DEDs and tandem DEDs. (a) A ribbon diagram of the tandem DED structure of MC159. (b) A stereo diagram of superimposed known DED structures. Green: DED1 of MC159; Blue: DED2 of MC159; Pink: FADD DED and Yellow: PEA-15 DED. (c) Charge triad motif. (d) Surface features of MC159, showing the hydrophobic patch and charge triad surfaces that are conserved in almost all DEDs. (e) MC159 DED1: DED2 interaction. Panels c, d and e are modified from Yang et al. (116).
In contrast, the structure of MC159 DED1 showed that it is structurally more divergent from the other known DED structures (116, 120). In particular, helix H3 is missing and replaced by a short loop connecting helices H2 and H4. Two additional helices are present, helix H0 at the N-terminus and helix H7 that brings the chain to the beginning of DED2. Only about half of the residues in DED1 were aligned within 3Å in Cα distance to DED2, FADD DED and PEA-15 DED.
Surface features
Two prominent conserved surface features are present on DEDs, which distinguish DEDs from other members of the DD superfamily. The first feature is a conserved hydrogen-bonded charge triad revealed by the high resolution structure of MC159 (116). The charge triad is formed by the E/D-RxDL motif and involves the Arg and Asp residues in the RxDL motif in helix H6 and the preceding loop, and an acidic residue in helix H2. Extensive hydrogen bonding interactions are observed among the charged side chains with the Arg residue situated in between the two acidic residues (Figure 4c, 4d). These hydrogen bonds likely help to maintain a precise organization of the side chains, which may be functionally important. It is also possible that they play a local structural role in maintaining the conformation of this region of the DEDs. The charge triad is highly conserved in most single and tandem DEDs. With the exception of the p75 DD, this motif is not present in other members of the DD superfamily, suggesting that it is a characteristic feature of DEDs alone. As shown below, this surface may be used for FADD DED self-association and for the interaction of FADD DED with MC159 and MC160.
The second surface feature is the conserved hydrophobic patch formed mostly by residues on H2 (Figure 4d). This was first observed in the NMR structure of FADD DED (25) and later shown to be conserved in most tandem DEDs as well (116). In caspase-8, caspase-10, c-FLIP and herpesvirus v-FLIPs, the two central residues at this patch are strictly identical with those in FADD DED. In MC159 and MC160, these two residues are still hydrophobic, but are replaced by conserved substitutions. As shown below, this surface is used extensively in DED: DED interactions such as the MC159 DED1: DED2 interaction and the interaction of FADD DED with caspase-8, caspase-10, c-FLIP and herpesvirus v-FLIPs.
DED: DED interaction as seen in tandem DED
The structure of MC159 revealed the first glimpse of a DED: DED interaction (116). Instead of beads on a string, DED1 and DED2 interact with each other intimately to form a rigid, dumbbell shaped structure. The two DEDs are related approximately by a translation across the contact interface so that one side of DED1 is contacting the equivalently opposite side of DED2. The translational relationship between DED1 and DED2 is made possible by helix H7 of DED1. The DED1: DED2 interface is extensive, burying approximately 1380Å2 surface area.
The interaction at the DED1: DED2 interface is mostly hydrophobic, mediated by helices H2 and H5 of DED1 and helices H1 and H4 of DED2 (Figure 4e). There are a total of 195 interfacial atomic contacts, among which 117 are between non-polar atoms. The H1 and H4 surface of DED2 is equivalent to the non-conserved hydrophobic patch identified in FADD DED structure on the opposite side of the conserved hydrophobic patch and does not appear to be important in FADD signaling (25). The interfacial residues, especially those that are completely buried at the DED1: DED2 interface and contribute large surface areas, such as F30, L31, F92, L93 and R97, are mostly conserved in tandem DEDs. This suggests that all known tandem DEDs form a similar rigid compact structure as MC159. This interaction between DED1 and DED2 differs from both the known Pelle DD: Tube DD interaction (above) and the Apaf-1 CARD: caspase-9 CARD interaction (below).
FADD DED: self-association and interaction with tandem DEDs
There are two main functions of FADD DED, self-association and interaction with tandem DED proteins such as caspase-8 and MC159. FADD DED-mediated self-association is recently shown to be important for the signaling competency of the DISC (117, 119, 121). Two separate regions of FADD DED however appear to be important for this self-association. The first region is the charge triad region of FADD DED (121) and the other region is the hydrophobic patch of FADD DED (117, 119). For the latter, the self-association interface will have to be close to or at the caspase-8 interaction surface (117, 119). It is possible that one of the mapped FADD self-association interfaces only indirectly affects FADD self-association due to structural alteration or another aspect of the FADD function.
The structure of MC159 provided a template for all tandem DED proteins, which include caspase-8, caspase-10, c-FLIP, herpesvirus v-FLIPs and the poxvirus v-FLIP MC159 and MC160 (116). Structure-based sequence analysis and mutagenesis divided tandem DEDs into two subclasses with regard to their mode of interaction with FADD DED. First, sequence analysis showed that the two core residues at the hydrophobic patch are strictly identical in FADD DED and tandem DEDs including caspase-8, caspase-10, c-FLIP and herpesvirus v-FLIPs. Mutational studies of FADD DED showed that this hydrophobic patch is involved in caspase-8 recruitment (25). Conversely, mutations on the conserved hydrophobic patch of DED2 of caspase-8 also abolished the interaction of caspase-8 with FADD. This suggests a mutual hydrophobic FADD DED: caspase-8 tandem DED interaction (116). Similarly, c-FLIP and herpesvirus v-FLIPs compete with caspase-8 recruitment to the DISC and interact with FADD DED similarly using the hydrophobic patch.
Second, consistent with the lack of strict conservation on the hydrophobic patch, mutations on the exposed hydrophobic patch of MC159 on DED2 did not affect its interaction with FADD DED. Instead, mutational studies on a large number of exposed residues of MC159 showed that MC159 interacts with FADD DED via a region encompassing its conserved charge triad on DED1 (116). Conversely, the charge triad region of FADD DED appears to be important for both its interaction with MC159 (116) and for its self-association (121). MC159 thus interferes with FADD self-association. Since FADD self-association is important for DISC clustering and signaling, MC159 inhibits DISC-mediated caspase activation.
These studies showed that tandem DEDs are divided into two classes with regards to interaction with FADD DED, those that are MC159-like, including MC159 and MC160, and those that are caspase-8-like, including caspase-8, caspase-10, c-FLIP and herpesvirus v-FLIPs. The former do not compete with caspase-8 recruitment to FADD, and likely inhibit caspase activation via disruption of FADD self-association. In contrast, the latter directly compete with caspase-8 recruitment and therefore caspase activation at the DISC.
STRUCTURAL AND BIOCHEMICAL STUDIES OF THE CARD SUBFAMILY
CARD structures
CARD containing proteins may be classified into two classes, those at the prodomains of caspases and those that act as adapters in the assembly of apoptotic and inflammatory signaling complexes. Currently, NMR structures are available for RAIDD CARD (26) and ICEBERG CARD (87). A crystal structure is available for the isolated CARD of Apaf-1 (122). In addition, the CARD structure of Apaf-1 is available in the context of the entire N-terminal fragment of Apaf-1 containing the CARD and the NOD (123) and in a complex with the caspase-9 CARD (124). The structure of CED-4 CARD is available as a complex of CED-4 and CED-9 (125).
Although the topology of these CARDs is identical with the conserved six-helical bundle fold of the DD superfamily, the structures of CARDs is unique in that helix H1 tends to be either bent or broken into two closely separated H1a and H1b helices. In addition, the orientations and lengths of several helices may be somewhat different among the different CARDs (Figure 5a). Superposition of the Apaf-1 CARD structures in isolation, in complex with caspase-9 CARD and in the context of its NOD domain show that there is limited structural plasticity in this domain (Figure 5b). One important feature is that the surfaces of these CARDs are invariantly polarized with both basic and acidic surfaces, which may be used for protein-protein interactions (Figure 5c).
Figure 5.

Structural features of CARDs and CARD: CARD interactions. (a) A stereo diagram of superimposed known CARD structures. Red: Apaf-1 CARD, Blue: ICEBERG CARD, Green: RAIDD CARD; Cyan: caspase-9 CARD; Violet: CED4 CARD. (b) Superimposed Apaf-1 CARD structures when determined alone (green), in complex with caspase-9 (violet) and in WD-deleted Apaf-1 (red). (c) Surface features of CARDs, showing the charged surfaces. Two orientations are shown, one similar to the caspase-9 interaction surface of Apaf-1 (left) and the other similar to the Apaf-1 interaction surface of caspase-9 (right). (d) Apaf-1 CARD: caspase-9 CARD structure. Apaf-1 CARD is shown in violet and caspase-9 CARD is in cyan.
CARD: CARD interaction
The only structure of a CARD: CARD complex is provided by the crystal structure of the complex between Apaf-1 CARD and caspase-9 CARD (124) (Figure 5d). The interaction is mediated by the mutual recognition of the slightly concave surface of caspase-9 CARD formed by the positively charged H1a, H1b and H4 helices and the convex surface of Apaf-1 CARD formed by the negatively charged H2 and H3 helices. Three positively charged residues in caspase-9 CARD, R13, R52, and R56 and two negatively charged residues in Apaf-1 CARD, D27 and E40, are crucial for this interaction (124). While this study confirmed the ionic nature of the Apaf-1 CARD: caspase-9 CARD interaction (Figure 5c), it remains to be determined whether this holds true for other CARD: CARD interactions, such as the ICEBERG: caspase-1 and the RAIDD: caspase-2 interactions.
Apparent lack of self-association of CARDs
Unlike DDs and DEDs, it is unclear at the moment whether CARDs also self-associate. In the EM structure of the apoptosome, the CARD of Apaf-1 appears to form a RING near the center of the assembly (126). In the presence of full-length caspase-9, even the isolated Apaf-1 CARD appears to undergo some level of oligomerization (127). However, the isolated Apaf-1 CARD and the complex of Apaf-1 CARD: caspase-9 CARD are both monomeric (122, 124). It is possible that the CARDs, at least those that are present with an oligomerization NOD and those that are at the prodomains of caspases, do not possess the ability for stable self-association, but rather are mostly involved in interaction with other CARDs.
STRUCTURAL AND BIOCHEMICAL STUDIES OF THE PYD subfamily
The recent NMR structures of the NALP1 PYD (27) and the ASC PYD (128) provided the definitive evidence that PYDs should be a subfamily of the DD superfamily (129) (Figure 6a). The structural information is consistent with previous suggestions based on sequence alignments and secondary structure predictions (130–132). Despite having the classical six-helical bundle fold of the DD superfamily, PYDs have an altered H3. In the case of the PYD from NALP1, H3 is completely replaced by a flexible loop. For the PYD from ASC, only a short helix of 4 residues remains, which is coupled with a long flexible loop preceding the helix. Whether this region rearranges to form a more extended H3 upon binding to a partner is unknown, but is a possibility. The flexible, elongated loop preceding or at H3 seems to be a common feature in many PYD sequences. Since H3 seems to play a critical role in protein-protein interactions in the DD superfamily (133, 134), the PYD: PYD interactions could be significantly different from classical modes of interactions in the DD superfamily.
Figure 6.

Structural features of PYDs. (a) A stereo diagram of superimposed PYDs. Cyan: NALP1 PYD; Magenta: ASC PYD. (b) Electrostatic surface features of PYDs. Left: the two opposite sides of NALP1; Right: the two opposite sides of ASC.
Although presently the PYD subfamily is the least well characterized of the four subfamilies, it is clear that like other subfamilies, proteins with PYDs are involved in inflammation, apoptosis and NF-κB signaling such as the formation of the inflammasome (99, 135). The mode of PYD: PYD interactions are presently completely unknown, but molecular modeling studies seem to suggest the involvement of charged residues and electrostatic interactions (Figure 6b). Acidic residues from H1 and H4 may interact with basic residues from H2 and H3 or the equivalent loops (128). In addition, site-directed mutational studies have suggested that H2, H3 and H4, but not H1, are involved in ASC self-association (136).
Hereditary mutations in some genes of the PYD subfamily have been implicated in a number of hyperinflammatory fever syndromes such as the Muckle-Wells syndrome and the familial Mediterranean fever (FMF), supporting the role of these proteins in regulating inflammatory responses (137, 138). One FMF associated mutation is located at the equivalent H3 region of the PYD (139) and another known mutation is located at H6 (140). It is possible that these mutations either disrupt a PYD: PYD interaction required for negative regulation of inflammation or are gain of function of a PYD: PYD interaction required for pro-inflammatory responses. The molecular basis of PYD: PYD interactions and disease mutations will have to wait for more structural studies.
STRUCTURAL COMPARISONS AMONG THE HOMOTYPIC INTERACTIONS
Altogether, three homotypic complex structures in the DD superfamily are known, the Pelle DD: Tube DD complex (109), the Apaf-1 CARD: caspase-9 CARD complex (124) and the DED1: DED2 interaction in the tandem DED of MC159 (116, 120). These structures raise the question of whether there are conserved modes of interaction common to all members of the DD superfamily. This question is especially interesting because it has been proposed that perhaps a conserved binding epitope inherent to the common fold of the DD superfamily exists (118).
Structural comparison of these three pairs of interactions by superimposing DED1 with any of the four proteins in the Pelle DD: Tube DD complex and in the Apaf-1 CARD: caspase-9 CARD complex shows that they all represent different modes of interaction. When DED1 is superimposed with either Pelle DD or Tube DD, the partner of the complex falls onto a very different place relative to DED2 (Figure 7a and 7b). The same is observed when DED1 is superimposed with caspase-9 CARD (Figure 7c). This means that different surfaces are used in each of these complexes. The closest structural similarity is seen when DED1 is superimposed with Apaf-1 CARD (Figure 7d) or when DED2 is superimposed with caspase-9 CARD. For both DED2 and caspase-9 CARD, it is the H1 and H4 helices that mediate the interaction with the partner. For DED1, the corresponding surface is formed by H2 and H5 helices. For Apaf-1 CARD, it is the H2 and H3 helices, which is similar but not identical with the surface used on DED1. These observations seem to suggest that interactions in the DD superfamily may occur in different modes. However, there may be certain “hot” spots for interaction on the surface of the DD fold. More structures are required to further elucidate this.
Figure 7.
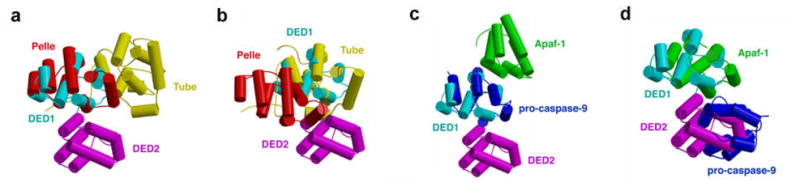
Comparison of the three pairs of known interactions in the DD superfamily, the Pelle DD: Tube DD complex, the Apaf-1 CARD: caspase-9 CARD complex and the DED1: DED2 in the tandem DED MC159. DED1 of MC159 is superimposed respectively with Pelle DD in the complex (a), Tube DD in the complex (b), caspase-9 CARD in the complex (c) and Apaf-1 CARD in the complex (d). This figure was modified from Yang et al. (116).
NON-HOMOTYPIC INTERACTIONS
Given the high structural similarity among the subfamilies of the DD superfamily, it is interesting and perhaps surprising to note that almost all known interactions in the DD superfamily are homotypic interactions between members of the same subfamily. Currently, we only know two cases of potential heterotypic interactions and it is not yet known whether these interactions are the exception or the rule.
One case of heterotypic interaction is present in the PEA-15 DED protein. PEA-15 is unusual because it has been suggested to interact homotypically with DED-containing proteins such as FADD and caspase-8 and heterotypically with non-DED proteins such as the MAP kinase ERK (118). Another case of heterotypic interaction is present in the CARD-containing protein ARC, which appears to interact heterotypically with both the DDs of Fas and FADD and the C-terminus of the Bcl-2 family protein Bax (141). These interactions underlie the ability of ARC to inhibit both the death receptor and the mitochondria cell death pathway.
EVOLUTIONARY REMARKS
The DD superfamily can be found in many multicellular organisms and is evolutionarily conserved. However, fewer members are present in lower organisms such as C. elegans and Drosophila than in mammals. This expansion of DD superfamily members is consistent with the expansion of apoptotic and inflammatory signaling apparatus in mammals and may reflect the much higher complexity of the mammalian signaling system in general.
Given the similar fold of the DD superfamily, one would be curious about the potential evolutionary relationship among these subfamilies and whether the different subfamilies arose from a common ancestor. To investigate this question, we constructed phylogenetic trees among members of the DD superfamily using either sequence or structural similarity (Figure 8). Strikingly, members of each subfamily are clustered together without a single anomaly and a similar evolutionary relationship was predicted based both on sequence and on structure. This suggests that although these proteins show low sequence homology even within each subfamily and share a similar fold across the different subfamilies, sequence and structural signatures are clearly both present to distinguish the subfamilies.
Figure 8.

Sequence and structural comparisons. (a) Phylogenetic tree of the DD superfamily constructed based on sequence similarity. Only those members with known structures are used. The calculation was performed using the multiple sequence alignment program MAFFT (http://timpani.genome.ad.jp/%7Emafft/server/). b) Phylogenetic tree of the DD superfamily constructed based on structural similarity. The calculation was performed using the program TraceSuite II (http://www-cryst.bioc.cam.ac.uk/~jiye/evoltrace/evoltrace.html).
Interestingly, both phylogenetic trees placed PYDs and DEDs as more related to each other than to other subfamilies of the DD superfamily. One small difference exists between the sequence-based and structure-based phylogenetic trees. The sequence-based phylogenetic tree assigned two large branches consisting of DDs in one and CARDs, DEDs and PYDs in the other. The structure-based tree assigned three equal branches, the DDs in one, the CARDs in another and the DEDs and the PYDs in the last.
The clear distinction between the subfamilies of the DD superfamily may be the underlying reason that these proteins almost always interact homotypically with other members of the same subfamily. One possible evolutionary scenario may be as the following. First, a primordial DD superfamily member may have arisen and this member may or may not self-associate. Then an interaction pair may have arisen between two duplicated modules. This interaction pair then further duplicated and co-evolved into the different subfamilies of the DD superfamily. As such, because the evolution takes place on the interaction pair, the homotypic nature of the interactions is preserved.
SUMMARY AND FUTURE PERSPECTIVES
The DD superfamily is one of the largest and most widely distributed domain superfamily. One important function of these domains is to participate in homotypic protein-protein interactions in the assembly of oligomeric signaling complexes of apoptosis and inflammation. Evolutionarily, it seems that the ever-expanding DD superfamily may have evolved by inserting into various signal transduction proteins such as caspases, kinases and adapter proteins. In this regard, it is amazing that almost all oligomeric signaling complexes in apoptosis and inflammation contain domains of the DD superfamily. Through self-associations and homotypic interactions with other members of each of the subfamilies, these proteins often form the platform of these oligomeric assemblies to allow “proximity” induced caspase and kinase activation.
Many biochemical and structural studies have been performed on these domains. These studies have revealed a conserved six-helical bundle fold of the DD superfamily. Almost all known protein-protein interactions in the superfamily are either self-association or homotypic interactions with other members of the same subfamily. This is somewhat surprising given the structural similarity among the different subfamilies, but may reflect evolutionary circumstances.
Currently there are only three known structures of homotypic complexes in the entire DD superfamily. Interestingly, they are all asymmetric, in contrast to what might have been expected for homotypic interactions. They all also differ in the surfaces used for the interactions. Given the paucity of structural information on these complexes, the question remains whether common modes of interactions may be observed from either within the subfamily or within the entire DD superfamily. More biochemical and structural studies are required to address this question and to understand the molecular basis of these interactions in the assembly of apoptotic and inflammatory signaling complexes.
Table 2.
NMR and crystal structures in the DD superfamily
| Technique | References | |
|---|---|---|
| Fas DD | NMR | (24) |
| p75 NGFR DD | NMR | (104) |
| FADD DD | NMR | (106, 107) |
| TNFR1 DD | NMR | (103) |
| IRAK4 DD | Crystallography | (105) |
| RAIDD DD | Crystallography | (108) |
| Tube DD: Pelle DD | Crystallography | (109) |
| FADD DED+DD | NMR | (119) |
| FADD DED | NMR | (25) |
| PEA-15 DED | NMR | (118) |
| MC159 tandem DED | Crystallography | (116, 120) |
| Apaf-1 CARD: Caspase-9 CARD | Crystallography | (124) |
| Apaf-1 CARD | Crystallography | (122) |
| Apaf-1 CARD+NOD | Crystallography | (123) |
| RAIDD CARD | NMR | (26) |
| ICEBERG CARD | NMR | (87) |
| CED4: CED-9 | Crystallography | (125) |
| ASC PYD | NMR | (128) |
| NALP1 PYD | NMR | (27) |
Acknowledgments
We thank Upendra Maddineni for careful readings of the manuscript. This work was supported by the National Institute of Health (AI45937 and AI50872). Y. C. L and S. C. L. are postdoctoral fellows of the Cancer Research Institute. We apologize to all whose work has not been appropriately reviewed or cited due to space limitations.
LITERATURE CITED
- 1.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duvall E, Wyllie AH. Death and the cell. Immunol today. 1986;7:115–19. doi: 10.1016/0167-5699(86)90152-0. [DOI] [PubMed] [Google Scholar]
- 3.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(Suppl):S97–107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 4.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–61. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 5.Salvesen GS. Caspases and apoptosis. Essays Biochem. 2002;38:9–19. doi: 10.1042/bse0380009. [DOI] [PubMed] [Google Scholar]
- 6.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 7.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 8.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–52. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 9.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–83. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 10.Krappmann D, Hatada EN, Tegethoff S, Li J, Klippel A, Giese K, Baeuerle PA, Scheidereit C. The I kappa B kinase (IKK) complex is tripartite and contains IKK gamma but not IKAP as a regular component. J Biol Chem. 2000;275:29779–87. doi: 10.1074/jbc.M003902200. [DOI] [PubMed] [Google Scholar]
- 11.Stancovski I, Baltimore D. NF-kB activation: The IkB kinase revealed? Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 12.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–4. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 13.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κb activation prevents cell death. Cell. 1996;87:565–76. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 14.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 15.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Reed JC, Doctor KS, Godzik A. The domains of apoptosis: a genomics perspective. Sci STKE. 2004;2004:re9. doi: 10.1126/stke.2392004re9. [DOI] [PubMed] [Google Scholar]
- 17.Kohl A, Grutter MG. Fire and death: the pyrin domain joins the death-domain superfamily. C R Biol. 2004;327:1077–86. doi: 10.1016/j.crvi.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56. doi: 10.1146/annurev.cellbio.21.012704.131040. [DOI] [PubMed] [Google Scholar]
- 19.McEntyre JR, Gibson TJ. Patterns and clusters within the PSM column in TiBS, 1992–2004. Trends Biochem Sci. 2004;29:627–33. doi: 10.1016/j.tibs.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Hu S, Vincenz C, Buller M, Dixit VM. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J Biol Chem. 1997;272:9621–4. doi: 10.1074/jbc.272.15.9621. [DOI] [PubMed] [Google Scholar]
- 21.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:1172–6. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 23.Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricuttio D, Wang G, McFadden G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23:587–98. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 1996;384:638–41. doi: 10.1038/384638a0. [DOI] [PubMed] [Google Scholar]
- 25.Eberstadt M, Huang B, Chen Z, Meadows RP, Ng S-C, Zheng L, JLM, Fesik SW. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature. 1998;392:941–5. doi: 10.1038/31972. [DOI] [PubMed] [Google Scholar]
- 26.Chou JJ, Matsuo H, Duan H, Wagner G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell. 1998;94:171–80. doi: 10.1016/s0092-8674(00)81417-8. [DOI] [PubMed] [Google Scholar]
- 27.Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, Tschopp J, Grutter MG, Wuthrich K. NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure (Camb) 2003;11:1199–205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–45. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 29.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J. 1995;14:5579–88. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845–53. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 31.Itoh N, Nagata S. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J Biol Chem. 1993;268:10932–7. [PubMed] [Google Scholar]
- 32.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 33.Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH, Wallach D. Self-association of the “death domains” of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J Biol Chem. 1995;270:387–91. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 34.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 35.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–27. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 36.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–15. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 37.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A. 1999;96:10964–7. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) Embo J. 1997;16:2794–804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117:855–8. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 41.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–8. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- 42.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 43.Shu HB, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–63. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 44.Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, Armstrong RC, Alnemri ES. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–5. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 45.Inohara N, Koseki T, Hu Y, Chen S, Nunez G. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc Natl Acad Sci U S A. 1997;94:10717–22. doi: 10.1073/pnas.94.20.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D. CASH, a novel caspase homologue with death effector domains. J Biol Chem. 1997;272:19641–4. doi: 10.1074/jbc.272.32.19641. [DOI] [PubMed] [Google Scholar]
- 47.Han DK, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, Baskin DG, Schwartz SM, Hood L. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc Natl Acad Sci U S A. 1997;94:11333–8. doi: 10.1073/pnas.94.21.11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–7. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 49.Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–88. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 50.Garvey TL, Bertin J, Siegel RM, Wang GH, Lenardo MJ, Cohen JI. Binding of FADD and caspase-8 to molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J Virol. 2002;76:697–706. doi: 10.1128/JVI.76.2.697-706.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shisler JL, Moss B. Molluscum contagiosum virus inhibitors of apoptosis: The MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology. 2001;282:14–25. doi: 10.1006/viro.2001.0834. [DOI] [PubMed] [Google Scholar]
- 52.Searles RP, Bergquam EP, Axthelm MK, Wong SW. Sequence and genomic analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J Virol. 1999;73:3040–53. doi: 10.1128/jvi.73.4.3040-3053.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poppema S, Maggio E, van den Berg A. Development of lymphoma in Autoimmune Lymphoproliferative Syndrome (ALPS) and its relationship to Fas gene mutations. Leuk Lymphoma. 2004;45:423–31. doi: 10.1080/10428190310001593166. [DOI] [PubMed] [Google Scholar]
- 54.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–33. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 55.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middelton LA, Lin AY, Strober W, Lenardo MJ, MPJ Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lyphoproliferative syndrome. Cell. 1995;81:935–46. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 56.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE. Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–8. [PubMed] [Google Scholar]
- 57.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–9. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 58.Kasahara Y, Wada T, Niida Y, Yachie A, Seki H, Ishida Y, Sakai T, Koizumi F, Koizumi S, Miyawaki T, Taniguchi N. Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol. 1998;10:195–202. doi: 10.1093/intimm/10.2.195. [DOI] [PubMed] [Google Scholar]
- 59.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 60.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 61.Adams JM, Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–20. doi: 10.1016/s0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]
- 62.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–50. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 63.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 64.Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, Yeh WC, Mak TW, Benchimol S. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc Natl Acad Sci U S A. 2005;102:14314–20. doi: 10.1073/pnas.0506475102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet. 2000;26:122–7. doi: 10.1038/79102. [DOI] [PubMed] [Google Scholar]
- 66.Duan H, Dixit VM. RAIDD is a new ‘death’ adaptor molecule. Nature. 1997;385:86–9. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 67.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–4. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 68.Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005;123:1079–92. doi: 10.1016/j.cell.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 69.Fitzgerald KA, Chen ZJ. Sorting out Toll signals. Cell. 2006;125:834–6. doi: 10.1016/j.cell.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 70.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun H, Bristow BN, Qu G, Wasserman SA. A heterotrimeric death domain complex in Toll signaling. Proc Natl Acad Sci U S A. 2002;99:12871–6. doi: 10.1073/pnas.202396399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–83. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 73.Letsou A, Alexander S, Orth K, Wasserman SA. Genetic and molecular characterization of tube, a Drosophila gene maternally required for embryonic dorsoventral polarity. Proc Natl Acad Sci U S A. 1991;88:810–4. doi: 10.1073/pnas.88.3.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–59. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 75.Gaide O, Martinon F, Micheau O, Bonnet D, Thome M, Tschopp J. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-kappaB activation. FEBS Lett. 2001;496:121–7. doi: 10.1016/s0014-5793(01)02414-0. [DOI] [PubMed] [Google Scholar]
- 76.Gaide O, Favier B, Legler DF, Bonnet D, Brissoni B, Valitutti S, Bron C, Tschopp J, Thome M. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:836–43. doi: 10.1038/ni830. [DOI] [PubMed] [Google Scholar]
- 77.Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, Nunez G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. 2001;276:19012–9. doi: 10.1074/jbc.M009984200. [DOI] [PubMed] [Google Scholar]
- 78.Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell. 2000;6:961–7. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 79.Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol. 2006;6:196–204. doi: 10.1038/nri1787. [DOI] [PubMed] [Google Scholar]
- 80.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 81.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Nunez G. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274:14560–7. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 82.Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- 83.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–31. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 84.Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–8. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- 85.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–8. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 86.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 87.Humke EW, Shriver SK, Starovasnik MA, Fairbrother WJ, Dixit VM. ICEBERG: a novel inhibitor of interleukin-1beta generation. Cell. 2000;103:99–111. doi: 10.1016/s0092-8674(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 88.Lee SH, Stehlik C, Reed JC. Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J Biol Chem. 2001;276:34495–500. doi: 10.1074/jbc.M101415200. [DOI] [PubMed] [Google Scholar]
- 89.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 90.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 91.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 92.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 93.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–40. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 94.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264–9. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 96.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 97.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 98.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–8. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 99.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 100.Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S, Godzik A, Reed JC. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J Biol Chem. 2001;276:9239–45. doi: 10.1074/jbc.M006309200. [DOI] [PubMed] [Google Scholar]
- 101.Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM, Vincenz C, Ward PA. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J Biol Chem. 2001;276:9230–8. doi: 10.1074/jbc.M009853200. [DOI] [PubMed] [Google Scholar]
- 102.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 103.Sukits SF, Lin LL, Hsu S, Malakian K, Powers R, Xu GY. Solution structure of the tumor necrosis factor receptor-1 death domain. J Mol Biol. 2001;310:895–906. doi: 10.1006/jmbi.2001.4790. [DOI] [PubMed] [Google Scholar]
- 104.Liepinsh E, Ilag LL, Otting G, Ibanez CF. NMR structure of the death domain of the p75 neurotrophin receptor. Embo J. 1997;16:4999–5005. doi: 10.1093/emboj/16.16.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lasker MV, Gajjar MM, Nair SK. Cutting edge: Molecular structure of the IL-1R-associated kinase-4 death domain and its implications for TLR signaling. J Immunol. 2005;175:4175–9. doi: 10.4049/jimmunol.175.7.4175. [DOI] [PubMed] [Google Scholar]
- 106.Jeong EJ, Bang S, Lee TH, Park YI, Sim WS, Kim KS. The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD. J Biol Chem. 1999;274:16337–42. doi: 10.1074/jbc.274.23.16337. [DOI] [PubMed] [Google Scholar]
- 107.Berglund H, Olerenshaw D, Sankar A, Federwisch M, McDonald NQ, Driscoll PC. The three-dimensional solution structure and dynamic properties of the human FADD death domain. J Mol Biol. 2000;302:171–88. doi: 10.1006/jmbi.2000.4011. [DOI] [PubMed] [Google Scholar]
- 108.Park HH, Wu H. Crystal structure of RAIDD death domain implicates potential mechanism of PIDDosome assembly. J Mol Biol. 2006;357:358–64. doi: 10.1016/j.jmb.2005.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao T, Towb P, Wasserman SA, Sprang SR. Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell. 1999;99:545–55. doi: 10.1016/s0092-8674(00)81542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun H, Towb P, Chiem DN, Foster BA, Wasserman SA. Regulated assembly of the Toll signaling complex drives Drosophila dorsoventral patterning. Embo J. 2004;23:100–10. doi: 10.1038/sj.emboj.7600033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, Jackson CE, Puck JM, Dale J, Straus SE, Peter ME, Krammer PH, Fesik S, Lenardo MJ. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci U S A. 1999;96:4552–7. doi: 10.1073/pnas.96.8.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hill JM, Morisawa G, Kim T, Huang T, Wei Y, Werner MH. Identification of an expanded binding surface on the FADD death domain responsible for interaction with CD95/Fas. J Biol Chem. 2004;279:1474–81. doi: 10.1074/jbc.M304996200. [DOI] [PubMed] [Google Scholar]
- 113.Park A, Baichwal VR. Systematic mutational analysis of the death domain of the tumor necrosis factor receptor-1-associated protein TRADD. J Biol Chem. 1996;271:9858–62. doi: 10.1074/jbc.271.16.9858. [DOI] [PubMed] [Google Scholar]
- 114.Telliez JB, Xu GY, Woronicz JD, Hsu S, Wu JL, Lin L, Sukits SF, Powers R, Lin LL. Mutational analysis and NMR studies of the death domain of the tumor necrosis factor receptor-1. J Mol Biol. 2000;300:1323–33. doi: 10.1006/jmbi.2000.3899. [DOI] [PubMed] [Google Scholar]
- 115.Weber CH, Vincenz C. A docking model of key components of the DISC complex: death domain superfamily interactions redefined. FEBS Lett. 2001;492:171–6. doi: 10.1016/s0014-5793(01)02162-7. [DOI] [PubMed] [Google Scholar]
- 116.Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ, Wu H. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005;20:939–49. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sandu C, Morisawa G, Wegorzewska I, Huang T, Arechiga AF, Hill JM, Kim T, Walsh CM, Werner MH. FADD self-association is required for stable interaction with an activated death receptor. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401966. [DOI] [PubMed] [Google Scholar]
- 118.Hill JM, Vaidyanathan H, Ramos JW, Ginsberg MH, Werner MH. Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. Embo J. 2002;21:6494–504. doi: 10.1093/emboj/cdf641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carrington PE, Sandu C, Wei Y, Hill JM, Morisawa G, Huang T, Gavathiotis E, Werner MH. The structure of FADD and its mode of interaction with procaspase-8. Mol Cell. 2006;22:599–610. doi: 10.1016/j.molcel.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 120.Li FY, Jeffrey PD, Yu JW, Shi Y. Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J Biol Chem. 2006;281:2960–8. doi: 10.1074/jbc.M511074200. [DOI] [PubMed] [Google Scholar]
- 121.Muppidi JR, Lobito AA, Ramaswamy M, Yang JK, Wang L, Wu H, Siegel RM. Homotypic FADD interactions through a conserved RXDLL motif are required for death receptor-induced apoptosis. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401855. [DOI] [PubMed] [Google Scholar]
- 122.Vaughn DE, Rodriguez J, Lazebnik Y, Joshua-Tor L. Crystal structure of Apaf-1 caspase recruitment domain: an alpha-helical Greek key fold for apoptotic signaling. J Mol Biol. 1999;293:439–47. doi: 10.1006/jmbi.1999.3177. [DOI] [PubMed] [Google Scholar]
- 123.Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y. Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature. 2005;434:926–33. doi: 10.1038/nature03465. [DOI] [PubMed] [Google Scholar]
- 124.Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–57. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 125.Yan N, Chai J, Lee ES, Gu L, Liu Q, He J, Wu JW, Kokel D, Li H, Hao Q, Xue D, Shi Y. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature. 2005;437:831–7. doi: 10.1038/nature04002. [DOI] [PubMed] [Google Scholar]
- 126.Yu X, Acehan D, Menetret JF, Booth CR, Ludtke SJ, Riedl SJ, Shi Y, Wang X, Akey CW. A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure (Camb) 2005;13:1725–35. doi: 10.1016/j.str.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 127.Shiozaki EN, Chai J, Shi Y. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci U S A. 2002;99:4197–202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liepinsh E, Barbals R, Dahl E, Sharipo A, Staub E, Otting G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol. 2003;332:1155–63. doi: 10.1016/j.jmb.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 129.Eliezer D. Folding pyrin into the family. Structure. 2003;11:1190–1. doi: 10.1016/j.str.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 130.Bertin J, DiStefano PS. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 2000;7:1273–4. doi: 10.1038/sj.cdd.4400774. [DOI] [PubMed] [Google Scholar]
- 131.Martinon F, Hofmann K, Tschopp J. The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr Biol. 2001;11:R118–20. doi: 10.1016/s0960-9822(01)00056-2. [DOI] [PubMed] [Google Scholar]
- 132.Pawlowski K, Pio F, Chu Z, Reed JC, Godzik A. PAAD - a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem Sci. 2001;26:85–7. doi: 10.1016/s0968-0004(00)01729-1. [DOI] [PubMed] [Google Scholar]
- 133.Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci. 2001;26:475–81. doi: 10.1016/s0968-0004(01)01905-3. [DOI] [PubMed] [Google Scholar]
- 134.Kaufmann M, Bozic D, Briand C, Bodmer JL, Zerbe O, Kohl A, Tschopp J, Grutter MG. Identification of a basic surface area of the FADD death effector domain critical for apoptotic signaling. FEBS Lett. 2002;527:250–4. doi: 10.1016/s0014-5793(02)03146-0. [DOI] [PubMed] [Google Scholar]
- 135.Gumucio DL, Diaz A, Schaner P, Richards N, Babcock C, Schaller M, Cesena T. Fire and ICE: the role of pyrin domain-containing proteins in inflammation and apoptosis. Clin Exp Rheumatol. 2002;20:S45–53. [PubMed] [Google Scholar]
- 136.Moriya M, Taniguchi S, Wu P, Liepinsh E, Otting G, Sagara J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry. 2005;44:575–83. doi: 10.1021/bi048374i. [DOI] [PubMed] [Google Scholar]
- 137.Consortium TFF. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 138.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kastner DL, O’Shea JJ. A fever gene comes in from the cold. Nat Genet. 2001;29:241–2. doi: 10.1038/ng1101-241. [DOI] [PubMed] [Google Scholar]
- 140.Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183–95. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- 141.Nam YJ, Mani K, Ashton AW, Peng CF, Krishnamurthy B, Hayakawa Y, Lee P, Korsmeyer SJ, Kitsis RN. Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol Cell. 2004;15:901–12. doi: 10.1016/j.molcel.2004.08.020. [DOI] [PubMed] [Google Scholar]