

Abstract
Central dopaminergic and noradrenergic systems play essential roles in controlling several forebrain functions. Consequently, perturbations of these neurotransmissions may contribute to the pathophysiology of neuropsychiatric disorders. For many years, there was a focus on the serotonin (5‐HT) system because of the efficacy of selective serotonin reuptake inhibitors (SSRIs), the most prescribed antidepressants in the treatment of major depressive disorder (MDD). Given the interconnectivity within the monoaminergic network, any action on one system may reverberate in the other systems. Analysis of this network and its dysfunctions suggests that drugs with selective or multiple modes of action on dopamine (DA) and norepinephrine (NE) may have robust therapeutic effects. This review focuses on NE‐DA interactions as demonstrated in electrophysiological and neurochemical studies, as well as on the mechanisms of action of agents with either selective or dual actions on DA and NE. Understanding the mode of action of drugs targeting these catecholaminergic neurotransmitters can improve their utilization in monotherapy and in combination with other compounds particularly the SSRIs. The elucidation of such relationships can help design new treatment strategies for MDD, especially treatment‐resistant depression.
Keywords: Dopamine, Major depressive disorder, Norepinephrine, Serotonin
Introduction
Given that the therapeutic efficacy of the tricyclic drugs was based on their ability to inhibit norepinephrine (NE) and serotonin (5‐HT) transporters, the role of brain 5‐HT and NE neurotransmissions in the mechanism of action of antidepressant drugs prompted extensive studies [1, 2, 3, 4]. Dopamine (DA) on the other hand, despite evidence of its involvement in the action of certain antidepressants, attracted less attention [5]. First, reserpine which depletes catecholamines (NE, DA, and epinephrine) results in lowering mood; second, the monoamine oxidase inhibitors (MAOIs), which increase the synaptic availability of catecholamines, have clinical efficacy in depression [6, 7]. Furthermore, it was reported that the catecholamine synthesis inhibitor α‐methylparatyrosine produced a resurgence of depressive symptoms in patients improved by the NE reuptake inhibitor (NRI) desipramine, but not by the SSRI fluoxetine [8]. This suggests that not all antidepressants work via a single monoamine‐related mechanism. While dietary depletion of the DA precursors phenylalanine and tyrosine does not result in the relapse of formerly depressed patients off their medication [9], an inhibition of tyrosine hydroxylase by α‐methylparatyrosine decreases catecholamine metabolites levels and induces a worsening of depressive symptoms in patients being treated with catecholamine reuptake inhibitors [8]. The fact that pure dopaminergic drugs, such as pramipexole, are effective antidepressants suggests that enhancing DA function may underlie, at least in part, a therapeutic response in major depressive disorder (MDD) [10, 11].
Mounting evidence indicates that acting on different systems may have a greater therapeutic effect in depression [12, 13]. Understanding the relationship between the NE and DA systems, and how therapeutic drugs modulate them, may open new avenues to treat depression, particularly treatment‐resistant depression.
DA Modulation of the Noradrenergic Transmission
Although retrograde tracer studies showed few projections from ventral tegmental area (VTA) neurons to the locus coeruleus (LC) [14, 15], dense expression of D2/3 mRNA and high density of binding sites using [125I]iodosulpiride have been documented [16, 17, 18, 19]. Further evidence for the projection of VTA neurons to the LC comes from the observation that the DA content of the LC is decreased following a 6‐hydroxydopamine (6‐OHDA) lesion of the VTA [20].
Experiments have been carried out to determine the role of DA and NE in their projection areas (Figure 1). Iontophoretic studies showed that local application of DA suppressed the firing activity of NE neurons in the LC [21, 22, 23]. Given this preponderant inhibitory role of DA input to the LC, the selective lesioning of VTA DA neurons resulted in a significant increase of the mean firing rate of LC NE neurons with a greater increase in neurons displaying bursting activities [24]. An initial study aimed at characterizing the receptor involved in this effect showed that DA has an inhibitory effect on cell firing in the LC, through stimulation of α2‐adrenoceptors [23]. Furthermore, the inhibitory effect of intravenously administered (+)‐3‐PP, a partial D2 receptor agonist, which was shown to inhibit substantia nigra DA neurons, was partially blocked by the α2‐adrenoceptor antagonist yohimbine, but not by the α1‐adrenoceptor antagonist prazosin, or the D2 receptor antagonist haloperidol [22]. Although the selective D2‐like receptor antagonist haloperidol has been reported to enhance the spontaneous firing activity of NE neuron in the LC [25, 26], another study has shown that systemic administration of haloperidol affected neither LC NE neuronal firing nor the inhibitory action of DA [21]. Furthermore, the effectiveness of iontophoretically applied NE and DA to suppress NE neuronal firing was blocked by the α2‐adrenoceptor antagonist idazoxan, but not by the D2‐like receptor antagonist raclopride. This suggests that only α2‐adrenoceptors are involved in the effect of DA in the LC [21]. This type of overlap in function for DA and NE is not unexpected as the molecules differ by only one hydroxy group in the β‐carbon of their side chain (Figure 2). It is interesting that the D2/3 receptor agonist pramipexole and the catecholamine releaser bupropion significantly inhibited LC NE discharge after 2 days of sustained administration [27, 28, 29]. This effect was reported to be through stimulation of α2‐adrenoceptor, since idazoxan increased LC NE neuron firing to the same level as in controls and in rats treated subcutaneously with either compound [27, 29, 30]. Furthermore, DA inhibits paradoxical sleep (likely through an action on NE neurons) in the LC via activation of α2‐adrenoceptors since this effect was mimicked by the α2‐adrenoceptor agonist clonidine and blocked by α2‐adrenoceptor antagonist RX821002 [31].
Figure 1.
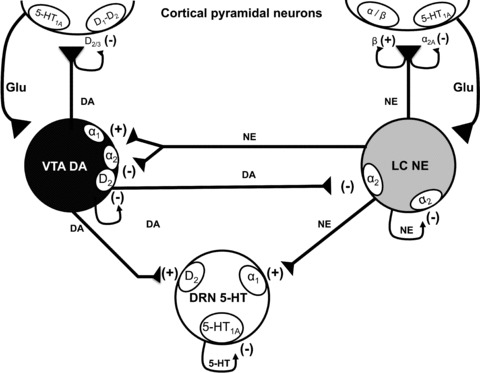
Schematic representation of the reciprocal interaction between serotonin (5‐HT) neurons in the dorsal raphe nucleus, norepinephrine (NE) neurons in the locus coeruleus and dopamine (DA) neurons in the ventral tegmental area. It also shows the nature of modulation of different neurotransmitters on diverse classes of auto‐ and heteroreceptors. (+) signs indicate an agonism or stimulatory effect and (–) signs indicate an antagonism or an inhibitory effect.
Figure 2.

Illustration of the conversion of DA to NE by β‐hydroxylase. Note that the difference between these two molecules resides only in one hydroxy group, thus imparting these two transmitters with only small differential three‐dimentional configurations.
Norepinephrine Modulation of the DA Transmission
Several studies using tracing techniques have shown that NE neurons in the LC project to the VTA and that immunoreactivity for NE transporters (NET) is present within the VTA region [32, 33, 34, 35, 36]. The presence of such projections was recently confirmed as being bilateral, but they were shown to also originate from the caudal medulla [37]. Moreover, an ultrastructural study has demonstrated that some NE terminals make close contacts onto VTA neurons [38]. Because the majority of contacts were not in direct synaptic apposition, it was suggested that most NE actions on DA neurons in the VTA are through extrasynaptic modulation. Furthermore, using quantitative autoradiography, α1‐adrenoceptors have also been detected in the VTA [39] and immunoreactivity for α2C‐adrenoceptors was also on DA neurons of VTA [40]. Finally, the selective NRI desipramine increased NE concentrations in VTA [41].
The lesion of the LC NE neurons, using local injection of 6‐OHDA, resulted in a significant decrease in brain NE and an increase in the discharge rate of VTA DA neurons attributable to a higher number of bursts and action potentials per burst [24] (see Figure 1). Despite these results being consistent with an inhibitory action of NE on VTA DA neurons, contradictory data have also been reported. For example, systemic administration of idazoxan or the selective NRI reboxetine, which raises extracellular NE levels in the VTA [41], increased the burst firing activity of DA neurons in the VTA [42, 43, 44, 45]. It has been demonstrated that the local application of the α2‐adrenoceptor agonist clonidine in the VTA did not inhibit DA neurons [46, 47]. As a result, it is unlikely that the inhibitory effect of NE involved postsynaptic α2‐adrenoceptors. However, recent data from Guiard et al. [21] indicate that idazoxan attenuates the inhibitory effect of iontophoretically applied NE on VTA DA neurons. Furthermore, electrophysiological studies have shown that the D2‐like receptor antagonist sulpiride prevented the inhibitory effect of NE on VTA DA neurons [46, 47, 48, 49]. It thus seems possible that this neurotransmitter may act through activation of both α2‐adrenoceptors and D2 receptors [21, 46, 49]. On the other hand, electrophysiological studies demonstrated that microiontophoretic application of DA in the VTA reduced the firing of DA neurons while this effect was blocked by sulpiride and also by the nonselective α‐adrenoreceptor antagonist piperoxane [46]. This is concordant with results showing that DA inhibits the discharge of DA neurons by acting both on D2 and α2‐adrenoceptors, since this inhibitory effect was prevented by idazoxan [21]. It is worth noting that intravenous injection of a high dose of clonidine increases both firing and burst activity of VTA DA neurons [50, 51]. It is thus possible that clonidine suppressed the firing rate of NE and 5‐HT neurons, as well as directly decreasing NE and 5‐HT release, through action on terminal α2‐adrenoceptors. Such action would remove inhibitory tones exerted by NE and 5‐HT on DA neurons.
Further evidence for a synergy between NE and DA was provided by recent electrophysiological studies showing that concurrent inhibition of NE and DA reuptake with intravenous injection of nomifensine produced a complete inhibition of firing of DA neurons, whereas the selective DA reuptake inhibitor GBR12909 failed to do so. In addition, nomifensine compromises the inhibitory potential of SSRI escitalopram on 5‐HT neuronal firing [52] (Figure 3). Because growing evidence demonstrates that both NE and DA exert an excitatory action on dorsal raphe nucleus (DRN) 5‐HT neuronal firing [13, 53, 54], the capacity of NET and DAT to alter the inhibitory effect of SSRI escitalopram on 5‐HT neuronal firing was investigated using the selective NRI reboxetine, the selective DA reuptake inhibitor GBR12909, and the dual NE and DA reuptake inhibitor nomifensine. It was found that neither pretreatment with reboxetine nor with GBR12909, at doses previously shown to elevate extracellular NE [55] or DA levels [56], respectively, attenuated the escitalopram‐induced decrease in DRN 5‐HT firing rates. However, when NE and DA levels were simultaneously increased by systemic administration of the dual‐acting reuptake inhibitor nomifensine, an upward shift of the dose‐response curve of escitalopram was observed demonstrating that both catecholamines were required to counteract the inhibitory effect of escitalopram on 5‐HT neurons [52]. It was therefore concluded that when the excitatory influence of both NE and DA is enhanced, the inhibitory effect of 5‐HT1A autoreceptor on 5‐HT neuronal firing is dampened. This is strengthened by the results of electrophysiological studies of the triple reuptake inhibitor SEP225289, showing that it was more potent at inhibiting NE neurons than 5‐HT neurons, despite apparent identical affinity of this compound for all three reuptake transporters [57].
Figure 3.

Effect of single‐ and dual‐acting catecholaminergic reuptake inhibitors on escitalopram‐induced decrease in DRN 5‐HT neuronal activity. (A, B): Examples of integrated firing histograms showing the effects of cumulative intravenous doses of escitalopram on the spontaneous activity of DRN 5‐HT neurons in presence of vehicle (A), or of the DA/NE reuptake inhibitor nomifensine (5 mg/kg; iv; B). The arrows indicate the compounds administered and the time at which the injection of the specified doses was completed. The escitalopram‐induced inhibition of firing rate was reversed with the 5‐HT1A receptor antagonist WAY100635. (C) Symbols represent the mean (± SEM) of percent of baseline firing rate of DRN 5‐HT neurons observed at each dose of escitalopram after the administration of vehicle; GBR 12909, reboxetine and nomifensine. These means were calculated on the 60 second‐period preceding each drug administration. ***P < 0.001. Statistical significance was taken as P < 0.05.
NE‐DA Interactions in the Forebrain
Several lines of evidence point out to an intricate relationship between NE and DA not only at the somatodendritic level as described above, but at the terminal areas as well. Electrophysiological interactions between NE and DA were mainly studied in the hippocampus. It was found that partial and total inhibition of CA3 pyramidal neuronal activity obtained with, respectively, iontophoretic application of DA and NE was not blocked by systemic injection of the D2 receptor antagonist haloperidol nor by local application of the D2 receptor antagonist raclopride [21]. However, as in the VTA and LC, idazoxan prevented the inhibitory effect of DA as well as NE on CA3 pyramidal cells [21]. To understand better the physiological importance of these effects, the possibility that the NE neurons themselves could be the main source of DA in the hippocampus was addressed. Indeed, the observation that the selective NRI, desipramine, but not the DA reuptake inhibitor GBR12909, prolonged the inhibitory effects of microiontophoretic applied‐DA strongly suggests that the clearance of DA in the hippocampus is mediated by the NET. This is consistent with previous data showing that DA reuptake by NE terminals occurs in the prefrontal cortex (PFC), the nucleus accumbens shell, and the bed nucleus of stria terminalis [58, 59]. NE and DA neurons converge in the medial PFC where NE terminals regulate DA release in this brain region. Microdialysis studies first suggested that DA in frontal cortex is elevated not only by blockade of DA uptake sites on DA terminals, but also by NET located on NE terminals [59, 60, 61, 62, 63], where NET is known to have a higher affinity for DA than DA transporter (DAT) [63, 64, 65]. Indeed, in the presence of blockade of NET by desipramine, GBR12909 further increased the extracellular concentrations of cortical DA [66]. Using NET knock‐out mice, it was shown that DA uptake into frontal cortex synaptosomes is the result of NET and not DAT blockade, because a selective concentration of GBR12909 did not block DA uptake into frontal cortex synaptosomes from NET knock‐out mice [67]. While controversial [68], it was also hypothesized that DA in the cerebral cortex may be released from noradrenergic neurons [59], since after 6‐OHDA lesion of VTA, there was no change in the concentration of extracellular DA in cerebral cortex, while there was a marked decrease in the ipsilateral nucleus accumbens. Furthermore, the administration of haloperidol failed to modify extracellular levels of DA in cortex while increasing it in nucleus accumbens [69].
The PFC network activity is fundamental in processing information in the brain [70] and malfunctioning of this structure can underlie a variety of symptoms common to several psychiatric illnesses [71], including mood disorder [72]. The PFC circuits are modulated by NE and DA which play a complementary and critical role in PFC function, where their depletion has been shown to be as detrimental as removing the cortex itself [73]. The action of NE through α2A‐adrenoceptors and DA through D1 receptors is key to PFC function [74, 75, 76]. These receptors regulate incoming glutamate signals at the level of dendritic spines on pyramidal cells in PFC. Indeed, these signals are sorted at the level of the head of a dendritic spine where it can pass to the apical dendrite.
In normal condition, where neurons efficiently process information, NE is released to strengthen signal detected as desirable, while in the case of neurons receiving inputs considered as noise, DA is released to weaken these inappropriate connections [77]. Under optimal neurochemical conditions, moderate levels of NE engage α2A‐adrenoceptors and increase signal in form of responses to preferred special directions, whereas moderate levels of D1 receptor stimulation decrease “noise” measured as responses to nonpreferred spatial directions. PFC working memory function is improved by α2A‐adrenoceptor stimulation and moderate levels of D1 receptor stimulation, but impaired by high levels of D1, α1, and β1 receptor stimulation [78, 79]. Stress exposure impairs working memory function through excessive stimulation of DA and NE receptors in PFC [78]. Optimal levels of D1 stimulation appear to focus signal transmission, conveying only large or temporally coincident signals to the cell body. However, higher concentration of DA or a D1 receptor agonist effectively impedes information transfer from dendrite to soma [76, 80]. This interruption of information transfer may underlie the working memory impairment seen at high levels of D1 receptor stimulation [76]. When moderate doses of atomoxetine and methylphenidate, which increase endogenous NE and DA levels in PFC [81, 82] were administered, there was an improvement of PFC function via actions on α2A‐adrenoceptor and D1 receptors [83, 84]. PFC functioning is also improved through stimulation of postsynaptic α2A‐adrenoceptors, as with guanfacine [74]. Indeed, animal studies have shown that guanfacine improves a variety of PFC functions, including working memory, response inhibition, attention regulation and conditional motor responding [78, 85]. Altogether, these data indicate that NE and DA act in complementary fashion to regulate the processing of information in the PFC pyramidal cells and that this processing is improved by optimal doses of drugs acting on NE and DA systems, thus reducing related psychiatric symptoms.
Single and Dual DA and NE Acting Antidepressants
In this section, we will discuss the effects of different drugs acting on NE or DA or both, via an action on DA and/or NE receptors or transporters. Understanding the mode of action of drugs targeting these catecholaminergic neurotransmitters can improve their utilization in monotherapy and in combination with other compounds particularly the SSRIs. The elucidation of such relationships can help design new treatment strategies for MDD, especially treatment‐resistant depression.
Pramipexole
Several antidepressants enhance DA neurotransmission [5, 86] and it is interesting to examine results obtained with the D2/D3 receptor agonist pramipexole, a drug with no known affinity for either NE or 5‐HT neuronal elements. In a randomized double‐blind study, Corrigan et al. [87] showed that by week 8, patients with MDD receiving pramipexole alone had significant improvement over baseline compared to the placebo group. Several other studies reported the efficacy of pramipexole as an augmentation strategy in resistant unipolar and bipolar depression [88, 89, 90, 91, 92, 93].
While subacute administration of pramipexole significantly decreases the spontaneous firing rate of VTA DA neurons and the extracellular concentration of DA in striatum [27, 94], sustained administration over two weeks resulted in recovery of discharge to normal level [27] (Figure 4) due to desensitization of D2 autoreceptor [27, 95] (Figure 5). While the percentage of bursts was decreased at 14 days, it was compensated by a return to the normal level of the number of bursts per minute. Restoration of firing rate of DA neurons, despite a decrease in its bursting activity, may lead to a net enhancement of the DA transmission in postsynaptic brain targets. Assessment of the tonic activation of DA receptors in postsynaptic regions is deemed crucial to confirm this possibility and is currently underway in our laboratory.
Figure 4.
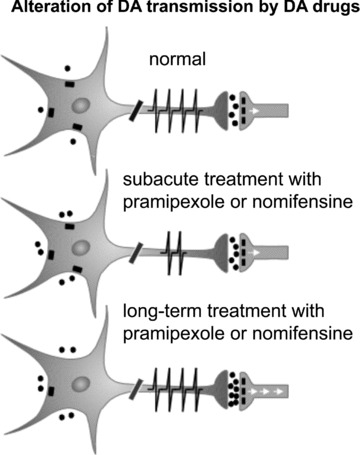
Diagrams of DA neurons representing their response and adaptations to the activation of D2 receptor by the D2/3 receptor agonist pramipexole or the inhibition of the NE and DA transporters with nomifensine. The spikes on the axon represent the firing activity of DA neurons. The dots around the DA neurons represent DA molecules. These neurons have D2/3 autoreceptors that inhibit firing and release when activated by an excess amount of DA. The effects of DA on postsynaptic neurons are mediated by several subtypes of DA receptors.
Figure 5.
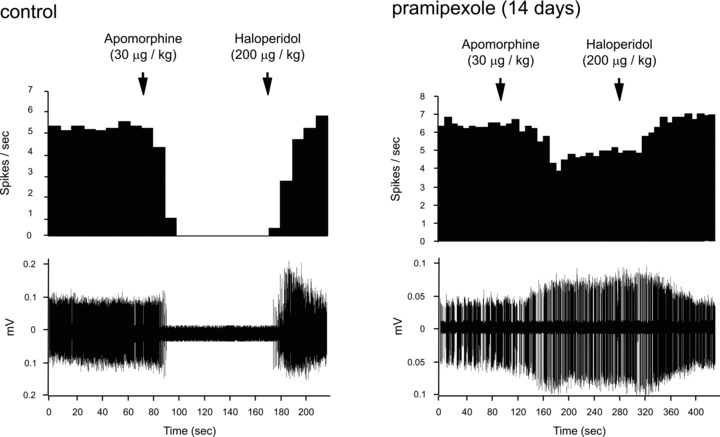
Assessment of the sensitivity of the VTA DA D2/3 autoreceptor: Integrated firing rate histogram illustrating the effect of apomorphine administration on VTA DA firing activity in controls and rats treated with pramipexole for 14 days. The apomorphine‐induced inhibition of firing rate was reversed with the D2 receptor antagonist haloperidol. Note that the inhibitory effect of apomorphine in control rats is dampened in rats treated with pramipexole, indicating a desensitization of D2/3 receptors.
Given the reciprocal interactions between DA, NE, and 5‐HT neurons, it is important to consider the effects of pramipexole on all three systems. It has been shown that short‐term administration of pramipexole resulted in significant reduction of LC NE firing activity. Despite the previously reported lack of affinity of pramipexole for NE neuronal elements, this effect may still happen through a direct activation of α2‐adrenergic autoreceptors, since idazoxan effectively reversed this inhibition [27]. Indeed, after long‐term administration of pramipexole, NE neurons regained their normal firing rate, due to desensitization of the α2‐adrenergic autoreceptors [27]. Importantly, a recent study showed that while the sensitivity of postsynaptic α2‐adrenoceptors and 5‐HT1A receptors are not changed after 14 days of administration of pramipexole, there was an enhancement of tonic activation of the 5‐HT1A receptors in hippocampus, and an increase of firing of 5‐HT neurons in the DRN [27, 96]. This confirms the increase in net 5‐HT transmission following long‐term administration of pramipexole.
These data revealed that in addition to its effects on the DA system, pramipexole exerts a facilitating action on 5‐HT neurotransmission. Drugs with dopaminergic properties may thus act, at least in part, through an action on the 5‐HT system. Altogether, these observations strengthen the importance of the interactions between DA, NE, and 5‐HT systems.
Nomifensine
Nomifensine, an antidepressant with potent NE and DA reuptake inhibiting properties, was as effective as traditional antidepressants. A comprehensive assessment of the pooled studies provided strong evidence for equal efficacy of nomifensine and imipramine [97, 98]. It was removed from the market because of aplastic anemia, unrelated to its reuptake inhibitory actions. Pooled analyses undertaken to explore the generalizability of treatment efficacy provided statistical evidence that more severely depressed patients showed enhanced response to nomifensine relative to placebo.
Animal studies showed that nomifensine markedly decreased the firing rate of NE neurons in the LC after 2 days, with no recovery of the firing rate of these neurons after 14 days [52] (Figure 6), similar to previous results found with the NRI reboxetine and MAOIs [99]. The absence of a recovery in the firing rate of these neurons is likely due to a lack of desensitization of somatodendritic α2‐adrenoceptors, which are known to inhibit NE neuronal firing [99]. These α2‐adrenoceptors have nevertheless the capacity to desensitize and allow a recovery of firing of NE neurons. This has initially been documented using the SSRI/5‐HT2A receptor antagonist, YM992, a drug that enhances NE release [100, 101].
Figure 6.
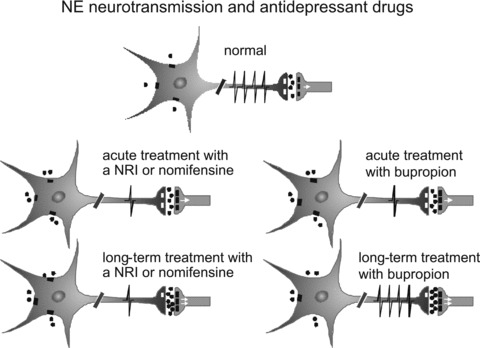
Diagrams of NE neurons representing their response and adaptations to the inhibition of the NE and DA transporters with nomifensine or release of DA and NE by bupropion. The spikes on the axons represent the firing activity of NE neurons. The dots around NE neuron represent NE molecules. These neurons have α2 autoreceptors that inhibit firing and release when activated by an excess amount of NE. The effects of NE on postsynaptic neurons are mediated by several subtypes of α‐ and β ‐adrenergic receptors. NRI, norepinephrine reuptake inhibitor.
Similar to NE neurons, the firing rate of DA neurons in the VTA was significantly decreased with the short‐term nomifensine regimen (Figure 4). Unlike NE neurons, however, both the firing rate and burst activity of DA neurons completely recovered to control levels following 14‐day of nomifensine administration (Figure 4), due to the desensitization of the somatodendritic D2 autoreceptor, as was reported for a 14‐day regimen of the D2 agonist quinpirole [52, 95]. Interestingly, nomifensine induced a significant increase in the firing rate of 5‐HT neurons after 2 days, which remained significantly elevated after the 14‐day regimen. It is well documented that increases in synaptically available NE and DA cause an excitation of DRN 5‐HT neurons via α1‐adrenoceptors [102] and D2 receptors [53, 103], respectively. Although it has been previously reported that acute administration of the NRI reboxetine increases 5‐HT firing [42], sustained administration of reboxetine is without effect [99]. Prolonged administration of desipramine and reboxetine desensitize α2‐adrenergic receptors present on 5‐HT terminals, leading to an increase in synaptic availability of endogenous 5‐HT in the rat hippocampus [104, 105]. Nevertheless, as for pramipexole, the increase in 5‐HT firing following nomifensine administration indicates that the dopaminergic property of nomifensine likely contributes to increase 5‐HT neuronal firing.
The increase in 5‐HT neuronal firing following 2 days of nomifensine administration implies a synergistic effect of the dual action of this drug since GBR 12909 and selective NRIs produced no such effect [52, 99]. However, the elevation of 5‐HT neuronal firing following 2 days of nomifensine administration was abolished by lesion of NE neurons, but not by the administration of the D2 receptor antagonist paliperidone [52, 106, 107]. This indicates on the one hand that NE plays a significant role in this increase, and on the other hand, that enhanced DA levels may also play a role but may not be sufficient on its own to induce such an increase. A similar increase in the firing rate of 5‐HT neurons was obtained following a 2‐day administration of aripiprazole, a D2 and 5‐HT1A receptor partial agonist; this increase was reversed by paliperidone, indicating again that it was mediated by D2 receptors [107]. These data show that nomifensine increases DA, NE and 5‐HT transmission. Interestingly, a similar increase in DRN 5‐HT neurons firing was obtained following an action through α1‐adrenoceptors in the case of nomifensine and through D2 receptors with aripiprazole.
Bupropion
Bupropion is an effective antidepressant when used alone or in combination with SSRIs as an augmentation strategy [108, 109]. It has been shown that bupropion has no significant affinity for various types of receptors such as α‐ or β‐adrenoceptors, 5‐HT, DA, or nicotinic receptors [110, 111, 112]. Although the mechanism of action of bupropion is not fully understood, DAT inhibition is unlikely because four positron emission tomography (PET) scan studies have reported that clinically effective doses of bupropion produce very low occupancy of dopamine reuptake sites [113, 114, 115, 116]. It is unlikely that such low DAT occupancy has an effect on DA transmission since the value obtained is hardly different from baseline [113]. Moreover, a lack of NE reuptake inhibition is indicated by its lack of inhibitory effect on the tyramine pressor response, contrarily to NRIs [117]. In preclinical studies, bupropion was hypothesized to be a NE releaser in LC and at the level of NE terminals in DRN [29]. Indeed, administration of subacute bupropion increases firing of 5‐HT neurons whereas NRIs do not. This effect was no longer present in NE‐lesioned rats. Finally, it is thought to be mediated through activation of α1‐adrenoceptor, which exerts an excitatory action on 5‐HT neurons activity [29, 118, 119]. Administration of bupropion for two days decreased the firing activity of LC neurons (Figure 6) through an overactivation of α2‐adrenoceptors, as the selective α2‐adrenoceptor antagonist idazoxan reversed this effect [29]. In addition, it was shown that NE neuronal firing recovers after 14 days of bupropion administration [28] (Figure 6), a phenomenon that does not occur with reboxetine, desipramine, or even MAOIs [99, 120]. Importantly, the recovery of the firing of LC NE neurons and the desensitization of the autoreceptors after administration of bupropion for 14 days (Figure 7) imply that there could be a sustained increase in NE neurotransmission in brain target areas. Interestingly, the tonic activation of α1‐, α2‐adrenenergic and 5‐HT1A receptors by endogenous NE and 5‐HT, respectively, was increased following 14 days of bupropion administration [121]. The bupropion regimen that alters NE and 5‐HT neuronal firing had no effect on the firing of DA neurons. Previous studies showed that only high intravenous doses of bupropion dose‐dependently reduced firing of brainstem dopamine neurons in the rat [30], therefore suggesting that this effect does not constitute a basis for its clinical effects. In vivo brain microdialysis studies demonstrated that after both acute and chronic administration, there was an enhancement of bupropion‐induced increase in extracellular DA in the nucleus accumbens and hippocampus regions, but not in the striatum [122, 123, 124]. Taken together, these data indicate that the increase in DA release is independent of the firing activity of VTA DA neurons, during not only subacute but also long‐term administration of bupropion [28, 29]. It is difficult to dissociate changes in DA release from changes in DA neuronal activity. However, in vivo studies have shown that a bupropion‐induced sensitization is rather due to an increase in the ability of bupropion to release DA [125, 126]. Nevertheless, unlike bupropion, the selective DA reuptake inhibitor GBR12909, also known to increase extracellular levels of DA in the cortex [127], decreases both the firing and burst activity of DA neurons in the VTA following a 2‐day administration [52]. In summary, it was shown that bupropion has the capacity to enhance synaptic availability of NE and DA in some brain regions, as well as to promptly increase the firing activity of 5‐HT neurons. These effects combined with the gradual normalization of NE neurotransmission following long‐term administration, may thus be the mechanisms whereby bupropion exerts its delayed therapeutic effect in MDD.
Figure 7.
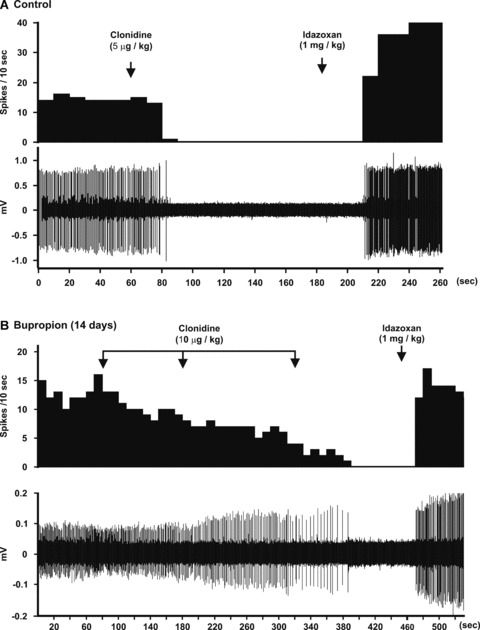
(A) The upper panel represents the integrated histogram of the firing activity of a LC NE neuron (lower panel) that was inhibited by the selective α2‐adrenoceptor agonist clonidine and reversed by the selective α2‐adrenoceptor receptor antagonist idazoxan. (B) Integrated histogram of the firing activity of a LC NE neuron (lower panel) illustrating the decreased responsiveness to three consecutive intravenous injections of clonidine in a rat treated with bupropion for 14 days, thus indicating a desensitization of α2‐autoreceptors.
Atypical Antipsychotics
Atypical antipsychotics, despite being D2 receptor antagonists, are even more potent 5‐HT2A receptor antagonists [128]. These two properties are believed to underlie their therapeutic action in psychosis, while producing minimal motor side effects. They also have affinities for receptors other than the D2 and the 5‐HT2A receptors. Quetiapine apparently differs from other typical and atypical antipsychotic drugs by its antidepressant activity and its proven efficacy in unipolar and bipolar disorders, as well as generalized anxiety disorder [129, 130, 131]. Its antidepressant activity could well stem from its α2‐adrenoceptor antagonistic activity, which would then be akin to that of mirtazapine, an α2‐adrenergic and 5‐HT2A receptor antagonist [132, 133]. Systemic administration of quetiapine also enhances the extracellular levels of NE and DA in the rat PFC as for mirtazapine [132, 134]. Some atypical antipsychotics may thus increase NE and 5‐HT transmission by blocking α2‐adrenoceptors on LC NE cell body as well as antagonizing α2‐adrenoceptors on NE and 5‐HT terminals in projection areas [104]. However, not all atypical antipsychotics have activity at α2‐adrenoceptors, like olanzapine, which was shown to have a beneficial therapeutic effect in MDD resistant patients to SSRIs [135, 136, 137]. This effect is thought to be through action on 5‐HT2A receptors located on GABA neurons controlling NE neuronal firing [100]. Indeed, because of their ability to block 5‐HT2A receptors, atypical antipsychotics reverse the SSRI‐induced inhibition of the firing rate and burst activity of NE neurons, as it was demonstrated for the combination of SSRIs fluoxetine and escitalopram with olanzapine and risperidone, respectively [136, 137, 138]. In addition, an important metabolite of quetiapine in humans, norquetiapine, appears to be a blocker of NET (Ki = 58 nM; [139]). Previous studies have shown that blockade of NET together with α2‐adrenoceptor antagonism leads to a synergistic effect on extracellular levels of NE [140]. Sustained treatment with NET blockers results in a decrease of NE firing activity without a recovery due to the absence of α2‐adrenoceptor desensitization [99]. Antagonism of the α2‐adrenoceptors could thus potentiate the effect of NET inhibitors, as will be discussed below when the SNRI venlafaxine is combined with mirtazapine in treatment‐resistant patients.
Antidepressant Combinations Acting on NE and/or DA
As mentioned above, the use of a dual DA and NRI, nomifensine, was shown to exert a robust antidepressant effect. This strategy has not been exploited since, although triple reuptake inhibitors (5‐HT, NE, and DA) are currently in development. Most of these agents, however, have a low affinity for DAT. Nevertheless, combinations of antidepressants resulting in at least a dual action on NE and DA have been used with success not only in treatment‐resistant depression, but also in drug‐naïve patients [141].
For instance, the combination of bupropion and mirtazapine has been shown to nearly double the remission rate in MDD after 6 weeks of concomitant administration from treatment initiation in a double‐blind randomized trial. In the same study, a noradrenergic regimen of venlafaxine combined with mirtazapine produced a 58% remission rate [141]. Similarly, addition of bupropion to the SNRI duloxetine has been reported in an open labeled study to be an effective strategy [142]. Trials examining the antidepressant potential of the addition of the atypical antipsychotics risperidone (an α2‐adrenoceptor antagonist) and aripiprazole (a D2 receptor partial agonist) at subtherapeutic regimens for psychosis, therefore not functionally antagonizing D2 receptors, have been shown to be effective with the SNRI venlafaxine [143, 144, 145].
Conclusion
In summary, agents that act selectively on DA or NE neuronal elements to enhance net transmission of these catecholamines can produce a clear antidepressant action. In addition, these two neuronal systems have important reciprocal anatomical and physiologically important interactions. Some strategies acting on both systems have been shown to be effective, not only in drug naïve patients, but also in treatment‐resistant depression. Finally, because the DA and NE systems also have reciprocal interactions with the 5‐HT system, drugs impacting the DA and NE systems also end up increasing 5‐HT transmission (Figure 1, Table 1). On such premises, it can be proposed that combinations of catecholamine actions could help further improve the treatment of MDD.
Table 1.
Effect of long‐term administration of various antidepressant medications with affinities for NE and DA elements on DA, NE and 5‐HT neurotransmissions
| Cell body α2 autoreceptor | Cell body D2 autoreceptor | Cell body 5‐HT1A autoreceptor | Net NE transmission | Net DA transmission | Net 5‐HT transmission | |
|---|---|---|---|---|---|---|
| Pramipexole | ↓ | ↓ | ↓ | N.D. | ↑ | ↑ |
| NRI | Ø | N.D. | Ø | ↑ | ↑* | ↑ |
| Nomifensine | Ø | ↓ | ↓ | ↑* | ↑* | ↑* |
| Bupropion | ↓ | N.D. | ↓ | ↑ | ↑* | ↑ |
| Mirtazapine | Ø# | N.D. | ↓ | ↑ | ↑ | ↑ |
| SSRI | N.D. | N.D. | ↓ | ↓ | ↓ | ↑ |
N.D., not determined; ø, no change; ↑, increased; ↓, decreased; *, presumed from their acute effect; #, these experiments were carried out after a washout, but in the presence of mirtazapine this receptor is antagonized.
Conflict of Interest
P. Blier has received grants, honoraria for serving on advisory boards and/or making presentation for Astra‐Zeneca, Biovail, Eli Lilly, Janssen, Lundbeck, Organon, Pharmacia and Wyeth.
Acknowledgment
The authors would like to thank Dr. Arnsten AF for her insight in part of the manuscript.
References
- 1. Blier P, De Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci 1994;15:220–226. [DOI] [PubMed] [Google Scholar]
- 2. Maes M, Meltzer HY. The serotonin hypothesis of major depression In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The fourth generation of progress. New York : Raven Press, 1995;933–944. [Google Scholar]
- 3. Heninger GR, Charney DS. Mechanisms of action of antidepressant treatments: Implications for the ethiology and treatment of depression disorders In: Meltzer HY, editor. Psychopharmacology: The third generation of progress. New York : Raven Press, 1987;535–544. [Google Scholar]
- 4. Delgado PL. Monoamine depletion studies: Implications for antidepressant discontinuation syndrome. J Clin Psychiatry 2006;67(Suppl 4):22–26. [PubMed] [Google Scholar]
- 5. Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psych 2007;64:327–337. [DOI] [PubMed] [Google Scholar]
- 6. Schildkraut JJ. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am J Psychiatry 1965;122:509–522. [DOI] [PubMed] [Google Scholar]
- 7. Bunney WE Jr., Davis JM. Norepinephrine in depressive reactions. A review. Arch Gen Psychiatry 1965;13:483–494. [DOI] [PubMed] [Google Scholar]
- 8. Miller HL, Delgado PL, Salomon RM, Berman R, Krystal JH, Heninger GR, Charney DS. Clinical and biochemical effects of catecholamine depletion on antidepressant‐induced remission of depression. Arch Gen Psychiatry 1996;53:117–128. [DOI] [PubMed] [Google Scholar]
- 9. Roiser JP, McLean A, Ogilvie AD, et al The subjective and cognitive effects of acute phenylalanine and tyrosine depletion in patients recovered from depression. Neuropsychopharmacology 2005;30:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kapur S, Mann JJ. Role of the dopaminergic system in depression. Biol Psychiatry 1992;32:1–17. [DOI] [PubMed] [Google Scholar]
- 11. Ordawy GA, Mann JJ. Neurocircuitry of mood disorders. In: Davis KL, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The fifth Generation of Progress Philadelphia : Lippinkott Williams & Willkins, 2002;1051–1064. [Google Scholar]
- 12. Blier P, Szabo ST. Potential mechanisms of action of atypical antipsychotic medications in treatment‐resistant depression and anxiety. J Clin Psychiatry 2005;66(Suppl 8):30–40. [PubMed] [Google Scholar]
- 13. Trivedi MH, Hollander E, Nutt D, Blier P. Clinical evidence and potential neurobiological underpinnings of unresolved symptoms of depression. J Clin Psychiatry 2008;69:246–258. [DOI] [PubMed] [Google Scholar]
- 14. Swanson LW. The projections of the ventral tegmental area and adjacent regions: A combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull 1982;9:321–353. [DOI] [PubMed] [Google Scholar]
- 15. Geffard M, Buijs RM, Seguela P, Pool CW, Le Moal M. First demonstration of highly specific and sensitive antibodies against dopamine. Brain Res 1984;294:161–165. [DOI] [PubMed] [Google Scholar]
- 16. Bouthenet ML, Martres MP, Sales N, Schwartz JC. A detailed mapping of dopamine D2 receptors in rat central nervous system by autoradiography with [125I]iodosulpride. Neuroscience 1987;20:117–155. [DOI] [PubMed] [Google Scholar]
- 17. Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: Comparison with dopamine D2 receptor mRNA. Brain Res 1991;564:203–219. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki M, Hurd YL, Sokoloff P, Schwartz JC, Sedvall G. D3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res 1998;779:58–74. [DOI] [PubMed] [Google Scholar]
- 19. Yokoyama C, Okamura H, Nakajima T, Taguchi J, Ibata Y. Autoradiographic distribution of [3H]YM‐09151–2, a high‐affinity and selective antagonist ligand for the dopamine D2 receptor group, in the rat brain and spinal cord. J Comp Neurol 1994;344:121–136. [DOI] [PubMed] [Google Scholar]
- 20. Ornstein K, Milon H, McRae‐Degueurce A, Alvarez C, Berger B, Wurzner HP. Biochemical and radioautographic evidence for dopaminergic afferents of the locus coeruleus originating in the ventral tegmental area. J Neural Transm 1987;70:183–191. [DOI] [PubMed] [Google Scholar]
- 21. Guiard BP, El Mansari M, Blier P. Crosstalk between dopaminergic and noradrenergic systems in the rat ventral tegmental area, locus coeruleus, and dorsal hippocampus. Mol Pharmacol 2008;74:1463–1475. [DOI] [PubMed] [Google Scholar]
- 22. Elam M, Clark D, Svensson TH. Electrophysiological effects of the enantiomers of 3‐PPP on neurons in the locus coeruleus of the rat. Neuropharmacology 1986;25:1003–1008. [DOI] [PubMed] [Google Scholar]
- 23. Cedarbaum JM, Aghajanian GK. Catecholamine receptors on locus coeruleus neurons: Pharmacological characterization. Eur J Pharmacol 1977;44:375–385. [DOI] [PubMed] [Google Scholar]
- 24. Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: An in‐vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol 2008;11:625–639. [DOI] [PubMed] [Google Scholar]
- 25. Piercey MF, Smith MW, Lum‐Ragan JT. Excitation of noradrenergic cell firing by 5‐hydroxytryptamine1A agonists correlates with dopamine antagonist properties. J Pharmacol Exp Ther 1994;268:1297–1303. [PubMed] [Google Scholar]
- 26. Nilsson LK, Schwieler L, Engberg G, Linderholm KR, Erhardt S. Activation of noradrenergic locus coeruleus neurons by clozapine and haloperidol: Involvement of glutamatergic mechanisms. Int J Neuropsychopharmacol 2005;8:329–339. [DOI] [PubMed] [Google Scholar]
- 27. Chernoloz O, El Mansari M, Blier P. Sustained administration of pramipexole modifies the spontaneous firing of dopamine, norepinephrine, and serotonin neurons in the rat brain. Neuropsychopharmacology 2009;34:651–661. [DOI] [PubMed] [Google Scholar]
- 28. El Mansari M, Ghanbari R, Janssen S, Blier P. Sustained administration of bupropion alters the neuronal activity of serotonin, norepinephrine but not dopamine neurons in the rat brain. Neuropharmacology 2008;55:1191–1198. [DOI] [PubMed] [Google Scholar]
- 29. Dong J, Blier P. Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacology (Berl) 2001;155:52–57. [DOI] [PubMed] [Google Scholar]
- 30. Cooper BR, Wang CM, Cox RF, Norton R, Shea V, Ferris RM. Evidence that the acute behavioral and electrophysiological effects of bupropion (Wellbutrin) are mediated by a noradrenergic mechanism. Neuropsychopharmacology 1994;11:133–141. [DOI] [PubMed] [Google Scholar]
- 31. Crochet S, Sakai K. Dopaminergic modulation of behavioral states in mesopontine tegmentum: A reverse microdialysis study in freely moving cats. Sleep 2003;26:801–806. [DOI] [PubMed] [Google Scholar]
- 32. Jones BE, Halaris AE, McIlhany M, Moore RY. Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurons. Brain Res 1977;127:1–21. [DOI] [PubMed] [Google Scholar]
- 33. Phillipson OT. Afferent projections to the ventral tegmental area of Tsai and interfascicular nucleus: A horseradish peroxidase study in the rat. J Comp Neurol 1979;187:117–143. [DOI] [PubMed] [Google Scholar]
- 34. Simon H, Le Moal M, Stinus L, Calas A. Anatomical relationships between the ventral mesencephalic tegmentum – A 10 region and the locus coeruleus as demonstrated by anterograde and retrograde tracing techniques. J Neural Transm 1979;44:77–86. [DOI] [PubMed] [Google Scholar]
- 35. Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD. Immunolocalization of the cocaine‐ and antidepressant‐sensitive l‐norepinephrine transporter. J Comp Neurol 2000;420:211–232. [PubMed] [Google Scholar]
- 36. Geisler S, Zahm DS. Afferents of the ventral tegmental area in the rat‐anatomical substratum for integrative functions. J Comp Neurol 2005;490:270–294. [DOI] [PubMed] [Google Scholar]
- 37. Mejías‐Aponte CA, Drouin C, Aston‐Jones G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: Prominent inputs from medullary homeostatic centers. J Neurosci 2009;29:3613–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liprando LA, Miner LH, Blakely RD, Lewis DA, Sesack SR. Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse 2004;52:233–244. [DOI] [PubMed] [Google Scholar]
- 39. Jones LS, Gauger LL, Davis JN. Anatomy of brain alpha 1‐adrenergic receptors: In vitro autoradiography with [125I]‐heat. J Comp Neurol 1985;231:190–208. [DOI] [PubMed] [Google Scholar]
- 40. Lee A, Wissekerke AE, Rosin DL, Lynch KR. Localization of alpha2C‐adrenergic receptor immunoreactivity in catecholaminergic neurons in the rat central nervous system. Neuroscience 1998;84:1085–1096. [DOI] [PubMed] [Google Scholar]
- 41. Reith ME, Li MY, Yan QS. Extracellular dopamine, norepinephrine, and serotonin in the ventral tegmental area and nucleus accumbens of freely moving rats during intracerebral dialysis following systemic administration of cocaine and other uptake blockers. Psychopharmacology (Berl). 1997;134:309–317. [DOI] [PubMed] [Google Scholar]
- 42. Linner L, Endersz H, Ohman D, Bengtsson F, Schalling M, Svensson TH. Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J Pharmacol Exp Ther 2001;297:540–546. [PubMed] [Google Scholar]
- 43. Shi WX, Pun CL, Smith PL, Bunney BS. Endogenous DA‐mediated feedback inhibition of DA neurons: Involvement of both D(1)‐ and D(2)‐like receptors. Synapse 2000;35:111–119. [DOI] [PubMed] [Google Scholar]
- 44. Grenhoff J, Svensson TH. Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur J Pharmacol 1993;233:79–84. [DOI] [PubMed] [Google Scholar]
- 45. Grenhoff J, Svensson TH. Clonidine modulates dopamine cell firing in rat ventral tegmental area. Eur J Pharmacol 1989;165:11–18. [DOI] [PubMed] [Google Scholar]
- 46. White FJ, Wang RY. Pharmacological characterization of dopamine autoreceptors in the rat ventral tegmental area: Microiontophoretic studies. J Pharmacol Exp Ther 1984;231:275–280. [PubMed] [Google Scholar]
- 47. Aghajanian GK, Bunney BS. Pharmacological characterization of dopamine “autoreceptors” by microiontophoretic single‐cell recording studies. Naunyn Schmiedebergs Arch Pharmacol 1977;297:1–7. [DOI] [PubMed] [Google Scholar]
- 48. Aghajanian GK, Bunney BS. Pharmacological characterization of dopamine “autoreceptors” by microiontophoretic single‐cell recording studies. Adv Biochem Psychopharmacol 1977;16:433–438. [PubMed] [Google Scholar]
- 49. White FJ, Wang RY. A10 dopamine neurons: Role of autoreceptors in determining firing rate and sensitivity to dopamine agonists. Life Sci 1984;34:1161–1170. [DOI] [PubMed] [Google Scholar]
- 50. Gobbi G, Muntoni AL, Gessa GL, Diana M. Clonidine fails to modify dopaminergic neuronal activity during morphine withdrawal. Psychopharmacology (Berl) 2001;158:1–6. [DOI] [PubMed] [Google Scholar]
- 51. Georges F, Aston‐Jones G. Prolonged activation of mesolimbic dopaminergic neurons by morphine withdrawal following clonidine: Participation of imidazoline and norepinephrine receptors. Neuropsychopharmacology 2003;28:1140–1149. [DOI] [PubMed] [Google Scholar]
- 52. Katz N, Guiard BP, El Mansari M, Blier P. Effects of acute and sustained administration of the catecholamine reuptake inhibitor nomifensine on the firing activity of monoaminergic neurons. J Psychopharmacol 2009; [Epub ahead of print]. doi: 10.1177/0269881109348178. [DOI] [PubMed] [Google Scholar]
- 53. Aman TK, Shen RY, Haj‐Dahmane S. D2‐like dopamine receptors depolarize dorsal raphe serotonin neurons through the activation of nonselective cationic conductance. J Pharmacol Exp Ther 2007;320:376–385. [DOI] [PubMed] [Google Scholar]
- 54. Guiard BP, El Mansari M, Blier P. Prospect of a dopamine contribution in the next generation of antidepressant drugs: The triple reuptake inhibitors. Curr Drug Targets 2009;10:1069–1084. [DOI] [PubMed] [Google Scholar]
- 55. Page M, Lucki I. Effects of acute and chronic reboxetine treatment on stress‐induced monoamine efflux in the rat frontal cortex. Neuropsychopharmacology 2002;27:237–247. [DOI] [PubMed] [Google Scholar]
- 56. Baumann, MH , Char GU, De Costa BR, Rice KC, Rothman RB. GBR12909 attenuates cocaine‐induced activation of mesolimbic dopamine neurons in the rat. J Pharmacol Exp Ther 1994;271:1216–1222. [PubMed] [Google Scholar]
- 57. Guiard B, Chenu F, El Mansari M, Blier P. Characterization of the electrophysiological properties of triple reuptake inhibitors on monoaminergic neurons. Int J Neuropsychopharmacol, in press. doi: 10.1017/S1461145710000076. [DOI] [PubMed] [Google Scholar]
- 58. Carboni E, Silvagni A. Dopamine reuptake by norepinephrine neurons: Exception or rule? Crit Rev Neurobiol 2004;16:121–128. [DOI] [PubMed] [Google Scholar]
- 59. Devoto P, Flore G. On the origin of cortical dopamine: Is it a co‐transmitter in noradrenergic neurons? Curr Neuropharmacol 2006;4:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carboni E, Tanda GL, Frau R, Di Chiara G. Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: Evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem 1990;55:1067–1070. [DOI] [PubMed] [Google Scholar]
- 61. Di Chiara G, Tanda GL, Frau R, Carboni E. Heterologous monoamine reuptake: Lack of transmitter specificity of neuron‐specific carriers. Neurochem Int 1992;20:231S–235S. [PubMed] [Google Scholar]
- 62. Tanda G, Carboni E, Frau R, Di Chiara G. Increase of extracellular dopamine in the prefrontal cortex: A trait of drugs with antidepressant potential? Psychopharmacology (Berl) 1994;115:285–288. [DOI] [PubMed] [Google Scholar]
- 63. Yamamoto BK, Novotney S. Regulation of extracellular dopamine by the norepinephrine transporter. J Neurochem 1998;71:274–280. [DOI] [PubMed] [Google Scholar]
- 64. Giros B, Wang YM, Suter S, McLeskey SB, Pifl C, Caron MG. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine‐norepinephrine transporters. J Biol Chem 1994;269:15985–15988. [PubMed] [Google Scholar]
- 65. Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem 1994;269:7124–7130. [PubMed] [Google Scholar]
- 66. Pozzi L, Invernizzi R, Cervo L, Vallebuona F, Samanin R. Evidence that extracellular concentrations of dopamine are regulated by noradrenergic neurons in the frontal cortex of rats. J Neurochem 1994;63:195–200. [DOI] [PubMed] [Google Scholar]
- 67. Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: Evidence from knock‐out mouse lines. J Neurosci 2002;22:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kawahara H, Kawahara Y, Westerink BH. The noradrenaline‐dopamine interaction in the rat medial prefrontal cortex studied by multi‐probe microdialysis. Eur J Pharmacol 2001;27;418:177–186. [DOI] [PubMed] [Google Scholar]
- 69. Devoto P, Flore G, Saba P, et al 6‐Hydroxydopamine lesion in the ventral tegmental area fails to reduce extracellular dopamine in the cerebral cortex. J Neurosci Res 2008;86:1647–1658. [DOI] [PubMed] [Google Scholar]
- 70. Goldman‐Rakic PS. Cellular and circuit basis of working memory in prefrontal cortex of nonhuman primates. Prog Brain Res 1990;85:325–335. [DOI] [PubMed] [Google Scholar]
- 71. Goldman‐Rakic PS. Cellular basis of working memory. Neuron 1995;14:477–485. [DOI] [PubMed] [Google Scholar]
- 72. Clark L, Chamberlain SR, Sahakian BJ. Neurocognitive mechanisms in depression: Implications for treatment. Annu Rev Neurosci 2009;32:57–74. [DOI] [PubMed] [Google Scholar]
- 73. Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 1979;205:929–932. [DOI] [PubMed] [Google Scholar]
- 74. Wang M, Ramos BP, Paspalas CD, et al Alpha2A‐adrenoceptors strengthen working memory networks by inhibiting cAMP‐HCN channel signaling in prefrontal cortex. Cell 2007;129:397–410. [DOI] [PubMed] [Google Scholar]
- 75. Seamans JK, Floresco SB, Phillips AG. D1 receptor modulation of hippocampal‐prefrontal cortical circuits integrating spatial memory with executive functions in the rat. J Neurosci 1998;18:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted‐U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 2007;10:376–384. [DOI] [PubMed] [Google Scholar]
- 77. Arnsten AF. Toward a new understanding of attention‐deficit hyperactivity disorder pathophysiology: An important role for prefrontal cortex dysfunction. CNS Drugs 2009;23(Suppl 1):33–41. [DOI] [PubMed] [Google Scholar]
- 78. Arnsten AF. Catecholamine and second messenger influences on prefrontal cortical networks of “representational knowledge”: A rational bridge between genetics and the symptoms of mental illness. Cereb Cortex 2007;17(Suppl 1):i6–15. [DOI] [PubMed] [Google Scholar]
- 79. Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 2009;10:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V‐VI rat prefrontal cortex neurons in vitro: Modulation of dendritic‐somatic signal integration. J Neurosci 1996;16:1922–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Swanson CJ, Perry KW, Koch‐Krueger S, Katner J, Svensson KA, Bymaster FP. Effect of the attention deficit/hyperactivity disorder drug atomoxetine on extracellular concentrations of norepinephrine and dopamine in several brain regions of the rat. Neuropharmacology 2006;50:755–760. [DOI] [PubMed] [Google Scholar]
- 82. Berridge CW, Devilbiss DM, Andrzejewski ME, et al Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry 2006;60:1111–1120. [DOI] [PubMed] [Google Scholar]
- 83. Arnsten AF, Dudley AG. Methylphenidate improves prefrontal cortical cognitive function through alpha2 adrenoceptor and dopamine D1 receptor actions: Relevance to therapeutic effects in Attention Deficit Hyperactivity Disorder. Behav Brain Funct 2005;1:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Biederman J, Arnsten AF, Faraone SV, et al New developments in the treatment of ADHD. J Clin Psychiatry 2006;67:148–159. [DOI] [PubMed] [Google Scholar]
- 85. Robbins TW, Arnsten AF. The neuropsychopharmacology of fronto‐executive function: Monoaminergic modulation. Annu Rev Neurosci 2009;32:267–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Willner P. The mesolimbic dopamine system as a target for rapid antidepressant action. Int Clin Psychopharmacol 1997;12(Suppl 3):S7–S14. [DOI] [PubMed] [Google Scholar]
- 87. Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety 2000;11:58–65. [DOI] [PubMed] [Google Scholar]
- 88. Lattanzi L, Dell’Osso L, Cassano P, et al Pramipexole in treatment‐resistant depression: A 16‐week naturalistic study. Bipolar Disord 2002;4:307–314. [DOI] [PubMed] [Google Scholar]
- 89. Goldberg JF, Frye MA, Dunn RT. Pramipexole in refractory bipolar depression. Am J Psychiatry 1999;156:798–803. [DOI] [PubMed] [Google Scholar]
- 90. Perugi G, Toni C, Ruffolo G, Frare F, Akiskal H. Adjunctive dopamine agonists in treatment‐resistant bipolar II depression: An open case series. Pharmacopsychiatry 2001;34:137–141. [DOI] [PubMed] [Google Scholar]
- 91. Sporn J, Ghaemi SN, Sambur MR, et al Pramipexole augmentation in treatment of unipolar and bipolar depression: A retrospective chart review. Ann Clin Psychiatry 2000;12:137–140. [DOI] [PubMed] [Google Scholar]
- 92. DeBattista C, Solvason HB, Breen JA, Schatzberg AF. Pramipexole augmentation of a selective serotonin reuptake inhibitor in the treatment of depression. J Clin Psychopharmacol 2000;20:274–275. [DOI] [PubMed] [Google Scholar]
- 93. Cassano P, Lattanzi L, Soldani F, Navari S, Battistini G, Gemignani A, Cassano GB. Pramipexole in treatment‐resistant depression: An extended follow‐up. Depress Anxiety 2004;20:131–138. [DOI] [PubMed] [Google Scholar]
- 94. Carter AJ, Müller RE. Pramipexole, a dopamine D2 autoreceptor agonist, decreases the extracellular concentration of dopamine in vivo. Eur J Pharmacol 1991;200:65–72. [DOI] [PubMed] [Google Scholar]
- 95. Pitts DK, Wang L, Kelland MD, Freeman AS, Chiodo LA. Repeated stimulation of dopamine D2‐like receptors: Reduced responsiveness of nigrostriatal and mesoaccumbens dopamine neurons to quinpirole. J Pharmacol Exp Ther 1995;275:412–421. [PubMed] [Google Scholar]
- 96. Chernoloz O, El Mansari M, Blier P. Effects of pramipexole administration on monoaminergic neurotransmission. European Neuropsychopharmacol 2009;19(Suppl 1):S8. [Google Scholar]
- 97. Gillings D, Grizzle J, Koch G, et al Pooling 12 nomifensine studies for efficacy generalizability. J Clin Psychiatry 1984;45:78–84. [PubMed] [Google Scholar]
- 98. Bremner JD, Abrahams LM, Crupie JE, McCawley A, Proctor RC, Sathananthan GL. Multicenter double‐blind comparison of nomifensine and imipramine for efficacy and safety in depressed outpatients. J Clin Psychiatry 1984;45:56–59. [PubMed] [Google Scholar]
- 99. Szabo ST, Blier P. Effect of the selective noradrenergic reuptake inhibitor reboxetine on the firing activity of noradrenaline and serotonin neurons. Eur J Neurosci 2001;13:2077–2087. [DOI] [PubMed] [Google Scholar]
- 100. Szabo ST, Blier P. Effects of serotonin (5‐hydroxytryptamine, 5‐HT) reuptake inhibition plus 5‐HT(2A) receptor antagonism on the firing activity of norepinephrine neurons. J Pharmacol Exp Ther 2002;302:983–991. [DOI] [PubMed] [Google Scholar]
- 101. Hatanaka K, Yatsugi S, Yamaguchi T. Effect of acute treatment with YM992 on extracellular norepinephrine levels in the rat frontal cortex. Eur J Pharmacol 2000;395:31–36. [DOI] [PubMed] [Google Scholar]
- 102. Baraban JM, Aghajanian GK. Suppression of firing activity of 5‐HT neurons in the dorsal raphe by alpha‐adrenoceptor antagonists. Neuropharmacology 1980;19:355–363. [DOI] [PubMed] [Google Scholar]
- 103. Martin‐Ruiz R, Ugedo L, Honrubia MA, Mengod G, Artigas F. Control of serotonergic neurons in rat brain by dopaminergic receptors outside the dorsal raphe nucleus. J Neurochem 2001;77:762–775. [DOI] [PubMed] [Google Scholar]
- 104. Mongeau R, Blier P, De Montigny C. The serotonergic and noradrenergic systems of the hippocampus: Their interactions and the effects of antidepressant treatments. Brain Res Brain Res Rev 1997;23:145–195. [DOI] [PubMed] [Google Scholar]
- 105. Szabo ST, Blier P. Effects of the selective norepinephrine reuptake inhibitor reboxetine on norepinephrine and serotonin transmission in the rat hippocampus. Neuropsychopharmacology 2001;25:845–857. [DOI] [PubMed] [Google Scholar]
- 106. Dremencov E, El Mansari M, Blier P. Distinct electrophysiological effects of paliperidone and risperidone on the firing activity of rat serotonin and norepinephrine neurons. Psychopharmacology (Berl) 2007;194:63–72. [DOI] [PubMed] [Google Scholar]
- 107. Chernoloz O, El Mansari M, Blier P. Electrophysiological studies in the rat brain on the basis for aripiprazole augmentation of antidepressants in major depressive disorder. Psychopharmacology (Berl) 2009;206:335–344. [DOI] [PubMed] [Google Scholar]
- 108. Zisook S, Rush AJ, Haight BR, Clines DC, Rockett CB. Use of bupropion in combination with serotonin reuptake inhibitors. Biol Psychiatry 2006;59:203–210. [DOI] [PubMed] [Google Scholar]
- 109. Trivedi MH, Fava M, Wisniewski SR, et al Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006;354:1243–1252. [DOI] [PubMed] [Google Scholar]
- 110. Ascher JA, Cole JO, Colin JN, et al Bupropion: A review of its mechanism of antidepressant activity. J Clin Psychiatry 1995;56:395–401. [PubMed] [Google Scholar]
- 111. Ferris RM, Beaman OJ. Bupropion: A new antidepressant drug, the mechanism of action of which is not associated with down‐regulation of postsynaptic beta‐adrenergic, serotonergic (5‐HT2), alpha 2‐adrenergic, imipramine and dopaminergic receptors in brain. Neuropharmacology 1983;22:1257–1267. [DOI] [PubMed] [Google Scholar]
- 112. Dwoskin LP, Rauhut AS, King‐Pospisil KA, Bardo MT. Review of the pharmacology and clinical profile of bupropion, an antidepressant and tobacco use cessation agent. CNS Drug Reviews 2006;12:178–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Meyer JH, Goulding VS, Wilson AA, Hussey D, Christensen BK, Houle S. Bupropion occupancy of the dopamine transporter is low during clinical treatment. Psychopharmacology (Berl) 2002;163:102–105. [DOI] [PubMed] [Google Scholar]
- 114. Learned‐Coughlin SM, Bergstorm M, Savitcheva I, Ascher J, Schmith VD, Langstorm B. In vivo activity of bupropion at the human dopamine transporter as measured by positron emission tomography. Biol Psychiatry 2003;54:800–805. [DOI] [PubMed] [Google Scholar]
- 115. Kugaya A, Seneca NM, Snyder PJ, et al Changes in human in vivo serotonin and dopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology 2003;28:413–420. [DOI] [PubMed] [Google Scholar]
- 116. Argyelan M, Szabo Z, Kanyo B, Tanacs A, Kovacs Z, Janka Z, Pavics L. Dopamine transporter availability in medication free and in bupropion treated depression: A 99mTc‐TRODAT‐1 SPECT study. J Affect Disord 2005;89:115–123. [DOI] [PubMed] [Google Scholar]
- 117. Gobbi G, Slater S, Boucher N, Debonnel G, Blier P. Neurochemical and psychotropic effects of bupropion in healthy male subjects. J Clin Psychopharmacol 2003;23:233–239. [DOI] [PubMed] [Google Scholar]
- 118. Baraban JM, Wang RY, Aghajanian G. Reserpine suppression of dorsal raphe neuronal firing: Mediation by adrenergic system. Eur J Pharmacol 1978;52:27–36. [DOI] [PubMed] [Google Scholar]
- 119. Svensson TH, Bunney BS, Aghajanian GK. Inhibition of both noradrenergic and serotonergic neurons in brain by the alpha‐adrenergic agonist clonidine. Brain Res 1975;92:291–306. [DOI] [PubMed] [Google Scholar]
- 120. Blier P, De Montigny C, Azzaro AJ. Modification of serotonergic and noradrenergic neurotransmissions by repeated administration of monoamine oxidase inhibitors: Electrophysiological studies in the rat central nervous system. J Pharmacol Exp Ther 1986;237:987–994. [PubMed] [Google Scholar]
- 121. Ghanbari R, El Mansari M, Blier P. Electrophysiological effects of sustained administration of bupropion on serotonergic and noradrenergic neurotransmission in the rat hippocampus. Biol Psychiatry 2009;65(8S):545. [DOI] [PubMed] [Google Scholar]
- 122. Nomikos GG, Damsma G, Wenkstern D, Fibiger HC. Effects of chronic bupropion on interstitial concentrations of dopamine in rat nucleus accumbens and striatum. Neuropsychopharmacology 1992;7:7–14. [PubMed] [Google Scholar]
- 123. Li SX, Perry KW, Wong DT. Influence of fluoxetine on the ability of bupropion to modulate extracellular dopamine and norepinephrine concentrations in three mesocorticolimbic areas of rats. Neuropharmacology 2002;42:181–190. [DOI] [PubMed] [Google Scholar]
- 124. Piacentini MF, Clinckers R, Meeusen R, Sarre S, Ebinger G, Michotte Y. Effect of bupropion on hippocampal neurotransmitters and on peripheral hormonal concentrations in the rat. J Appl Physiol 2003;95:652–656. [DOI] [PubMed] [Google Scholar]
- 125. Nomikos GG, Damsma G, Wenkstern D, Fibiger HC. In vivo characterization of locally applied dopamine uptake inhibitors by striatal microdialysis. Synapse 1990;6:106–112. [DOI] [PubMed] [Google Scholar]
- 126. Stamford JA, Kruk ZL, Millar J. Dissociation of the actions of uptake blockers upon dopamine overflow and uptake in the rat nucleus accumbens: In vivo voltammetric data. Neuropharmacology 1989;28:1383–1388. [DOI] [PubMed] [Google Scholar]
- 127. Caruana DA, Sorge RE Stewart J, Chapman CA. Dopamine has bidirectional effects on synaptic responses to cortical inputs in layer II of the lateral entorhinal cortex. J Neurophysiol 2006;96:3006–3015. [DOI] [PubMed] [Google Scholar]
- 128. Schotte A, Janssen PF, Gommeren W, et al Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996;124:57–73. [DOI] [PubMed] [Google Scholar]
- 129. Calabrese JR. Efficacy of quetiapine monothreapy in bipolar I and II depression: A double‐blind, placebo‐controlled study (the BOLDER II study). J Clin Psychopharmacol 2006;26:600–609. [DOI] [PubMed] [Google Scholar]
- 130. Thase ME, Macfadden W, Weisler RH, et al A randomized, double‐blind, placebo‐controlled trial of quetiapine in the treatment of bipolar I and II depression. Am J Psychiatry 2005;162:1351–1360. [DOI] [PubMed] [Google Scholar]
- 131. Young A, McElroy S, Chang W, Olausson B, Paulsson B, Brecher M, Nordenhem A. A double‐blind, placebo‐controlled study of quetiapine and paroxetine as monotherapy in adults with bipolar depression (EMBOLDEN II). Int J Neuropsychopharmacol 2008;11(Suppl 1):S186. [DOI] [PubMed] [Google Scholar]
- 132. Millan MJ, Gobert A, Rivet JM, et al Mirtazapine enhances frontocortical dopaminergic and corticolimbic adrenergic, but not serotonergic, transmission by blockade of alpha2‐adrenergic and serotonin2C receptors: A comparison with citalopram. Eur J Neurosci 2000;12:1079–1095. [DOI] [PubMed] [Google Scholar]
- 133. Haddjeri N, Blier P, De Montigny C. Acute and long‐term actions of the antidepressant drug mirtazapine on central 5‐HT neurotransmission. J Affect Disord 1998;51:255–266. [DOI] [PubMed] [Google Scholar]
- 134. Pira L, Mongeau R, Pani L. The atypical antipsychotic quetiapine increases both noradrenaline and dopaime release in the rat prefrontal cortex. Eur J Pharmacol 2004;504:61–64. [DOI] [PubMed] [Google Scholar]
- 135. Shelton RC, Williamson DJ, Corya SA, et al Olanzapine/fluoxetine combination for treatment‐resistant depression: A controlled study of SSRI and nortriptyline resistance. J Clin Psychiatry 2005;66:1289–1297. [DOI] [PubMed] [Google Scholar]
- 136. Seager MA, Huff KD, Barth VN, Phebus LA, Rasmussen K. Fluoxetine administration potentiates the effect of olanzapine on locus coeruleus neuronal activity. Biol Psychiatry 2004;55:1103–1109. [DOI] [PubMed] [Google Scholar]
- 137. Seager MA, Barth VN, Phebus LA, Rasmussen K. Chronic coadministration of olanzapine and fluoxetine activates locus coeruleus neurons in rats: Implications for bipolar disorder. Psychopharmacology (Berl) 2005;181:126–133. [DOI] [PubMed] [Google Scholar]
- 138. Dremencov E, El Mansari M, Blier P. Noradrenergic augmentation of escitalopram response by risperidone: Electrophysiologic studies in the rat brain. Biol Psychiatry 2007;61:671–678. [DOI] [PubMed] [Google Scholar]
- 139. Jensen NH, Rodriguez RM, Caron MG, Wetsel WC, Rothman RB, Roth BL. N‐desalkylquetiapine, a potent norepinephrine reuptake inhibitor and partial 5‐HT1A agonist, as a putative mediator of quetiapine's antidepressant activity. Neuropsychopharmacology 2008;33:2303–2312. [DOI] [PubMed] [Google Scholar]
- 140. Dennis T, L’Heureux R, Carter C, Scatton B. Presynaptic alpha‐2 adrenoceptors play a major role in the effects of idazoxan on cortical noradrenaline release (as measured by in vivo dialysis) in the rat. J Pharmacol Exp Ther 1987;241:642–649. [PubMed] [Google Scholar]
- 141. Blier P, Ward P, Trembley P, Laberge L, Hebert C, Bergeron . Combination of antidepressant medications from treatment initiation for major depressive disorder: A double‐blind randomized study. Am J Psychiatry 2010;167:281–288. [DOI] [PubMed] [Google Scholar]
- 142. Papakostas GI, Worthington JJ 3rd., Iosifescu DV, et al The combination of duloxetine and bupropion for treatment‐resistant major depressive disorder. Depress Anxiety 2006;23:178–181. [DOI] [PubMed] [Google Scholar]
- 143. Berman RM, Fava M, Thase ME, et al Aripiprazole augmentation in major depressive disorder: A double‐blind, placebo‐controlled study in patients with inadequate response to antidepressants. CNS Spectr 2009;14:197–206. [DOI] [PubMed] [Google Scholar]
- 144. Mahmoud RA, Pandina GJ, Turkoz I, Kosik‐Gonzalez C, Canuso CM, Kujawa MJ, Gharabawi‐Garibaldi GM. Risperidone for treatment‐refractory major depressive disorder: A randomized trial. Ann Intern Med 2007;147:593–602. [DOI] [PubMed] [Google Scholar]
- 145. Keitner GI, Garlow SJ, Ryan CE, Ninan PT, Solomon DA, Nemeroff CB, Keller MB. A randomized, placebo‐controlled trial of risperidone augmentation for patients with difficult‐to‐treat unipolar, non‐psychotic major depression. J Psychiatr Res 2009;43:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]