

Abstract
Mutations in intermediate filament protein genes are responsible for a number of inherited genetic diseases including skin blistering diseases, corneal opacities, and neurological degenerations. Mutation of the arginine (Arg) residue of the highly conserved LNDR motif has been shown to be causative in inherited disorders in at least four different intermediate filament (IF) proteins found in skin, cornea, and the central nervous system. Thus this residue appears to be broadly important to IF assembly and/or function. While the genetic basis for these diseases has been clearly defined, the inability to determine crystal structure for IFs has precluded a determination of how these mutations affect assembly/structure/function of IFs. To investigate the impact of mutation at this site in IFs, we have mutated the LNDR to LNDS in vimentin, a Type III intermediate filament protein, and have examined the impact of this change on assembly using electron paramagnetic resonance. Compared with wild type vimentin, the mutant shows normal formation of the coiled coil dimer, with a slight reduction in the stability of the dimer in rod domain 1. Probing the dimer-dimer interactions shows the formation of normal dimer centered on residue 191 but a failure of dimerization at residue 348 in rod domain 2. These data point toward a specific stage of assembly at which a common disease-causing mutation in IF proteins interrupts assembly.
The intermediate filament (IF)1 protein gene family consists of about 60 members at present. While primary sequence among the family members shows a considerable degree of sequence variation, the vast majority of IF proteins show conservation of a predicted domain structure. This structure consists of a central rod domain whose predicted secondary structure is well conserved, and head and tail domains, where both size and primary sequence, are more variable. While the predicted secondary structure of the central rod domain is conserved, there is much primary sequence variability except at two small motifs located at either end of the central rod domain. At these sites, sequence conservation has been quite strong. These two motifs have been referred to as the “rod initiation” and “rod termination” motifs. Not surprisingly, a disproportionate fraction of human disease-causing mutations in IF proteins are found in these highly conserved motifs (1–11).
In the early 1990s, three lines of evidence independently identified IF protein genes as the site of mutations leading to epidermolysis bullosa simplex (EBS) and other skin blistering diseases in humans such as epidermolysis hyperkeratosis (3, 5, 12–19). First, Fuchs and co-workers (20) working in cell culture and mouse systems showed that cytokeratin mutations gave rise to EBS-like defects in mice. Second, genetic linkage analysis in humans indicated that keratin genes were involved in skin blistering diseases (1). Third, keratin mutants were identified on the basis of abnormal antibody binding, caused by changes in the primary sequence of epidermal IF protein (13). Subsequent characterization of additional EBS cases revealed a hotspot for mutations at the conserved motif LNDR, located at the beginning of the central rod domain. Commonly, a point mutation in the IF gene led to an Arg → His (12) or Arg → Cys mutation in this motif (12, 21). Subsequently, mutations in the cornea-specific keratins K3 and K12 at the same LNDR sequence were shown to segregate with a corneal dystrophy phenotype (22–24). Most recently, the same region in GFAP has been shown to be the site of mutations leading to the neurodegenerative Alexander disease (25). Thus, the fourth residue of this LNDR motif appears to be of critical importance to several IF proteins, from multiple classes of IF.
The mechanism of these genetic mutations seems clear. Alteration of the arginine codon (CGN) is consistent with the hypothesis that CpG dinucleotides are sites of methylation-induced deamidation of cytosine, leading to a Cys → Thr transition (CGY → TGY cysteine codon) (26). However, the structural impact of this Arg → Cys substitution on IF assembly and structure remains poorly defined, as IF proteins and IFs have not been crystallized. Whether the result of in vitro mutagenesis or random chance, mutations in keratin genes typically have been studied in vitro by 1) analysis of the assembly characteristics of the mutant proteins and 2) the ability of the mutant proteins to assemble into intermediate filament networks in transfected cells. In general, there is a good correlation between the severity of skin blistering seen in a clinically affected individual and the magnitude of assembly abnormalities seen when the mutant protein is analyzed in vitro (19). Thus, mutants that fail to form filaments in vitro and fail to integrate into cellular IF networks in cell culture produce the worst cases of skin blistering. Analysis of the effect of specific mutations on keratin assembly has been described by Steinert and co-workers (27, 28) who designed an experimental protocol based on the cross linking between proteins at different urea concentrations. Cross-linking between proteins reflects the proximity of the cross linking moieties and thus can be used to establish whether normal interactions have/have not occurred during assembly. Specifically, comparison of cross-links in mutant proteins to those formed in wild type proteins can be used to infer whether distances between cross-linking moieties have changed as a result of the mutation. Since IF proteins will undergo stepwise assembly when dialyzed out of high concentrations of urea or guanidine, these cross-linking studies can be performed on intermediates of assembly, providing data relevant to the stage of assembly affected by the mutation (29).
Mehrani et al. (27) showed specifically that K14 LNDR mutated to LNDL was unable to form IFs, unable to assemble into existing IF networks in cells, and was significantly altered in the tetramer configuration. Lys14 LNAR was similarly unable to form filaments in vitro and unable to integrate into IF networks but exhibited slightly less stability than the wild type. They summarize the characterization of these substitutions in the Asp and Arg locations of LNDR as significantly destabilizing the two-molecule entities (tetramer).
Following the demonstration of the applicability of site-directed spin labeling electron paramagnetic resonance (SDSL EPR) to the study IF structure (30, 31), we began a series of experiments designed to test the hypothesis that EPR can be used to identify the effects of mutations on IF assembly. To perform these experiments, EBS-like mutations (LNDR → LNDS and NDR → LNDC) were engineered into human vimentin (1). In vitro assembly verifies that assembly is aborted very early in the process with both mutants failing to assemble into 10-nm filaments. Because the spin labeling protocol that we employ targets cysteine residues, we have used a vimentin LNDR → LNDS mutant to study the impact of mutations at this site on assembly. In previous work we have identified specific “reporter” residues within vimentin that, when spin-labeled, reveal whether specific stages of assembly have occurred and when in the course of assembly they occur. We can therefore discriminate between mutations that affect initial coiled coil dimer formation, or subsequent dimer-dimer interactions. Using this approach we report the impact of EBS-like mutations on the assembly of the Type III IF protein vimentin.
MATERIALS AND METHODS
Vimentin characterization, mutation, cloning, expression, purification, and spin labeling were described in detail in a previous reports (30, 31). In brief, the spin label is ultimately attached to cysteine residues that are targeted to specific sites in vimentin. Cysteine codons were introduced into the vimentin expression construct (generously provided by Dr. Roy Quinlan, University of Durham, Durham, UK) using the Stratagene QuikChange kit (Stratagene, La Jolla, CA). Combinations of mutations (for example, Ser113 plus Cys191) were created, where possible, by cloning of restriction fragments, each containing one mutation, together into an expression vector. If not possible, then mutagenesis was performed in a sequential fashion. Sequence changes were verified by DNA sequencing. Mutant vimentin was produced by bacterial expression and purified from inclusion bodies using high/low salt washes, and chromatography. Spin labeling was accomplished by incubation of the purified vimentin in 100 μM tris-(2-carboxyethyl)phosphine (Molecular Probes, Eugene, OR) followed by 500 μM O-87500 (Toronto Research Chemicals, Toronto, Canada). Unincorporated spin label was removed by CM-Sepharose chromatography using a GE Healthcare FPLC (30, 31). Labeled proteins were stored long term at −80 °C.
Filament assembly was performed either as a single step dialysis against filament assembly buffer or as a stepwise process following the protocol of Carter et al. (32) Briefly, labeled proteins were solubilized in 8 M urea, which was then removed by dialysis, either in a single step procedure, or where indicated, in a stepwise process through progressively reduced concentrations of urea, followed by low ionic strength Tris, followed by the addition of NaCl and MgCl2. Filament assembly was verified by electron microscopy of negatively stained samples.
EPR was conducted on a JEOL X-band spectrometer equipped with a loop gap resonator. Approximately 4 μl of sample, at a concentration of 25–75 μM protein, were placed in a sealed quartz capillary tube. Spectra were acquired at 20–22 °C with a single 60-s scan over 100 G at a microwave power of 2 milliwatts and a modulation amplitude optimized to the natural line width of the attached nitroxide.
RESULTS
Electron microscopy cysteine-free vimentin (Cys328 → Ser) used as a starting point for subsequent studies shows long uniform filaments, which are morphologically indistinguishable from wild type vimentin (Fig. 1A). In contrast, vimentin bearing the LNDR → LNDS mutation failed to form recognizable filaments (Fig. 1B). In these preparations, assembly was aborted at a very early stage, yielding uniformly small “rodlets.” Vimentin bearing the LNDC mutation similarly caused a very early failure in the assembly process, yielding aggregates of small particles (not shown).
Fig. 1. Electron microscopy of wild type and mutant vimentin assembled in vitro.
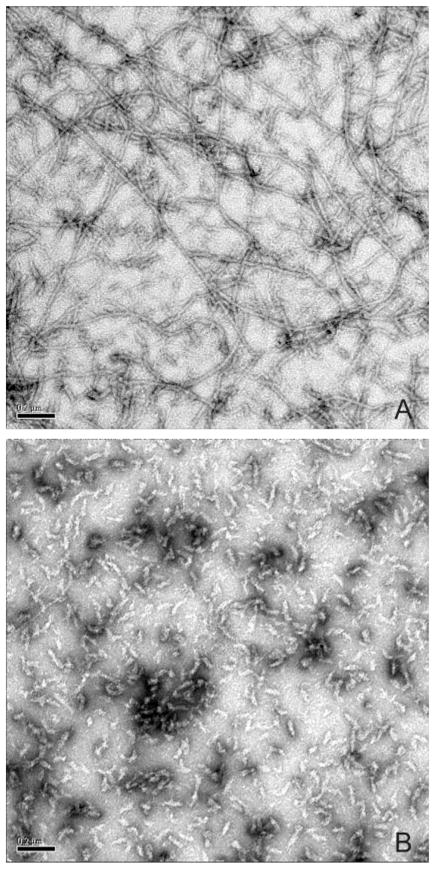
A, control showing a negatively stained preparation of cysteine-free vimentin, used as a starting point for subsequent studies. Long, uniformly shaped 10-nm filaments are evident. B, negatively stained preparation of vimentin bearing the LNDS mutation. Filament assembly is blocked at a very early stage, resulting in a population of small, uniformly sized particles.
We then used site-directed spin labeling followed by EPR spectroscopy to examine the structural and assembly consequences of the R113S (LNDR to LNDS) mutation (Fig. 2). We have previously shown that SDSL EPR yields structural information at several levels in intact IFs, as well as in assembly intermediates that form during in vitro assembly. Depending on the specific site that is labeled, EPR can monitor the assumption of α helical structure, the formation of coiled coil dimers, and the formation of subsequent higher order interactions between dimers, either in intact filaments or at specific stages during in vitro assembly that occur as filament protein is dialyzed out of 8 M urea. We approached this by creating the R113S mutation and then placing spin labels at different locations within the mutant protein to monitor the impact that this mutation produced at the spin labeled site (a schematic indicating the relative locations of these mutants is presented in Fig. 3).
Fig. 2. EPR spectra of spin-labeled vimentin at selected positions.

The residue location of the spin label is indicated for each row of spectra. A, spectra from the indicated position when vimentin contains the native residue at position 113 (Arg113). B, spectra of the spin-labeled residues in combination with the LNDR mutation Arg113 → Ser. In A and B, each spectrum represents the signal in the presence of 2–6 M urea (see key for colored lines), and all spectra are normalized to an identical concentration of spin-labeled protein. C, comparison of the EPR spectra at 2 M urea when protein contains either native Arg113 (black trace) or the LNDR mutation Arg113 → Ser (red trace). To facilitate a comparison, the spectra in C are magnified by a factor of 2 over the spectra in A and B.
Fig. 3. Schematic of vimentin predicted domain structure and spin label placement.

A, coils prediction (44) of human vimentin. The boundaries of the central rod domain, as well as the linker regions, are based on predictions of coiled coil structure and yield a domain structure depicted in A. B, schematic of a vimentin monomer, with LNDS motif, and the specific spin label reporter residues indicated at their approximate locations. Note that residues 131, 178, and 333 are a,d positions in the heptad and are located at the interface between two monomers. Residues 191 and 348 are non a,d positions and are located on the exterior surface of a dimer. C, schematic indicating dimer formation. D, schematic indicating that in the LNDS mutant, the 191: 191 dimer forms, while the 348:348 dimer does not, as assessed by EPR using these residues as reporters for activity at these sites.
To examine the impact of the R113S mutation on α helical, coiled coil dimer formation, we first evaluated the spectral changes as a function of urea concentration of protein containing the wild type Arg113, with the spin label located at either position 178 (a d position in rod domain 1) or 333 (a d position in rod domain 2). Spin labels at these sites provide data about 1) secondary structure (α helix formation) and 2) coiled coil formation, since these two residues would be within 2 nm of each other in a coiled coil dimer. As demonstrated previously (30, 31), a,d positions within a coiled coil generate a characteristic immobilized and dipolar-broadened spectrum when the urea concentration is lowered to 2 M. In the wild type vimentin, a tightly packed environment at position 178 (rod 1) occurs earlier and is almost completely achieved at 4 M urea, whereas a strongly broadened spectrum from position 333 (rod 2) is not achieved until 2 M urea (Fig. 2a). The magnified spectra for spin labeled 178 and 333 at 2 M urea are shown in Fig. 2c. This observation suggests that in wild type vimentin, coiled coil formation occurs more readily in rod domain 1B in the region surrounding position 178 than in rod domain 2 near position 333.
We then repeated these studies but now using vimentin bearing the R113S mutation. In the presence of the R113S mutation, position 178 remains flexible at 4 M urea (Fig. 2b), indicating the LNDS mutation causes a slight perturbation in coiled coil formation in rod 1B. In contrast, the increased broadening at position 333 in rod 2 occurs just as readily in the LNDS mutant as it does in vimentin that contains the wild type Arg113 (Fig. 2b). This suggests that the presence of the mutation at residue 113 (rod domain 1) has little impact on the formation of the coiled coil dimer in rod domain 2 but does affect the ability of rod domain 1 to undergo normal α helical coiled coil assembly.
To further investigate the impact of this mutation on early assembly, we placed a spin label closer to the LNDR motif at position 131. Position 131 is predicted to be a (d) position at the end of rod 1A. As shown in Fig. 2A, vimentin with a label located at 131 does not display the characteristic broad spectrum of an a/d position (see “Discussion”). A slight effect of the LNDS mutation is observed at position 131. As shown in Fig. 2C, the spectrum of spin-labeled 131 in 2 M urea is somewhat sharper, suggesting that the R113S mutation may impart further destabilization to this region of the protein.
We have shown in previous work that residues 191 (rod 1) and 348 (rod), both of which are non a,d, positions and therefore on the outside of the coiled coil dimer (see Fig. 3), can be used to monitor the formation of interactions between vimentin dimers during filament assembly studies (30, 31). These studies established that dimers interact at residue 191 (rod domain 1) before interactions occur at residue 348 (rod domain 2) thus establishing a sequence of dimer-dimer interactions during in vitro assembly. To evaluate the impact of the R113S mutation on these interactions, we spin-labeled proteins individually at both 191 and 348. Examination of mutant protein labeled at residue 348 (an e position in rod 2) shows that, in the range of 6–3 M urea, the R113S mutant behaves similarly to the wild type protein (Fig. 2, A versus B). This is consistent with the data derived from mutants labeled at residue 333, indicating normal secondary structure in rod domain 2. However, as described below, the R113S mutation produces a loss of the dipolar broadening of spin label placed at residue 348 that arises at 2 M urea in wild type protein. This suggests that the approximation of two dimers centered around residue 348 fails to occur in a normal manner.
While the LNDS mutation causes a small fraction of the spin labels at 191 to remain highly mobile at low urea levels, the level of dipolar broadening compared with protein with native Arg113 is unchanged, indicating that a magnetic interaction occurs between labels located at 191, as R113S vimentin dimers assemble into higher ordered structures. Thus the A11 alignment, as measured by dipolar interaction at 191, is not detectably perturbed by the R113S mutation. In contrast, evidence for A22 alignment, as reported by labels placed at position 348, is completely eliminated in vimentin containing the R113S mutation. This difference is highlighted in Fig. 2C, where the normalized 2 M urea spectra of spin-labeled 348 with and without the LNDS mutations are compared. From this we conclude that the R113S mutation permits the formation of coiled coil dimers in rod 2 but that the mutation has a demonstrable impact on the dimer-dimer interactions that should occur in rod 2.
DISCUSSION
The helix initiation and termination motifs of IF proteins have proven to be “hotspots” for mutations that result in a disease phenotype. In the helix initiation motif (LNDR) a frequent cause of disease is mutation of the Arg residue, commonly to a Cys or His. Mutation of this site has proven causative for human diseases in the Type I cytokeratins (e.g. skin blistering and corneal dystrophies) but also in the Type III IF protein GFAP as well (1, 3, 5, 14, 16, 19, 21, 22, 25, 33). Thus this specific site appears to be critical to normal assembly in multiple IF proteins from more than one class, suggesting that the data derived from study of this mutation may be broadly applicable. An interesting side note is that the lens-specific intermediate filament protein, CP49, shows striking divergence at this motif (LGGC) that includes a Cys in the fourth position (LGGC) (34, 35).
While the exact nucleic acid changes resulting in these diseases have been identified, the impact of each mutation on assembly, structure, and function of the IFs has proven far more difficult to define. This difficulty largely derives from the inability to develop crystals of intact IF proteins or of intact IFs. While progress has been made in determining crystal structure of fragments of the IF protein vimentin (36, 37), the capacity to study structure in intact proteins, or in intact IFs, remains elusive. Thus, the means by which a specific disease-causing mutation alters or aborts assembly of IFs has remained difficult to approach experimentally. Because IFs must integrate into the biology of the cell, EBS-type mutations may result from changes that are permissive to filament assembly and/or from mutations that block the linkage between filaments and other cellular structures. Plectin, for example, links IFs to the plasma membrane. Mutations in the plectin gene that alter interactions with IFs have been identified that also result in skin blistering phenotypes (38–40). Another example is the LNDR → LNDH mutation that has also been identified as an EBS causing mutation (12). In vitro this mutant readily assembles into long, normal IFs, perhaps better than wild type filaments, suggesting the disease phenotype results from another mechanism and not the failure to form filaments (41).
In the absence of the crystal structure of IFs, our understanding of the structural changes caused by these many disease causing mutations is likely to result from the integration of data from several different approaches such as in vitro assembly, cross-linking, analytical ultracentrifugation, x-ray crystallography, and SDSL EPR.
Coulombe and co-workers (29) used cross-linking and chromatography under different denaturing conditions to characterize a proline mutant of keratin 16 and concluded that the effect of the mutant was to destabilize keratin tetramers. Steinert and co-workers (28) have developed cross-linking protocols that measure differences in the cross-linking behavior between mutant and wild type cytokeratins. These allow for the inference of how assembly and structure may be affected by these mutations (28).
Analysis of the LNDR → LNDK mutation in K14 suggested a decrease in the stability of the keratin A11 tetramer in urea (27). When assembled in vitro, this mutant forms shorter than normal filaments. Taken together, the authors conclude that decreased A11 tetramer stability is responsible for filament abnormalities although they leave open the possibility that these residues are important for A12 or Acn interactions (27).
Molecular modeling of rod 1A structures and the effect of known mutations leading to EBS have suggested that LNDR → LNDS mutation in Lys14 produces a much greater distortion than the typical EBS mutant Lys14 LNDR → LNDC (42). The computer program predicts a decreased helix stability for LNDS mutation, and LNDC remains essentially unchanged. The net effect of these structural changes on higher order structures (dimers or tetramers) or filament assembly were not elucidated by the modeling. Our EM observations of vimentin show similarities between the small aggregates formed by vim Cys113 and vim Ser113, though they were not identical.
We previously showed the ability of site directed spin labeling and electron paramagnetic resonance (SDSL EPR) to contribute to the study of IFs and IF assembly (30, 31). Using this approach we have defined specific regions of IF proteins that are involved in coiled coil dimer formation and defined the proximity of specific residues between monomers and between higher order multimers. The approach has also proven successful in defining the progression of assembly steps that occur during in vitro assembly, sequencing the formation of α helix, coiled coil dimer, and the sequence of specific dimer-dimer interactions.
In this report we sought to determine whether SDSL EPR could be used to define changes in IF architecture and assembly that are induced by disease-causing mutations. We elected to study the mutation of the fourth residue of the well conserved LNDR helix initiation motif, largely because mutation at this site has proven to be disease causing in several IF proteins from at least two IF classes. This suggests that this specific site is of relatively broad importance to IF assembly. We introduced the mutation in vimentin as it has been the protein from which we have gathered all initial SDSL EPR data.
In earlier work (30, 31) we showed that EPR spectra of spin labels placed at residues 178, 191, 333, and 348 display line shapes indicative of their positions within the heptad repeat, consistent with their proximity to one another in a coiled coil dimer. These residues thus serve as good indicators for the assumption/presence of coiled coil dimer at these sites. To evaluate the consequences of the LNDR → LNDS mutation we targeted spin labels to these characterized sites, as well as a site closer to the mutation in rod 1A (residue 131), and compared the spectra between wild type and R113S mutants.
The EPR spectra suggest that coiled coil dimer formation appears to proceed almost identically in the wild type and R113S mutation as monitored by spin labels at residues 178 (rod domain 1), 191 (rod domain 1), 333 (rod domain 2), and 348 (rod domain 2). A subtle difference in the EPR spectra of spin labels placed at residues 178 suggests that the transition to fully ordered structure is slightly delayed in the mutant protein, occurring somewhat later in the dialysis process than seen for the wild type. This slower attainment normal structure may be the result of decreased stability of the coiled coil dimer in this region of the mutant protein. However, upon completion of dialysis, the spectra for both mutant and wild type proteins were indistinguishable from one another, despite dramatic differences in the outcome of filament assembly between wild type and mutant proteins.
In previous work we identified several residues that serve as reporters of interactions between a given vimentin dimer and its nearest neighbors in intact filaments. Residues 191 and 348, for example, are sites of anti-parallel overlap between one vimentin dimer and other adjacent dimers in intact filaments (30, 31). We therefore compared the EPR spectra of wild type and R113S mutant vimentin spin-labeled at these sites. No differences were seen between the EPR spectra from wild type and mutant vimentin for spin labels placed at residue 191. This suggested that the mutation did not alter the correct positioning of adjacent dimers centered at this residue in rod domain 1.
In contrast, comparison of wild type and mutant proteins labeled at residue 348, an indicator of anti-parallel overlap centered around this site in rod 2, revealed marked differences between wild type and R113S mutants. EPR spectra of the mutant showed no indication that proper alignment of these regions had occurred. Whether this failure to align was drastic or simply sufficient to displace the residue beyond the 2-nm limits of sensitivity for EPR is not yet known. In previous work we showed that the association between IF dimers at residue 191 (rod domain 1) occurs prior to that which occurs at 348 (rod domain 2), at least during in vitro assembly. Thus the data reported here would suggest that the initial lateral association between dimers centered around rod 1 is not detectably disturbed but that the association centered around rod 2 is. It is important to note, however, that these data are snapshots of small regions and cannot be used to make predictions on the behavior of large segments of the molecule.
Also worth noting was the analysis of wild type and mutant proteins labeled at residue 131, a d position, in rod 1 closer to the site of the mutation. In both the wild type and R113S mutant, the 2 M urea spectra of spin-labeled residue 131 do not display the characteristic broad line shape of a a/d positions elsewhere in the molecule. The spectra are consistent with the assumption of α helical secondary structure at this site but not with the formation of coiled coil dimer. This observation is in agreement with the observations of Strelkov et al. (37), who examined crystals of vimentin sequence 102–138 of rod domain 1. Their analysis of rod 1A showed the presence of α helices but not coiled coils (37). These investigators postulated that this region might well form coiled coil in the native protein and that the absence of coiled coil in the crystals may have resulted from the fact that these fragments were removed from the context of the whole protein/filament. The data presented here would support an absence of coiled coil at this site, although spectra from additional sites need to be studied to establish a higher level of confidence. It is also worth noting that algorithms designed to predict secondary structure also show a greater degree of variability in the confidence with which coiled coil structure is predicted for this region of residues 106–139, predictions that are also consistent with no or minimized coiled coil structure (43, 44).
EPR spectra of spin-labeled IF proteins provide data that reveal structure at very specific sites within intact intermediate filaments. While such views lack the context and resolution that would be provided by crystal structure of intact filaments, they do offer data that directly addresses local structure in intact filaments under physiologic conditions. The data reported here suggest that the human vimentin R113S mutation, within the limits of resolution offered by SDSL EPR, does not affect either coiled coil formation, or the initial dimerization centered around rod 1, but does demonstrably alter the dimerization centered around rod 2. These data thus provide groundwork for continued studies on the normal and pathological structure of IFs.
Footnotes
The abbreviations used are: IF, intermediate filament; EBS, epidermolysis bullosa simplex; SDSL-EPR, site-directed spin labeling electron paramagnetic resonance.
This work was supported by Grant DAMD017-02-1-0664 (to J. H.) from the United States Army Medical Research and Material Command, National Institutes of Health Grant RO1 NEI EY08747 (to P. G. F.), and March of Dimes Grant 5-FY02-202 (to J. C. V.).
References
- 1.Bonifas JM, Rothman AL, Epstein EH., Jr Science. 1991;254:1202–1205. doi: 10.1126/science.1720261. [DOI] [PubMed] [Google Scholar]
- 2.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 3.Chan YM, Yu QC, Fine JD, Fuchs E. Proc Natl Acad Sci U S A. 1993;90:7414–7418. doi: 10.1073/pnas.90.15.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Bonifas JM, Matsumura K, Ikeda S, Leyden WA, Epstein EH., Jr J Invest Dermatol. 1995;105:629–632. doi: 10.1111/1523-1747.ep12323846. [DOI] [PubMed] [Google Scholar]
- 5.Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E. Cell. 1992;70:811–819. doi: 10.1016/0092-8674(92)90314-3. [DOI] [PubMed] [Google Scholar]
- 6.Coleman CM, Hannush S, Covello SP, Smith FJ, Uitto J, McLean WH. Am J Ophthalmol. 1999;128:687–691. doi: 10.1016/s0002-9394(99)00317-7. [DOI] [PubMed] [Google Scholar]
- 7.Corden LD, Swensson O, Swensson B, Rochels R, Wannke B, Thiel HJ, McLean WH. Br J Ophthalmol. 2000;84:527–530. doi: 10.1136/bjo.84.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulombe PA, Fuchs E. Semin Dermatol. 1993;12:173–190. [PubMed] [Google Scholar]
- 9.Fuchs E. Biochem Soc Trans. 1991;19:1112–1115. doi: 10.1042/bst0191112. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs E, Coulombe PA. Cell. 1992;69:899–902. doi: 10.1016/0092-8674(92)90607-e. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs E. J Cell Biol. 1994;125:511–516. doi: 10.1083/jcb.125.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Syder AJ, Yu QC, Paller AS, Giudice G, Pearson R, Fuchs E. J Clin Invest. 1994;93:1533–1542. doi: 10.1172/JCI117132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lane EB, Rugg EL, Navsaria H, Leigh IM, Heagerty AH, Ishida-Yamamoto A, Eady RA. Nature. 1992;356:244–246. doi: 10.1038/356244a0. [DOI] [PubMed] [Google Scholar]
- 14.Shemanko CS, Mellerio JE, Tidman MJ, Lane EB, Eady RA. J Invest Dermatol. 1998;111:893–895. doi: 10.1046/j.1523-1747.1998.00388.x. [DOI] [PubMed] [Google Scholar]
- 15.Corden LD, Mellerio JE, Gratian MJ, Eady RA, Harper JI, Lacour M, Magee G, Lane EB, McGrath JA, McLean WH. Hum Mutat. 1998;11:279–285. doi: 10.1002/(SICI)1098-1004(1998)11:4<279::AID-HUMU5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Leigh IM, Lane EB. Arch Dermatol. 1993;129:1571–1577. [PubMed] [Google Scholar]
- 17.Rugg EL, Morley SM, Smith FJ, Boxer M, Tidman MJ, Navsaria H, Leigh IM, Lane EB. Nat Genet. 1993;5:294–300. doi: 10.1038/ng1193-294. [DOI] [PubMed] [Google Scholar]
- 18.Chan Y, Anton-Lamprecht I, Yu QC, Jackel A, Zabel B, Ernst JP, Fuchs E. Genes Dev. 1994;8:2574–2587. doi: 10.1101/gad.8.21.2574. [DOI] [PubMed] [Google Scholar]
- 19.Letai A, Coulombe PA, McCormick MB, Yu QC, Hutton E, Fuchs E. Proc Natl Acad Sci U S A. 1993;90:3197–3201. doi: 10.1073/pnas.90.8.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs E. Cell. 1991;64:365–380. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- 21.Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS, Fuchs E. Cell. 1991;66:1301–1311. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- 22.Nishida K, Honma Y, Dota A, Kawasaki S, Adachi W, Nakamura T, Quantock AJ, Hosotani H, Yamamoto S, Okada M, Shimomura Y, Kinoshita S. Am J Hum Genet. 1997;61:1268–1275. doi: 10.1086/301650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irvine AD, Corden LD, Swensson O, Swensson B, Moore JE, Frazer DG, Smith FJ, Knowlton RG, Christophers E, Rochels R, Uitto J, McLean WH. Nat Genet. 1997;16:184–187. doi: 10.1038/ng0697-184. [DOI] [PubMed] [Google Scholar]
- 24.Gupta SK, Hodge WG. Curr Opin Ophthalmol. 1999;10:234–241. doi: 10.1097/00055735-199908000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez D, Gauthier F, Bertini E, Bugiani M, Brenner M, N’Guyen S, Goizet C, Gelot A, Surtees R, Pedespan JM, Hernandorena X, Troncoso M, Uziel G, Messing A, Ponsot G, Pham-Dinh D, Dautigny A, Boespflug-Tanguy O. Am J Hum Genet. 2001;69:1134–1140. doi: 10.1086/323799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper DN, Youssoufian H. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 27.Mehrani T, Wu KC, Morasso MI, Bryan JT, Marekov LN, Parry DA, Steinert PM. J Biol Chem. 2001;276:2088–2097. doi: 10.1074/jbc.M007260200. [DOI] [PubMed] [Google Scholar]
- 28.Wu KC, Bryan JT, Morasso MI, Jang SI, Lee JH, Yang JM, Marekov LN, Parry DA, Steinert PM. Mol Biol Cell. 2000;11:3539–3558. doi: 10.1091/mbc.11.10.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wawersik M, Paladini RD, Noensie E, Coulombe PA. J Biol Chem. 1997;272:32557–32565. doi: 10.1074/jbc.272.51.32557. [DOI] [PubMed] [Google Scholar]
- 30.Hess JF, Budamagunta MS, Voss JC, FitzGerald PG. J Biol Chem. 2004;279:44841–44846. doi: 10.1074/jbc.M406257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess JF, Voss JC, FitzGerald PG. J Biol Chem. 2002;277:35516–35522. doi: 10.1074/jbc.M206500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter JM, Hutcheson AM, Quinlan RA. Exp Eye Res. 1995;60:181–192. doi: 10.1016/s0014-4835(95)80009-3. [DOI] [PubMed] [Google Scholar]
- 33.Ma L, Yamada S, Wirtz D, Coulombe PA. Nat Cell Biol. 2001;3:503–506. doi: 10.1038/35074576. [DOI] [PubMed] [Google Scholar]
- 34.Hess JF, Casselman JT, FitzGerald PG. Curr Eye Res. 1993;12:77–88. doi: 10.3109/02713689308999499. [DOI] [PubMed] [Google Scholar]
- 35.Hess JF, Casselman JT, FitzGerald PG. J Biol Chem. 1996;271:6729–6735. doi: 10.1074/jbc.271.12.6729. [DOI] [PubMed] [Google Scholar]
- 36.Strelkov SV, Herrmann H, Geisler N, Lustig A, Ivaninskii S, Zimbelmann R, Burkhard P, Aebi U. J Mol Biol. 2001;306:773–781. doi: 10.1006/jmbi.2001.4442. [DOI] [PubMed] [Google Scholar]
- 37.Strelkov SV, Herrmann H, Geisler N, Wedig T, Zimbelmann R, Aebi U, Burkhard P. EMBO J. 2002;21:1255–1266. doi: 10.1093/emboj/21.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLean WH, Pulkkinen L, Smith FJ, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, McGrath JA, McMillan JR, Eady RA, Leigh IM, Christiano AM, Uitto J. Genes Dev. 1996;10:1724–1735. doi: 10.1101/gad.10.14.1724. [DOI] [PubMed] [Google Scholar]
- 39.Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG. Journal of Neuropathology and Exp Neurol. 1999;58:832–846. doi: 10.1097/00005072-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Andra K, Lassmann H, Bittner R, Shorny S, Fassler R, Propst F, Wiche G. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrmann H, Wedig T, Porter RM, Lane EB, Aebi U. J Struct Biol. 2002;137:82–96. doi: 10.1006/jsbi.2002.4466. [DOI] [PubMed] [Google Scholar]
- 42.Smith TA, Steinert PM, Parry DA. Proteins. 2004;55:1043–1052. doi: 10.1002/prot.20089. [DOI] [PubMed] [Google Scholar]
- 43.Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS. Proc Natl Acad Sci U S A. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lupas A, Van Dyke M, Stock J. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]