

Abstract
By using structure-based drug design and isosteric replacement, diarylaniline and 1,5-diarylbenzene-1,2-diamine derivatives were synthesized and evaluated against wild type HIV-1 and drug-resistant viral strains, resulting in the discovery of diarylaniline derivatives as a distinct class of next-generation HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI) agents. The most promising compound 37 showed significant EC50 values of 0.003-0.032 μM against HIV-1 wild-type strains and of 0.005-0.604 μM against several drug-resistant strains. Current results also revealed important structure-activity relationship (SAR) conclusions for diarylanilines and strongly support our hypothesis that an NH2 group on the central benzene ring ortho to the aniline moiety is crucial for interaction with K101 of the NNRTI binding site in HIV-1 RT, likely by forming H-bonds with K101. Furthermore, molecular modeling studies with molecular mechanism/general born surface area (MM/GBSA) technology demonstrated the rationality of our hypothesis.
Introduction
According to UNAIDS statistics, more than 60 million people worldwide have been infected by the human immunodeficiency virus (HIV), and about 25 million patients have died of AIDS. In the absence of an effective vaccine, there is a need to develop effective anti-HIV therapeutics to prolong the lives of HIV-infected individuals. Thus far, more than 20 anti-HIV drugs have been approved by the U.S. FDA (www.fda.gov/oashi/aids/virals.html) including reverse transcriptase inhibitors (RTIs), protease inhibitors (PIs), fusion inhibitors, integrase inhibitors, and entry inhibitors (CCR5 co-receptor antagonist). Highly active antiretroviral therapy (HAART), which uses a combination of three to four drugs, can significantly reduce the morbidity and mortality of HIV-1 infected patients. However, as a result of emerging drug-resistant HIV mutants, increasing numbers of HIV-infected patients fail to respond to HAART. Thus, the development of new anti-HIV drugs is urgently required.
To address this need, we have synthesized compounds targeting HIV-1 reverse transcriptase (RT), one of the most important enzymes in the HIV-1 life cycle. It has two known drug-target sites, the substrate binding site and an allosteric site, which is distinct from, but closely located to, the substrate binding site.1,2 Specifically, we focused on non-nucleoside reverse transcriptase inhibitors (NNRTIs) that interact with the allosteric binding site, a highly hydrophobic cavity, in a noncompetitive manner to cause distortion of the three-dimensional structure of the enzyme and thus inhibit RT catalytic function. NNRTIs currently approved for AIDS therapy include delavirdine (1), nevirapine (2), efavirenz (3), and etravirine (TMC125, 4) (Figure 1).3 In general, NNRTIs exhibit high inhibitory potency and low toxicity, but drug resistance to NNRTIs has emerged rapidly as a result of mutations in amino acid residues that are in or surround the NNRTI binding site. Compound 4 is the most recently approved NNRTI and is active against many drug-resistant HIV-1 strains. The related riplivirine (TMC278, 5)4 is now undergoing phase III clinical trials as a promising new drug candidate. Compounds 4, 5, and TMC120 (6),5 a prior clinical candidate, belong to the diarylpyrimidine (DAPY) family (Fig 1), and all are very potent against wild-type and many drug-resistant HIV-1 strains with nanomolar EC50 values. They have excellent pharmacological profiles, which has encouraged more research to explore next-generation NNRTI agents.6-8 In this study, we used isosteric replacements to synthesize new NNRTIs, and consequently discovered a series of diarylaniline compounds with high potency against both wild-type and RT-resistant viral strains.
Figure 1.

HIV-1 NNRTI agents (1-6).
Design
Prior studies4,9 on DAPY derivatives have resulted in informative SAR conclusions, including (1) a “U” or horseshoe binding conformation in contrast to the typical butterfly-like binding shape of 1-3, (2) a proper positioning of two phenyl rings in the eastern and western wings of the NNRT binding pockets, (3) a para-substituted aniline moiety in the eastern wing, and (4) two hydrogen bonds between K101 of HIV-1 RT to the NH linker and the nitrogen atom on the pyrimidine ring.10 Crystal structures of complexes K103N HIV-1 RT/4 [protein data base (PDB) code: 1sv5], HIV-1 RT/5 (PDB: 2zd1), K103N/Y181C HIV-1 RT/5 (PDB: 3bgr) and HIV-1 RT/6 (PDB: 1s6q)11,12 further indicate that two flexible linkers between rings allow the inhibitors to adopt bound conformations with either wild-type or mutant HIV-1 RT, resulting in high potency against both wild-type and a wide range of drug-resistant mutant HIV-1 RT. Therefore, the molecular flexibility of DAPY compounds is considered to be very crucial for next-generation NNRTI drugs.
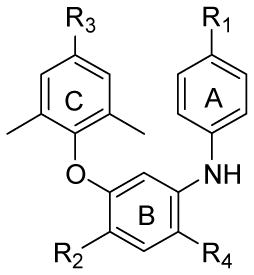
Based on these SAR data, our strategy was to synthesize new compounds by using isosteric replacement in the central B-ring of DAPY compounds. Unlike the reported DAPY derivatives,13-15 the new analogs designed in this study (Figure 2) have a central benzene ring rather than the pyrimidine in DAPY compounds. The new compounds also retain two phenyl rings in the eastern and western wings, but have different para-substituents from the DAPY compounds. Because the presence of a central benzene ring does not change molecular topology and flexibility, these new compounds are expected to have similar binding orientations and conformations to those of DAPYs. However, they will lose the H-bond between K101 and the nitrogen on the pyrimidine ring of DAPY family compounds.10 To compensate for this loss, we introduced a nitro or amino group (R4) on the central benzene ring at the ortho-position to the aniline ring to serve as H-bond acceptor or donor, respectively, and to interact with K101 on the binding site. Furthermore, we investigated how an additional nitro or amino group (R2) on the opposite side of the central phenyl ring would affect anti-HIV activity.
Figure 2.

Design of Target Compounds.
Chemistry
All target compounds were synthesized via the short routes detailed in Schemes 1 and 2. The starting material, 1,3-dichloro-4,6-dinitrobenzene or 2,4-dichloro-1-nitrobenzene, is inexpensive and commercially available. The dichloronitrobenzene was reacted with a para-substituted aniline in a 1:1.1 molar ratio in DMF at room temperature for less than 40 min in the presence of triethylamine to provide corresponding N-aryl-5-chloro- 2,4-dinitroanilines (7-11) or in the presence of potassium tert-butoxide (t-BuOK) to provide corresponding N-aryl-5-chloro-2-nitroanilines (12). The nucleophilic substitution took place between the aromatic amine and the chloride at the ortho-position to the nitro group in 71-94% yields. Compounds 7-12 were then coupled with a substituted phenol by using microwave irradiation in DMF (or DMSO) in the presence of potassium carbonate with stirring at 190 °C for 15 min to afford diaryl-dinitroanilines (13-23) and diaryl-mononitroanilines (24-28). 16, 17 Some compounds could also be prepared by the traditional method of heating at 130 °C for 5 h with yields ranging from 74-94%. As shown in Scheme 2, di-nitro (19-22) and mono-nitro (24, 26-27) compounds were reduced completely by using hydrazine hydrate in isopropanol in the presence of FeCl3·6H2O and activated carbon at reflux for 20-30 min to prepare corresponding di-amino (32-35) or mono-amino (29-31) compounds, respectively, in 83-95% yields. Alternatively, 19, 21, and 22 were reduced selectively by formic acid in the presence of Pd-C (10%) and triethylamine in acetonitrile under reflux for 1 h to give corresponding 1,5-diaryl-4-nitrobenzene-1,2-diamines 36-38, respectively.18 Next, 37 was reacted with triethyl orthoformate under acidic conditions to provide the benzoimidazole product 39. This reaction validated that the nitro group ortho to the NH-linked aniline was reduced selectively in the prior step. Furthermore, active compound 36 was converted to hydrochloride salt 40 (shown in Scheme 2) in acetone to investigate the effect of improved molecular water-solubility on anti-HIV activity.
Scheme 1.

Synthesis of target compounds 13-28. “c or d” indicated two different reaction conditions, the former is under microwave irradiation and the later is a traditional heating method. a) Et3N/DMF, r.t. 40 min; b) t-BuOK/DMF, r.t. 1 h; c) K2CO3/DMF or DMSO, 190 °C, MW, 10-15 min; d) K2CO3/DMF, 130 °C, 5h.
Scheme 2.

a) FeCl3·6H2O/C, N2H4·H2O, (CH3)2CHOH, reflux, 20-30 min; b) Et3N/HCOOH, Pd/C, CH3CN, reflux, 1 h; c) triethyl orthoformate, HCl (1N in diethyl ether), r.t. 3 h; d) CH3COCH3, HCl (18% in diethyl ether).
Results and Discussion
All target compounds were first tested against wild-type HIV-1 (IIIB strain) replication in the H9 cell line, and the results are summarized in Table 1. The most potent compound 37 had an EC50 value of 0.003 μM and an extremely high selective index (SI) of 20,887 in this initial assay. Compound 36 and its hydrochloride salt 40 were also very potent with EC50 values of 0.016 μM and 0.012 μM, respectively; SI values of both compounds were >2800. Other active compounds 27, 29-31, 34-35, and 38 had EC50 values ranging between 0.030–0.073 μM and SI values between 520–1698. In addition, compounds 19, 20, 22, 24, 26 and 32 showed potency at the sub-micromolar level and had a SI range of >113–313. These promising results demonstrated that the isosteric replacement of carbon for nitrogen in the central B-ring of DAPY compounds was successful and resulted in the discovery of a series of 1,5-diarylbenzene-1,2-diamine analogs as potent NNRTI agents.
Table 1.
Antiviral and cytotoxicity data of 13-38 and 40*
| Compound | R1 | R2 | R3 | R4 | HIV-1 IIIB in H9 cells |
||
|---|---|---|---|---|---|---|---|
| EC50a (μM) | CC50b (μM) | SIc | |||||
| 13 | OMe | NO2 | H | NO2 | 3.840 | >61.12 | >16 |
| 14 | OMe | NO2 | Me | NO2 | 2.990 | >59.10 | >20 |
| 15 | OMe | NO2 | Br | NO2 | 3.630 | 51.23 | 14 |
| 16 | Me | NO2 | Br | NO2 | 4.310 | 52.97 | 12 |
| 17 | Cl | NO2 | Br | NO2 | NA | — | — |
| 18 | NO2 | NO2 | Br | NO2 | >49.7 | >49.70 | >1 |
| 19 | CN | NO2 | Br | NO2 | 0.172 | >51.76 | >301 |
| 20 | CN | NO2 | H | NO2 | 0.545 | >61.88 | >113 |
| 21 | CN | NO2 | CN | NO2 | 4.190 | >58.28 | >14 |
| 22 | CN | NO2 | Me | NO2 | 0.280 | >59.81 | >214 |
| 23 | CN | NO2 | CHO | NO2 | 1.530 | 57.87 | 38 |
| 24 | CN | H | Br | NO2 | 0.317 | >57.08 | >180 |
| 25 | CN | H | H | NO2 | 3.147 | 69.64 | 22 |
| 26 | CN | H | CN | NO2 | 0.208 | >65.10 | >313 |
| 27 | CN | H | Me | NO2 | 0.067 | >67.02 | >1,000 |
| 28 | CN | H | CHO | NO2 | 2.190 | 14.20 | 6 |
| 29 | CN | H | Br | NH2 | 0.047 | 44.61 | 949 |
| 30 | CN | H | CN | NH2 | 0.070 | 49.77 | 711 |
| 31 | CN | H | Me | NH2 | 0.073 | 51.02 | 699 |
| 32 | CN | NH2 | Br | NH2 | 0.161 | 38.38 | 238 |
| 33 | CN | NH2 | H | NH2 | 3.226 | 53.20 | 16 |
| 34 | CN | NH2 | CN | NH2 | 0.030 | 50.95 | 1,698 |
| 35 | CN | NH2 | Me | NH2 | 0.070 | 50.00 | 714 |
| 36 | CN | NO2 | Br | NH2 | 0.016 | 45.47 | 2,842 |
| 37 | CN | NO2 | CN | NH2 | 0.003 | 62.66 | 20,887 |
| 38 | CN | NO2 | Me | NH2 | 0.062d | 32.22d | 520 |
| 40 | Hydrochloride salt of 36 | 0.012 | 36.42 | 3,035 | |||
| AZT | 0.052 | 1873 | 36,019 | ||||
Assays using H9 cells were performed at Panacos Pharmaceuticals, Inc., Gaithersburg, Maryland, USA. Results are averages of three independent assays. Standard deviations were not provided by Panacos.
Concentration of a compound that causes 50% inhibition of HIV-1 IIIB replication.
Concentration of a compound that causes cytotoxicity to 50% cells.
SI (selective index) = CC50/EC50.
Data from Duke University, USA. NA: not active.
Some SAR results for this compound series can be concluded from the data in Table 1. 1) For the para-substituent on the A-ring (R1 moiety), the cyano (CN) group appears to be necessary at this position for increased anti-HIV activity. Among compounds 13-19, only 19 (R1 = CN) exhibited high anti-HIV-1 activity with an EC50 value of 0.172 μM and SI of >301, while 13-18 (R1 = MeO, Me, Cl, or NO2) were much less active (EC50 >2.99 μM) or inactive. Except for compounds 21, 23, 28, and 33, all the other compounds containing the active p-cyanoanilino A-ring (19-20, 22, 24-27, 29-32, 34-38, and 40) exhibited potent inhibitory activity against wild-type HIV-1 replication. 2) The presence of NH2 (R4 moiety) on the central B-ring ortho to anilino A-ring position in 29-38 and 40 generally enhanced the anti-HIV-1 activity of the analogs (EC50: 0.003-0.161 μM), except for 33. The NH2-substituted compounds (32, 34, 35, 29, 30, 31) were as or more potent than the corresponding NO2-substituted compounds (19, 21, 22, 24, 26, 27), respectively. This result is consistent with that in our previous report on diarylpyridine compounds,19 and therefore, supports our hypothesis that an amino group at the R4 position of the central B-ring is crucial for interaction with K101 on the NNRTI binding site and can form additional H-bonds to the protein. 3) On the central B-ring ortho to the phenoxy C-ring position (R2 substituent), NO2 moiety (36-38) is more favorable compared with H (29-31) or NH2 (32, 34-35) for achieving high anti-HIV-1 potency. 4) Br, CN, and CH3 para-substituent (R3 moiety) on the C-ring showed better anti-HIV activity, but CHO and H (23, 28, and 33) did not. In addition, compound 40, the hydrochloride salt of 36, displayed more potent inhibitory activity than 36, suggesting a possibility to improve water-solubility in active compounds.
The most active compounds 34, 36, and 37 were further evaluated in comparison with the anti-HIV-1 NNRTI drugs 1 and 4 against wild-type HIV-1 and several RTI-resistant viral strains in the MT-2 cell line. These data are summarized in Tables 2 and 3. As shown in Table 2, we found that both 37 and 36 showed better EC50 values (0.005 and 0.008 μM, respectively) against HIV-1IIIB wild-type virus than 1. While drug 1 lost its antiviral replication activity completely against multi-RTI-resistant mutants, compounds 34, 36, and 37 retained their high potency towards these viruses. Impressively, compound 37 still showed the most potent EC50 value of 0.005 μM against multi-RTI-resistant mutants 8605MR and 6005MR. Compound 36 also was highly active against these mutants, with similar or slightly lower potency than 37. Compound 37 kept some of its antiviral activity against NNRTI-resistant mutant Y181C and A17 (Y181C/K103N) with EC50 values of 0.504 and 0.231 μM, respectively, while 1 completely lost its potency. Compound 36 also showed moderate inhibition activity towards NNRTI-resistant mutants, although it is less potent than 37.
Table 2.
Data against HIV-1IIIB and multi-resistant viral strains*
| Compound | EC50 (μM) in MT-2 cell line |
||||
|---|---|---|---|---|---|
| Wild type | NNRTI-resistant mutanta | Multi-RTI-resistant mutanta | |||
| IIIB | Y181C | A17 (Y181C/K103N) | 8605MR | 6005MR | |
| 34 | 0.065 | 1.734 | 1.244 | 0.138 | 0.033 |
| 36 | 0.008 | 1.298 | 0.254 | 0.020 | 0.007 |
| 37 | 0.005 | 0.504 | 0.231 | 0.005 | 0.005 |
| 1 | 0.053 | >3.700 | >3.700 | >3.700 | >3.700 |
Data provided by Panacos Pharmaceuticals Inc, US. Results are average of three independent assays. Standard deviations were not provided by Panacos. Compound 1 was used as control due to the fact that 4 was not approved for market at the testing time.
NNRTI-resistant mutant and multi-mutated viral strains were from Panacos Inc. 8605MR: multi-mutated-RT with M41L, D67N, L210W, T215Y, M184V, K103N; 6005MR: multi-mutated-RT with M41L, L74V, M184V, L210W, T215Y, ins SS, A98G..
Table 3.
Data against HIV-1IIIB and A17 in the MT-2 cell line*
| Compound | EC50 (μM) in MT-2 cell line |
RF | |
|---|---|---|---|
| IIIB (wild-type) | A17 (Y181C/K103N) a | ||
| 34 | 0.211 ± 0.053 | 15.469 ± 9.035 | 73.3 |
| 36 | 0.052 ± 0.005 | 1.055 ± 0.088 | 20.3 |
| 37 | 0.032 ± 0.005 | 0.604 ± 0.179 | 18.9 |
| 4 | 0.058 ± 0.001 | 0.575 ± 0.093 | 9.9 |
Data provided by Dr. Shibo Jiang, New York Blood Center, US. Results are average of three independent assays.
The multi-NRTI-resistant strain A17 from NIH with mutations at amino acids 103 (K → N) and 181 (Y → C) in the viral RT domain is highly resistant to NRTIs21.
The antiviral activity of 34, 36, and 37 were further tested against wild-type virus and A17 resistant mutant in parallel with drug 4 after it was approved by US FDA and became available on the market (Table 3). The viral inhibition activity of 37 was slightly better than that of 4 against HIV-1IIIB (wild-type) in this screening, which double-confirmed our results in Table 2. Compounds 37 and 4 showed similar activity (EC50: 0.604 and 0.575 μM, respectively) against A17 (from NIH) with mutations in the viral RT domain that is highly resistant to NRTIs. These results provide further support that the DAPY pyrimidine ring can be replaced by a benzene ring to generate new potent compounds as a distinct class of next-generation NNRTIs. Meanwhile, although compound 34 (R2 = NH2) was not as potent as 4 and 37, it had a higher resistant fold change (RF >73, ratio of EC50 against mutant strain/EC50 against wild-type strain) against A17 compared with other compounds, suggesting that the R2 substituent on the central B-ring may play an important role in the anti-HIV-1 activity against drug-resistant virus.
To confirm RT is the biological target of the newly synthesized diarylaniline analogs, compounds 34 and 37 were evaluated against viral enzyme RT (from HIV-1DH012 lysate) and compared with 4. From Figure 3, we can see all three compounds potently inhibited RT activity. Inhibition of DH012 RT activity by 34 and 37 suggests that HIV-1 RT is a target of the compounds. However, the anti-RT potency of these two compounds was much weaker than 4 even though compound 37 was more potent than 4 against HIV-1 replication (Table 3). The discrepancy in potency between anti-HIV-1 replication and anti-RT activity could be due to the fact that different viral strains were used in the assays. The T cell-adapted virus HIV-1 IIIB was used in the virus replication assay (Table 3), whereas the primary isolate DH012 was used for the RT assay. It is also possible that 34 and 37 inhibited HIV-1 RT in a different manner than 4. In addition to inhibition of the RNA-dependent DNA polymerase activity of HIV-1 RT, compound 37 might also inhibit DNA-dependent DNA polymerase activity that could not be optimally detected by the RT assay used in this study.
Figure 3.

RT inhibition activity of diarylanaline analogs 34 and 37*
* Data provided by Dr. Chin-Ho Chen, Duke University Medical Center, US. Results are average of three independent assays. The RT used in the assays is from HIV-1DH012 lysate.
Molecular Modelling Studies
Computational studies of three compounds 21, 34 and 37 were carried out to understand how the substituents on the central B-ring of these diarylaniline analogues might interact with HIV-1 RT. Due to the high structural similarity with DAPY, the ligand/protein complexes of the three compounds were created with the help of the experimental crystal structure of HIV-1 in interaction with DAPY compound (PDB code: 1s6q).11
After energetic optimizations, the ligand positions in the NNRTI binding pocket were carefully checked. To understand the so-called “induced fit effect,” all complexes were compared to HIV-RT alone, which was computed by the same protocol. The Root Mean Square Deviation (RMSD) permits the determination of geometrical variations between the free and complex protein; all values are compiled in Table 4. From this table, it appears that, globally, the ligand/protein structure is kept and that only small adaptations are made by the protein. Comparison of the backbone and side-chain RMSDs shows that the side-chain residues have moved much more in the optimization process than the peptidic chain, indicating that the secondary enzyme structure was conserved and only small adaptations of residues occurred. In comparison, the less biologically active compound 21 induced more “deformation” (i.e., a higher RMSD) of the protein than either 34 or 37, denoting that the protein needs a more significant deformation for adaptation to 21, which is energetically unfavorable for binding.
Table 4.
RMSD values of the HIV-1 RT protein interacting with 21, 34 and 37, when compared to the protein alone*.
| Ligand | RMSD (Å) |
||
|---|---|---|---|
| Global | Backbone | Side-chain | |
| 21 | 0.666 | 0.501 | 0.738 |
| 34 | 0.654 | 0.493 | 0.725 |
| 37 | 0.635 | 0.480 | 0.703 |
Values are divided into Global (all protein atoms), Backbone (peptidic bonds) and Side-chain (only side-chain atoms).
The Molecular Mechanics/General Born Surface Area (MM/GBSA) technique is one of the best computational methods to evaluate free energy of binding, generally from molecular dynamics simulations, for ligands in biological macromolecules.20,21 Recently, it has been shown on a large number of enzymatic systems (including HIV-1 RT) that the MM/GBSA method is able to rescore docking results, even with only one minimized structure.22 Therefore, we employed this technique on our three compounds, and the results are compiled in Table 5. To take into account the protein adaptation to the ligands, the calculations were made by considering the three species individually (protein, ligand and complex). The computed free energies of binding paralleled the experimental antiviral activity data, i.e., the ranking of EC50 values. Furthermore, the energetic decompositions indicated which contributions are the most important for the binding affinity and which are not. Comparison of values in Table 5 indicated that the differences of binding free energies come mainly from the gas phase energy and, more precisely, from the electrostatic contribution of the ligand to the protein. However, the non-polar solvent effect is almost identical for the three compounds. This result may reflect the fact that, in the X-ray structure, the NNRTI binding pocket is deeply inserted into the HIV-1 RT enzyme, and no water molecules are close to the DAPY ligand. Indeed, non-polar solvent free energy of binding comes from the entropic desolvation of water around the ligand interacting site.
Table 5.
MM/GBSA Energetic values of the three ligand/HIV-1 RT complexes*.
| Ligand | 21 | 34 | 37 |
|---|---|---|---|
| Energy | kcal/mol | ||
| ΔEelec | 31.4 | −23.8 | −56.5 |
| ΔEvdw | −53.8 | −61.3 | −55.5 |
| ΔEint | 14.4 | 7.2 | −8.3 |
| ΔEgas | −8.0 | −77.9 | −120.3 |
| ΔGsolvent_np | −6.2 | −6.9 | −6.4 |
| ΔGsolvent_p | 1.5 | 35.3 | 54.6 |
| ΔGsolvent | −4.7 | 28.4 | 48.2 |
| ΔG | −12.7 | −49.6 | −72.1 |
Eelec, Evdw and Eint signify: electrostatic, van der Waals and internal (bonds + angles + dihedrals) energies in gas phase (in vacuo), respectively. Egas is the sum of the first three energy terms. Gsolvent_p, Gslovent_np, and Gsolvent are the polar and non-polar contributions of the solvent free energy, and the sum of these two terms (solvent free energy), respectively; G is the free energy of binding (Egas + Gsolvent).
Based on the above computational studies, the docking of three ligands with HIV-1 RT is illustrated more clearly in Figure 4. Compounds 21, 34 and 37 differ structurally only in the identity of the R2 and R4 substituents on the central aromatic ring (21: NO2, NO2; 34: NH2, NH2; 37: NO2, NH2). Therefore, our analyses focused on these moieties. As shown in Figure 4, a hydrogen bond between the NH linker and the peptidic carbonyl oxygen of K101, like that for DAPY compounds, is observed with all three compounds. However, the neighbouring amino group (R4) present in 34 and 37 provides two more hydrogen bonds with K101: one is between the peptidic carbonyl oxygen of the protein residue and one NH vector of the ligand NH2 group and the second one involves the NH atoms of K101 and targets the nitrogen atom of the ligand NH2 group. The most active compound 37 also has a nitro group as the R2 substituent on the central ring. This group provides a small electrostatic interaction with the positively charged K172 (ligand nitrogen – protein nitrogen distance of 6.68 Å; see Figure 3, bottom). When the nitro group in 37 was replaced by an amino group in 34, the corresponding ligand-protein electrostatic interaction decreased (see Figure 3, middle). Indeed, the distance between the nitrogen atom of the amino moiety of 34 and the K101 nitrogen increased from 6.68 Å to 7.04 Å, indicating that repulsion has occurred. Moreover, this electrostatic effect was also observed computationally (Table 5) with electrostatic energy values of -56.5 kcal/mol and -23.8 kcal/mol for 37 and 34, respectively. In comparison with 34 and 37, the least active compound 21 has two nitro groups on the central B-ring (see Figure 3, top). Thus, the rightmost (R2) nitro group of 21 has the same electrostatic interaction with K172 found in 37, and the ligand nitrogen – protein nitrogen distance shortens to 6.56 Å. However, the presence of the leftmost (R4) nitro group in 21 completely disrupts the hydrogen bond network with K101. The NH atoms of K101 face the nitrogen atom of the nitro moiety identically as for the amino group, but the nitrogen of a nitro group is electropositive, while that of an amino group is electronegative. Thus, for 21, electrostatic repulsion occurs between the R4 nitro group and the NH of K101, which is also observed in the computational electrostatic energy value of 31.4 kcal/mol (Table 5). Because a hydrogen bond is mainly electrostatic, the two supplementary H-bonds found with 34 and 37 could explain the experimental EC50 differences, which correlated with the computed free energies of binding. Therefore, these molecular modeling results also strongly support our hypothesis and demonstrate why the amino-substituted compounds 29-38 are more potent than nitro-substituted compounds 19-28.
Figure 4.
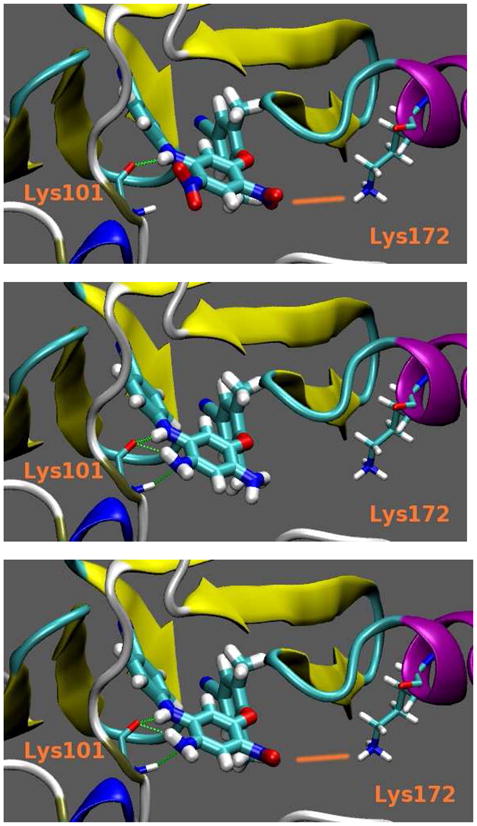
21 (top), 34 (middle) and 37 (bottom) in interaction with HIV-1 RT.
Conclusion
Based on our isosteric replacement strategy, diarylaniline derivatives were discovered as a distinct class of HIV-1 NNRTI agents. Current results supported our hypothesis that the NH2 group on the central B-ring ortho to the anilino ring position is crucial for interaction with K101 on the NNRTI binding site by forming additional H-bonds. In the series of new compounds, active diarylaniline derivatives are highly potent against both wild-type and drug resistant HIV-1 strains. Among them, the most promising compound is 37 with low EC50 values against HIV-1 wild-type (0.003 – 0.005 μM) and several multi-drug-resistant strains (0.005 - 0.604 μM). Generally speaking, 36 and 37 showed slightly better EC50 values (0.052 and 0.032 μM, respectively) than 4 (EC50 0.058 μM), a newly marketed HIV-1 NNRTI drug. In addition, they can be synthesized more conveniently than the DAPY derivatives 4 and 5.23, 24 The present biological and modeling studies further revealed some important SARs for this series of diarylanilines: 1) the new scaffold can maintain a similar binding orientation and conformation to those of DAPY derivatives; 2) a para-cyanoaniline moiety is necessary for anti-HIV activity; 3) an amino group on the central B-ring ortho to the aniline moiety position is crucial for achieving high potency, likely by forming H-bonds with K101; 4) an additional nitro group (R2) on the central aromatic ring is helpful for enhancing affinity of the inhibitor by interaction with K172; and 5) the para-substituent (R3) on the phenoxy ring is changeable but can greatly affect the anti-HIV activity.
Experimental Section
Chemistry
Melting points were measured with an RY-1 melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as the internal standard. The solvent used was DMSO-d6, unless otherwise indicated. Mass spectra (MS) were measured on an ABI Perkin-Elmer Sciex API-150 mass spectrometer with electrospray ionization, and the relative intensity of each ion peak is presented as percent (%). The purities of target compounds were ≥95% measured by HPLC analyses, which were performed by Agilent 1100 HPLC system with a UV detector, using a Grace Alltima HP C18 column (100 × 2.1 mm, 3 μm) eluting with a mixture of solvents A and B (condition 1: acetonitrile/water 80:20, flow rate 0.2 mL/min; condition 2: MeOH/water 80:20, flow rate 0.2 mL/min; UV 254 nm). The microwave reactions were performed on a microwave reactor from Biotage, Inc. Thin-layer chromatography (TLC) was performed on silica gel GF254 plates. Silica gel GF254 (200–300 mesh) from Qingdao Haiyang Chemical Company was used for TLC, preparative TLC, and column chromatography. Medium-pressure column chromatography was performed using a CombiFlash® CompanionTM purification system. All chemicals were obtained from Beijing Chemical Works or Sigma-Aldrich, Inc.
General Procedure for the Preparation of N1-Aryl-5-chloro-2-nitroanilines (7-12)
Method A. A para-sustituted aniline (1.1 equiv) was added slowly into a solution of 2,4-dichloronitrobenzene (1 equiv) in DMF (3 mL) and triethylamine (1.5 mL, excess). The yellow mixture was stirred at rt for 40 min. The mixture was then poured into water (ca. 30 mL) and stirred for an additional 30 min. The solid product was collected and purified by recrystallization from EtOH. Method B. To a solution of 2,4-dichloronitrobenzene (1 equiv) and a substituted aniline (1.1 equiv) in DMF (3 mL), was slowly added potassium tert-butoxide (2 equiv) at ice-water bath temperature, then stirred at rt for about 1 h until the reaction was completed as monitored by TLC. The mixture was poured into ice-water, and pH was adjusted to 6 with 5% aq HCl The solid was collected, washed with water three times, and purified by recrystallization from EtOH.
5-Chloro-N-(4-methoxyphenyl)-2, 4-dinitroaniline (7)
Method A, yield: 82%, starting with 237 mg (1 mmol) of 1,3-dichloro-4,6-dinitrobenzene and 136 mg (1.1 mmol) of 4-methoxyaniline to afford 226 mg of 7, yellow solid, mp 150 – 151 °C. 1H NMR (CDCl3) δ ppm 3.88 (3H, s, OCH3), 7.03 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.05 (1H, s, ArH-6), 7.21 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 9.09 (1H, s, ArH-3), 9.74 (1H, s, NH). MS m/z (%) 324 (M+1, 100), 326 (M+3, 30).
5-Chloro-N-(4-methylphenyl)-2,4-dinitroaniline (8)
Method A, yield: 94%, starting with 237 mg (1 mmol) of 1,3-dichloro-4,6-dinitrobenzene and 118 mg (1.1 mmol) of 4-methylaniline to afford 324 mg of 8, yellow solid, mp 148 – 150 °C. 1H NMR (CDCl3) δ ppm 2.43 (3H, s, CH3), 7.12 (1H, s, ArH-6), 7.17 (2H, d, J = 8.0 Hz, ArH-3′, 5′), 7.32 (2H, d, J = 8.0 Hz, ArH-2′, 6′), 9.09 (1H, s, ArH-3), 9.80 (1H, br. NH). MS m/z (%): 308 (M+1, 100), 310 (M+3, 31).
5-Chloro-N-(4-chlorophenyl)-2,4-dinitroaniline (9)
Method A, yield: 80%, starting with 237 mg (1 mmol) of 1,3-dichloro-4,6-dinitrobenzene and 166 mg (1.3 mmol) of 4-chloroaniline at 80 °C for 20 min to afford 263 mg of 9, yellow solid, mp 136 – 138 °C. 1H NMR (CDCl3) δ ppm 7.11 (1H, s, ArH-6), 7.25 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.50 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 9.08 (1H, s, ArH-3), 9.77 (1H, s, NH). MS (negative charge) m/z (%): 326 (M-1, 100), 328 (M+1, 80).
5-Chloro-N-(4-nitrophenyl)-2,4-dinitroaniline (10)
Method B. yield: 83%, starting with 237 mg (1 mmol) of 1,3-dichloro-4,6-dinitrobenzene and 166 mg (1.2 mmol) of 4-nitroaniline to afford 280 mg of 10, yellow solid, mp 140 – 141 °C. 1H NMR δ ppm 7.55 (1H, s, ArH-6), 7.60 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 8.29 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.90 (1H, s, ArH-3), 10.19 (1H, s, NH). MS m/z (%): 308 (M+1, 100), 310 (M+3, 30).
5-Chloro-N-(4-cyanophenyl)-2,4-dinitroaniline (11)
Method B. yield: 89%, starting with 237 mg (1 mmol) of 1,3-dichloro-4,6-dinitrobenzene and 130 mg (1.1 mmol) of 4-cyanoaniline to afford 284 mg of 11, yellow solid, mp 174 – 176 °C. 1H NMR (CDCl3) δ ppm 7.41 (1H, s, ArH-6), 7.57 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.91 ( 2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.90 (1H, s, ArH-3), 10.07 (1H, s, NH). MS m/z (%): 319 (M+1, 100), 321 (M+3, 23).
5-Chloro-N-(4-cyanophenyl)-2-nitroaniline (12)
Method B. yield 71%, starting with 576 mg (3 mmol) of 2,4-dichloro-1-nitrobenzene and 425 mg (3.6 mmol) of 4-cyanoaniline to afford 583 mg of 12, red-yellow solid, mp 123 – 124 °C. 1H NMR δ ppm 7.17 (1H, d, J = 9.0 Hz, ArH-4), 7.40 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.45 (1H, s, ArH-6), 7.78 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.14 (1H, d, J = 9.0 Hz, ArH-3), 9.48 (1H, s, NH). MS m/z (%): 272 (M-1, 100), 321 (M+1, 22).
General Preparation of Diphenylether (13-28). Method C (microwave)
A mixture of N-aryl-5-chloro-2-nitroaniline (1 equiv) and 2,6-dimethylphenol (1.2 equiv) in DMF (3 mL) in the presence of anhydrous potassium carbonate (2 equiv) was irradiated under microwave with stirring at 190 °C for 15 min. The mixture was poured into ice-water, pH adjusted to neutral with 5% HCl aq, and the mixture extracted with EtOAc three times. After removal of organic solvent in vacuo, crude product was purified by PTLC or a silica column (eluent: petroleum ether/EtOAc). Method D (traditional): A mixture of N-aryl-5-chloro-2-nitroaniline (11 or b, 1 equiv) and a para-substituted 2,6-dimethylphenol (1.2 equiv) in DMF in the presence of anhydrous potassium carbonate (excess) was heated at 130 °C for 5 h. Work-up was the same as above, and crude product was purified by a silica gel column.
5-(2″,6″-Dimethylphenoxy)-N-(4-methoxyphenyl)-2,4-dinitroaniline (13)
Method C: yield 75%, starting with 129 mg (0.4 mmol) of 7 and 59 mg (0.48 mmol) of 2,6-dimethylphenol to afford 123 mg of 13, yellow solid, mp 169-170 °C. 1H NMR (CDCl3) δ ppm 2.06 (6H, s, 2×CH3), 3.78 (3H, s, OCH3), 5.91 (1H, s, ArH-6), 6.75 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.89 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.11 (1H, t, J = 6.8 Hz, ArH-4′′), 7.69 (2H, d, J = 6.8 Hz, ArH-3″, 5″), 8.96 (1H, s, ArH-3), 9.70 (1H, s, NH). MS m/z (%):410 (M+1, 100); HPLC Purity: 99.4%.
N1-(4′-Methoxyphenyl)-5-(2″,4″,6″-trimethylphenoxy)-2,4-dinitroaniline (14)
Method C: yield 33%, starting with 162 mg (0.5 mmol) of 7 and 82 mg (0.6 mmol) of 2,4,6-trimethylphenol to afford 70 mg of 14, red solid, mp 155-158 °C. 1H NMR (CDCl3) δ ppm 1.98 (6H, s, 2×CH3), 2.16 (3H, s, CH3), 3.73 (3H, s, OCH3), 5.82 (1H, s, ArH-6), 6.69 (2H, s, ArH), 6.72 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.84 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 9.11 (1H, s, ArH-3), 9.60 (1H, s, NH). MS m/z (%):424 (M+1, 100); HPLC Purity: 98.3%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-methoxyphenyl)-2,4-dinitroaniline (15)
Method C: yield 53%, starting with 647 mg (2 mmol) of 7 and 422 mg (2.1 mmol) of 4-bromo-2,6-dimethylphenol to afford 517 mg of 15, yellow solid, mp 221 – 223 °C. 1H NMR (CDCl3) δ ppm 2.07 (6H, s, 2×CH3), 3.78 (3H, s, OCH3), 5.91 (1H, s, ArH-6), 6.75 (2H, d, J = 9.0 Hz, ArH-2′, 6′), 6.89 (2H, d, J = 9.0 Hz, ArH-3′, 5′), 7.34 (2H, s, ArH-3″, 5″), 9.17 (1H, s, ArH-3), 9.68 (1H, s, NH). MS m/z (%): 488 (M+1, 100), 490 (M+3, 98); HPLC Purity: 95.8%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-methylphenyl)-2,4-dinitroaniline (16)
Method C: yield 52%, starting with 154 mg (0.5 mmol) of 8 and 121 mg (0.6 mmol) of 4-bromo-2, 6-dimethylphenol to afford 123 mg of 16, yellow solid, mp 200 – 202 °C. 1H NMR (CDCl3) δ ppm 2.08 (6H, s, 2×CH3), 2.36 (3H, s, CH3), 5.89 (1H, s, ArH-6), 6.86 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.10 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.16 (2H, s, ArH-3″, 5″), 9.17 (1H, s, ArH-3), 9.71 (1H, s, NH). MS m/z (%):472 (M+1, 100), 474 (M+3, 92); HPLC Purity: 97.5%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-chlorophenyl)-2,4-dinitroaniline (17)
Method C: yield: 64%, starting with 328 mg (1.0 mmol) of 9 and 201 mg (1.0 mmol) of 4-bromo-2,6-dimethylphenol to afford 315 mg of 17, yellow solid, mp 206 – 208 °C. 1H NMR δ ppm 2.01 (6H, s, 2×CH3), 5.70 (1H, s, ArH-6), 7.15 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.35 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.37 (2H, s, ArH-3″, 5″), 8.96 (1H, s, ArH-3), 10.01 (1H, s, NH). MS m/z (%): 492 (M+1, 70), 494 (M+3, 100); HPLC Purity: 96.3%. 5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-nitrophenyl)-2,4-dinitroaniline (18)
Method C: yield 78%, starting with 154 mg (0.44 mmol) of 10 and 107 mg (0.53 mmol) of 4-bromo-2, 6-dimethylphenol to afford 173 mg of 18, yellow solid, mp 190 – 191 °C. 1H NMR δ ppm 2.05 (6H, s, 2×CH3), 6.06 (1H, s, ArH-6), 7.31 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.41 (2H, s, ArH-3″, 5″), 8.13 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.96 (1H, s, ArH-3), 10.13 (1H, s, NH). MS m/z (%): 503 (M+1, 100), 505 (M+3, 98); HPLC Purity: 97.7%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-cyanophenyl)-2,4-dinitrsoaniline (19)
Method D: yield 85%, starting with 3.18 g (10 mmol) of 11 and 2.4 g (12 mmol) of 2,6-dimethyl-4-bromophenol in the presence of K2CO3 (excess, 3.0 g, 21.7 mmol) to afford 4.3 g of 19, yellow solid, mp 276 – 278 °C. 1H NMR δ ppm 2.05 (6H, s, 2×CH3 ), 5.93 (1H, s, ArH-6), 7.28 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.42 (2H, s, ArH-3″, 5″), 7.73 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.96 (1H, s, ArH-3), 10.07 (1H, s, NH). MS (negative charge) m/z (%): 481 (M-1, 100), 483 (M+1, 99); HPLC Purity: 98.5%.
N-(4′-Cyanophenyl)-5-(2″,6″-dimethylphenoxy)-2,4-dinitroaniline (20)
Method C: yield: 81%, starting with 319 mg (1.0 mmol) of 11 and 146.4 mg (1.2 mmol) of 2,6-dimethylphenol to afford 327 mg of 20, orange solid, mp 229 } 230 °C. 1H NMR δ ppm 2.06 (6H, s, 2×CH3), 6.01 (1H, s, ArH-6), 7.09 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.11 (1H, t, J = 6.8 Hz, ArH-4″), 7.28 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.69 (2H, d, J = 6.8 Hz, ArH-3″, 5″), 8.96 (1H, s, ArH-3), 10.07 (1H, s, NH). MS m/z (%):405 (M+1, 100); HPLC Purity: 99.9%.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N-(4′-cyanophenyl)-2,4-dinitroaniline (21)
Method D: yield 94%, starting with 1.27 g (4.0 mmol) of 11 and 705 mg (4.8 mmol) of 2,6-dimethyl-4-cyanophenol in DMF (4 mL) in the presence of anhydrous potassium carbonate (966 mg, 7 mmol) to afford 1.61g of 21, orange solid, mp >300°C. 1H NMR δ ppm 2.08 (6H, s, 2×CH3), 5.90 (1H, s, ArH-6), 7.29 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.73 (2H, s, ArH-3″, 5″), 7.74 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.97 (1H, s, ArH-3), 10.07 (1H, s, NH). MS m/z (%): 430 (M+1, 100); HPLC Purity: 98.1%.
N-(4′-Cyanophenyl)-2,4-dinitro-5-(2″,4″,6″-trimethylphenoxy)aniline (22)
Method C: yield 70%, starting with 637 mg (2 mmol) of 11 and 286 mg (2.1 mmol) of 2,4,6-trimethylphenol to afford 580 mg of 22, yellow solid, mp 223 – 225 °C. 1H NMR δ ppm 2.00 (6H, s, 2×CH3), 2.22 (3H, s, CH3), 5.92 (1H, s, ArH-6), 6.95 (2H, s, ArH-3″, 5″), 7.27 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.72 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.95 (1H, s, ArH-3), 10.04 (1H, s, NH). MS m/z (%):419 (M+1, 100); HPLC Purity: 98.3%.
N-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-formylphenoxy)-2,4-dinitroaniline (23)
Method C: yield 74%, starting with 160 mg (0.5 mmol) of 11 and 113 mg (0.6 mmol) 4-hydroxy-3,5-dimethylbenzaldehyde to afford 144 mg of 23, yellow solid, mp 260 – 263 °C. 1H NMR (CDCl3) δ ppm 2.23 (6H, s, 2×CH3), 6.24 (1H, s, ArH-6), 7.10 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.54 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.67 (2H, s, ArH-3″, 5″), 9.20 (1H, s, ArH-3), 9.97 (2H, s, NH & CHO), MS m/z (%)433 (M+1, 100); HPLC Purity: 98.7%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N-(4′-cyanophenyl)-2-nitroaniline (24)
Method C: yield: 93%, starting with 12 (100 mg, 0.36 mmol) and potassium 4-bromo-2,6-dimethylphenoxide (105 mg, 0.44 mmol) in DMF (2 mL) were irradiated under microwave with stirring at 192 °C for 10 min to get 147 mg of 24, light yellow solid, mp 143 °C. 1H NMR (CDCl3) δ ppm 2.03 (6H, s, 2×CH3), 6.19 (1H, dd, J = 9.2 & 2.4 Hz, ArH-6), 6.74 (1H, d, J = 2.4 Hz, ArH-4), 7.21 (2H, d, J = 8.8 Hz, ArH-2′, 6′, 7.30 (2H, s, ArH-3″, 5″), 7.57 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.13 (1H, d, J = 9.2 Hz, ArH-3), 9.69 (1H, s, NH). MS m/z (%): 438 (M+1. 100), 440 (M+3, 98); HPLC Purity: 95.7%.
N-(4′-Cyanophenyl)-5-(2″,6″-dimethylphenoxy)-2-nitroaniline (25)
Method C: yield: 31%, starting with 12 (50 mg, 0.18 mmol) and 2,6-dimethylphenol (31 mg, 0. 25 mmol) in the presence of potassium carbonate (58 mg, 0.42 mmol) in DMSO (2 mL) to afford 20 mg of 25, yellow solid, mp 158-60 °C. 1H NMR (CDCl3, 400 MHz) d 2.12 (6H, s, 2×CH3), 6.35 (1H, q, J1 = 9.2 Hz, J2 = 2.4 Hz, ArH-4), 6.73 (1H, d, J = 2.4 Hz, ArH-6), 7.10 (3H, m, ArH-3″, 4″, 5″), 7.24 (2H, d, J = 8.4 Hz, ArH-2′, 6′), 7.59 (2H, d, J = 8.4 Hz, ArH-3′, 5′), 8.21 (1H, d, J = 9.2 Hz, ArH-3), 9.76 (1H, s, NH). MS m/z (%): 360 (M+, 100); HPLC Purity: 96.6%.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N-(4′-cyanophenyl)-2-nitroaniline (26)
Method D: yield 79%, starting with 821 mg (3 mmol) of 12 and 530 mg (3.6 mmol) of 2,6-dimethyl-4-cyanophenol in the presence of K2CO3 (excess, 1.45 g, 10.5 mmol) to afford 0.92 g of 26, yellow solid, mp186 – 188 °C. 1H NMR (CDCl3) δ ppm 2.16 (6H, s, 2×CH3), 6.15 (1H, dd, J = 9.2 & 2.4 Hz, ArH-4), 6.87 (1H, d, J = 2.4 Hz, ArH-6), 7.31 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.45 (2H, s, ArH-3″, 5″), 7.66 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.21 (1H, d, J = 9.2 Hz, ArH-3), 9.75 (1H, s, NH). MS m/z (%):385 (M+1, 100); HPLC Purity: 99.0%.
N-(4′-Cyanophenyl)-5-(2″,4″,6″-trimethylphenoxy)-2-nitroaniline (27)
Method D: yield 74%, starting with 274 mg (1 mmol) of 12 and 241 mg (1.2 mmol) of 2,4,6-trimethylphenol in the presence of excess K2CO3 to afford 225 mg of 27, yellow solid, mp 168 – 170 °C; 1H NMR (CDCl3) δ ppm 2.00 (6H, s, 2×CH3), 2.23 (3H, s, CH3), 6.25 (1H, d, J = 9.2 Hz, ArH-4), 6.69 (1H, s, ArH-6), 6.84 (2H, s, ArH-3″, 5″), 7.18 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.53 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.12 (1H, d, J = 9.2 Hz, ArH-3), 9.67 (1H, s, NH); MS m/z (%) 474 (M+1, 100); HPLC Purity: 95.9%.
N-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-formylphenoxy)-2-nitroaniline (28)
Method C: yield 70%, starting with 137 mg (0.5 mmol) of 12 and 90 mg (0.6 mmol) of 4-hydroxy-3,5- dimethylbenzaldehyde to afford 135 mg of 28, yellow solid, mp 152 – 155 °C. 1H NMR (CDCl3) δ ppm 2.21 (6H, s, 2×CH3), 6.23 (1H, dd, J = 8.8 & 2.4 Hz, ArH-4), 6.84 (1H, d, J = 2.4 Hz, ArH-6), 7.30 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.64 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.67 (2H, s, ArH), 8.22 (1H, d, J = 8.8 Hz, ArH-3), 9.76 (1H, br. s, NH), 9.97 (1H, s, CHO). MS m/z (%): 338 (M+1, 100); HPLC Purity: 98.6%.
General Preparation of 1,5-Diarylbenzene-1,2-diamines or 1,2,4-Triamines (29-38)
To a solution of a diaryl-nitrobenzene (24, 26-27 or 19-22, 1 equiv) in 15 mL of isopropanol was added FeCl3·6H2O (1 equiv), activated carbon (2 equiv), and N2H4·H2O (10 equiv), successively. The mixture was refluxed for 20-30 min. After removal of carbon and solvent, the residue was purified by a silica gel column (eluent: CHCl3/MeOH 40:1) to obtain corresponding diarylbenzene-1,2-diamine or -1,2,4-triamines respectively.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)benzene-1,2-diamine (29)
Yield 83%, starting with 150 mg (0.34 mmol) of 24 to afford 115 mg of 29, brown solid, mp 154 – 155 °C. 1H NMR d ppm 2.07 (6H, s, 2×CH3), 4.55 (2H, s, ArNH2), 6.34 (1H, s, ArH-6), 6.47 (1H, d, J = 8.8 Hz, ArH-4), 6.65 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.73 (1H, d, J = 8.8 Hz, ArH-3), 7.34 (2H, s, ArH-3″, 5″), 7.49 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.08 (1H, s, NH); MS m/z (%) 408 (M+1, 100), 410 (M+3, 98); HPLC Purity: 96.8%.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)benzene-1,2-diamine (30)
Yield 83%, starting with 130 mg (0.34 mmol) of 26 to afford 100 mg of 30, brown solid, mp 154 – 156 °C. 1H NMR (CDCl3) δ ppm 2.18 (6H, s, 2×CH3), 3.0-4.0 (2H, NH2), 5.67 (1H, s, NH), 6.47 (1H, q, J = 8.8 & 2.8 Hz, ArH-4), 6.58 (1H, d, J = 2.8 Hz, ArH-6), 6.70 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.74 (1H, d, J = 8.8 Hz, ArH-3), 7.40 (2H, s, ArH-3″, 5″), 7.47 (2H, d, J = 8.8 Hz, ArH-3′, 5′). MS m/z (%): 355 (M+1, 100); HPLC Purity: 99.2%.
N1-(4′-Cyanophenyl)-5-(2″,4″,6″-trimethylphenoxy)benzene-1,2-diamine (31)
Yield 84%, starting with 150 mg (0.4 mmol) of 27 to afford 116 mg of 31, mp152 – 154 °C. 1H NMR δ ppm 2.02 (6H, s, 2×CH3), 2.22 (3H, s, CH3), 4.48 (2H, s, NH2), 6.29 (1H, d, J = 2.8 Hz, ArH-6), 6.48 (1H, dd, J = 8.4 & 2.8 Hz, ArH-4), 6.64 (1H, d, J = 8.4 Hz, ArH-3), 6.72 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.48 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.07 (1H, s, NH). MS m/z (%): 344 (M+1, 100); HPLC Purity: 99.8%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)benzene-1,2,4-triamine (32)
Yield 95%, starting with 150 mg (0.31 mmol) of 19 to afford 125 mg of 32, brown solid, mp 181 – 183 °C. 1H NMR δ ppm 2.08 (6H, s, 2×CH3), 4.28 (2H, s, NH2), 4.91 (2H, s, NH2), 5.70 (1H, s, ArH-3), 6.22 (1H, s, ArH-6), 6.43 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.30 (2H, s, ArH-3″, 5″), 7.38 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.71 (1H, s, NH). MS m/z (%): 423 (M+1, 100), 425 (M+3, 98); HPLC Purity: 96.1%.
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethylphenoxy)benzene-1,2,4-triamine (33)
Yield 30%, starting with 152 mg (0.31 mmol) of 20 to afford 39 mg of 33, brown solid, mp 156-8 °C. 1H NMR δ ppm 2.08 (6H, s, 2×CH3), 4.22 (2H, s, NH2-4), 4.84 (2H,s, NH2-2), 5.70 (1H, s, ArH-3), 6.23 (1H, s, ArH-6), 6.43 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.99 (2H, d, J = 6.8 Hz, ArH-3″, 5″), 7.07 (1H, t, J = 6.8 Hz, ArH-4”), 7.36 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.67 (1H, s, NH). MS m/z (%): 345 (M+, 100); HPLC Purity: 99.6%.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)benzene-1,2,4-triamine (34)
Yield 86%, starting with 300 mg (0.7 mmol) of 21 to afford 223 mg of 34, brown solid, mp 144 °C; 1H NMR δ ppm 2.08 (6H, s, 2×CH3), 4.33 (2H, s, NH2), 4.97 (2H, s, NH2), 5.71 (1H, s, ArH-3), 6.23 (1H, s, ArH-6), 6.43 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.38 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.61 (2H, s, ArH-3″, 5″), 7.71 (1H, s, NH); MS m/z (%) 370 (M+1, 100); HPLC Purity: 100.0%.
N1-(4′-Cyanophenyl)-5-(2″,4″,6″-trimethylphenoxy)benzene-1,2,4-triamine (35)
Yield 85%, starting with 150 mg (0.36 mmol) of 22 to afford 110 mg of 35, light brown solid, mp 152 – 154 °C. 1H NMR (CDCl3) δ ppm 2.08 (6H, s, 2×CH3), 2.26 (3H, s, CH3), 3.50 (2H, s, NH2), 4.0 (2H, s, NH2), 5.33 (1H, s, ArH-6), 5.97 (1H, s, ArH-3), 6.29 (1H, s, NH), 6.47 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.86 (2H, s, ArH-3″, 5″), 7.36 (2H, d, J = 8.8 Hz, ArH-3′, 5′). MS m/z (%): 359 (M+1, 100); HPLC Purity: 100.0%.
5-(4″-Bromo-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-nitrobenzene-1,2-diamine (36)
Pd-C (10%, 30 mg) was added to a solution of 19 (338 mg, 0.7 mmol) in acetonitrile (6 mL) and triethylamine (6 mL). The mixture was cooled to -15 °C and then formic acid (95%, 1 mL, excess) in acetonitrile (4 mL) was slowly added at the low temperature. The mixture was then heated to reflux for 1 h, following which the Pd-C and solvent were removed. The residue was purified by PTLC (petroleum ether/EtOAc 4:1) to obtain 269 mg of 36, yield 85%, deep red solid, mp 218 – 220 °C. 1H NMR δ ppm 2.07 (6H, s, 2×CH3), 5.13 (2H, s, NH2), 6.16 (1H, s, ArH-6), 6.82 (2H, d, J = 8.4 Hz, ArH-2′,6′), 7.40 (2H, s, ArH-3″, 5″), 7.55 (2H, d, J = 8.4 Hz, ArH-3′,5′), 7.95 (1H, s, ArH-3), 8.45 (1H, s, NH). MS m/z (%) 453 (M+1, 95), 455 (M+3, 100); HPLC Purity: 97.0%.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-nitrobenzene-1,2-diamine (37)
The preparation was the same as that of 36. Yield 86%, starting with 150 mg (0.35 mmol) of 21 to afford 237 mg of 37, red solid, mp 224 – 226 °C. 1H NMR δ ppm 2.07 (6H, s, 2×CH3), 5.17 (2H, s, NH2), 6.15 (1H, s, ArH-6), 6.81 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.55 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.71 (2H, s, ArH), 8.06 (1H, s, ArH-3), 8.45 (1H, s, NH). MS m/z (%): 400 (M+1, 100); HPLC Purity: 96.8%.
N1-(4′-Cyanophenyl)-5-(2″,4″,6″-trimethylphenoxy)-4-nitrobenzene-1,2-diamine (38)
The preparation was the same as that of 36. Yield 79%, starting with 300 mg (0.72 mmol) of 22 to afford 220 mg of 38, red solid, mp 110 – 112 °C. 1H NMR (CDCl3) δ ppm 2.07 (6H, s, 2×CH3), 2.28 (3H, s, CH3), 6.36 (1H, s, ArH-6), 6.76 2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.89 (2H, s, ArH-3″, 5″), 7.45 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.62 (1H, s, ArH-3). MS m/z (%): 389 (M+1, 100); HPLC Purity: 98.3%.
6-(4-Cyano-2,6-dimethylphenoxy)-1-(4-cyanophenyl)-5-nitro-1H-benzoimidazole (39)
Triethyl orthoformate (1 mL, excess) and HCl in diethyl ether (1N, 1 mL) were added successively to a solution of 37 (50 mg, 0.125 mmol) in DMF (3 mL) under N2. The mixture was stirred at rt for 3 h, poured into water with pH adjusted to 6-7, and left to stand overnight. The resulting yellow solid was collected and washed with water. The crude product was purified by a silica gel column (eluent: CH2Cl2/EtOAc 10:1) to afford 42 mg of 39, yield 88%, light yellow solid, mp 250 – 252 °C. 1H NMR (CDCl3) δ ppm 2.21 (6H, s, 2×CH3), 6.43 (1H, s, ArH-7), 7.43 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.46 (2H, s, ArH-3″, 5″), 7.86 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 8.16 (1H, s, ArH-2), 8.54 (1H, s, ArH-4). MS m/z (%): 410 (M+1, 100); HPLC Purity: 96.0%.
(4″-Cyano-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-nitrobenzene-1,2-diamine hydrochloride (40)
To a solution of 36 (100 mg) in acetone (10 mL) was slowly added HCl diethyl ether solution (18%, 3 mL), and a light yellow solid appeared. The solid was filtered out and recrystallized from anhydrous MeOH to afford 52 mg of 40, yield 48%, yellow solid, mp 170 – 171 °C; 1H NMR δ ppm 2.07 (6H, s, 2×CH3), 6.17 (1H, s, ArH-6), 6.83 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.40 (2H, s, ArH-3″, 5″), 7. 54 (1H, s, ArH-3), 7.56 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.50 (1H, s, NH); MS m/z (%) 453 (M+1, 100), 455 (M+3, 100); HPLC Purity: 97.6%.
HIV Growth Inhibition Assay in H9 Lymphocytes from Panacos, Inc
The evaluation of HIV-1 inhibition was carried out as follows according to established protocols. The human T-cell line, H9, was maintained in continuous culture with complete medium (RPMI 1640 with 10% fetal calf serum supplemented with L-glutamine at 5% CO2 and 37 °C. Test samples were first dissolved in dimethyl sulfoxide at a concentration of 10 mg/mL to generate master stocks with dilutions made into tissue culture media to generate working stocks. The following drug concentrations were routinely used for screening: 100, 20, 4 and 0.8 μg/mL. For agents found to be active, additional dilutions were prepared for subsequent testing so that an accurate EC50 value could be determined. Test samples were prepared and to each sample well was added 90 μL of media containing H9 cells at 3 × 105 cells/mL and 45 μL of virus inoculum (HIV-1 IIIB isolate) containing 125 TCID50. Control wells containing virus and cells only (no drug) and cells only (no virus or drug) were also prepared. A second set of samples were prepared identical to the first and were added to cells under identical conditions without virus (mock infection) for toxicity determinations (IC50 defined below). In addition, AZT was also assayed during each experiment as a positive drug control. On days 1 and 4 post-infection (PI), spent media was removed from each well and replaced with fresh media. On day 6 PI, the assay was terminated and culture supernatants were harvested for analysis of virus replication by p24 antigen capture. Compound toxicity was determined by XTT using the mock-infected samples.
Assay for Measuring the Inhibitory Activity of Compounds on HIV-1 IIIB Replication in MT-2 Cells
The inhibitory activity of compounds on HIV-1 IIIB replication was determined as previously described. In brief, 1 × 104 MT-2 cells were infected with an HIV-1 strain (100 TCID50) in 200 μL RPMI 1640 medium containing 10% FBS in the presence or absence of a test compound at graded concentrations overnight. Then, the culture supernatants were removed and fresh media containing no test compounds were added. On the fourth day post-infection, 100 μL of culture supernatants were collected from each well, mixed with equal volumes of 5% Triton X-100 and assayed for p24 antigen, which was quantitated by ELISA. Briefly, wells of polystyrene plates (Immulon 1B, Dynex Technology, Chantilly, VA) were coated with HIV immunoglobulin (HIVIG) in 0.085 M carbonate-bicarbonate buffer (pH 9.6) at 4 °C overnight, followed by washes with washing buffer (0.01M PBS containing 0.05% Tween-20) and blocking with PBS containing 1% dry fat-free milk (Bio-Rad, Inc., Hercules, CA). Virus lysates were added to the wells and incubated at 37 °C for 1 h. After extensive washes, anti-p24 mAb (183-12H-5C), biotin-labeled anti-mouse IgG1 (Santa Cruz Biotech., Santa Cruz, CA), streptavidin-labeled horseradish peroxidase (Zymed, S. San Francisco, CA), and the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma Chemical Co., St. Louis, MO) were added sequentially. Reactions were terminated by addition of 1N H2SO4. Absorbance at 450 nm was recorded in an ELISA reader (Ultra 386, TECAN, Research Triangle Park, NC). Recombinant protein p24 purchased from US Biological (Swampscott, MA) was included for establishing standard dose response curves. Each sample was tested in triplicate. The percentage of inhibition of p24 production was calculated as previously described.25 The effective concentrations for 50% inhibition (EC50) were calculated using a computer program, designated CalcuSyn,26 kindly provided by Dr. T. C. Chou (Sloan-Kettering Cancer Center, New York, New York).
HIV-1 Infectivity Assay with Mutant Viral Strains in MT-2 Cell Line
A diluted drug resistant HIV-1 stock, mutated viral strain (8605MR or 6005MR from Panacos, Inc), at a multiplicity of infection (MOI) of 0.001 TCID50/cell was used to infect MT-2 cells. Twenty μL of the virus and 20 μL of compounds at various concentrations in RPMI 1640 containing 10% fetal bovine serum were added to 20 μL of MT4 cells at 6 × 105 cells/mL in a 96-well microtiter plate. The cell/virus /compound mixture was then incubated at 37°C in a humidified CO2 incubator. Fresh medium (180 μL) containing an appropriate concentration of the compound was added to each well of the cultures on day 2. On day 4 post-infection, supernatant samples were harvested and assayed for P24 using an ELISA kit from ZeptoMetrix Corporation, Buffalo, New York.
Assessment of In Vitro Cytotoxicity in MT-2 Cells
The in vitro cytotoxicity of compounds on MT-2 cells was measured by XTT assay.27 Briefly, 100 μL of the test compound at graded concentrations were added to equal volumes of cells (5 × 105/mL) in wells of a 96-well plate. After incubation at 37 °C for 4 days, 50 μL of XTT solution (1 mg/mL) containing 0.02 μM of phenazine methosulphate (PMS) was added. After 4 h, the absorbance at 450 nm was measured with an ELISA reader. The CC50 (concentration for 50% cytotoxicity) values were calculated using the CalcuSyn computer program as described above.
Assay for RT Enzymatic Inhibition
The RT activity of HIV-1DH012 (a primary isolate viral strain) was determined in the presence of various concentrations of the tested compounds using a Roche colorimetric HIV-1 RT assay kit following the protocol provided by the manufacturer.
Computational Study
Three-dimensional constructions of compounds 21, 34 and 37 were achieved by using the Sybyl version 7.2 software with the tripos force-field and Gasteiger-Hückel atomic partial charges.28 In order to get the correct orientation of the three compounds inside the HIV-1 RT enzyme, the constructions were made by modifying the experimental coordinates of the DAPY compound in the HIV-1 RT enzyme (PDB code 1s6q). Then, the compounds were optimized with 20 simplex iterations followed by 1000 steps of Powell algorithm.
Energy minimizations were obtained with the Amber 9 software.29 Ligand parameters were created according to the antechamber procedure and the partial charges were evaluated following the AM1 bond charge correction (AM1-BCC) method.30-32 The general amber force field33 (gaff) was set for the ligand, whereas the ff03 force field34 was used for the protein. Minimizations were done with 1000 steps of steepest descent followed by 4000 steps of conjugate gradient. Solvent effects were taken into account through general born model implicit solvent model.35-38 The Molecular Mechanics/General Born Surface Area (MM/GBSA) method39-41 permitted the evaluation of the ligand/protein interaction free energies, and calculation details may be found elsewhere.42 Finally, VMD software43 was employed to visualize and analyze the 3D structures resulting from AMBER calculations.
Supplementary Material
Acknowledgments
We would like to thank Ms. Nicole Kilgore at Panacos Pharmaceuticals, Inc. for providing data listed in Tables 1 and 2. HIV-1 mutated viral strains 8605MR (multi-mutated-RT: M41L, D67N, L210W, T215Y, M184V, K103N) and 6005MR (multi-mutated-RT: M41L, L74V, M184V, L210W, T215Y, ins SS, A98G) were from Panacos, Inc and HIV-1 strains IIIB, A17 were provided by the NIH AIDS Reagent & Reference Program, by contributions from Drs. R. Gallo, E. Emini, and Trimeris, Inc. This investigation was supported by grants 20472114 and 7052057 from the National Natural Science Foundation of China and Beijing government, respectively, awarded to L. Xie, as well as US NIH grants awarded to K. H. Lee (AI33066), S. Jiang (AI46221), and C. H. Chen (AI65310).
Abbreviations
- CC50
concentration for 50% cytotoxicity
- DAPY
diarylpyrimidine
- EC50
effective concentration for 50% inhibition
- HAART
highly active antiretroviral therapy
- HIV
human immunodeficiency virus
- MM/GBSA
molecular mechanism/general born surface area
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- NRTI
nucleoside reverse transcriptase inhibitor
- PDB
protein data base
- PI
protease inhibitor
- RF
resistant fold
- RMSD
root mean square deviation
- RT
reverse transcriptase
- RTI
reverse transcriptase inhibitor
- SAR
structure-activity relationship
- SI
selective index (ratio of CC50/EC50)
Footnotes
Supporting Information Available: HPLC conditions and summary of HPLC purity data for final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tantillo C, Ding JP, Jacobo-Molina A, Nanni RG, Boyer PL, Hughes SH, Pauwels R, Andries K, Janssen PAJ, Arnold E. Locations of anti-AIDS drug binding sites and resistance mutations in the three-dimensional structure of HIV-1 reverse transcriptase: implications for mechanisms of drug inhibition and resistance. J Mol Biol. 1994;243:369–387. doi: 10.1006/jmbi.1994.1665. [DOI] [PubMed] [Google Scholar]
- 2.Pauwels R. New non-nucleoside reverse transcriptase inhibitors (NNRTIs) in development for the treatment of HIV infections. Curr Opin Pharmacol. 2004;4:437–446. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Andries K, Azijn H, Thielemans T, Ludovici D, Kukla M, Heeres J, Janssen P, De Corte B, Vingerhoets J, Pauwels R, de Béthuneet MP. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against non-nucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicro Agents Chemother. 2004;48:4680–4686. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl] amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Tucker TJ, Sisko JT, Tynebor RM, Williams TM, Felock PJ, Flynn JA, Lai MT, Liang YX, Gaughey GM, Liu MQ, Miller M, Moyer G, Munshi V, Perlow-Poehnelt R, Prasad S, Reid JC, Sanchez R, Torrent M, Vacca JP, Wan BL, Yan YW. Discovery of 3-{5-[(6-Amino-1H-pyrazolo [3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem. 2008;51:6503–6511. doi: 10.1021/jm800856c. [DOI] [PubMed] [Google Scholar]
- 7.Romines KR, Freeman GA, Schaller LT, Cowan JR, Gonzales SS, Tidwell JH, Andrews CW, Stammers DK, Hazen RJ, Ferris RG, Short SA, Chan JH, Boone LR. Structure–activity relationship studies of novel benzophenones leading to the discovery of a potent, next generation HIV non-nucleoside reverse transcriptase inhibitor. J Med Chem. 2006;49:727–739. doi: 10.1021/jm050670l. [DOI] [PubMed] [Google Scholar]
- 8.Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: a new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 9.Das K, Lewi PJ, Hughes SH, Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 10.De Corte BL. From 4,5,6,7-Tetrahydro-5-methylimidazo[4,5,1-jk](1.4)benzodiazepin-2(1H)-one (TIBO) to Etravirine (TMC125): Fifteen Years of Research on Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase. J Med Chem. 2005;48:1689–1696. doi: 10.1021/jm040127p. [DOI] [PubMed] [Google Scholar]
- 11.Das K, Clark AD, Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, De Béthune M-P, Boyer PL, Clark P, Hughes SH, Janssen PA, Arnold E. Roles of conformational and positional adaptability in structure based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 12.Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci US A. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heeres J, de Jonge M, Koymans LMH, Daeyaert FFD, Vinkers M, Van Aken KJA, Arnold E, Das K, Kilonda A, Hoornaert GJ, Compernolle F, Cegla M, Azzam RA, Andries K, de Béthune MP, Azijn H, Pauwels R, Lewi PJ, Janssen PAJ. Design, synthesis, and SAR of a novel pyrazinone series with non-nucleoside HIV-1 reverse transcriptase inhibitory activity. J Med Chem. 2005;48:1910–1918. doi: 10.1021/jm040829e. [DOI] [PubMed] [Google Scholar]
- 14.Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. Optimization of azoles as anti-human immunodeficiency virus agents guided by free-energy calculations. J Am Chem Soc. 2008;130:9492–9499. doi: 10.1021/ja8019214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweeney ZK, Dunn JP, Li Y, Heilek G, Dunten P, Elworthy TR, Han XC, Harris SF, Hirschfeld DR, Hogg JH, Huber W, Kaiser AC, Kertesz DJ, Kim W, Mirzadegan T, Roepel MG, Saito YD, Silva TMPC, Swallow S, Tracy JL, Villasenor A, Vora H, Zhou AS, Klumpp K. Discovery and optimization of pyridazinone non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2008;18:4352–4354. doi: 10.1016/j.bmcl.2008.06.072. [DOI] [PubMed] [Google Scholar]
- 16.Frlan R, Kikelj D. Recent progress in diaryl ether synthesis. Synth. 2006;14:2271–2285. [Google Scholar]
- 17.Li F, Wang QR, Ding ZB, Tao FG. Microwave-assisted synthesis of diaryl ethers without catalyst. Org Lett. 2003;5:2169–2171. doi: 10.1021/ol0346436. [DOI] [PubMed] [Google Scholar]
- 18.Piersanti G, Giorgi L, Bartoccini F, Tarzia G, Minetti P, Gallo G, Giorgi F, Castorina M, Ghirardi O, Carminati P. Synthesis of benzo[1,2-d;3,4-d]diimidazole and 1H-pyrazolo[4,3-b]pyridine as putative A2A receptor antagonists. Org Biomol Chem. 2007;5:2567–2571. doi: 10.1039/b707599e. [DOI] [PubMed] [Google Scholar]
- 19.Tian XT, Qin BJ, Lu H, Lai WH, Jiang S, Lee KH, Chen CH, Xie L. Discovery of diarylpyridine derivatives as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem Lett. 2009;19:5482–5485. doi: 10.1016/j.bmcl.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 21.Obiol-Pardo C, Rubio-Martinez J. Comparative evaluation of MMPBSA and XSCORE to compute binding free energy in XIAP-peptide complexes. J Chem Inf Model. 2007;47:134–142. doi: 10.1021/ci600412z. [DOI] [PubMed] [Google Scholar]
- 22.Guimaraes CR, Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J Chem Inf Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- 23.Ludovici DW, De Corte BL, Kukla MJ, Ye H, Ho CY, Lichtenstein MA, Kavash RW, Andries K, de Bethune MP, Azijn H, Pauwels R, Lewi PJ, Heeres J, Koymans LMH, de Jonge MR, Van Aken KJA, Daeyaert FFD, Das K, Arnold E, Janssen PAJ. Evolution of anti-HIV drug candidates. part 3: diarylpyrimidine (DAPY) analogues. Bioorg Med Chem Lett. 2001;11:2235–2239. doi: 10.1016/s0960-894x(01)00412-7. [DOI] [PubMed] [Google Scholar]
- 24.Mordant C, Schmitt B, Pasquier E, Demestre C, Queguiner L, Masungi C, Peeters A, Smeulders L, Bettens E, Hertogs K, Heeres J, Lewi P, Guillemont J. Synthesis of novel diarylpyrimidine analogues of TMC278 and their antiviral activity against HIV-1 wild-type and mutant strains. Eur J Med Chem. 2007;42:567–579. doi: 10.1016/j.ejmech.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N′-(2,2,6,6-tetramethylpiperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 27.Dupnik KM, Gonzales MJ, Shafer RW. Most multidrug-resistant HIV-1 reverse transcriptase clones in plasma encode functional reverse transcriptase enzymes. Antivir Ther. 2001;6:41–46. [Google Scholar]
- 28.SYBYL 70. Tripos Inc; 1699 South Hanley Rd., St Louis, Missouri, 63144 USA: [Google Scholar]
- 29.Case D, Darden T, Cheatham T, Simmerling C, Wang J, Duke R, Luo R, Merz K, Pearlman D, Crowley M, Walker R, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong K, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews D, Schafmeister C, Ross W, Kollman P. AMBER, 90. University of California; San Francisco: 2006. [Google Scholar]
- 30.Dewar M, Zoebisch E, Healy E, Stewart J. AM1: A new general purpose quantum mechanical molecular model. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 31.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 32.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 34.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 35.Beroza P, Case D. Calculations of proton-binding thermodynamics in proteins. Methods Enzymol. 1998;295:170–189. doi: 10.1016/s0076-6879(98)95040-6. [DOI] [PubMed] [Google Scholar]
- 36.Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 37.Gilson MK. Theory of electrostatic interactions in macromolecules. Curr Opin Struct Biol. 1995;5:216–223. doi: 10.1016/0959-440x(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 38.Madura JD, Davis ME, Gilson MK, Wade RC, Luty BA, Mc Cammon JA. Biological applications of electrostatic calculations and brownian dynamics simulations. Rev Comput Chem. 1994;5:229–267. [Google Scholar]
- 39.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Morin P, Wang W, Kollman PA. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J Am Chem Soc. 2001;123:5221–5230. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Kollman PA. Computational study of protein specificity: the molecular basis of HIV-1 protease drug resistance. Proc Natl Acad Sci USA. 2001;98:14937–14942. doi: 10.1073/pnas.251265598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Y, Barbault F, Gourmala C, Zhang Y, Maurel F, Hu Y, Fan B. Cellular interaction through LewisX cluster: theoretical studies. J Mol Model. 2008;14:901–910. doi: 10.1007/s00894-008-0325-9. [DOI] [PubMed] [Google Scholar]
- 43.Humphrey W, Dalke A, Schulten K. VMD - visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.