

Abstract
Pemphigus vulgaris (PV) is an autoimmune bullous disease in which autoantibodies against proteins of the desmosomal adhesion complex perturb desmosomal function, leading to intercellular adhesion defects in the oral mucosa and skin. Previous studies have demonstrated a central role for downregulation of the desmosomal cadherin desmoglein 3 (DSG3) in the pathogenesis of PV. However, the effects of non-cadherin desmosomal proteins in modulating the cellular manifestations of PV remain poorly understood. Here, we characterize the expression and functional importance of Perp, a newly discovered tetraspan desmosomal protein, in PV. Our data demonstrate that PV autoantibodies disrupt Perp expression at the membrane and trigger its internalization along with DSG3 into the endosomal pathway, where it is ultimately targeted to the lysosome for degradation. We further show that Perp deficiency exacerbates the pathogenic effects of PV autoantibodies on keratinocytes by enhancing both the depletion of desmosomal DSG3 and intercellular adhesion defects. Together, our findings highlight the importance of non-cadherin desmosomal proteins in modulating PV phenotypes and provide new insight into Perp’s role in the desmosome.
INTRODUCTION
Desmosomes are cell–cell adhesion complexes that maintain tissue integrity in epithelia exposed to mechanical stress, such as the skin (Yin and Green, 2004). The core of the desmosome comprises three classes of proteins: cadherins, armadillo proteins, and plakins. Desmosomal cadherins include desmogleins (DSGs) 1–4 and desmocollins 1–3, which are single-span transmembrane proteins whose ectodomains engage in homotypic and heterotypic interactions between membranes of apposing cells (Getsios et al., 2004). The cytoplasmic surface of the desmosomal cadherin tail interacts with the armadillo proteins plakoglobin (PG), which bridges DSGs and desmocollins to the intermediate filament linker protein desmoplakin (DP), and plakophilins, which cluster desmosomal proteins through lateral interactions (Hatzfeld, 1999, 2007; Garrod et al., 2002). Defects in any of these classes of desmosomal components can lead to impaired intercellular adhesion and increased skin fragility and blistering (Yin and Green, 2004).
Pemphigus vulgaris (PV) is an autoimmune blistering disease in which autoantibodies against desmosomal proteins, especially DSG3, perturb their function, leading to suprabasilar blistering of the oral mucosa and the skin (Stanley and Amagai, 2006). The functional importance of anti-DSG3 antibodies in the pathogenesis of PV has been demonstrated by their necessity and sufficiency in producing the blistering phenotype. Specifically, intraperitoneal injection of purified IgG, monoclonal antibodies, or single chain variable region fragments (ScFv) directed against DSG3 into neonatal mice recapitulates the suprabasilar blistering characteristic of PV (Amagai et al., 1992; Tsunoda et al., 2003; Payne et al., 2005). Conversely, absorption of autoantibodies from PV patient sera with extracellular domain fragments of DSG3 prevents blister formation in the same neonatal mouse model (Amagai et al., 1994a). The critical importance of DSG3 dysfunction in blistering is further supported by the finding that Dsg3-null mice develop acantholysis and blistering of the oral mucosa similar to that observed in PV patients (Koch et al., 1997).
Although it is clear that PV autoantibodies target DSG3, how this interaction leads to disrupted cell–cell adhesion is not understood completely (Sharma et al., 2007). It appears that exposure of cells to PV autoantibodies causes depletion of DSG3 and keratin retraction, with consequent intercellular adhesion defects (Ishii et al., 2005; Yamamoto et al., 2007). Epitope mapping experiments have suggested that PV autoantibodies target the extracellular domain of DSG3 to disrupt its intercellular adhesive function, thus directly facilitating cell–cell separation and blister formation (Amagai et al., 1994b; Futei et al., 2000). However, PV autoantibodies also trigger a series of intracellular events, including internalization of DSG3 and PG and eventual degradation of DSG3 in lysosomes (Calkins et al., 2006). Moreover, recent studies implicate specific signaling cascades in provoking the desmosome dissolution observed in PV. Specifically, inhibition of p38MAPK abolishes blistering in neonatal mice injected with PV autoantibodies, despite histological evidence of intact antibody–antigen interactions (Berkowitz et al., 2006). In addition, PG-deficient mice display a surprising resistance to PV autoantibodies, manifested as a failure to exhibit the hallmark PV phenotypes of keratin retraction and defective cell–cell adhesion, supporting a possible signaling role for PG that is independent of its structural function within the desmosome (Caldelari et al., 2001). Non-cadherin proteins therefore appear to actively modulate the response to PV autoantibodies.
Perp is a recently discovered tetraspan membrane protein that plays an important role in epithelial integrity through a function at the desmosome (Ihrie et al., 2005). Perp-deficient mice exhibit blistering in the oral mucosa and skin reminiscent of that seen in PV patients or in either humans or mice carrying mutations in genes encoding specific desmosomal components (Holthofer et al., 2007). Loss of Perp leads to clear ultrastructural defects in desmosomes, with desmosomes in Perp-deficient tissues appearing wider and less electron dense than in their wild-type counterparts—an indication of improper desmosome assembly (Ihrie et al., 2005). In addition, Perp deficiency leads to enhanced solubility of DSG3 and PG in the detergent Triton X-100, reflective of improper incorporation of these components into the desmosome. Together, these findings suggest a defect in the proper assembly or maintenance of desmosomes in the absence of Perp.
Given that Perp deficiency affects the solubility of DSG3, the key antigen targeted by PV autoantibodies, and that mice lacking Perp develop severe blistering of the oral mucosa similar to that observed with PV, we hypothesize that Perp might play an important role in PV pathogenesis. Here, we test this idea by characterizing the behavior of Perp after exposure of keratinocytes to PV autoantibodies and by examining the consequences of Perp deficiency for the manifestation of PV-associated phenotypes. Our results suggest a role for Perp in modulating the PV phenotype and thereby provide insight into PV pathogenesis as well as Perp’s function in desmosomal adhesion.
RESULTS
Characterizing Perp localization in response to pemphigus vulgaris autoantibodies
To investigate the role of Perp in PV, we first characterized the effects of PV autoantibodies on Perp localization. We purified the IgG fraction from normal human serum and PV patient serum, then verified the activity of isolated PV IgG with several assays. First, immunofluorescence analysis revealed that these antibodies stained the plasma membrane of human keratinocyte monolayers in a pattern indistinguishable from that of DSG3, the antigen targeted in PV (Figure 1a). Second, Western blot analysis showed that both PV serum and purified PV IgG, but not normal human serum or purified IgG, recognize a protein of ~130 kDa, the size of DSG3 (Amagai et al., 1991; Figure 1b). Third, immunoprecipitation–western blot experiments demonstrated that PV IgG can immunoprecipitate DSG3 and its interacting partner PG (Figure 1c). In contrast, the cytoskeletal linker protein DP was not detected in this immunoprecipitated protein complex. Finally, PV IgG reduced the intercellular adhesive strength of human keratinocyte monolayers, as evidenced by the increased fragmentation of these monolayers in response to mechanical stress (Figure 1d). These studies confirm the ability of purified PV IgG to bind to DSG3 in human keratinocytes, as well as to induce intercellular adhesion defects, the cellular hallmark of PV.
Figure 1. Characterization of PV IgG.
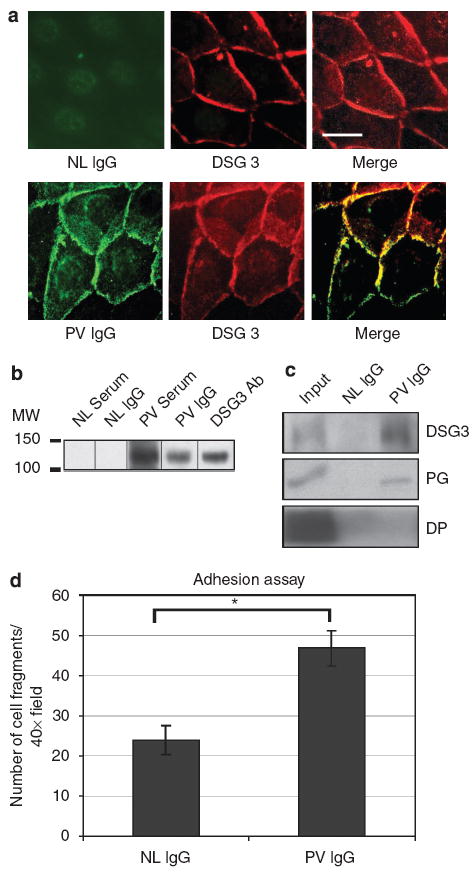
Studies in (a), (b), and (c) were conducted using human keratinocytes cultured in 0.5mm calcium for 18 hours before experimentation. (a) Representative immunofluorescence images show that PV IgG, but not normal (NL) IgG, recognizes a plasma membrane antigen with a localization pattern similar to that of DSG3. Scale bar = 10 μm. (b) Western blot analysis examining reactivity of human sera and antibodies against human keratinocyte protein lysates. PV and NL sera, purified PV and NL IgG fractions, and DSG3 antibodies were tested. (c) Immunoprecipitation–Western blot analysis of proteins immunoprecipitated by NL and PV IgG and probed for the desmosome components DSG3, PG, and DP. 10% of the input is shown. (d) Mechanical dissociation assay performed on monolayers of wild-type mouse keratinocytes treated with PV IgG or NL IgG for 24 hours. The numbers of fragments generated were counted in four 40 × fields in each of three experiments, and results represent the mean±SEM (n = 12). Statistical significance (*) was determined using the unpaired Student’s t-test (P<0.05).
To determine if Perp localization is affected by PV antibodies, we induced desmosome formation in human keratinocytes with high calcium, then treated the cells with either normal or PV IgG for varying amounts of time. In cells treated with normal IgG, Perp remained at the plasma membrane throughout the 6-hour time course, consistent with its role in cell–cell adhesion (Figure 2a). In contrast, in PV IgG-treated cells, Perp membrane staining was disrupted, with numerous Perp-containing puncta localizing to the cytoplasm by 3 hours. By 6 hours, Perp membrane staining was further diminished whereas Perp-containing puncta coalesced around the nucleus. These data suggest that PV autoantibodies trigger Perp internalization in human keratinocytes.
Figure 2. Perp is internalized with DSG3 and PG upon PV IgG exposure.
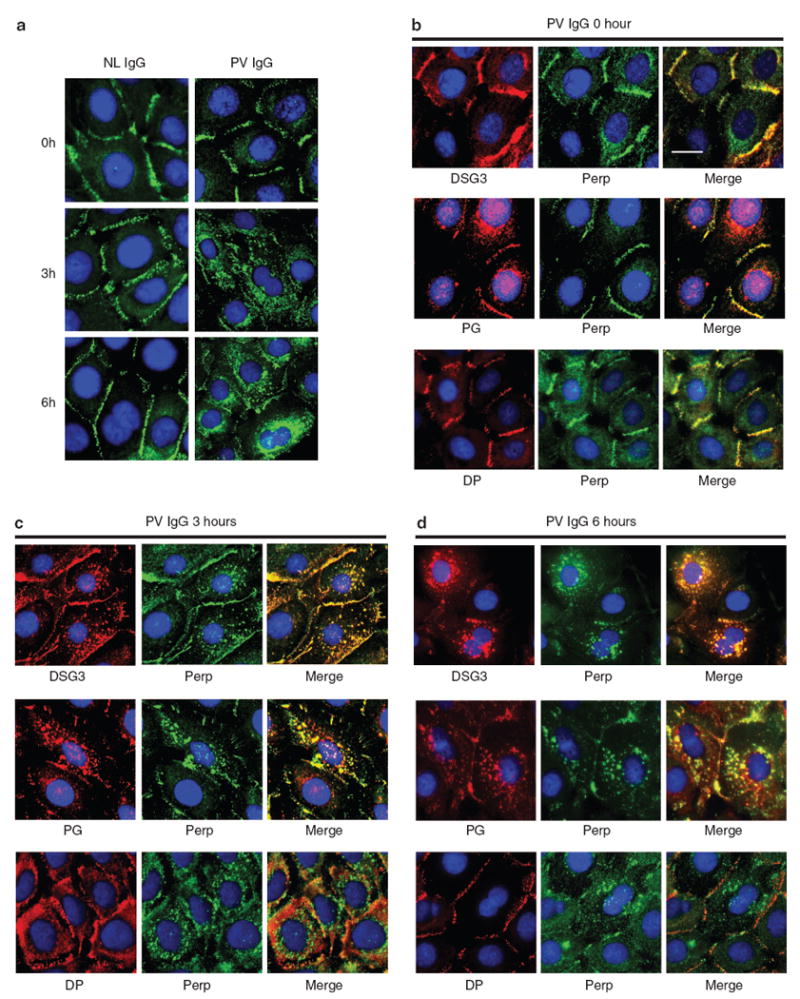
(a) Representative immunofluorescence images of Perp in human keratinocyte monolayers treated with NL (left) or PV (right) IgG for 0, 3, or 6 hours. (b–d) Representative immunofluorescence images of Perp and DSG3, PG, or DP in human keratinocyte monolayers treated with PV IgG for 0, 3, or 6 hours. Nuclei are stained with DAPI. Scale bar = 10 μm and applies to all photomicrographs in the figure.
To determine whether Perp traffics with other desmosomal proteins during the internalization process, we examined Perp colocalization with either DSG3 or PG, both of which have been shown previously to undergo internalization in response to PV autoantibodies (Calkins et al., 2006). Before PV autoantibody exposure, Perp staining showed significant overlap with DSG3, PG, and DP staining at the plasma membrane (Figure 2b). In cells exposed to PV autoantibodies, numerous cytoplasmic puncta containing DSG3 or PG appeared by 3 hours after treatment, and accumulated in the perinuclear region by 6 hours, consistent with previous reports (Calkins et al., 2006; Figure 2c and d). Interestingly, at both time points, DSG3- and PG-containing vesicles showed extensive colocalization with cytoplasmic Perp-containing puncta. In contrast, DP did not colocalize with Perp-positive puncta. These data suggest that Perp traffics with DSG3 and PG, and that Perp might associate with either DSG3 or PG independently or form a DSG3–PG–Perp complex as these proteins undergo internalization in response to PV autoantibodies.
PV IgG induces Perp internalization via the endosomal pathway in a fashion similar to PG
Recent studies have shown that within several hours of PV IgG exposure, DSG3 enters the endosomal pathway, eventually getting degraded after fusion with lysosomes (Calkins et al., 2006). As Perp colocalizes with both DSG3 and PG during the internalization process, we hypothesized that it too would enter the endosomal pathway in response to PV autoantibodies. We examined Perp and PG localization in direct comparison to that of EEA-1, an early endosome marker (Mu et al., 1995), in keratinocyte monolayers treated with PV IgG for 0 and 3 hours. At 0 hour, neither PG nor Perp colocalized with EEA-1, as both desmosomal proteins localized to the membrane whereas EEA-1-positive endosomes were dispersed throughout the cytoplasm (Figure 3a). However, after 3 hours of PV IgG treatment, a significant fraction of PG now costained with EEA-1-labeled endosomes, consistent with the entrance of PG into the endosomal compartment (Figure 3b). Moreover, in keeping with our previous observation that Perp colocalizes with PG, we observed a significant overlap between Perp-containing puncta and EEA-1-labeled endosomes 3 hours after initiating PV IgG treatment (Figure 3b). These findings confirm that similarly to PG, Perp is internalized via the endosomal pathway.
Figure 3. Perp is internalized into endosomes in a similar pattern to PG upon PV IgG exposure.
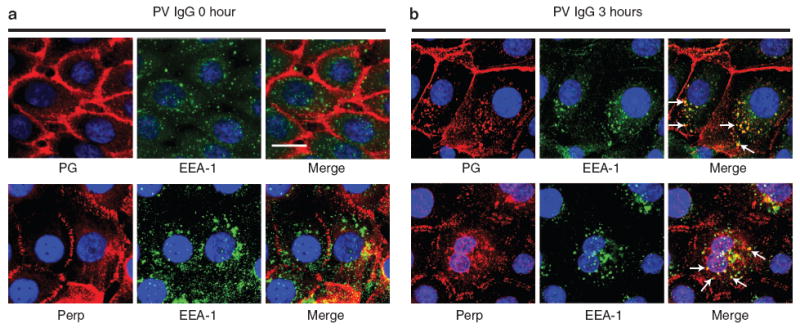
Representative immunofluorescence images for the endosome marker EEA-1 and either Perp or PG in human keratinocyte monolayers treated with PV IgG for 0 (a) or 3 hours (b). Nuclei are stained with DAPI. Arrows indicate several puncta in which Perp or PG colocalizes with EEA-1. Scale bar = 10 μm and applies to all photomicrographs in the figure.
Following internalization via endosomes, Perp might be recycled back to the membrane for reassembly into new desmosomes or undergo degradation through fusion with lysosomes, or both. To determine if a portion of cellular Perp might be depleted via lysosomes in response to PV autoantibodies, we costained PV IgG-treated human keratinocytes for Perp and CD63, a lysosome marker (Metzelaar et al., 1991). At 0 hour, as with PG, Perp failed to colocalize with perinuclear aggregates of CD63-positive lysosomes (Figure 4a). By 3 hours after PV IgG exposure, however, a portion of PG appeared in lysosomes (Figure 4b). Similarly, colocalization of Perp and CD63 was apparent 3 hours after treatment, indicating that Perp is delivered to lysosomes for degradation during this time frame (Figure 4b). Although DSG3 was previously shown to internalize and sequentially transit from the endosome to the lysosome in response to PV IgG (Calkins et al., 2006), the visualization of Perp in both endosomes and lysosomes at the 3 hour time point here likely reflects temporal heterogeneity in the internalization process across the population of cells. Together, our data indicate that Perp is internalized into the endocytic pathway in the setting of PV, as previously reported for DSG3 and PG (Calkins et al., 2006).
Figure 4. Perp enters lysosomes in response to PV IgG exposure.
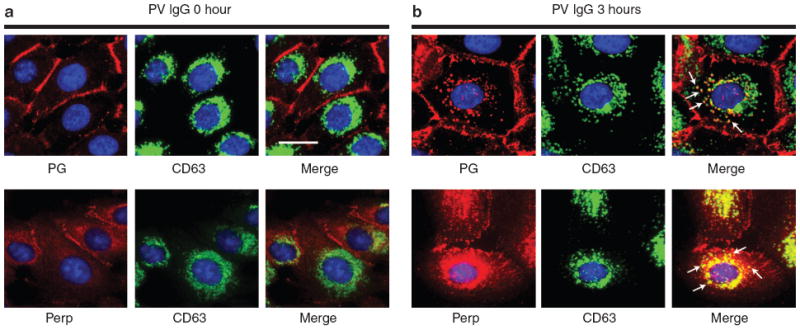
Representative immunofluorescence images for the lysosome marker CD63 and either Perp or PG in human keratinocyte monolayers treated with PV IgG for 0 (a) or 3 hours (b). Nuclei are stained with DAPI. Arrows indicate several puncta in which Perp or PG colocalizes with CD63. Scale bar = 10 μm and applies to all photomicrographs in the figure.
Loss of Perp cooperates with PV autoantibodies to enhance DSG3 and PG depletion from desmosomes
In epithelial monolayers, proteins stably incorporated into desmosomes can only be solubilized by chaotropic agents such as urea, reflecting the inherent stability of desmosomal complexes at the membrane and their connection to keratin intermediate filaments (Franke et al., 1983; Kapprell et al., 1985; Bornslaeger et al., 2001; Vasioukhin et al., 2001). Increased solubility of desmosomal constituents in milder agents such as the detergent Triton X-100 indicates either improper assembly into mature desmosomes or disrupted connections to the intermediate filament network (South et al., 2003). To determine whether Perp solubility is affected by PV autoantibodies, we examined Perp levels in Triton X-100-soluble and urea fractions from mouse keratinocytes treated with normal or PV serum. Although the amount soluble in the Triton X-100 fraction was not clearly changed, Perp levels were diminished in the urea fraction of cells treated with PV serum, suggesting that exposure to PV autoantibodies causes depletion of Perp from mature desmosomes (Figure 5). This observation is consistent with the idea that desmosomal Perp is being internalized and ultimately degraded in the lysosome.
Figure 5. Loss of Perp cooperates with PV serum to induce depletion of DSG3 and PG.
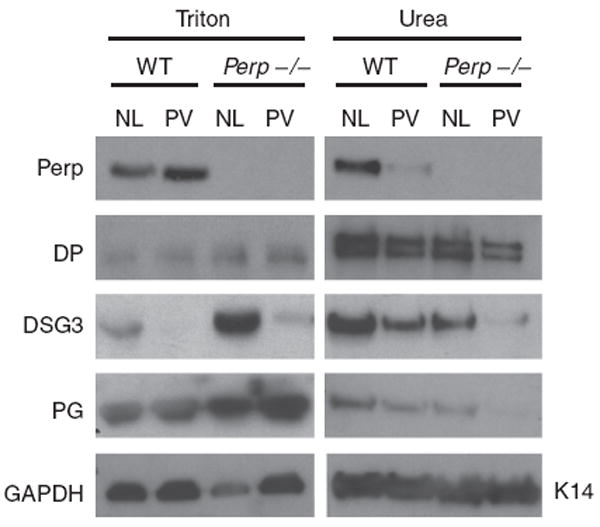
Western blot analysis of Triton X-100 and urea fractions from wild-type (WT) and Perp−/− mouse keratinocyte monolayers treated with normal (NL) or PV serum (PV) for 24 hours. An examination of Perp solubility and levels as well as the effects of Perp loss on the solubility profiles of key desmosomal proteins (DP, DSG3, and PG) is shown. GAPDH and Keratin 14 (K14) serve as loading controls for the Triton-soluble and urea fractions, respectively.
To examine the combined effects of Perp deficiency and PV autoantibody treatment on desmosomal complexes, we analyzed the solubility of other desmosomal proteins under these conditions. Consistent with our previous studies showing that Perp deficiency leads to increased Triton X-100 solubility of DSG3 and PG (Ihrie et al., 2005), we found that Perp−/− keratinocytes treated with normal serum displayed dramatically increased DSG3 levels in the Triton X-100 soluble fraction and reduced DSG3 levels in the urea fraction relative to those in wild-type mouse keratinocytes treated with normal serum (Figure 5). The same pattern was observed with PG. In contrast, the solubility profile of DP remained relatively unaffected by Perp deficiency. The significant titration of DSG3 and PG from the urea to Triton X-100 soluble pool suggests a defect in the stable assembly of these proteins into the desmosomes of Perp-deficient cells. To determine if there is a cooperative effect of PV autoantibodies and Perp loss, we compared the solubility profiles of DSG3 and PG in wild-type and Perp−/− mouse keratinocytes treated with PV serum. In wild-type cells, PV serum treatment resulted in decreased DSG3 in both the Triton X-100 and urea fractions compared to the normal serum control, suggesting a depletion of DSG3 following PV serum exposure (Figure 5). The levels of PG in the urea fraction also declined, likely reflecting its decreased incorporation into desmosomes. Loss of Perp further augmented the effects of PV autoantibodies on DSG3 and PG levels in the urea fraction. Although some DSG3 and PG remained in the urea fraction of wild-type cells exposed to PV autoantibodies, very little of either protein was detected in the urea fraction of treated Perp-deficient cells, suggesting a cooperative effect of Perp loss and PV IgG treatment in depletion of these proteins from mature desmosomes. In contrast, Perp deficiency did not significantly affect DP levels in the urea fraction of PV autoantibody-treated keratinocytes. Together, these data suggest that Perp facilitates proper assembly of DSG3 and PG into mature desmosomes, and that Perp deficiency cooperates with PV autoantibodies to induce desmosomal DSG3 and PG depletion.
Loss of Perp cooperates with PV autoantibodies to impair intercellular adhesion
To examine how the enhanced solubility of desmosomal DSG3 and PG due to Perp deficiency affects the physiologic response central to PV pathology, we compared the intercellular adhesive strength of wild-type and Perp-deficient mouse keratinocytes treated with PV IgG using a quantitative mechanical dissociation assay (Huen et al., 2002), where increased fragmentation of the cell monolayer reflects a decrease in intercellular adhesive strength. Previous studies have demonstrated a positive correlation between the degree of monolayer fragmentation produced by sera from different PV patients and the severity of clinical symptoms (Ishii et al., 2005). First, we showed that compared to wild-type keratinocytes, significantly more fragments were observed in Perp−/− keratinocytes on treatment with control normal IgG, indicating that loss of Perp significantly reduces the baseline adhesive strength of keratinocytes (Figure 6). This finding is in agreement with our previous observations demonstrating Perp’s important role in desmosomal adhesion at the basal state in mouse epithelia (Ihrie et al., 2005). As expected, exposure of wild-type keratinocyte monolayers to PV IgG resulted in the generation of greater numbers of fragments than in normal IgG-treated counterparts. Interestingly, PV IgG treatment also led to increased fragmentation of Perp-deficient monolayers relative to control IgG-treated Perp−/− keratinocytes. Furthermore, PV IgG treatment of Perp-deficient keratinocytes resulted in enhanced dissociation relative to wild-type counterparts on PV IgG treatment. These findings demonstrate that Perp deficiency exacerbates the intercellular adhesive defect of keratinocytes in response to PV autoantibodies, highlighting a cooperativity between Perp loss and DSG3 loss.
Figure 6. Loss of Perp enhances the intercellular adhesion defects induced by PV IgG.
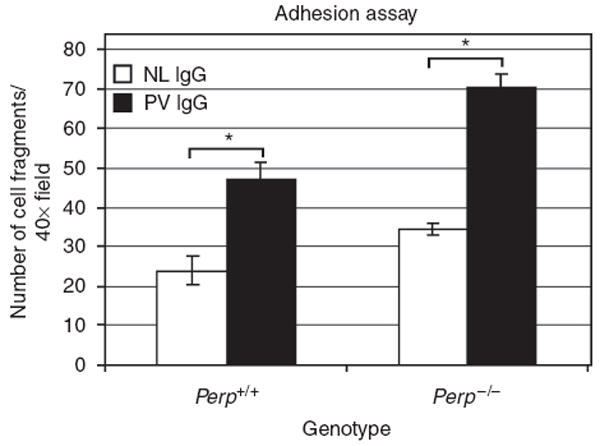
A mechanical dissociation assay on wild-type and Perp−/− mouse keratinocyte monolayers treated with normal (NL) or PV IgG for 24 hours was performed, and the numbers of resulting cell monolayer fragments per 40 × field were counted. Results represent the mean±SEM (n = 12). Statistical significance (*) was determined using the unpaired Student’s t-test (P<0.05).
DISCUSSION
Although many studies have implicated the effects of PV autoantibodies on DSG3 as central to the disease pathogenesis, few have examined the contributions of non-cadherin desmosomal proteins to the manifestations of PV. Here, we examine the effects of PV autoantibodies on a recently discovered desmosomal protein, Perp. By characterizing the pattern of Perp expression in keratinocytes after exposure to PV antibodies, and by examining the consequences of Perp deficiency for PV phenotypes, we gain insight into PV pathogenesis as well as Perp’s role in desmosomal adhesion.
To understand the basis for PV pathology, previous studies have characterized the process of desmosome dissolution in response to PV antibodies. Binding of PV autoantibodies leads to the formation of a DSG3-PV Ig complex, which dissociates from DP but remains associated with PG as it is internalized into endosomes (Calkins et al., 2006). The complex eventually gets degraded in lysosomes, causing depletion of DSG3 at the cell surface and consequent intercellular adhesion defects. To determine whether Perp expression is similarly perturbed by PV autoantibodies, we tracked Perp localization relative to DSG3 and PG. Before PV autoantibody exposure, Perp colocalizes with DSG3 and PG at the plasma membrane, consistent with its role as a desmosomal protein. After PV autoantibody treatment, Perp membrane expression diminishes, and Perp localizes to cytoplasmic puncta with DSG3 and PG. By staining for EEA-1 and CD63, we show that these Perp-containing puncta are endosomes and lysosomes, respectively. These colocalization data are consistent with previous studies showing DSG3 and PG colocalization with EEA-1 and CD63 within several hours of PV autoantibody treatment (Calkins et al., 2006). Our results suggest a model in which, on PV autoantibody binding, the desmosomal complex disassembles, with Perp separating from DP but remaining closely associated with DSG3 and PG and entering the endosomal pathway, where it can ultimately undergo lysosomal degradation. Given that Perp deficiency is sufficient to compromise desmosomal adhesion, PV-induced internalization and downregulation of Perp may contribute to the intercellular adhesion defects seen in the disease.
To clarify Perp’s contribution to PV pathology, we took advantage of keratinocytes derived from Perp−/− mice (Ihrie et al., 2005). We hypothesized that Perp loss could potentially either block or exacerbate the effects of PV autoantibodies. An example of the former phenomenon comes from studies demonstrating a central role for PG in the pathogenesis of PV. In the absence of PG, PV autoantibodies fail to induce keratin retraction or intercellular adhesion defects, two cellular hallmarks of the disease, supporting a signaling function for PG outside of its canonical structural role in the desmosome (Caldelari et al., 2001). However, we determined that Perp-deficient cells do not display a defect in their response to PV autoantibodies, suggesting that Perp is not required for relaying a signal to trigger desmosome disassembly in PV. Instead, we observed a cooperative effect of Perp deficiency and PV autoantibody exposure, manifested as increased DSG3 depletion and augmented intercellular adhesion defects. These findings suggest that Perp, through its role in adhesion, can dampen the pathogenic effects of PV antibodies.
The observed cooperativity between Perp loss and PV antibodies could reflect different potential modes of action for Perp in desmosomal adhesion. For example, as a tetraspan membrane protein, Perp might act by providing intercellular structural connections through homotypic interactions of its extracellular loops. The disruption of these interactions as Perp is internalized following PV autoantibody exposure could contribute to the observed adhesion defects. Indeed, disruption of DSG homotypic and heterotypic interactions by direct binding of PV antibodies has long been proposed as an important event in PV pathogenesis (Amagai et al., 1994b; Futei et al., 2000; Sharma et al., 2007). Thus, loss of both sets of contacts might result in an exacerbated adhesion defect. Alternatively, given that Perp appears to colocalize with DSG3 and PG—the two key participants in PV—and that Perp deficiency leads to increased solubility of DSG3 and PG, Perp might act to promote interactions between DSG3 and PG, or to facilitate proper incorporation of the DSG3–PG complex into the desmosome. In fact, disruption of the DSG3 and PG interaction via deletion of the armadillo-binding domain on DSG3 inhibits proper incorporation of DSG3 into desmosomes (Troyanovsky et al., 1994; Andl and Stanley, 2001). Thus, impaired incorporation of DSG3 into desmosomal complexes resulting from Perp deficiency may somehow enhance the ability of DSG3 to be targeted by PV autoantibodies. However, the exact mechanism by which PV IgG mediates DSG3 internalization is controversial. For example, time-lapse microscopy studies tracking desmosome assembly have shown that DSG3 is initially delivered to nondesmosomal simple membrane clusters without connections to the intermediate filament network before being incorporated into mature desmosomes, and that PV IgG induces internalization of these clusters before targeting established desmosomes (Sato et al., 2000). In contrast, immunoelectron microscopy experiments on skin from a PV mouse model have suggested that PV autoantibodies only target desmosomal DSG3 (Shimizu et al., 2004). Whether in simple membrane clusters or at the desmosome, if Perp deficiency renders DSG3 more susceptible to PV autoantibodies, it could translate into the enhanced intercellular adhesion defects observed in response to both Perp deficiency and PV autoantibody exposure.
Our data implicate Perp in modulating cellular responses to PV autoantibodies. Specifically, the combined effects of Perp loss and PV autoantibody exposure suggest that Perp normally plays a protective role against the pathogenic effects of PV autoantibodies, possibly through structural support, or by facilitating assembly of DSG3 and PG into desmosomes. These studies suggest the possibility of ultimately manipulating Perp to mitigate some of the clinical manifestations of PV. In future investigations we will examine this possibility as well as distinguishing the models through which Perp promotes desmosomal adhesion.
MATERIALS AND METHODS
All studies were conducted in accordance with the Declaration of Helsinki Principles for Human Tissue Research and were approved by the Institutional Review Board of Stanford University.
Mouse keratinocyte culture
Mouse keratinocytes were isolated from the epidermis of 2-day-old wild-type or Perp-null mice as described (Hennings et al., 1980). The skin was digested in epidermal keratinocyte media (CellNTec, Billerica, MA) containing 0.5% trypsin (Invitrogen, Carlsbad, CA) and 0.5% dispase (BD Transduction Labs, Franklin Lakes, NJ) at 4 °C for 48 hours. Keratinocytes were recovered from the epidermis and plated in a rich media (DMEM (Invitrogen) containing 25% F12 media (Invitrogen), 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA), 5 μgml−1 insulin (Sigma Chemical Corp., St Louis, MO), 0.4 μgml−1 hydrocortisone (Sigma Chemical Corp.), 10−10m cholera toxin (MP Biomedicals, Solon, OH), 10 ngml−1 EGF (Sigma Chemical Corp.), and antibiotics) onto collagen-fibronectin-coated tissue culture dishes. After 24 hours, the media was changed to low calcium growth media consisting of EMEM (Cambrex, East Rutherford, NJ) plus 8% dialyzed fetal bovine serum, 0.05mm calcium chloride (Sigma Chemical Corp.), and antibiotics.
Human keratinocyte culture
Human keratinocytes were isolated as described (Halbert et al., 1992). Cells were grown on collagen-fibronectin-coated dishes in keratinocyte serum-free media (Invitrogen). Keratinocyte monolayers were made from cells that were switched to keratinocyte serum-free media containing 0.5mm calcium for 18 hours before the start of the experiments.
IgG purification
PV serum came from plasmapheresis fluid that was collected from a single patient diagnosed with PV as confirmed by direct immunofluorescent microscopy of perilesional skin showing intercellular IgG. This patient had both cutaneous and mucosal involvement. The serum titer was measured by immunofluorescence analysis on monkey esophagus sections (Immco Diagnostics, Buffalo, NY) and determined to be 1:40. Normal human serum was purchased from Cambrex. Both normal and PV serum were heat inactivated for 30 minutes at 55 °C and coarsely filtered. To purify IgG, serum was diluted 1:10 v/v with 1.0 m Tris pH 8.0 and incubated with Protein G Sepharose 4 Fast Flow beads (Amersham Biosciences, Piscataway, NJ). Beads were then washed three times each with 100 and 10mm Tris pH 8.0. Bound IgG was eluted with 50mm glycine pH 3.0, and neutralized with 1 m Tris pH 8.0 at 1:10 v/v. Fractions were analyzed by SDS–PAGE and Coomassie blue staining. Antibody-containing fractions were dialyzed against phosphate-buffered saline (PBS) and concentrated using centrifugal filters (Millipore, Billerica, MA). The final concentration of isolated IgG was determined using the BCA protein assay (Pierce Biotechnology, Rockford, IL). The titer of the purified IgG at a working concentration of 2mgml−1 was determined to be 1:1280. Cells were typically treated with 2mgml−1 IgG or 1:4 v/v heat-inactivated serum in 0.5mm calcium keratinocyte media for 0–24 hours.
Immunofluorescence analysis
Keratinocytes were cultured to ~75% confluence, then switched to 0.5mm calcium-containing media for 18 hours, and then treated with 2mgml−1 normal or PV IgG for up to 6 hours. Cells were then fixed in ice-cold methanol for 7 minutes or in 4% paraformaldehyde for 10 minutes, treated with 10 μgml−1 proteinase K (Sigma Chemical Corp.) for 90 seconds and permeabilized with 0.5% Triton X-100 (Fisher Scientific, Pittsburgh, PA) for 7 minutes if necessary. The cells were washed in PBS and blocked in PBS containing 5% normal goat serum (Sigma Chemical Corp.), 1% bovine serum albumin (Sigma Chemical Corp.), and 0.01% Triton X-100 (Fisher Scientific) for 30 minutes at room temperature, then incubated with the following primary antibodies for 30 minutes at 37 °C: anti-human Fc (gift of Dr Ronald Levy, Stanford University), rabbit anti-Perp (Ihrie et al., 2005), mouse anti-human DSG3 (Zymed Laboratories, South San Francisco, CA), rabbit anti-mouse DSG3 (gift of Dr John R. Stanley, University of Pennsylvania), 1407 chicken anti-PG (gift of Dr Kathleen J. Green, Northwestern University), mouse anti-DP I and II (Fitzgerald Industries, Concord, MA), mouse anti-EEA-1 (BD Transduction Labs), and H5C6 mouse anti-CD63 (University of Iowa Developmental Studies Hybridoma Bank, Iowa City, IA). After several brief washes in PBS+0.01% Tween-20 (Fisher Scientific), the fluorescently conjugated secondary antibodies and a 1 μgml−1 DAPI solution (Sigma Chemical Corp.) were applied to the coverslips for 30 minutes at 37 °C. Coverslips were then washed in PBS-Tween and mounted onto slides with Mowiol mounting medium (Calbiochem, San Diego, CA). Cells were examined by widefield fluorescence using a Leica DM6000B fluorescence microscope (Leica Microsystems, Bannockburn, IL), and image acquisition was performed using a Retiga Exi camera (Qimaging, Surrey, British Columbia, Canada) and Image Pro 6.2 software from Media Cybernetics (Silver Spring, MD).
Western blot analysis
Triton-soluble and urea-soluble protein fractions, as well as whole cell lysates were prepared as described (Ihrie et al., 2005). Western blot analysis was performed according to standard protocols using the following antibodies: mouse anti-human DSG3 (Zymed Laboratories), rabbit anti-DP (Fitzgerald Industries), 11E4 mouse anti-PG (Zymed Laboratories), mouse anti-GAPDH (Fitzgerald Industries), rabbit anti-mouse keratin 14 (Covance, Princeton, NJ), rabbit anti-Perp (Ihrie et al., 2005), PV serum, normal serum (Cambrex), and purified IgG from PV and normal serum.
Coimmunoprecipitation
Confluent human keratinocytes were cultured in keratinocyte serum-free media containing 0.5mm calcium for 4 hours, then lysed with CoIP buffer (10% glycerol, 0.5% Triton X-100, 145mm NaCl, 5mm EDTA, 2mm EGTA, 1mm PMSF, 10mm Tris-HCl pH 7.4) on ice. Lysate was mixed, cleared of insoluble material, and incubated with 200 μg PV or normal IgG overnight at 4 °C. Protein G beads were added the following day and mixed for several hours at 4 °C. Beads were washed several times with lysis buffer and analyzed by western blot using standard methods.
Adhesion assays
Mechanical dissociation assays were performed as described (Huen et al. 2002) on mouse keratinocytes grown to confluence in 0.05mm calcium media, then switched to 0.5mm calcium for 18 hours, and treated with 2mgml−1 normal or PV IgG for 24 hours. Cell monolayer fragments in four different 40 × fields were counted using a Leica MX6 dissecting microscope (Leica Microsystems). Triplicates for each treatment group were included in the analysis.
Acknowledgments
We thank K. Green for plakoglobin antibodies, J.R. Stanley for desmoglein 3 antibodies, and R. Levy for human Fc antibodies. This work was supported by a Howard Hughes Medical Institute Research Training Fellowship and a Stanford University School of Medicine Medical Scholar Research Fellowship (to B.N.), the Office of Research and Development, Palo Alto VA Medical Center (to M.P.M.) and by the National Institutes of Health (to L.D.A. and M.P.M.).
Abbreviations
- DP
desmoplakin
- DSG
desmoglein
- PBS
phosphate-buffered saline
- PG
plakoglobin
- PV
pemphigus vulgaris
Footnotes
CONFLICT OF INTEREST The authors state no conflict of interest.
References
- Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest. 1994a;94:59–67. doi: 10.1172/JCI117349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M, Karpati S, Klaus-Kovtun V, Udey MC, Stanley JR. Extracellular domain of pemphigus vulgaris antigen (desmoglein 3) mediates weak homophilic adhesion. J Invest Dermatol. 1994b;102:402–8. doi: 10.1111/1523-1747.ep12372164. [DOI] [PubMed] [Google Scholar]
- Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992;90:919–26. doi: 10.1172/JCI115968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M, Klaus-Kovtun V, Stanley JR. Anutoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–77. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- Andl CD, Stanley JR. Central role of the plakoglobin-binding domain for desmoglein 3 incorporation into desmosomes. J Invest Dermatol. 2001;117:1068–74. doi: 10.1046/j.0022-202x.2001.01528.x. [DOI] [PubMed] [Google Scholar]
- Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci USA. 2006;103:12855–60. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, et al. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci. 2001;114:727–38. doi: 10.1242/jcs.114.4.727. [DOI] [PubMed] [Google Scholar]
- Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, et al. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 2001;153:823–34. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 2006;281:7623–34. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- Franke WW, Kapprell HP, Mueller H. Isolation and symmetrical splittleing of desmosomal structures in 9M urea. Eur J Cell Biol. 1983;32:117–30. [PubMed] [Google Scholar]
- Futei Y, Amagai M, Sekiguchi M, Nishifuji K, Fujii Y, Nishikawa T. Use of domain-swapped molecules for conformational epitope mapping of desmoglein 3 in pemphigus vulgaris. J Invest Dermatol. 2000;115:829–34. doi: 10.1046/j.1523-1747.2000.00137.x. [DOI] [PubMed] [Google Scholar]
- Garrod DR, Merritt AJ, Nie Z. Desmosomal cadherins. Curr Opin Cell Biol. 2002;14:537–45. doi: 10.1016/s0955-0674(02)00366-6. [DOI] [PubMed] [Google Scholar]
- Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271–81. doi: 10.1038/nrm1356. [DOI] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–34. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M. The armadillo family of structural proteins. Int Rev Cytol. 1999;186:179–224. doi: 10.1016/s0074-7696(08)61054-2. [DOI] [PubMed] [Google Scholar]
- Hatzfeld M. Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochem Biophys Acta. 2007;1773:69–77. doi: 10.1016/j.bbamcr.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980;19:245–54. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- Holthofer B, Windhoffer R, Troyanovsky S, Leube RE. Structure and function of desmosomes. Int Rev Cytol. 2007;264:65–163. doi: 10.1016/S0074-7696(07)64003-0. [DOI] [PubMed] [Google Scholar]
- Huen AC, Park JK, Godsel LM, Chen X, Bannon LJ, Amargo EV, et al. Intermediate filament-membrane attachments function synergistically with actin-dependent contacts to regulate intercellular adhesive strength. J Cell Biol. 2002;159:1005–17. doi: 10.1083/jcb.200206098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120:843–56. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol. 2005;124:939–46. doi: 10.1111/j.0022-202X.2005.23714.x. [DOI] [PubMed] [Google Scholar]
- Kapprell HP, Cowin P, Franke WW, Ponstingl H, Opferkuch HJ. Biochemical characterization of desmosomal proteins isolated from bovine muzzle epidermis: amino acid and carbohydrate composition. Eur J Cell Biol. 1985;36:217–29. [PubMed] [Google Scholar]
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzelaar MJ, Wijngaard PL, Peters PJ, Sixma JJ, Nieuwenhuis HK, Clevers HC. CD63antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J Biol Chem. 1991;266:3239–45. [PubMed] [Google Scholar]
- Mu F-T, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton R, Campbell PL, et al. EEA1, an early endosome associated protein. J Biol Chem. 1995;270:13503–11. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–99. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Aoyama Y, Kitajima Y. Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest. 2000;80:1583–92. doi: 10.1038/labinvest.3780168. [DOI] [PubMed] [Google Scholar]
- Sharma P, Mao X, Payne AS. Beyond steric hindrance: the role of adhesion signaling pathways in the pathogenesis of pemphigus. J Dermatol Sci. 2007;48:1–14. doi: 10.1016/j.jdermsci.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol. 2004;122:1145–53. doi: 10.1111/j.0022-202X.2004.22426.x. [DOI] [PubMed] [Google Scholar]
- South AP, Wan H, Stone MG, Dopping-Hepenstal PJ, Purkis PE, Marshall JF, et al. Lack of plakophilin 1 increases keratinocyte migration and reduces desmosome stability. J Cell Sci. 2003;116:3303–14. doi: 10.1242/jcs.00636. [DOI] [PubMed] [Google Scholar]
- Stanley JR, Amagai M. Pemphigus, bullous impetigo, and staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355:1800–10. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM, Troyanovsky RB, Eshkind LG, Krutovskikh VA, Leube RE, Franke WW. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol. 1994;127:151–60. doi: 10.1083/jcb.127.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–8. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E. Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 2001;3:1076–85. doi: 10.1038/ncb1201-1076. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem. 2007;282:17866–76. doi: 10.1074/jbc.M607963200. [DOI] [PubMed] [Google Scholar]
- Yin T, Green KJ. Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol. 2004;15:665–77. doi: 10.1016/j.semcdb.2004.09.005. [DOI] [PubMed] [Google Scholar]