

Abstract
The chemotaxis pathway of Escherichia coli and Salmonella typhimurium is the paradigm for the ubiquitous class of 2-component signaling pathways in prokaryotic organisms. Chemosensing begins with the binding of a chemical attractant to a transmembrane receptor on the cell surface. The resulting transmembrane signal regulates a cytoplasmic, multiprotein signaling complex that controls cellular swimming behavior by generating a diffusible phosphoprotein. The minimal functional unit of this signaling complex, termed the core complex, consists of the transmembrane receptor, the coupling protein CheW, and the histidine kinase CheA. Though the structures of individual components are largely known and the core complex can be functionally reconstituted, the architecture of the assembled core complex has remained elusive. To probe this architecture, the present study has utilized an enhanced version of the protein-interactions-by-cysteine-modification method (PICM-β) to map out docking surfaces on CheA essential for kinase activity and for core complex assembly. The approach employed a library of 70 single, engineered cysteine residues, scattered uniformly over the surfaces of the five CheA domains in a cysteine-free CheA background. These surface Cys residues were further modified by the sulfhydryl-specific alkylating agent, 5-fluorescein-maleimide (5FM). The functional effects of individual Cys and 5FM-Cys surface modifications were measured by kinase assays of CheA activity in both the free and core complex-associated states, and by direct binding assays of CheA associations with CheW and the receptor. The results define (i) two mutual docking surfaces on the CheA substrate and catalytic domains essential for the association of these domains during autophosphorylation, (ii) a docking surface on the CheA regulatory domain essential for CheW binding, and (iii) a large docking surface encompassing regions of the CheA dimerization, catalytic, and regulatory domains proposed to bind the receptor. To test the generality of these findings, a CheA sequence alignment was analyzed, revealing that the newly identified docking surfaces are highly conserved among CheA homologues. These results strongly suggest that the same docking sites are widely utilized in prokaryotic sensory pathways. Finally, the results provide new structural constraints allowing the development of improved models for core complex architecture.
The homodimeric, histidine kinase CheA is the central processing unit of the conserved signaling pathway that controls chemotaxis in Escherichia coli, Salmonella typhimurium, and many other bacteria (for reviews see refs 1–7). This pathway is the prototype for the large, ubiquitous class of two-component pathways that regulate a wide array of cell processes in prokaryotes. CheA and the other pathway components assemble to form large clusters of signaling complexes at the poles of the cell. The minimal signaling unit required for receptor-regulated kinase activity, termed the core complex, comprises the oligomeric transmembrane chemoreceptor, the coupling protein CheW, and CheA (8, 9). Current structural models propose that CheA and CheW dock to the cytoplasmic tip of the receptor oligomer as illustrated in Figure 1A. When assembled into the core complex, the trans-autophosphorylation activity of the CheA dimer is stimulated dramatically by the apo receptor, while attractant binding to the receptor strongly inhibits this autokinase activity.
Figure 1.
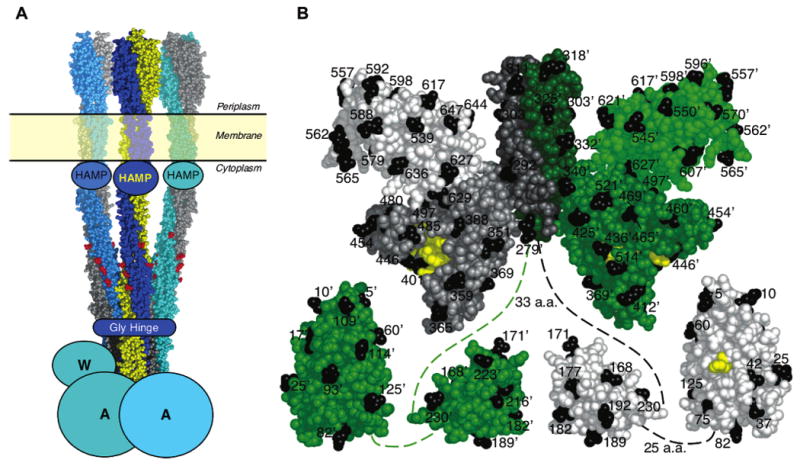
Summary of structural models from previous studies. (A) Model of the core complex consisting of the transmembrane receptor oligomer, CheA kinase, and CheW coupling protein. The receptors are shown as a trimer-of-dimers (49, 50) based on a composite structural model (reviewed in ref 6). HAMP is a conserved domain of unknown structure (51–53), while Gly Hinge is also a conserved element (54). Homodimeric CheA and monomeric CheW are schematic. (B) Structural model of CheA showing one subunit in shades of green and the other in shades of gray. Black identifies the 70 positions of Cys substitutions used in this study. Yellow marks catalytic residues including H48, the site of phosphorylation on the P1 domain, and the ATP binding residues on the P4 domain. The composite CheA structure is assembled from S. typhimurium P1 domain (12), E. coli P2 domain (10), and T. maritima P3+P4+P5 domain fragment (11). Dashed lines indicate extended flexible linkers of undetermined structure that connect the domains.
Each 73 kDa subunit of the CheA homodimer is composed of five structural domains with distinct functions. High-resolution structures exist for the two isolated N-terminal domains (P1 and P2), and for a dimeric fragment containing the three C-terminal domains (P3, P4, P5) (10–12). The N-terminal substrate domain, P1, is a five-helix bundle that contains His 48, the site of phosphorylation. The response regulator binding domain, P2, possesses high-affinity docking surfaces for CheY and CheB and is coupled at both termini to flexible linkers allowing free rotation relative to the other CheA domains (13–15). This mobility, together with the observation that P2 is not essential for chemotaxis (16, 17), suggests that P2 does not play a significant structural role in defining the architecture of the core complex. The dimerization domain, P3, is a four-helix bundle formed by the association of two helices from each subunit and contains most of the dimerization contacts of the homodimer. The catalytic domain, P4, possesses essential catalytic residues, including the ATP binding site, and binds the P1 substrate domain from the other subunit in the same dimer. The resulting interaction is essential for trans-autophosphorylation (18) and measurably enhances dimer stability (19). The regulatory domain, P5, interacts with the receptor and CheW and is essential for receptor-mediated kinase regulation (20, 21).
High-resolution structural information is also available for the other components of the signaling complex, including the coupling protein CheW and the cytoplasmic domain of the receptor (22, 23). Thus far, however, the fully assembled receptor–CheW–CheA core complex has been resistant to structural analysis, leaving the molecular interactions between core complex components poorly defined. Even the subunit stoichiometry of the core complex is still highly debated, with estimates ranging from 2:2:2 (24) to 6:4:1 (25, 26) to 6:2:2 (27) (polypeptide ratio receptor/CheW/CheA). Previous models for core complex architecture are based on relatively few experimental constraints (25, 28, 29).
Recent studies have defined protein–protein docking surfaces on CheW and the receptor essential to core complex assembly. Genetic suppressor analyses and competition binding experiments have identified CheW surfaces responsible for binding the receptor, while NMR chemical shift measurements and fluorescence anisotropy binding measurements have identified CheW surfaces that dock to CheA (22, 30–32). Suppressor and chemical mapping studies have identified receptor surfaces essential for binding CheA and CheW (30, 33, 34).
The present study uses an enhanced version of the protein-interactions-by-cysteine-modification (PICM)1 technique (33–35), termed PICM-β, to map out the essential docking sites on the surface of S. typhimurium CheA. In PICM, surface modifications are introduced into a target protein at selected locations by cysteine mutation followed by attachment of a bulky, sulfhydryl-specific probe. Previous PICM studies relied solely on enzyme activity measurements to determine which modifications disrupt essential docking sites on the receptor surface. In PICM-β, both enzyme activity and direct binding measurements are employed, allowing for a more rigorous identification of docking sites and, in favorable cases, the identification of the docking partner for a given site. The results identify four distinct docking surfaces on CheA and strongly constrain models of the core complex, enabling the development of new working models for core complex architecture. One of the four docking surfaces characterized herein, specifically the surface on CheA P5 that binds CheW, has been independently probed by X-ray crystallography and EPR in a report that appeared following completion of the present study (55). These methods identify the same CheW binding site on CheA P5 as reported here, strongly supporting the accuracy of the PICM-β approach.
Materials and Methods
Materials
Cysless CheA, single-Cys mutant CheA, and CheW proteins were expressed with N-terminal 6-His tags from plasmids pET6H-CheA and pET6H-CheW (constructed from pET28, Novagen; Bornhorst, Munger, and Falke, unpublished results) in E. coli strain BL21(DE3) (Stratagene), then were isolated by standard Ni–NTA affinity chromatography (36). Wild-type CheY was expressed and isolated from plasmid pRBB40 as described previously (37), while 6-His-tagged CheY was expressed from plasmid pVSCheY-6H (derived from pVS33, a gift from Victor Sourjik and Howard Berg, Harvard University) in strain Bl21(DE3). The transmembrane aspartate (Tar) or serine (Tsr) receptor was expressed from plasmid pSCF6 (38) or pJC3 (39), respectively, in strain RP3808 lacking the major receptors and soluble chemotaxis proteins, and then receptor-containing membranes were isolated as described previously (40, 41). Protein purity and concentration was determined by SDS–PAGE.
Reagents were obtained from the following sources: [γ-32P]-ATP from Perkin-Elmer; QuickChange site-directed mutagenesis kit from Stratagene; mutagenic oligonucleotides from Integrated DNA Technologies; restriction enzymes from New England Biolabs; sulfhydryl-specific probes 5-fluorescein-maleimede (5FM) and 5-tetramethyl-rhodamine-maleimide (5TMRM) from Molecular Probes Invitrogen; all other reagent grade chemicals from Sigma unless noted otherwise.
Mutagenesis
Site-directed mutagenesis was performed as previously described (42) using the PCR-based QuickChange mutagenesis kit to engineer point mutations into S. typhimurium CheA in plasmid pET6H-CheA. First, each of the three intrinsic cysteines was mutated to both alanine and serine. The most active of these mutations were combined to create a cysteine-free CheA construct C120S/C218A/C432A (Cysless), which when reconstituted into the core complex yielded a specific kinase activity of 1.1 ± 0.2 relative to wild-type CheA. Subsequently, Cysless CheA served as the background into which single cysteine mutations were incorporated. Silent restriction sites were designed into the mutagenic primers, where possible, allowing screening for Cys mutants by restriction. All resulting plasmids were sequenced to directly confirm the desired mutations, then expressed and purified as described above.
CheA Alkylation Reactions
Prior to PICM analysis, purified, single-cysteine CheAs were labeled with 5-fluoresceine-maleimide (5FM) as previously described (33, 34). CheA at a concentration of 3.3 μM (free CheA activity assay), 1.7 μM (complex activity assay), or 7.5 μM (complex binding assay) or up to 200 μM (CheW binding assay) was reacted with 500 μM 5FM (final concentration) in 50 mM Tris, pH 7.5, 1 mM EDTA, 10% glycerol, and 2.5% dimethylformamide (v/v). Unlabeled controls were generated at the same time by adding the same volume of dimethylformamide lacking 5FM. All samples were incubated for 10 min (or 20 min for CheW binding assay) at room temperature and then quenched by addition of DTT to 50 mM. The extent of labeling of each CheA mutant was determined as previously described (33, 34), and ranged between 80 and 100% for all but one mutant (S351C). Prior to CheW binding experiments, the unbound probe was removed by G25 Sephadex chromatography.
Free CheA Kinase Assay
Unlabeled and labeled free CheA was mixed with CheY to a final concentration of 2 μM CheA, 10 μM His6-CheY, and 15% PEG. The previously described (43) autophosphorylation reaction was started by addition of [γ-32P]-ATP and quenched after 10 s by the addition of 3 times the reaction volume of 2× Laemmli sample buffer containing 25 mM EDTA. The unheated sample was then loaded onto 15% acrylamide (acrylamide/bisacrylamide 40: 1.25), 20% urea (w/v) Laemmli SDS–PAGE gels. The resulting gels were dried and phosphorimaged (Molecular Dynamics) to quantitate the level of [γ-32P]-phospho-CheY, which is proportional to the initial rate of CheA autophosphorylation. All reactions were performed in triplicate.
Receptor-Stimulated Kinase Assay
The core complex was reconstituted in vitro by mixing CheA (0.5 μM unlabeled or labeled, final concentration), aspartate receptor (4 μM), CheW (2 μM), and wild-type CheY (10 μM) as previously described (8, 9, 40). The complex was allowed to assemble for 45 min before kinase reactions were carried out as described above. All reactions were performed in triplicate, in the presence and absence of 1 mM aspartate. The PICM parameter was calculated as the ratio of kinase rates in the labeled/unlabeled samples.
Core Complex Binding Assay
The core complex was reconstituted in vitro from serine receptor (10 μM), CheW (5 μM), and 5FM-labeled CheA (0.5 μM). Serine receptor (Tsr) was used instead of the aspartate receptor (Tar) because the more highly overexpressed Tsr was found to bind a significantly higher amount of CheA. After allowing the complex to form for 45 min, the amount of labeled CheA successfully incorporated into the membrane-bound complexes was quantitated by a modification of a previous protocol (25). Membranes in a 10 μL aliquot were pelleted by centrifugation in a Beckman TLA 100.3 rotor at 100 000 rpm (∼540 000g) for 15 min at 2 °C. Only 5 μL of the supernatant was saved in order not to contaminate the supernatant with pellet. The pellet was then rinsed carefully with 1 mL of water to remove any residual supernatant. After the pellet-containing tubes were air-dried at room temperature for about 30 min, the pellet was resuspended to the original volume of the sample with 2× Laemmli sample buffer by pump mixing with a pipet (9 μL of sample buffer + 1 μL of pellet = 10 μL of original volume). Equal volumes of supernatant and pellet were then run on SDS polyacrylamide gels. The resulting fluorescent bands of labeled CheA were imaged on a UV transilluminator using a digital camera and quantitated by densitometry (Alpha Innotech). The relative binding parameter was calculated using eq 1. Each experiment was performed in triplicate and was repeated at least once. Error bars are standard error of the mean (n ≥ 6).
CheA–CheW Binding Assay
Fluorescence anisotropy binding measurements were carried out as previously described (31, 32) with the following modifications. The CheW construct utilized possessed an N-terminal 6-His tag with a single cysteine residue four amino acids before the native N-terminal Met of Cysless CheW. This construct was labeled with 5-tetramethyl-rhodamine maleimide (5TMRM), and then excess probe was removed by G25-Sephadex chromatography. Subsequently, 5FM-labeled CheA (stock concentration of 250–800 μM) was titrated (0–100 μM final) into a cuvette containing 5TMRM-labeled CheW (1 μM). (Here, 5FM served only as a bulky modification, not as a fluorescent probe or FRET acceptor.) The fluorescence anisotropy of the 5TMRM-labeled CheW was measured using a PTI QM-2000-6SE Fluorimeter with two detection channels. The 5TMRM excitation and emission wavelengths were 565 and 580 nm, respectively, with band-pass 6 and 8 nm, respectively. Use of a 550 nm long pass filter on the excitation channel ensured that fluorescein was not excited. A computer-controlled polarizer was used on the excitation channel to excite the sample alternately with vertically (0°) and horizontally (90°) polarized light. Fixed vertical and horizontal polarizers were used on the A and B detection channels, respectively. Samples were maintained at 25 °C. All data were fit with KaleidaGraph.
Assignment of Threshold Values
To classify Cys substitutions and 5FM modifications as either nonperturbing or perturbing, a threshold level was assigned for each activity and binding assay. First, the threshold for inhibitory modifications was established by starting with a high threshold that classified most modifications as perturbing, yielding a delocalized pattern of perturbing sites over all regions of the CheA surface. Subsequently, the threshold was lowered until the perturbations resolved into localized clusters. Often, the clusters observed for different assays were located on different, nonoverlapping surfaces, since these assays were detecting different protein–protein interactions. The lowest reasonable threshold value was chosen, since the false negatives generated by setting the threshold too low (thereby excluding some true docking site residues) were preferable to the false positives generated by setting the threshold too high (thereby implicating nondocking site residues in a putative docking surface). Second, for some assays, a threshold for superactivating modifications was established using an analogous approach that started with a low threshold and moved it upward. Overall, the threshold approach is based on the assumption that each protein–protein interaction surface is localized in space to one contiguous region of the CheA surface. The approach can miss contact residues on the periphery of an interaction face, where a modified side chain can project out of the interaction zone. By contrast, the approach is well-suited for detection of the centrally located residues within a docking face, as well as side chains which are essential for docking.
Results
Experimental Strategy
The goals of this study were to define the protein–protein docking sites of CheA and to identify the docking partners of those sites. The experimental strategy employed a new, enhanced version of the protein-interactions-by-cysteine-modification method (PICM-β). First, we generated a fully functional, cysteine-free CheA mutant (C120S/C218A/C432A, see Materials and Methods) lacking all three native cysteine residues. This Cysless CheA was then used as a background for the creation of a library of 70 point mutants in which single cysteine substitutions were systematically distributed over all surfaces of the five CheA structural domains. Each single-cysteine CheA mutant was further modified by covalently coupling a bulky probe to its cysteine sulfhydryl. Subsequently, four functional assays were used to measure the effects of Cys substitution and probe coupling on (i) the kinase activity of free CheA, (ii) the kinase activity of the reconstituted core complex, (iii) the binding of CheA to the core complex, and (iv) the binding of CheW to free CheA. This enhanced PICM-β approach combines standard kinase activity measurements with direct binding measurements, thereby providing rigorous identification of essential docking sites and their binding partners.
Selection and Construction of Cysteine Substitutions
The 70 positions selected for cysteine substitutions were spaced as uniformly as possible over the surface of CheA to ensure maximal coverage as illustrated in Figure 1B and summarized in Table 1. Selection was guided by high-resolution structures: (i) the crystal structure of S. typhimurium P1 domain residues 1–131 (12); (ii) the NMR structure of E. coli P2 domain residues 159–227 (10), which is 82.1% identical and 89.6% equivalent to the S. typhimurium P2 domain; and (iii) the crystal structure of the dimeric Thermatoga maritima P3+P4+P5 domain fragment spanning residues 290–671 (11), which is 42.7% identical and 70.3% equivalent to the corresponding S. typhimurium fragment. In the structure of the P3+P4+P5 domain fragment, the relative positions and contacts between the domains, which are coupled by flexible hinges (11), could be significantly influenced by crystal packing forces. Thus, both solvent-exposed surfaces and domain–domain contact surfaces of these domains were targeted for cysteine substitutions. The only exception was the P3–P3′ contact at the dimer interface, which is known to be essential for dimer stability and was therefore not targeted. Because of the high degree of conservation, it was straightforward to align the three CheA sequences to accurately place Cys substitutions in S. typhimurium CheA corresponding to the selected surface positions in the E. coli and T. maritima structures.
Table 1.
Effects of Substitutions on Free CheA Autokinase Activitya
| kinase activity | kinase activity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| position | domain | 2° struct. | Cys | Cys-F1 | labeling efficiency | position | domain | 2° struct. | Cys | Cys-F1 | labeling efficiency |
| I5C | P1 | α1 | (+) | (+) | 0.88 | I388C | P4 | α11 | (+) | [-] | 0.88 |
| Q10C | P1 | α1 | [++] | (+) | 0.86 | H401C | P4 | α11 | (+) | (+) | 0.80 |
| D17C | P1 | α1 | (+) | (+) | 0.91 | A412C | P4 | α12 | (+) | (+) | 0.81 |
| Q25C | P1 | α1 | (+) | (+) | 0.80 | E425C | P4 | β8 | (+) | [++] | 0.89 |
| A37C | P1 | α2 | (+) | (+) | 0.97 | T436C | P4 | β9 | (+) | (+) | 0.87 |
| A42C | P1 | α2 | [-] | [-] | 0.88 | R446C | P4 | α13 | (+) | [-] | 0.86 |
| T60C | P1 | L | (+) | (+) | 1.02 | Q454C | P4 | L | (+) | (+) | 0.94 |
| E75C | P1 | α3 | (+) | (+) | 0.94 | E460C | P4 | L | (+) | (+) | 0.87 |
| Q82C | P1 | L | (+) | (+) | 1.03 | D465C | P4 | α14 | (+) | (+) | 0.80 |
| E93C | P1 | α4 | (+) | (+) | 1.10 | M469C | P4 | L | (+) | (+) | 0.97 |
| E109C | P1 | L | (+) | (+) | 1.11 | E480C | P4 | L | (+) | [-] | 0.99 |
| A114C | P1 | α5 | (+) | (+) | 0.85 | V485C | P4 | L | [-] | [-] | 1.01 |
| Q125C | P1 | α5 | (+) | (+) | 0.81 | R497C | P4 | α16 | [++] | (+) | 0.94 |
| V168C | P2 | β1 | (+) | (+) | 1.04 | S514C | P4 | L | (+) | (+) | 0.86 |
| R171C | P2 | L | (+) | (+) | 0.96 | L521C | P4 | β11 | (+) | (+) | 0.82 |
| V177C | P2 | α6 | (+) | (+) | 0.95 | E539C | P5 | β13 | (+) | (+) | 0.84 |
| E182C | P2 | α6 | (+) | (+) | 0.89 | L545C | P5 | α17 | (+) | (+) | 0.99 |
| T189C | P2 | L | (+) | (+) | 0.92 | E550C | P5 | β14 | (+) | (+) | 0.88 |
| D192C | P2 | L | (+) | (+) | 0.86 | E557C | P5 | L | (+) | (+) | 0.82 |
| V216C | P2 | α7 | (+) | (+) | 0.87 | L562C | P5 | β15 | (+) | (+) | 0.86 |
| V220C | P2 | α7 | (+) | (+) | 0.84 | G565C | P5 | L | (+) | (+) | 0.80 |
| A223C | P2 | L | (+) | (+) | 0.95 | E570C | P5 | β16 | (+) | (+) | 0.90 |
| K230C | P2 | β3 | (+) | (+) | 1.01 | V579C | P5 | β17 | (+) | (+) | 0.85 |
| T279C | P3 | L | (+) | [-] | 0.88 | D588C | P5 | L | (+) | (+) | 1.04 |
| I292C | P3 | α8 | (+) | [-] | 1.03 | T592C | P5 | L | (+) | (+) | 0.96 |
| S303C | P3 | α8 | (+) | (+) | 0.86 | Q596C | P5 | L | (+) | (+) | 0.93 |
| E311C | P3 | L | (+) | (+) | 0.81 | I598C | P5 | β18 | (+) | (+) | 0.95 |
| G318C | P3 | α9 | (+) | (+) | 0.90 | R607C | P5 | β19 | (+) | (+) | 0.80 |
| G325C | P3 | α9 | (+) | (+) | 0.87 | I617C | P5 | β19 | (+) | (+) | 1.03 |
| R332C | P3 | α9 | (+) | (+) | 0.84 | Q621C | P5 | β19 | (+) | (+) | 0.92 |
| S340C | P3 | α9 | (+) | (+) | 0.81 | L627C | P5 | L | (+) | (+) | 0.98 |
| S351C | P4 | α10 | (+) | [-] | 0.68 | S629C | P5 | α19 | (+) | (+) | 0.84 |
| D359C | P4 | α10 | (+) | [-] | 1.00 | G636C | P5 | L | (+) | (+) | 0.93 |
| G365C | P4 | L | [-] | [-] | 0.88 | G644C | P5 | L | (+) | (+) | 0.89 |
| E369C | P4 | β6 | (+) | (+) | 0.84 | S647C | P5 | L | (+) | (+) | 0.89 |
The autophosphorylation activity of each single Cys CheA was tested in vitro without (Cys) and with (Cys-F1) the attached 5-FM probe. The resulting kinase activities were operationally defined as (+), [-], or [++] corresponding to normal, inhibited (less than 25% normal), or super-activated (over 250% normal) relative to Cysless CheA, respectively. Labeling efficiency is the final extent of 5FM labeling and has an estimated error of ± 10%.
The resulting 70 single-Cys CheA mutants were constructed by PCR site-directed mutagenesis, overexpressed in E. coli, and purified using their N-terminal 6-His affinity tag. All 70 of the resulting mutants were stable, soluble proteins that labeled efficiently (Table 1) with 5-fluorescein-maleimide (5FM), a bulky, anionic, water-soluble, sulfhydryl-specific probe. The observation that all 70 positions are accessible to 5FM (including those buried between domains in the P3+P4+P5 crystal structure (11)) supports a dynamic picture of free CheA in which flexible interdomain hinges allow substantial domain movements, providing at least transient exposure of each domain surface to solvent.
Effects of Cysteine Substitutions and Bulky Probe Attachment on the Kinase Activity of Free CheA
To identify sites of protein–protein contacts, the effects of Cys substitution and probe coupling were first measured on the intrinsic autophosphorylation activity of free CheA kinase. This analysis was designed to identify modifications that perturb global CheA folding or that modify the assembly or geometry of the active complex formed by the P1 substrate and P4 catalytic domains during the autophosphorylation reaction. Thus, the initial autophosphorylation rate was measured for each free CheA mutant, in both its unlabeled and 5FM-labeled states, by monitoring accumulation of substrate phospho-CheY under conditions where CheA autophosphorylation was rate-limiting.
Table 1 summarizes the observed autophosphorylation activities of the modified CheA proteins. Altogether, 140 proteins were tested: 70 unmodified and 70 5FM-modified Cys mutants, the latter termed F1. Of these 140 proteins, 124 were observed to retain 25–250% of the Cysless kinase activity and were operationally defined as functional. By contrast, 13 modifications at 10 different positions retained less than 25% of the Cysless activity and were operationally defined as inhibitory (A42C, A42F1, T279F1, I292F1, S351F1, D359F1, G365C, G365F1, I388F1, R446F1, E480F1, V485C, V485F1). Interestingly, three modifications yielded kinase activities exceeding 250% that of Cysless and were operationally defined as superactivated (Q10C, R497C, E425F1).
Simple mechanistic explanations exist for the majority of the observed perturbations. For example, each of the 10 sites yielding inhibitory 5FM modifications lie in close proximity to the active site residues on the P1 substrate and P4 catalytic domains. At six of these sites, 5FM modifications retain core complex formation and thus do not disrupt global folding (A42F1, S351F1, D359F1, I388F1, R446F1, V485F1; see Figure 3A below). Instead, the simplest explanation is that these six surface positions lie on the docking faces of the P1 and P4 domains that must contact each other with the proper geometry and stability during the autophosphorylation event, such that the coupling of a bulky probe at these positions disrupts catalysis (see Discussion). At the four other sites, 5FM modifications appear to perturb CheA folding or essential interdomain contacts, since they prevent core complex formation (G365F1, E480F1; see Figure 3A below) or lie at the interface between the dimerization and catalytic domains (T279F1,I292F1). Finally, the three superactivating modifications (Q10C, R497C, E425F1) are proposed to interfere with an inhibitory interaction that normally down-regulates free CheA activity until it assembles into the core complex (see Discussion).
Figure 3.
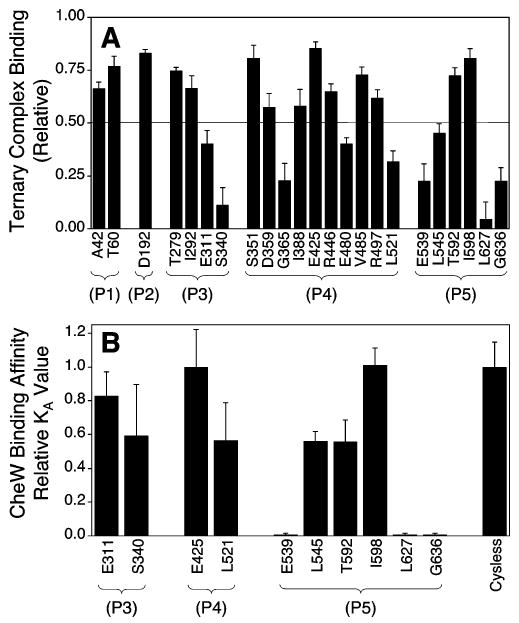
Summary of the effects of 5-fluorescein-maleimide (5FM) attachment on CheA binding to its docking partners. (A) Effects of 5FM attachment on relative incorporation of CheA into the core complex as defined by the binding parameter BC (see text, eq 1). The horizontal line indicates operational threshold of strong complex affinity. (B) Effects of 5FM attachment on free CheA binding to CheW. Values shown are the binding affinity (KA = 1/KD), relative to that of Cysless CheA.
Effects of Cysteine Substitutions and Bulky Probe Attachment on CheA Kinase ActiVity in the Membrane-Bound Core Complex
To identify surfaces of CheA that are required for CheW or receptor docking, or for receptor activation within the assembled core complex, the kinase activity of each CheA mutant was measured in the reconstituted, membrane-bound core complex. The 10 mutants for which Cys substitution or probe coupling inhibited free CheA kinase activity were excluded from further analysis, leaving 60 mutants to characterize. Briefly, the labeled and unlabeled forms of each mutant were reconstituted with CheW and the membrane-bound aspartate receptor. Subsequently, the kinase activity of the core complex-associated CheA was quantitated in a standard in vitro assay, both in the presence and absence of attractant. This assay monitored the initial rate of phospho-CheY production under conditions where the CheA autophosphorylation reaction was rate-limiting.
As summarized in Figure 2A, the 60 Cys substitutions tested yielded kinase activities in the reconstituted core complex ranging from 9% to 194% of that observed for reconstituted Cysless CheA. Altogether, 58 of the 60 Cys substitutions were operationally defined as functional, yielding kinase activities between 25% and 200% that of Cysless. For each of these mutants, addition of saturating aspartate reduced the kinase activity approximately 100-fold or more, confirming that they were functionally coupled to the aspartate receptor. The remaining two Cys substitutions (L627C, G636C) were operationally defined as inhibitory.
Figure 2.
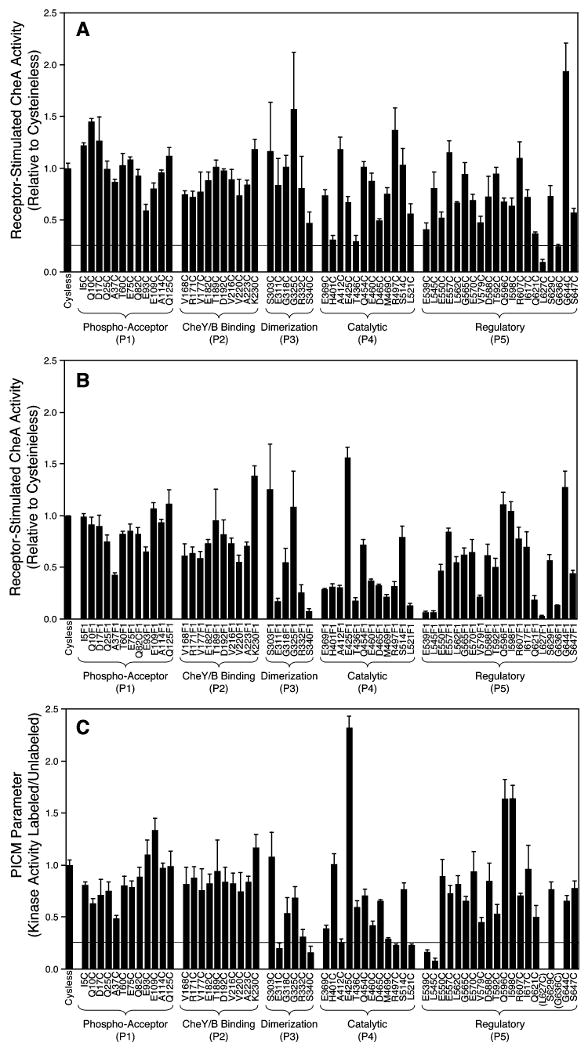
Summary of the effects of modifications on receptor-stimulated CheA kinase activity in the reconstituted signaling complex. (A) Effect of cysteine modification on receptor-stimulated CheA activity relative to that of Cysless CheA. Values below the indicated threshold are operationally defined as perturbed. (B) Effect of 5-fluorescein-maleimide (5FM) attachment on receptor-stimulated CheA activity relative to that of Cysless CheA. (C) PICM parameter (see text) for each cysteine position. Again, values below the indicated threshold are operationally defined as perturbed. (In panels A and C, five above-threshold modifications were ambiguous because their error bars crossed the threshold: H401C, T436C, R332F1, A412F1, M469F1. All five were found to be nonperturbing in the pull-down assay for core complex binding (56), confirming their identification as nonperturbing modifications and validating the indicated thresholds.)
As summarized in Figure 2B, covalent labeling of the 60 Cys mutants with 5FM yielded kinase activities in the reconstituted core complex ranging from 2% to 115% of that observed for reconstituted Cysless CheA. To determine the effect of probe coupling on the activity of each Cys mutant, the PICM parameter was calculated as the ratio of kinase activities in the labeled/unlabeled states. This PICM parameter is zero when probe coupling completely destroys kinase activity, is unity when the probe has no effect on kinase activity, and is greater than unity when the probe increases kinase activity. The two positions at which Cys substitution inhibited the kinase activity of the core complex prior to probe coupling (L627C, G636C) were omitted, leaving 58 positions for PICM analysis. Figure 2C summarizes the resulting 58 PICM parameters, which range from 0.08 to 2.3. Altogether, 51 of 58 positions exhibited PICM parameters from 0.25 to 2.0 and were operationally defined as positions at which probe coupling was nonperturbing. Of the remaining seven positions, six yielded PICM parameters under 0.25 and were defined as positions at which probe coupling was inhibitory (E311F1, S340F1, R497F1, L521F1, E539F1, L545F1). The final position (E425F1) yielded a PICM parameter of 2.3 and was defined as a position at which probe coupling was superactivating. Each of the mutants for which probe coupling was nonperturbing or superactivating exhibited normal kinase inhibition by aspartate (100-fold or more, not shown), confirming that each of the corresponding probe-modified CheA proteins was functionally coupled to the aspartate receptor in the reconstituted core complex.
Together, these findings narrowed the search for essential docking surfaces to eight positions (E311, S340, R497, L521, E539, L545, L627, G636). All eight of the corresponding Cys mutants were active kinases in the free state, both in their unlabeled and 5FM-labeled forms (Table 1), but were poor kinases when reconstituted with CheW and receptor in the core complex in their 5FM-labeled (E311F1, S340F1, R497F1, L521F1, E539F1, L545F1) or even their unlabeled (L627C, G636C) state (Figures 2A,C). It follows that side chain modifications at these eight positions either inhibit the assembly of the core complex by altering an essential docking surface or block transmission of the activating signal from receptor to CheA in the assembled core complex. To resolve these possibilities, the effects of modifications on the assembly of the core complex were measured directly.
Effects of Bulky Probe Attachment on CheA Binding to the Membrane-Bound Core Complex
To directly quantitate the abilities of modified CheAs to assemble into the reconstituted core complex, a membrane pull-down assay (25) was employed. The eight CheA positions implicated in core complex assembly were tested in this pull-down experiment, together with five other positions chosen as positive controls based on the lack of perturbation observed for modifications at these positions (Table 1, Figure 2). Also tested were 10 positions at which modifications inhibit the kinase activity of free CheA (see above). To maximize specificity, accuracy, and precision, all 23 selected CheA mutants were tested in their 5FM-labeled state, thereby enabling fluorescence quantitation of the CheA band during SDS–PAGE analysis of the membrane-bound core complex. Each pull-down experiment incubated a 5FM-labeled CheA with receptor-containing membranes in the absence and presence of CheW, then pelleted the membranes. A binding parameter (BC), representing the relative ability of a modified CheA to assemble into the membrane-bound core complex, was calculated as follows:
| (1) |
where M is the percentage of total CheA found in the membrane pellet either in the presence (+W) or absence (−W) of CheW. This parameter ranges between zero and unity for cases in which the presence of CheW yields no increase in receptor binding, or greatly increases receptor binding, respectively. Poor binders were operationally defined as those exhibiting a binding parameter in the bottom half of the range (BC < 0.5), while strong binders were defined as those in the upper half of the range (BC > 0.5, corresponding to at least 3-fold enhancement of membrane binding by CheW).
Figure 3A summarizes the core complex binding parameters of the 23 modified CheA proteins selected for analysis. The five positive control CheAs, which exhibited normal receptor-mediated kinase activation in the reconstituted core complex, were all classified as strong binders to the membrane-bound complex (BC values ranged from 0.73 to 0.85), confirming the accuracy of the assay. Similarly, one of the eight CheAs labeled with 5FM at a potential docking position (R497F1) was a strong binder (BC value was 0.62). It follows that this position is not essential for docking, but instead is involved in signal transmission from the receptor to CheA within the assembled core complex. The seven remaining CheAs labeled with 5FM at potential docking positions (E311F1, S340F1, L521F1, E539F1, L545F1, L627F1, and G636F1) were classified as poor binders (BC values ranged from 0.05 to 0.45). Thus, bulky probe labeling at these seven positions all caused defects in core complex assembly, providing strong evidence that these positions are located on critical docking surfaces that directly contact the receptor, or CheW, within the assembled core complex. Turning to the 10 5FM-labeled CheAs which exhibited low kinase activities in the free state, two were weak core complex binders (G365F1, E480F1) suggesting that their activity loss arises from a folding perturbation, while eight were strong binders (A42F1, T279F1, I292F1, S351F1, D359F1, I388F1, R446F1, V485F1) and thus are folded properly in the regions essential for docking.
Effects of Bulky Probe Attachment on CheA–CheW Binding
To positively identify the positions involved in CheW docking, a direct CheA–CheW binding measurement was carried out. A fluorescence anisotropy measurement (32, 44) was utilized to quantitate the binding of CheW to selected modified CheA proteins in the free state. The analysis compared CheW binding to Cysless CheA and to 10 bulky probe-labeled variants: the seven 5FM-CheAs identified as poor binders in core complex assembly, and three 5FM-CheAs serving as positive controls. The method monitored the tumbling of a fluorescent CheW protein labeled with tetramethyl-rhodamine maleimide (5TMRM) at a single Cys residue near its modified N-terminus. The binding of this 5TMRM-labeled CheW to free CheA yielded a detectable increase in the 5TMRM fluorescence anisotropy due to the decreased molecular tumbling of CheW. Addition of increasing concentrations of CheA to the sample yielded a fluorescence anisotropy titration curve for the 5TMRM-labeled CheW, which enabled the determination of the KA (= 1/KD) for the CheA–CheW interaction.
Figure 3B compares the KA values of the 10 modified CheA proteins to the KA value measured for Cysless CheA. Three of the variants that exhibited poor core complex assembly also failed to bind CheW (E539F1, L627F1, G636F1), thereby directly locating these three positions to the CheW docking surface. By contrast, the three positive controls (E425F1, T592F1, I598F1), as well as the four remaining variants that exhibited poor complex assembly (E311F1, S340F1, L521F1, L545F1), all bound CheW with near-native affinity (KA values within 2-fold of Cysless CheA). Interestingly, the latter four variants exhibit normal CheW binding but are poorly incorporated into the core complex. The simplest explanation is that these four positions (E311, S340, L521, L545) lie on the surface of CheA that contacts the receptor, but not CheW, in the assembled core complex (see Discussion).
Conservation of CheA Docking Residues
To determine whether the newly identified CheA docking sites are conserved, the sequences of 61 CheA proteins were compared. These sequences were chosen by carrying out a homology search using the sequence of S. typhimurium CheA as a probe, then selecting the 61 most homologous sequences from different genera to yield a diverse sample that ranged from eubacterial to archaeal CheAs. A sequence alignment (created using JalView (45)) was used to generate conservation scores varying from 0 (no conservation) to 11 (complete identity) for each sequence position (see Figure 4B and Supporting Information). Strikingly, the highly conserved positions on the CheA surface are localized to the same regions as the experimentally defined docking sites, while the unconserved surface positions are located outside the experimental docking sites (Figure 4 and Discussion). These findings indicate that the CheA surface regions used as docking sites for P1 domain, P4 domain, CheW, and the receptor are widely conserved across bacterial species.
Figure 4.
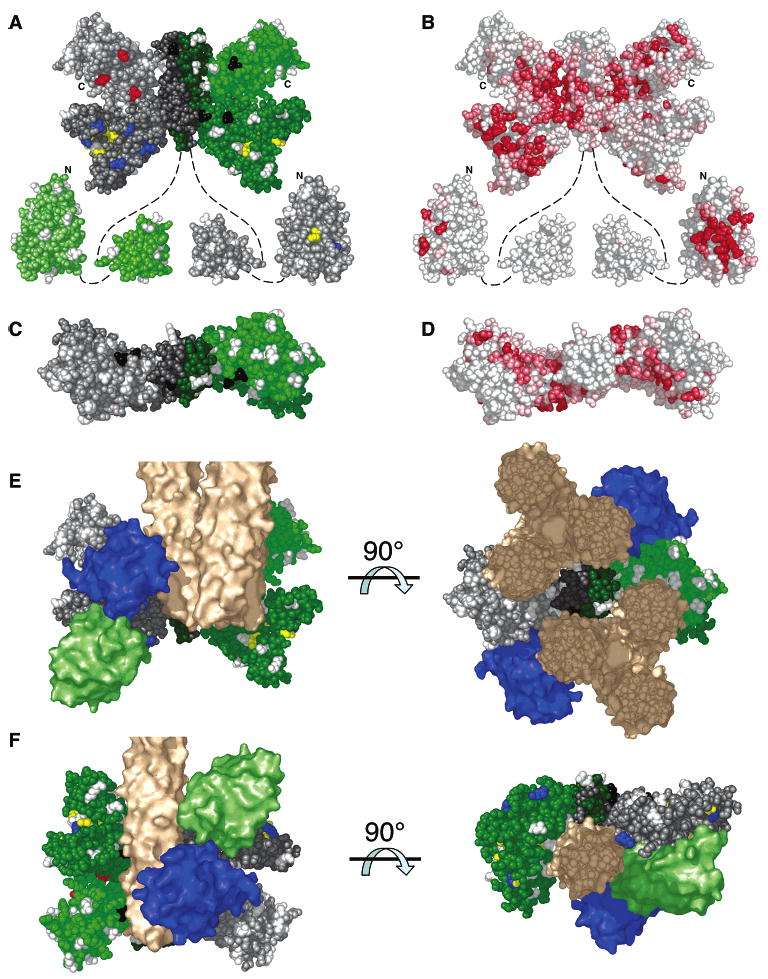
Spatial mapping of results, and working models for core complex architecture. (A) Experimental PICM parameters. Nonperturbing positions (white) lie on surfaces with no detected docking interactions. Perturbing positions assigned to the P1–P4 interaction faces (blue), to the CheW docking site (red), and to an essential surface proposed to bind the receptor (black) are also indicated. (B) Conserved surface residues, where the intensity of red is proportional to the degree of conservation (see Supporting Information). (C and D) Top views of panels A and B, respectively. (E and F) Two working models for core complex architecture. P1, CheW, and receptor are shown in light green, blue, and tan, respectively.
Discussion
The present study employs the PICM-β method to map out essential protein docking surfaces on the surface of the histidine kinase CheA. These surfaces are identified by the functional effects of two different modifications, Cys substitution and bulky probe attachment, at 70 solvent-exposed positions systematically distributed among the five CheA domains. At a subset of positions, termed sensitive positions, modifications are observed to disrupt one or more CheA functions: (i) kinase activity of free CheA, (ii) binding of CheW to CheA, (iii) docking of CheA to the core complex, or (iv) the activation of CheA by receptor in the assembled core complex. Significantly, these sensitive positions are strongly clustered in localized regions of the CheA surface. Within each cluster, modifications at sensitive positions exhibit similar, characteristic effects on CheA function. Such clustering effectively maps out docking surfaces for the P1 and P4 domains, CheW, and receptor.
Internal Controls
The known features of the P1 substrate domain and P2 response regulator binding domain make them useful as internal controls for the PICM-β method. The P2 domain serves as a negative control, since a modified CheA missing this domain is still an active kinase functional in phosphotransfer to CheY both in vitro and in vivo (16, 17). Thus, bulky probes on the known CheY docking face of the P2 domain (46) are predicted to have no effect on CheA kinase activity. The PICM-β method reveals that Cys substitution or 5FM-labeling at all 10 P2 domain positions examined indeed have little or no effect on CheA kinase activity, both in the free state and in the reconstituted core complex, under conditions where CheA autophosphorylation is rate-determining (Table 1). The P1 substrate domain is useful as a positive control because it contains the site of autophosphorylation, His 48. During the autophosphorylation reaction, the substrate face of P1 possessing H48 must contact the P4 catalytic domain and insert H48 into the catalytic pocket. Thus, bulky modifications on the substrate face of the P1 domain are predicted to block autophosphorylation. The PICM-β analysis reveals that Cys or 5FM modification of the A42 position, only 10 Å from H48, dramatically inhibits the autophosphorylation reaction (Table 1). Since A42 is fully solvent-exposed in the free domain and the 5FM-modified protein binds normally to the core complex (Figure 3A), it is unlikely that modification of this position misfolds the protein. Nor is the native Ala side chain involved in the hydrogen bond network that controls the side chain conformation of H48 (12). Instead, the simplest explanation is that the additional bulk of Cys and 5FM modifications at the A42 position introduces a steric block into the substrate face of P1 essential for docking to the P4 catalytic domain during autophosphorylation. By contrast, modifications at 11 positions on other faces of P1 have little or no effect on the autophosphorylation reaction (Table 1). Overall, the negative and positive internal controls provide validation of the PICM-β method and strongly constrain the location of the P1 substrate face that contacts the P4 domain as illustrated in Figure 4A.
Location of the P1 Domain Docking Site on the P4 Domain
While the location of the catalytic cleft on the P4 domain is known, the location and size of the docking surface that contacts the P1 domain during autophosphorylation is not well-defined. Additional strong constraints are provided by the present observation that modifications at a cluster of five positions on a single face of the P4 domain (S351, D359, I388, R446, V485) strongly inhibit the autophosphorylation reaction (Table 1), while modifications at 33 positions on other faces of the P4 and P5 domains have little or no effect. The five sensitive positions are surface-exposed, and their modifications retain normal core complex docking, thus, it is unlikely that the modifications perturb local or global folding (Figure 3A). Instead, the simplest explanation is that these positions define the essential docking site on the surface of the P4 domain that binds the P1 domain, as illustrated in Figure 4A. Thus, the PICM-β analysis of P1 and P4 identifies the functionally critical surfaces on both of these domains that contact one another during the autophosphorylation reaction.
Locations of the CheW and Receptor Docking Sites on the P3, P4, and P5 Domains
CheA also possesses docking sites for both CheW and the receptor, and these sites are essential to assembly of the core complex (20, 21, 24, 47). The PICM-β findings obtained for the reconstituted core complex place strong constraints on the locations of these sites. Probe modifications at seven positions on the P3, P4, and P5 domains of CheA (E311, S340, L521, E539, L545, L627, G636) disrupt both assembly of the core complex and receptor-mediated stimulation of CheA kinase activity (Figures 2 and 3A). By contrast, probe modifications at 53 other surface locations on CheA have little or no effect on receptor-mediated kinase activation in the core complex. We hypothesize that the seven sensitive positions define a large docking region, illustrated in Figure 4A, that contains both the CheW and receptor docking sites.
The effects of modifications on CheW binding to CheA, measured by a direct binding assay, rigorously define the CheW docking site. At three of the seven sensitive positions where modifications disrupt core complex assembly (E539, L627, G636), these same modifications block CheW binding to free CheA (Figure 4A,B). This cluster of three positions, located on a single face of the P5 regulatory domain, reveals the location of the CheW docking site as illustrated in Figure 4A. At the remaining four sensitive positions on the P3, P4, and P5 domains (E311, S340, L521, L545), bulky probe attachment has no effect on CheW binding to CheA, indicating these four positions lie outside the newly identified CheW docking site. Thus, PICM-β analysis confirms previous evidence, suggesting that CheW interacts primarily with the P5 domain (20, 21), and pinpoints the location of CheW docking on P5.
The remaining four sensitive positions (E311, S340, L521, L545) are not involved in CheW binding but are observed to be critical for the assembly of the core complex formed by CheA, CheW, and the receptor. By process of elimination the simplest explanation is that these positions lie within a distinct receptor docking site (Figure 4A). Interestingly, modifications at the sensitive positions within the CheW docking site typically yield greater losses in core complex stability than modifications within the putative receptor docking site (Figure 3B). This observation is consistent with the fact that the CheW site is considerably smaller than the receptor site, and thus is more easily perturbed by a single modification. Moreover, CheW is believed to play a dominant role in stabilizing the core complex by bridging CheA and the receptor, while the direct binding of CheA to the receptor is weaker (25). The present findings indicate that the CheW and putative receptor docking sites are both essential to core complex assembly and to receptor-stimulated activation of CheA kinase within the assembled core complex.
The newly defined CheW and receptor docking sites differ from those proposed by a previous study that employed a novel molecular modeling approach to predict docking interactions (28). Modifications at 12 positions predicted by the prior model to lie within the CheW or receptor docking sites are observed to retain receptor-mediated kinase stimulation in the reconstituted core complex, indicating that bulky probe attachment at these positions does not disrupt core complex assembly. Furthermore, modifications at six positions predicted by the prior model to lie outside the CheW and receptor docking sites are observed to disrupt core complex assembly and receptor-mediated kinase stimulation. It is more difficult to evaluate the predictions of two other models (25, 29), which do not specify CheA docking surfaces in detail.
By contrast, the newly proposed CheW docking site on CheA is in excellent agreement with the findings of an independent analysis of the CheW–CheA complex using X-ray crystallography and EPR, reported (55) after completion of the present study. This independent analysis utilized CheA and CheW from T. maritima, while the present work utilized S. typhimurium proteins. The close agreement between the observed CheW docking sites confirms the accuracy of the PICM-β approach and argues that the CheA–CheW interaction face is highly conserved. Overall, the present study provides the first experimental map of the docking sites for P1 domain, P4 domain, and the receptor on the surface of CheA (Figure 4). Moreover, together with the parallel high-resolution analysis of the Thermatoga CheW–CheA interaction, the present results pinpoint the location of the CheW docking site on CheA in distantly related bacteria.
The CheA Docking Surfaces Are Conserved
To further examine the conservation of the four newly identified docking sites, a sequence alignment of 61 distantly related CheA homologues spanning two superkindoms was carried out. The results clearly show that localized regions of the CheA surface are conserved, as illustrated in Figures 4B,D. These conserved regions exhibit striking overlap with the newly defined docking sites for P1 domain, P4 domain, CheW, and receptor. Moreover, the surfaces lying outside these docking sites are generally not conserved. Such close correspondence provides strong evidence that the targeted docking sites have been correctly identified by the PICM-β method and that distantly related CheA homologues share these essential sites. The P2 domain is a useful negative control in this analysis of conservation, since a previous study revealed that different CheA homologues use different surfaces of P2 to bind CheY (48). Thus, the surface of P2 is predicted to be unconserved, as is observed (Figure 4B).
New Working Models for the Architecture of the Core Complex
The newly identified docking sites, together with published evidence from other laboratories, enable the development of novel models for the architecture of the core complex. Two preliminary models, each proposing approximate docking geometries for the known structures of the component proteins within the assembled core complex, are illustrated in Figures 4E,F. Since the available data only weakly constrain the location of the mobile P2 domain, the models do not include this domain.
The two models were developed in five stages as follows. First, assuming that crystal packing forces perturb the relative positions of CheA domains in the structure of the P3+P4+P5 domain fragment, the domains were rotated about their flexible hinges to flatten the dimer into a planar configuration possessing a 180° axis of symmetry along the long axis of the P3 domain (Figure 4A). Second, the P1 domain was placed with H48 inserted into the catalytic cleft and with A42 contacting the P4 domain; then, while maintaining these constraints, P1 was rotated until it contacted the five positions on P4 defining the rest of the P1 docking site (S351, D359, I388, R446, V485) (Figure 4E). Third, CheW was positioned with loop L1 of its second domain, which is believed to dock to CheA (22, 31, 32), contacting the three P5 domain positions defining the CheW docking site (E539, L627, G636) (Figure 4E). Fourth, the receptor was introduced as either a trimer-of-dimers or as an isolated dimer (Figures 4E,F), since both binding modes have been proposed (1, 23, 29). In both cases, the receptor was positioned with its cytoplasmic tip, known to possess the CheA docking surface (30, 34), in contact with the four CheA residues defining the receptor docking site (E311, S340, L521, L545). In the case of the receptor trimer-of-dimers (Figure 4E), the proposed geometry allows 5FM probes coupled to positions G318, G325, and R332 to insert between receptor dimers, thereby explaining the minimal effects of these modifications on core complex assembly and function. In the case of the isolated receptor dimer (Figure 4F), a small rotation of the P4 and P5 domains is needed to optimize receptor contacts, and the current data do not resolve whether the receptor long axis is parallel or antiparallel to the long axis of the P3 domain. Fifth, the bound CheW molecule was rotated to place its known receptor docking face (30–32) in contact with the adjacent receptor, while maintaining its docking contacts to the CheW docking site on P5 (Figures 4E,F).
The two resulting models are consistent with extensive evidence and explain the observation that the fully assembled, core ternary complex is significantly more stable than the binary complexes between any pair of components. The models share both similarities and differences with recently proposed alternative models based on crystallographic and EPR data (55). Most of the disagreements among the models arise from different assumptions about the relative spatial positions of CheA domains P3, P4, and P5, emphasizing the sensitivity of current models to interdomain geometries that remain undetermined for the assembled core complex.
Notably, the present preliminary models (Figures 4E,F) suggest potential pathways for the transmission of regulatory signals from the receptor to CheA. Such signals could be transmitted by direct contacts between the receptor and the CheA P3, P4, and P5 domains. Alternatively, signals could be transmitted indirectly via CheW-mediated contacts between the receptor and the CheA P1 or P5 domain. Future studies will investigate the mechanism of signal transmission through the complex, as well as the mechanism of CheA regulation by these signals. Other studies will continue the analysis of core complex architecture.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Profs. Sandy Parkinson, Victor Sourjik, Howard Berg, and Bob Bourret for providing strains and constructs, as well as Profs. Art Pardi, Marcelo Sousa, and Mike Stowell for critical reading of the manuscript.
Footnotes
Abbreviations: PICM, Protein-Interactions-by-Cysteine-Modification; 5FM, 5-fluorescein maleimide; 5TMRM, 5-tetramethyl-rhodamine maleimide; EDTA, ethylenediaminetetraacetic acid; DTT, dithiotheitol.
Support provided by NIH R01 GM-040731 (to J.J.F.) and by NIH T32 GM-065103 (to A.S.M.)
Supporting Information Available: The sequence alignment of 61 CheA homologues used to generate the map in Figure 4A of conserved positions on the CheA surface. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Parkinson JS, Ames P, Studdert CA. Collaborative signaling by bacterial chemoreceptors. Curr Opin Microbiol. 2005;8:116–121. doi: 10.1016/j.mib.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Wadhams GH, Armitage JP. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol. 2004;5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 3.Wolanin PM, Stock JB. Bacterial chemosensing: cooperative molecular logic. Curr Biol. 2004;14:R486–487. doi: 10.1016/j.cub.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Bourret RB, Stock AM. Molecular information processing: lessons from bacterial chemotaxis. J Biol Chem. 2002;277:9625–9628. doi: 10.1074/jbc.R100066200. [DOI] [PubMed] [Google Scholar]
- 5.Spudich JL, Luecke H. Sensory rhodopsin II: functional insights from structure. Curr Opin Struct Biol. 2002;12:540–546. doi: 10.1016/s0959-440x(02)00359-7. [DOI] [PubMed] [Google Scholar]
- 6.Falke JJ, Hazelbauer GL. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem Sci. 2001;26:257–265. doi: 10.1016/s0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA. The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu Rev Cell Dev Biol. 1997;13:457–512. doi: 10.1146/annurev.cellbio.13.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borkovich KA, Kaplan N, Hess JF, Simon MI. Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc Natl Acad Sci USA. 1989;86:1208–1212. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ninfa EG, Stock A, Mowbray S, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J Biol Chem. 1991;266:9764–9770. [PubMed] [Google Scholar]
- 10.McEvoy MM, Zhou H, Roth AF, Lowry DF, Morrison TB, Kay LE, Dahlquist FW. Nuclear magnetic resonance assignments and global fold of a CheY-binding domain in CheA, the chemotaxis-specific kinase of Escherichia coli. Biochemistry. 1995;34:13871–13880. doi: 10.1021/bi00042a019. [DOI] [PubMed] [Google Scholar]
- 11.Bilwes AM, Alex LA, Crane BR, Simon MI. Structure of CheA, a signal-transducing histidine kinase. Cell. 1999;96:131–141. doi: 10.1016/s0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 12.Mourey L, Da Re S, Pedelacq JD, Tolstykh T, Faurie C, Guillet V, Stock JB, Samama JP. Crystal structure of the CheA histidine phosphotransfer domain that mediates response regulator phosphorylation in bacterial chemotaxis. J Biol Chem. 2001;276:31074–31082. doi: 10.1074/jbc.M101943200. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive binding to the kinase CheA. Biochemistry. 1995;34:14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, McEvoy MM, Lowry DF, Swanson RV, Simon MI, Dahlquist FW. Phosphotransfer and CheY-binding domains of the histidine autokinase CheA are joined by a flexible linker. Biochemistry. 1996;35:433–443. doi: 10.1021/bi951960e. [DOI] [PubMed] [Google Scholar]
- 15.McEvoy MM, de la Cruz AF, Dahlquist FW. Large modular proteins by NMR. Nat Struct Biol. 1997;4:9. doi: 10.1038/nsb0197-9. [DOI] [PubMed] [Google Scholar]
- 16.Stewart RC, Jahreis K, Parkinson JS. Rapid phosphotransfer to CheY from a CheA protein lacking the CheY-binding domain. Biochemistry. 2000;39:13157–13165. doi: 10.1021/bi001100k. [DOI] [PubMed] [Google Scholar]
- 17.Jahreis K, Morrison TB, Garzon A, Parkinson JS. Chemotactic signaling by an Escherichia coli CheA mutant that lacks the binding domain for phosphoacceptor partners. J Bacteriol. 2004;186:2664–2672. doi: 10.1128/JB.186.9.2664-2672.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilwes AM, Quezada CM, Croal LR, Crane BR, Simon MI. Nucleotide binding by the histidine kinase CheA. Nat Struct Biol. 2001;8:353–360. doi: 10.1038/86243. [DOI] [PubMed] [Google Scholar]
- 19.Kott L, Braswell EH, Shrout AL, Weis RM. Distributed subunit interactions in CheA contribute to dimer stability: a sedimentation equilibrium study. Biochim Biophys Acta. 2004;1696:131–140. doi: 10.1016/j.bbapap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Bourret RB, Davagnino J, Simon MI. The carboxy-terminal portion of the CheA kinase mediates regulation of autophosphorylation by transducer and CheW. J Bacteriol. 1993;175:2097–2101. doi: 10.1128/jb.175.7.2097-2101.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison TB, Parkinson JS. A fragment liberated from the Escherichia coli CheA kinase that blocks stimulatory, but not inhibitory, chemoreceptor signaling. J Bacteriol. 1997;179:5543–5550. doi: 10.1128/jb.179.17.5543-5550.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griswold IJ, Zhou H, Matison M, Swanson RV, McIntosh LP, Simon MI, Dahlquist FW. The solution structure and interactions of CheW from Thermotoga maritima. Nat Struct Biol. 2002;9:121–125. doi: 10.1038/nsb753. [DOI] [PubMed] [Google Scholar]
- 23.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 24.Gegner JA, Graham DR, Roth AF, Dahlquist FW. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;70:975–982. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 25.Levit MN, Grebe TW, Stock JB. Organization of the receptor-kinase signaling array that regulates Escherichia coli chemotaxis. J Biol Chem. 2002;277:36748–36754. doi: 10.1074/jbc.M204317200. [DOI] [PubMed] [Google Scholar]
- 26.Bornhorst JA, Falke JJ. Quantitative analysis of aspartate receptor signaling complex reveals that the homogeneous two-state model is inadequate: development of a heterogeneous two-state model. J Mol Biol. 2003;326:1597–1614. doi: 10.1016/s0022-2836(03)00026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M, Hazelbauer GL. Cellular stoichiometry of the components of the chemotaxis signaling complex. J Bacteriol. 2004;186:3687–3694. doi: 10.1128/JB.186.12.3687-3694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu TS, Le Novere N, Levin MD, Beavil AJ, Sutton BJ, Bray D. Molecular model of a lattice of signalling proteins involved in bacterial chemotaxis. Nat Cell Biol. 2000;2:792–796. doi: 10.1038/35041030. [DOI] [PubMed] [Google Scholar]
- 29.Francis NR, Wolanin PM, Stock JB, Derosier DJ, Thomas DR. Three-dimensional structure and organization of a receptor/signaling complex. Proc Natl Acad Sci USA. 2004;101:17480–17485. doi: 10.1073/pnas.0407826101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu JD, Parkinson JS. Genetic evidence for interaction between the CheW and Tsr proteins during chemoreceptor signaling by Escherichia coli. J Bacteriol. 1991;173:4941–4951. doi: 10.1128/jb.173.16.4941-4951.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boukhvalova MS, Dahlquist FW, Stewart RC. CheW binding interactions with CheA and Tar. Importance for chemotaxis signaling in Escherichia coli. J Biol Chem. 2002;277:22251–22259. doi: 10.1074/jbc.M110908200. [DOI] [PubMed] [Google Scholar]
- 32.Boukhvalova M, VanBruggen R, Stewart RC. CheA kinase and chemoreceptor interaction surfaces on CheW. J Biol Chem. 2002;277:23596–23603. doi: 10.1074/jbc.M202288200. [DOI] [PubMed] [Google Scholar]
- 33.Bass RB, Coleman MD, Falke JJ. Signaling domain of the aspartate receptor is a helical hairpin with a localized kinase docking surface: cysteine and disulfide scanning studies. Biochemistry. 1999;38:9317–9327. doi: 10.1021/bi9908179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehan RS, White NC, Falke JJ. Mapping out regions on the surface of the aspartate receptor that are essential for kinase activation. Biochemistry. 2003;42:2952–2959. doi: 10.1021/bi027127g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bass RB, Falke JJ. Detection of a conserved alpha-helix in the kinase-docking region of the aspartate receptor by cysteine and disulfide scanning. J Biol Chem. 1998;273:25006–25014. doi: 10.1074/jbc.273.39.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bornhorst JA, Falke JJ. Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 2000;326:245–254. doi: 10.1016/s0076-6879(00)26058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourret RB, Drake SK, Chervitz SA, Simon MI, Falke JJ. Activation of the phosphosignaling protein CheY. II. Analysis of activated mutants by 19F NMR and protein engineering. J Biol Chem. 1993;268:13089–13096. [PMC free article] [PubMed] [Google Scholar]
- 38.Chervitz SA, Lin CM, Falke JJ. Transmembrane signaling by the aspartate receptor: engineered disulfides reveal static regions of the subunit interface. Biochemistry. 1995;34:9722–9733. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danielson MA, Bass RB, Falke JJ. Cysteine and disulfide scanning reveals a regulatory alpha-helix in the cytoplasmic domain of the aspartate receptor. J Biol Chem. 1997;272:32878–32888. doi: 10.1074/jbc.272.52.32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster DL, Mowbray SL, Jap BK, Koshland DE., Jr Purification and characterization of the aspartate chemoreceptor. J Biol Chem. 1985;260:11706–11710. [PubMed] [Google Scholar]
- 42.Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, Falke JJ. C2 domain of protein kinase C alpha: elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42:1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tawa P, Stewart RC. Kinetics of CheA autophosphorylation and dephosphorylation reactions. Biochemistry. 1994;33:7917–7924. doi: 10.1021/bi00191a019. [DOI] [PubMed] [Google Scholar]
- 44.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd. Kluwer Academic/Plenum; New York: 1999. [Google Scholar]
- 45.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 46.McEvoy MM, Hausrath AC, Randolph GB, Remington SJ, Dahlquist FW. Two binding modes reveal flexibility in kinase/response regulator interactions in the bacterial chemotaxis pathway. Proc Natl Acad Sci USA. 1998;95:7333–7338. doi: 10.1073/pnas.95.13.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gegner JA, Dahlquist FW. Signal transduction in bacteria: CheW forms a reversible complex with the protein kinase CheA. Proc Natl Acad Sci USA. 1991;88:750–754. doi: 10.1073/pnas.88.3.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park SY, Beel BD, Simon MI, Bilwes AM, Crane BR. In different organisms, the mode of interaction between two signaling proteins is not necessarily conserved. Proc Natl Acad Sci USA. 2004;101:11646–11651. doi: 10.1073/pnas.0401038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai RZ, Manson JM, Bormans AF, Draheim RR, Nguyen NT, Manson MD. Cooperative signaling among bacterial chemoreceptors. Biochemistry. 2005;44:14298–14307. doi: 10.1021/bi050567y. [DOI] [PubMed] [Google Scholar]
- 50.Studdert CA, Parkinson JS. Crosslinking snapshots of bacterial chemoreceptor squads. Proc Natl Acad Sci USA. 2004;101:2117–2122. doi: 10.1073/pnas.0308622100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ames P, Parkinson JS. Transmembrane signaling by bacterial chemoreceptors: E. coli transducers with locked signal output. Cell. 1988;55:817–826. doi: 10.1016/0092-8674(88)90137-7. [DOI] [PubMed] [Google Scholar]
- 52.Butler SL, Falke JJ. Cysteine and disulfide scanning reveals two amphiphilic helices in the linker region of the aspartate chemoreceptor. Biochemistry. 1998;37:10746–10756. doi: 10.1021/bi980607g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward SM, Delgado A, Gunsalus RP, Manson MD. A NarX-Tar chimera mediates repellent chemotaxis to nitrate and nitrite. Mol Microbiol. 2002;44:709–719. doi: 10.1046/j.1365-2958.2002.02902.x. [DOI] [PubMed] [Google Scholar]
- 54.Coleman MD, Bass RB, Mehan RS, Falke JJ. Conserved glycine residues in the cytoplasmic domain of the aspartate receptor play essential roles in kinase coupling and on–off switching. Biochemistry. 2005;44:7687–7695. doi: 10.1021/bi0501479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park SY, Borbat P, Gonzalez-Bonet G, Bhatnagar J, Pollard AM, Freed JH, Bilwes AM, Crane BR. Reconstruction of the chemotaxis receptor-kinase assembly. Nat Struct Mol Biol. 2006;13:400–407. doi: 10.1038/nsmb1085. [DOI] [PubMed] [Google Scholar]
- 56.Miller AS. PhD Thesis. Department of Chemistry & Biochemistry, University of Colorado; Boulder, CO: 2006. Structural and mechanistic studies of the receptor-kinase complex of bacterial chemotaxis. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.