

Abstract
The information for correct localization of newly synthesized proteins in both prokaryotes and eukaryotes resides in self-contained, often transportable targeting sequences. Of these, signal sequences specify that a protein should be secreted from a cell or incorporated into the cytoplasmic membrane. A central puzzle is presented by the lack of primary structural homology among signal sequences, although they share common features in their sequences. Synthetic signal peptides have enabled a wide range of studies of how these “zipcodes” for protein secretion are decoded and used to target proteins to the protein machinery that facilitates their translocation across and integration into membranes. We review research on how the information in signal sequences enables their passenger proteins to be correctly and efficiently localized. Synthetic signal peptides have made possible binding and crosslinking studies to explore how selectivity is achieved in recognition by the signal sequence-binding receptors, signal recognition particle, or SRP, which functions in all organisms, and SecA, which functions in prokaryotes and some organelles of prokaryotic origins. While progress has been made, the absence of atomic resolution structures for complexes of signal peptides and their receptors has definitely left many questions to be answered in the future.
Keywords: signal sequence, protein export, synthetic signal peptides, NMR, CD, fluorescence, crosslinking, SecA, SRP
BACKGROUND
The 1999 Nobel Prize in Physiology or Medicine was awarded to Günter Blobel for his articulation of the “signal hypothesis,” which described the way proteins get targeted with fidelity and efficiency to noncytoplasmic locations, in particular the extracellular milieu.1 Central to this hypothesis were signal sequences whose role was akin to zipcodes, i.e., to specify the eventual destination of their passenger protein. Implicit in the signal hypothesis are “decoders” or zipcode readers, that recognize the targeting information and direct the nascent or newly synthesized polypeptide chain to the correct targeting machinery.2 The fact that signal sequences are self-contained, often transient (removed after their targeting duties are complete), and usually transportable from one protein to another, led us 25 years ago to launch a research program using synthetic signal peptides to explore the roles and interactions of signal sequences. This research program was feasible solely because of the enabling synthetic methodologies pioneered by R. Bruce Merrifield.3 In fact, a graduate student in our laboratory at that time, Martha Briggs, visited Rockefeller to learn the “tricks of the trade” from the master. Familiar to all who knew Bruce was the warm greeting she received and the rapid training in effective synthetic methods (of course, the wrist-action shaker!). We were on our way.
Several questions motivated our early studies. Some we now happily can answer, and some remain mysteries and topics of current research. This paper reviews briefly our past work on signal peptide conformations, including efforts to elucidate the functionally required structural properties of signal sequences and to understand how signal peptides interact with membranes. Then, in the last two sections, we describe current understanding of signal sequence recognition by the signal recognition particle (SRP) and by SecA, the protein complexes that “decode” the targeting information in signal sequences and bind to them in a selective and regulated manner, and we report our recent efforts to elucidate these key events.
Signal Peptide Conformations and Membrane Interactions
Signal sequences are highly variable and lack primary structural similarity. Nonetheless, they share common properties, viz. a charged N-terminal region (usually basic), a hydrophobic core of ~10 amino acids which often ends with a turn-forming residue, and a 6–8 amino acid-long, less hydrophobic (but usually uncharged) region that includes the cleavage site information.4,5 Despite the lack of primary structural homology, the features of signal sequences enable them to be identified in protein sequences with high confidence.6 The common features are also quite universal, characterizing signal sequences from prokaryotes, yeast, and mammals. In fact, signal sequences from one organism generally work to specify export in the other organisms. In addition, many different signal sequences within a given organism are recognized by each of the recognition species (SecA or SRP). These shared functions and abilities to interact with a variety of species, despite widely varying sequences, present a central puzzle of signal sequence structure–function correlation.
We began to tackle this puzzle by comparing the biophysical properties of synthetic signal peptides representing functional signal sequences from bacterial exported proteins with those of defective signal sequences. Our initial work focused on the signal sequence for the Escherichia coli outer membrane protein maltoporin, also called LamB (the λ-phage receptor), because extensive genetic studies in the Silhavy lab7–10 had yielded a number of defective mutants as well as so-called pseudorevertants, in which second site mutations led to restored function.10 These LamB signal sequences allowed us to correlate conformational behavior with ability to perform the required functions in targeting.
Signal Peptides Adopt Helical Conformations in Interfacial Environments
Our results on the LamB signal peptides and several other examples clearly demonstrated that functional signal peptides, while lacking significant structure in aqueous solution, have a high propensity to adopt an α-helical conformation in interfacial environments.11–14 The helix content is highest, and the helix is most stable in the hydrophobic core of the signal sequence.12,15,16 When the helical conformation required in the targeting pathway was not revealed by these studies, the intrinsic tendency of signal sequences to fold into an α-helix was established.
Signal Peptides Spontaneously Insert Into Lipid Bilayers
We utilized a number of assays, including surface tensiometry of peptide–lipid monolayers, to assess the tendency of signal peptides to insert into a lipid phase and to characterize the manner in which they interacted with a lipid monolayer or bilayer.13,17–21 Strikingly, functional signal sequences share the capacity to insert spontaneously and deeply into the acyl chain region of a lipid bilayer. Mutations that disrupt the ability to insert into a lipid phase led to loss of targeting function. Through the use of a broad set of signal peptides, we found that their spontaneous membrane insertion and in vivo function rely on a mean residue hydrophobicity in the hydrophobic core higher than 2.4 on the Kyte-Doolittle scale (which is halfway between the hydrophobicities of Ala and Leu).21 Completely consistent conclusions were reached by Kendall and coworkers,22 using a distinct approach: they idealized the hydrophobic core of the E. coli alkaline phosphatase (PhoA) signal peptide and showed that a mix of equal amounts of Ala and Leu was required for function. They further demonstrated that the optimal length core for secretion was 10 amino acids.
In a subsequent study, we determined that the properties we observed for isolated signal peptides were “dominant” over the adjacent sequence of the mature secreted protein, in the sense that the presence of the first part of the passenger protein C-terminal to the signal sequence does not alter its conformational and membrane-interactive properties.23 Intriguingly, mutations in the early region of a passenger protein can abolish correct targeting, implying that there are restrictions on the allowed sequences of secreted proteins (e.g., Ref. 24).
The Conformation of Signal Peptides in a Lipid Bilayer Reflects Their Unusual Pattern of Charged and Hydrophobic Amino Acids
We exploited the ability of signal peptides to spontaneously insert into model bilayers in lipid vesicles to determine their membrane-associated conformation, using transferred nuclear Overhauser effect (trNOE) NMR.25 We also studied the manner in which they associated with the membrane using paramagnetic broadening of the NMR signals arising from the bound conformations,25 EPR of spin-labeled lipids in the presence of signal peptides,26 and extensive mapping of fluorescently labeled signal peptides using a variety of exogenous aqueous or lipid-resident quenchers.20,27,28 The picture that emerged was consistent with all these data: The charged N-terminal region of the signal peptide associates with the lipid head groups of the cis leaflet of the bilayer in an extended conformation, while the hydrophobic core inserts well into the acyl chain region in a helical conformation. The C-terminus of a signal peptide remains accessible on the cis side of the membrane most of the time, but makes brief visits to the trans side.27 Thus, unlike a typical membrane anchor, a secretory signal peptide is not stable in a transmembrane conformation.
Probing the Targeting Pathway
The availability of peptides corresponding to functional and defective mutant signal sequences provided ideal probes of the export machinery. Validating this approach was our key observation that synthetic peptides corresponding to functional signal sequences inhibit the translocation of precursor proteins across E. coli membrane vesicles, while nonfunctional peptides do not.29 The inhibition seemed to be protein-mediated, and could be reversed, at least in part, by addition of an excess of the major E. coli signal sequence receptor, SecA, arguing that the peptides inhibit by binding to SecA. As described later, our later work indeed confirms that exogenously added synthetic signal peptides interact directly with SecA and modulate its activity. In early work, we also showed that synthetic peptides corresponding to functional signal sequences modulate the GTPase activity of SRP/SRP receptor, while nonfunctional peptides do not.30 Subsequent experiments in our lab31 brought these results into question because of artifactual interactions between the signal peptides and the RNA SRP component; nonetheless, this research study paved the way for our ongoing studies of SRP-signal peptide interaction (see later).
SRP: THE UNIVERSAL SIGNAL SEQUENCE RECEPTOR
The first step in SRP-mediated protein targeting is the recognition of the signal sequence on the nascent protein. Despite the diversity of signal sequences, SRP productively recognizes and selectively binds them, and this binding event serves as the critical sorting step in protein localization within the cell. The structural details that confer on SRP this distinctive ability are poorly understood. Crucial to answering this question is the direct atomic resolution structural information on an SRP-signal sequence complex, which remains elusive. Later, we briefly provide background on SRP functions and its recognition of signal sequences, and then describe the strategies used by our lab to study the interaction between SRP and signal sequences using synthetic signal peptides as a primary tool.
The SRP-Dependent Targeting Cycle in Eukaryotes and Prokaryotes
The mechanism by which SRP targets newly synthesized proteins to their final cellular destination is highly conserved and essential throughout all kingdoms of life.32,33 Ribosome-associated SRP recognizes and binds signal peptides of nascent secretory or membrane proteins (in eukaryotes) or integral plasma membrane proteins (in prokaryotes; Figure 1). The ribosome-nascent chain complex is then targeted by SRP to the SRP receptor, which is anchored to the membrane. Upon interaction, SRP and its receptor (both containing homologous GTP binding domains) mutually stimulate their GTP hydrolysis activities, the ribosome nascent chain is delivered to the translocation channel that spans the membrane, termed the “translocon,” and GDP-bound SRP is released from the complex to start another sorting cycle. In eukaryotes, translation is arrested or retarded until the ribosome-nascent chain complex is situated at the translocon.34–38
FIGURE 1.

Protein export to the endoplasmic reticulum (ER) and insertion into the ER membrane in eukaryotes, and to the plasma membrane or the periplasm in E. coli. SRP-mediated targeting: As a membrane protein (pink) is emerging from the ribosome (gray), SRP (light blue) recognizes and binds the signal sequence (red), and the entire nascent chain complex is targeted to the SRP receptor (dark blue) at the target membrane (yellow). In eukaryotes, translation is arrested or retarded at this step. The nascent protein is maintained in a conformation compatible with translocation. Upon interaction between the SRP and the SRP receptor, the ribosome nascent chain is delivered to the Sec translocon complex (green), which enables the nascent protein to cross or insert into the membrane. SRP is released and starts a new targeting cycle. SecA-mediated targeting: After a secretory protein is translated on the ribosome, the molecular chaperone SecB (orange) binds to the mature regions of the preprotein and keeps it in a conformation competent for translocation. This SecB-preprotein complex may be recognized by SecA (purple), which binds both the signal sequence and the mature part of the preprotein, either in its membrane-resident or its cytoplasmic form. The complex docks on the SecYEG translocon, and SecB is released. SecA undergoes major conformational changes as it inserts into the membrane and carries out its next roles as a “translocase,” facilitating translocation of the preprotein across the inner membrane via the translocon channel.
SRP Structure
The eukaryotic SRP is a cytoplasmic ribonucleoprotein composed of six polypeptides and a 7S RNA. Gram-positive bacteria and archea exhibit SRP assemblies of different complexity. The minimal functional unit of SRP, as found in gram-negative bacteria, consists of only one protein and a 4.5S RNA, (for a review see Ref. 32).
SRP RNA participates in the central step of protein targeting by catalyzing the interaction between SRP and the SRP receptor, and consequently is essential for SRP function in vivo. Interestingly, RNA enhances the GTPase activity of the SRP–SRP receptor complex in a manner analogous to GTPase-activating proteins.39 The importance of this role is underlined by the finding that RNA mutants unable to stimulate GTPase activity show severe growth phenotypes in in vivo complementation experiments.39,40 Recently, the role of SRP RNA in the communication between the individual domains of Ffh (see later) and the coordination of different steps of the targeting reaction has been described.41,42
The protein subunit present in E. coli SRP is termed Ffh, as it is the prokaryotic homolog to the fifty-four kDa subunit of the eukaryotic SRP (SRP54). Ffh (as well as SRP54) is the subunit responsible for signal sequence binding, and it is conserved in all the SRPs found in nature. Ffh consists of three domains: The N terminal (“N domain”) is a four-helix bundle structure43 responsible for interaction with the ribo-some.44 The “G domain,’ which is a conserved Ras-like GTPase domain, is closely associated to the N domain.43,45,46 The G domain contains the GTP-binding site and establishes interaction with its homologous domain in the SRP receptor. The 10-residue segment that links the N and G domains is part of the G domain but is closely packed to the N domain surface, yielding a single “NG” structural unit stable to proteolysis. The structural elements found at the N and G interface enable a dynamic communication between these domains. The C-terminal domain of Ffh, called the “M domain” because of its high methionine content, comprises the RNA-binding site,47,48 although some structural studies raise the possibility that the N domain may make contact with the RNA as well.49–52 The M domain is mostly helical and contains a highly conserved hydrophobic loop (so-called finger loop), which is disordered in some crystal structures.47,53 We found that the M domain is inherently flexible and binding of 4.5S RNA to Ffh stabilizes the M domain.48 Subsequently, in a study that involved the use of fragments of the E. coli protein component of SRP, our lab found evidence that this finger loop itself is detrimental to the stability of the M domain, and proposed that one of the functional requirements for the SRP RNA resides in its ability to counteract the destabilizing influence of the finger loop.54
Signal Sequence Binding by SRP
A substantial body of evidence implicates the M domain in signal-sequence binding.55,56 Crystal structures of the M domain reveal two features that are postulated to form the signal-sequence binding site: a deep, highly conserved hydrophobic groove lined with Met side-chains, and the finger loop, which may act as a lid for the cavity.47,53 Recent cryo-EM studies on complexes of ribosome, nascent chain, and SRP show an additional electron density near the M-domain of the SRP, which has been attributed to the signal sequence in the hydrophobic groove of the M-domain.57,58 These studies provide persuasive, yet largely indirect, evidence that the M domain is solely responsible for signal sequence binding. However, other evidence showed that the NG domain is required for efficient signal peptide binding.48,55,59
Our lab studied the interaction between signal peptides and SRP by using reconstituted fragments of SRP from E. coli and synthetic signal peptides.48 We found that specific binding of signal peptides causes a dramatic and global destabilization of the SRP protein. Intriguingly, this effect was also seen when signal peptides bound to isolated NG domains. Binding of 4.5S RNA inhibits this catastrophic effect of signal peptide binding to Ffh by locally stabilizing the M domain. The fact that the functional signal peptide-induced destabilizing effect can be observed for the isolated NG domain implies that the NG domain is directly involved in signal peptide binding.
We thus created an in vitro, purified system to assess the role of the NG domain in the interaction between SRP and signal sequences using reconstituted SRP from E. coli and synthetic signal peptides.60 Strikingly, our data clearly showed that the signal peptide can be directly attached to the NG domain by crosslinking, and, moreover, that the NG domain can independently bind the signal peptide (see later for details). More recently, we probed further the molecular details that underlie the interaction between signal sequences and SRP using photoinduced crosslinking approaches and exploring the positional dependence of the crosslinking products (A. Szymanska, E. M. Clerico, and L. M. Gierasch, manuscript in preparation). We used benzophenone-mediated (BPM) crosslinking61 and photoinduced crosslinking of unmodified proteins (PICUP).62,63 Here, both methods lead to a covalent linkage between the SRP (in vitro reconstituted using recombinant Ffh protein from E. coli and a 43-oligonu-cleotide RNA, representing the minimal element of 4.5S RNA, transcribed in vitro) and solid-phase synthesized variants of a Leu-enriched alkaline phosphatase signal sequence-based peptide (PhoA; see Table I). It had been shown previously in an in vitro translation system that these signal sequences can interact strongly with E. coli SRP.64 The synthetic signal peptides were biotinylated at the N-terminus to enable detection of the crosslinked products by enhanced chemiluminescence (ECL). Following crosslinking, the NG and M domains of Ffh were separated, and the domain of Ffh that was crosslinked to the biotinylated signal peptide was identified by ECL.60
Table I.
Signal Peptides Used for Crosslinking to SRP
| Peptide | Sequencea |
|---|---|
| BioPhoAb | Biotin-MKQKKKLALLLLLLLLTPVTKA-NH2 |
| BioPhoA C2 | Biotin-MCKQKKKLALLLLLLLLTPVTKA-NH2 |
| BioPhoA C10 | Biotin-MKQKKKLALCLLLLLLTPVTKA-NH2 |
| BioPhoA C11 | Biotin-MKQKKKLALLCLLLLLTPVTKA-NH2 |
| BioPhoA C12 | Biotin-MKQKKKLALLLCLLLLTPVTKA-NH2 |
| BioPhoA C13 | Biotin-MKQKKKLALLLLCLLLTPVTKA-NH2 |
| BioPhoA C14 | Biotin-MKQKKKLALLLLLCLLTPVTKA-NH2 |
| BioPhoA C23 | Biotin-MKQKKKLALLLLLLLLTPVTKACNH2 |
| BioPhoA10-Bpa | Biotin-MKQKKKLAL(Bpa)LLLLLLTPVTKA-NH2 |
| BioPhoA11-Bpa | Biotin-MKQKKKLALL(Bpa)LLLLLTPVTKA-NH2 |
| BioPhoA12-Bpa | Biotin-MKQKKKLALLL(Bpa)LLLLTPVTKA-NH2 |
| BioPhoA13-Bpa | Biotin-MKQKKKLALLLL(Bpa)LLLTPVTKA-NH2 |
| BioPhoA14-Bpa | Biotin-MKQKKKLALLLLL(Bpa)LLTPVTKA-NH2 |
Sequences of the PhoA-derived signal peptides used for crosslinking. The positions indicated in bold were replaced by Cys to create the BioPhoACn series of peptides or Bpa to create the BioPhoABpan series. The BioPhoACn-BPM series of peptides was created with the BioPhoACn set of peptides by alkylation of the free Cys with 4-maleimido-benzophenone.
Biotinylated wild-type alkaline phospatase (PhoA) signal peptide.
PICUP crosslinks two unmodified proteins and creates a direct linkage between side chains on the two crosslinking partners (here SRP and the signal peptide) without the intervention of a “spacer arm” (Figure 2). This approach thus offers the advantage, in principle, of mapping the binding site with high spatial fidelity. PICUP preferentially generates a radical from a Tyr, His, or Met side chain, which is then attacked by a neighboring nucleophile. Here, we “walked” Cys residues through the hydrophobic core of the synthetic signal sequence (positions 10–14) and made single Cys substitutions at the termini (positions 2 and 23) to enable mapping of the binding site (see Table I). The Cys sulfhydryl is a good nucleophile at the pH used, and thus is the likely candidate to attack the radical.
FIGURE 2.

Design of the signal peptides used for crosslinking to SRP. Three groups of signal peptides were used for crosslinking: the unmodified BioPhoACn series, which had Cys residues substituted at various positions (see Table I); the BioPhoACn-BPM series created by alkylation of the Cys groups by 4-maleimido-benzophenone; and the BioPhoAnBpa series created by incorporation of benzoylphenylalanine (Bpa) into the peptide sequence. In all cases, “n” indicates the position of the reactive probe. Here, one example of each group is shown with the reactive group at position 10 of the peptide, and the distance between the reactive group and the peptide backbone is depicted.
In BPM crosslinking, the benzophenone group introduced into the hydrophobic core of the signal peptide constitutes the reactive photoprobe. Benzophenone was introduced at positions 10–14 by modification of a Cys residue with 4-maleimido-benzophenone, which generates a relatively flexible linker with a length of 12 Å between the signal peptide backbone and the photoactivatable group. This distance is reduced to ~6 Å in the other series of peptides tested, where benzoylphenylalanine was directly incorporated into the peptide sequence at the same positions during synthesis (Table I and Figure 2). Upon irradiation with UV light, a benzophenone diradical is generated that inserts into an electron-rich σ bond, thus favoring crosslinking to nearby C—H bonds susceptible to facile hydrogen abstraction (e.g., hydrophobic residues like Met, Leu or Val).61
Given the differences between these two crosslinking methods (both the nature of the chemistry involved in the crosslinking reaction and the distance between the peptide backbone and the reactive group), it was not surprising that distinct patterns of crosslinked products between signal peptide and Ffh were obtained. But the differences were remarkable and speak to the complex nature of the SRP-signal sequence recognition process: By PICUP, regardless of the position of the reactive group, signal peptide was found linked to the NG domain of SRP, as observed previously in our lab.60 In striking contrast, BPM crosslinking attached the signal peptide preferentially to the M domain, confirming the results obtained by other groups that used analogous crosslinking chemistries with a similar length spacer arm, but in the context of the ribosome.56,65 A position-dependent pattern in the BPM crosslinking efficiency was noticeable, with the highest efficiency at the center of the hydrophobic core and a diminution in efficiency consistent with a helical conformation of the signal peptide bound at an M domain-resident binding site (Figure 3). Our observation thus supports previous suggestions that the signal peptide binds SRP in a helical conformation.57,58
FIGURE 3.

Schematic representation of the hydrophobic core of the signal peptide (seen from the top) bound to the M domain of Ffh in a helical conformation, as suggested by the positional dependence of BPM-mediated crosslinking. The intensity of the gray shading for different residues indicates the crosslinking efficiency at that position.
The same pattern was observed, regardless of the length of the spacer arm between the photoreactive probe and the peptide backbone (~6 or ~12 Å). As mentioned earlier, the M domain contains a hydrophobic cavity lined with Met side chains. The flexibility of the Met side chains lining the proposed signal peptide-binding site53,66 has been invoked to account for the ability of SRP to bind a wide variety of signal peptides with different primary structures. The fact that the benzophenone groups separated from the peptide backbone by spacer arms of different lengths can still bind to the M domain and generate the same crosslinking pattern can similarly be explained in terms of the plasticity of the binding site conferred by the Met side chains.
We conclude from our findings that both the M and the NG domains play a role in the binding of signal peptides by SRP. The M domain of Ffh contains a hydrophobic groove lined with Met side-chains, providing a sequestered, malleable signal peptide-binding site to accommodate the central hydrophobic core of the signal peptide, which is the hallmark of an SRP substrate. This pocket must also bring together more polar residues that would interact with polar ends of the peptide. We speculate that the interactions between the signal peptide and the NG domain can be captured using PICUP because of the spatial requirements of the reaction: only short-distance interaction between the side chains of interacting peptides/proteins that are close to each other can be observed by this method.62,63 Also, crosslinking by PICUP only happens between groups with the appropriate reactivity, and the reaction requires very short time (≤1 s). Our observations are consistent with a model in which the NG domain interacts closely with the signal peptide, at least part of the time, emphasizing the extensive dynamics of the recognition process. We fully recognize the intrinsic limitations of cross-linking as a method to deduce structural aspects of protein–ligand interactions, and we eagerly await direct determination of the so-far elusive high resolution structures of signal peptide-SRP complexes.
THE UNIQUELY BACTERIAL SIGNAL SEQUENCE RECEPTOR: SecA
Most proteins that are destined for the extracytoplasmic environment in bacteria utilize the so-called Sec pathway for targeting. This pathway is initiated when a signal sequence on a newly synthesized precursor protein is recognized by SecA, a protein that occurs only in prokaryotes and organelles of prokaryotic origin, such as the chloroplast. Like SRP, SecA has the capacity to selectively bind signal peptides based on their common features, despite their variability in primary sequence. Unlike SRP, SecA carries out critical steps in protein targeting beyond recognition of the “zipcode” for secretion, including acting as a processive ATP-driven motor to facilitate translocation of the preprotein across the membrane.67 Our understanding of signal sequence recognition and mechanistic aspects of preprotein translocation by SecA has been greatly enhanced through the use of synthetic signal peptides. In this section, we review how synthetic signal peptides have been exploited in a wide range of biochemical and biophysical assays to begin to understand these multiple roles of SecA as a gateway to secretion and as a preprotein translocase.
SecA is Central to the Bacterial Sec Pathway
After their translation and release from the ribosome, most secretory preproteins destined for the Sec pathway are bound by the molecular chaperone SecB via their mature regions to keep them in a translocationally component state (Figure 1).68 This SecB-preprotein complex is targeted to SecA, a 102-kDa protein that recognizes and binds SecB, the N-terminal signal sequence, and a mature part of the preprotein. The new ternary complex docks to the SecYEG translocon at the membrane via SecA-SecY interactions as well as SecA-membrane insertion, and SecB is released. Harnessing the energy of ATP hydrolysis and the proton-motive force, SecA undergoes dramatic conformational changes and translocates the preprotein across the inner membrane.67,68
Performing its many functions requires SecA to undergo substantial conformational gymnastics. Studies from a variety of groups over the past decade have revealed that SecA is a multidomain protein that can exist as a monomer or a dimer, and that its domain rearrangements can be triggered by binding to any of its several ligands.67,69–81 The key to the conformational rearrangements appears to be intramolecular interactions that stabilize a more tightly packed, “closed” form of the protein. Weakening these interactions relieves these intramolecular constraints, and the protein domains partially dissociate, creating a less tightly packed, “open” form of the protein. These conformational rearrangements convert SecA from a water-soluble dimeric protein to an integral membrane protein, whose oligomeric state is as yet unclear despite extensive effort.82
The complex overall architecture of the cytoplasmic form of SecA, which represents the closed form, was first revealed at atomic resolution in a crystal structure of Bacillus subtilis SecA reported by Hunt et al.81 The structure showed the unique multiple domain organization of SecA (Figure 4A). The motor domains of SecA (termed NBF in the figure) share homology with the super family II DEAD helicases,83 which bind nucleotide at the interface between the two motor domains.81 Subsequently, several other dimeric crystal structures from various bacteria have been reported.84–87 All of these structures have a similar monomer architecture, but their dimer interfaces are different. It is a formal possibility that SecA may make several different functional dimer interfaces, or, more likely, some of the dimer interfaces seen in crystals are artifacts of crystal packing.
FIGURE 4.
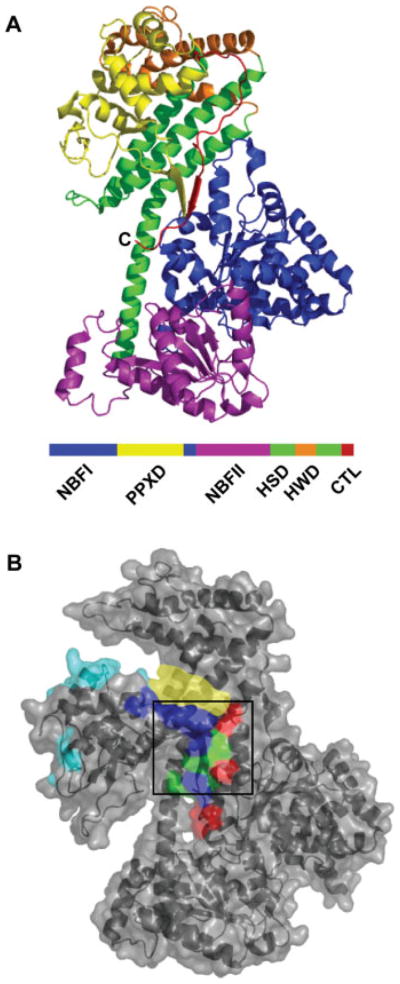
Crystal structures of cytoplasmic SecA. A. One monomer of the dimeric crystal structure of B. subtilis SecA81 (PDB: 1M6N) with the domains color-coded: blue, first nucleotide binding fold (NBFI); yellow, preprotein crosslinking domain (PPXD); purple, second nucleotide binding fold (NBFII); green, α-helical scaffold domain (HSD); orange, α-helical wing domain (HWD); and red, carboxy-terminal linker (CTL). The C-terminus is indicated (C). B. Surface representation and cartoon structure of the monomeric form of B. subtilis SecA91 (PDB: 1TF5) color-coded for the different sites proposed for signal sequence binding: red, Chou and Gierasch, 200592; blue, Papanikou et al., 200589; green, overlap between the latter two sites; cyan, Musial-Siwek et al., 200793; yellow, Osborne et al., 2004.91 The black box is the CTL Groove proposed as a possible binding site by Hunt et al., 2002.81 The last 60 residues of SecA (from the C-terminus) were not resolved in this structure. All images were created using PyMOL.
Unfortunately, none of the available SecA crystal structures provides insight into the open form, which has undergone the domain rearrangements that occur during preprotein translocation. Early work by several laboratories including our own showed that disruption or removal of the C-terminal domain of SecA led to elevation of its ATPase activity to levels near to those associated with translocation, increased signal sequence binding, and dissociation of the other domains.72,74,75,88 In parallel, this C-terminal region was shown to be the region inserted into the membrane as SecA became a membrane-resident protein.75,76,79,80 Thus, the picture that emerges is that the ligand-mediated triggering of domain dissociation leads to insertion of SecA into the membrane and activation of its translocase function. Economou’s group69,89 pointed to a specific region near the C-terminus, residues 783–795, that serves as an intramolecular regulator of ATPase activity (IRA1), and proposed that ligands (or deletion) caused the release of this IRA inhibition. They also identified another region that had synergistic effects, and termed it IRA2 and the first segment IRA1.69,70 IRA2, in fact, is the second helicase domain, which was clear once the crystal structure was available.
The tendency to shift to the open conformation can also be enhanced by temperature, supporting the idea that there are intramolecular interactions whose disruption leads to domain dissociation. Hunt et al.81 characterized this endothermic transition, showing intriguingly that mutations that lower the transition temperature also confer azide resistance on the host and show protein localization defects. A recent elegant NMR and calorimetry study of full-length and truncated SecA constructs revealed the domain dissociation upon removal of the C-terminal domain and pointed to the ways the remaining domains underwent increased dynamics in the open form.71,90
SecA-Signal Sequence Recognition
The key initial step in the SecA translocation cycle is the recognition and binding of the signal sequence. An early study used crosslinking of reconstituted SecA fragments and in vitro translated preproteins to identify residues 267–340 as the preprotein crosslinking domain (referred to as PPXD in Figure 4A).94 This region was proposed to be responsible for signal sequence binding. Today, over 15 years later, the exact signal sequence binding site on SecA still remains poorly understood despite numerous biophysical and biochemical experiments (discussed later).
The issue of signal sequence recognition by SecA is confounded by the major conformational changes undergone by SecA and the lack of clarity about which form of SecA binds the precursor protein. Synthesizing all of the literature to date suggests that the cytoplasmic form of SecA may form an initial complex with the precursor protein, but the open form must bind and deliver the signal sequence to the membrane export machinery. SecA translocase function likely requires several variations on the open, membrane-resident form, and during these steps the signal sequence must be released.
In order to understand the structural aspects of signal sequence recognition by cytoplasmic SecA, we employed NMR techniques including line broadening and trNOE to determine the structure and important binding residues of a solubility-enhanced variant of the LamB signal peptide (KRRLamB, sequence: MMITLRKRRKLPLAVAVAAGVMSAQAMA) bound to SecA.92 The observed trNOEs showed that the bound signal peptide adopts an α-helical structure in the h- and c-regions, and differential line broadening argued that one side of the helix in the h-region is more strongly bound to SecA (Figure 5). Additionally, the positive n-region and the hydrophobic h-region of the signal peptide were intimately involved in binding. These results suggested that the initial signal sequence-binding pocket on SecA diplays both electrostatic and hydrophobic character.
FIGURE 5.

Effects of SecA online-broadening of 1D proton NMR spectra of the KRR-LamB signal peptide. Selected regions of 1D 1H NMR spectra of 1 mM KRR-LamB signal peptide alone, signal peptide with 10 μM SecA, and signal peptide with 20 μM SecA. Note that the Leu and Val side chain resonances (~1.0 ppm) broaden appreciably, as do those of Arg6, while those of Lys7 do not (from Chou and Gierasch, 200592).
Considerable effort has been expended to determine where on soluble SecA the signal sequence binds (see Figure 4B for the various proposals depicted on a structure of SecA). From their crystal structure, Hunt et al. identified a region near the site where the C-terminal region (termed CTL in Figure 4A) docks onto the junction of the helicase domains and the helical scaffold domain (HSD in Figure 4A) as the most likely signal sequence binding site. In contrast, Rapoport and coworkers91 proposed as a potential binding site the cleft that opens between the PPXD and the C-terminal domain in the monomeric SecA crystal form they solved. Using fragments, deletional analysis, and chemical crosslinking of synthetic signal peptides, Economou’s group concluded that the region at the base of the PPXD (so-called stem region) serves to bind the signal sequence.69,89 Results of a FRET-based study with complementary crosslinking approaches also implicated the PPXD (specifically the third helix) in signal sequence binding,93,95 which is consistent with the early crosslinking studies from Mizushima’s lab.94 We have performed a sequence alignment of 550 SecA proteins, and the region proposed by Musial-Siwek et al.93 has a low conservation score (J. L. Maki and L. M. Gierasch, unpublished observations), raising doubt about whether it serves as a direct binding site for signal sequences. Taken together, it seems that the central region adjacent and perhaps including the PPXD is involved in initial signal sequence recognition, but the exact location of the signal sequence-binding site on cytoplasmic SecA remains elusive.
In addition to the less-than-clearcut picture of signal sequence binding by cytoplasmic SecA, the major conformational change in SecA after its initial encounter with preprotein and SecB points to the importance of signal sequence recognition by the open form of SecA. Supporting this was our finding using biotin-labeled LamB signal peptides that signal peptides bind more strongly to C-terminally truncated SecA64, which has properties similar to the open translocation-active state, than to full-length SecA.72 This observation supports the existence of two binding sites: a low-affinity binding site in cytosolic SecA and a high-affinity site in the activated form. IRA1 may be acting as a molecular switch to regulate signal sequence binding,67 since its deletion also increased binding.69 The PPXD is implicated in binding to open SecA by the finding that signal peptides stabilize an isolated construct corresponding to the PPXD in a distinct conformation.89
As expected for a complex interplay of ligand-modulated conformational transitions, the binding of signal peptides shifts the closed to open equilibrium in SecA and modulates its ATPase activity. The addition of signal peptide to SecA in solution and in phospholipids/liposomes causes a change in SecA fluorescence79,96 as well as in protease sensitivity.69,97 The elevated ATPase rate of translocation-active SecA arising either from C-terminal truncation to form SecA64 or from removal of IRA1 was quenched by functional signal peptides.69,72 These results support an interdependent role of IRA1 and signal peptides during the translocation cycle.69,89
A System to Explore How Signal Peptides Bind the Open Conformation of SecA
We have followed up on the observation that low concentrations of urea stimulate SecA ATPase activity and induce a conformational change that is likely analogous to that caused by IRA1 deletion or C-terminal truncation.88 Thus, we have used the urea-activated state of SecA (which we call u-SecA) as a model for the open, translocation-active form. The advantage of u-SecA is that it is soluble in aqueous solution, unlike other activated SecAs. We have correlated the activation of SecA by urea with an array of biophysical properties and demonstrated that the peak in ATPase activity of SecA at 2.2M urea represents a conformationally well-defined intermediate state (J. L. Maki and L. M. Gierasch, manuscript in preparation). At 22°C, we see an eightfold stimulation in the ATPase activity (Figure 6A), which is comparable to the activity observed for translocation-active SecA.71 At this concentration of urea, the far-UV CD of SecA indicates about a 25% loss of the α-helical content. The urea dependence of the CD signal shows a plateau, suggesting that a stable state is populated (Figure 6B). Trp fluorescence and near-UV CD experiments confirm that SecA undergoes a conformational change to a stable state in 2.2M urea, and limited proteolysis of u-SecA and cytosolic SecA shows that u-SecA is more pro-tease sensitive (data not shown). Therefore, SecA in 2.2M urea (u-SecA) likely represents a partially unfolded intermediate of SecA in which the intramolecular constraint (IRA1, most likely) is relieved, and u-SecA can be used to explore signal sequence binding to the open form of SecA.
FIGURE 6.

Effects of low concentrations of urea on SecA activity, conformation, and signal sequence binding. A. The relative ATPase activity of SecA in low concentrations of urea determined by the malachite green/ammonium molybdate ATPase assay.98 The ATPase activity of SecA in the absence of urea is set to 100%. The standard deviation for each sample was less than 10%. B. The far-UV CD spectra of SecA measured in several different concentrations of urea. SecA is completely unfolded in 8M urea. C. Detection by ECL of limited chymotryptic digested SecA and u-SecA cross-linked to Bio-KRRLamB19C-BPM.
Signal Peptide Crosslinking to u-SecA Points to a Second Binding Site
We have employed the BPM photoactivatable crosslinking approach to determine how signal peptides interact differently with u-SecA and compare the binding mode to that in cytoplasmic SecA (J. L. Maki and L. M. Gierasch, manuscript in preparation). Biotinylated Cys-containing KRR-LamB signal peptides were modified with BPM (as described in the SRP section) and photo-crosslinked to both SecA and u-SecA. Limited chymotryptic digestion of signal peptide-crosslinked SecA yielded a strikingly different pattern for the two different forms of SecA, supporting a different mode of binding (Figure 6C). We are currently mapping these sites more precisely to learn how the initial recognition of the signal sequence is altered as SecA undergoes activation that then leads to targeting to the translocon and translocation across the inner membrane. These data will complement future determinations of high resolution structures of SecA-signal peptide complexes.
ONGOING PUZZLES AND GAPS
Despite many years of effort, the central puzzle of signal sequence recognition—how selectivity is achieved yet sequence diversity accommodated—remains unsolved, largely due to the absence of atomic resolution structural data in both the SRP and SecA systems. Promising NMR studies on SecA look likely to yield new insights imminently,71 and increasingly detailed cryoEM images57,58 may solidify the SRP story. But understanding the functional interaction of signal sequences with their receptors in both systems will require much more than these new views of initial binding: The precursor protein must translocate across the membrane and fold to its native state, and for this to happen, the signal sequence must be released at the right time and place from its receptor. Therefore, a mechanism to regulate signal sequence binding is required in both cases. Also, these proteins do not work in isolation: SRP is associated with the ribosome and the signal sequence is presented on a tethered nascent chain. Both proteins hand the precursor off to the translocon, which also binds the signal sequence. So there is much ground to cover still. But it is abundantly clear that our current knowledge has been facilitated by the ability to synthesize signal peptides in many variants and with multiple modifications, and overcoming these future challenges will be made possible by the same tools. Without the power of solid phase peptide synthesis, it is hard to imagine where this field would be.
Acknowledgments
We thank Joanna Swain and Danny Schnell for critical reading of the manuscript.
Contract grant sponsor: NIH
Contract grant number: GM034962
References
- 1.Blobel G. Chembiochem. 2000;1:86–102. doi: 10.1002/1439-7633(20000818)1:2<86::AID-CBIC86>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 2.Briggs MS, Gierasch LM. Adv Protein Chem. 1986;38:109–180. doi: 10.1016/s0065-3233(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 3.Merrifield R. In: Nobel Lectures, Chemistry. Malmstrom B, editor. World Scientific Publishing; Singapore: 1992. pp. 1981–90. [Google Scholar]
- 4.von Heijne G. J Mol Biol. 1985;184:99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- 5.Gierasch LM. Biochemistry. 1989;28:923–930. doi: 10.1021/bi00429a001. [DOI] [PubMed] [Google Scholar]
- 6.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 7.Emr SD, Hedgpeth J, Clement JM, Silhavy TJ, Hofnung M. Nature. 1980;285:82–85. doi: 10.1038/285082a0. [DOI] [PubMed] [Google Scholar]
- 8.Emr SD, Hanley-Way S, Silhavy TJ. Cell. 1981;23:79–88. doi: 10.1016/0092-8674(81)90272-5. [DOI] [PubMed] [Google Scholar]
- 9.Benson SA, Hall MN, Silhavy TJ. Annu Rev Biochem. 1985;54:101–134. doi: 10.1146/annurev.bi.54.070185.000533. [DOI] [PubMed] [Google Scholar]
- 10.Emr SD, Silhavy TJ. Proc Natl Acad Sci USA. 1983;80:4599–4603. doi: 10.1073/pnas.80.15.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs MS, Gierasch LM. Biochemistry. 1984;23:3111–3114. doi: 10.1021/bi00309a001. [DOI] [PubMed] [Google Scholar]
- 12.Bruch MD, McKnight CJ, Gierasch LM. Biochemistry. 1989;28:8554–8561. doi: 10.1021/bi00447a043. [DOI] [PubMed] [Google Scholar]
- 13.McKnight CJ, Briggs MS, Gierasch LM. J Biol Chem. 1989;264:17293–17297. [PubMed] [Google Scholar]
- 14.Briggs MS, Gierasch LM. Biochemistry. 1991;88:5799–5803. [Google Scholar]
- 15.Hoyt DW, Gierasch LM. J Biol Chem. 1991;266:14406–14412. [PubMed] [Google Scholar]
- 16.Rizo J, Blanco FJ, Kobe B, Bruch MD, Gierasch LM. Biochemistry. 1993;32:4881–4894. doi: 10.1021/bi00069a025. [DOI] [PubMed] [Google Scholar]
- 17.Briggs MS, Gierasch LM, Zlotnick A, Lear JD, DeGrado WF. Science. 1985;228:1096–1099. doi: 10.1126/science.3158076. [DOI] [PubMed] [Google Scholar]
- 18.Briggs MS, Cornell DG, Dluhy RA, Gierasch LM. Science. 1986;233:206–208. doi: 10.1126/science.2941862. [DOI] [PubMed] [Google Scholar]
- 19.Cornell DG, Dluhy RA, Briggs MS, McKnight CJ, Gierasch LM. Biochemistry. 1989;28:2789–2797. doi: 10.1021/bi00433a008. [DOI] [PubMed] [Google Scholar]
- 20.Jones JD, McKnight CJ, Gierasch LM. J Bioenerg Biomembr. 1990;22:213–232. doi: 10.1007/BF00763166. [DOI] [PubMed] [Google Scholar]
- 21.Hoyt DW, Gierasch LM. Biochemistry. 1991;30:10155–10163. doi: 10.1021/bi00106a012. [DOI] [PubMed] [Google Scholar]
- 22.Laforet GA, Kendall DA. J Biol Chem. 1991;266:1326–1334. [PubMed] [Google Scholar]
- 23.McKnight CJ, Stradley SJ, Jones JD, Gierasch LM. Proc Natl Acad Sci USA. 1991;88:5799–5803. doi: 10.1073/pnas.88.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kajava AV, Zolov SN, Kalinin AE, Nesmeyanova MA. J Bacteriol. 2000;182:2163–2169. doi: 10.1128/jb.182.8.2163-2169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Jones JD, Rizo J, Gierasch LM. Biochemistry. 1993;32:13991–13999. doi: 10.1021/bi00213a032. [DOI] [PubMed] [Google Scholar]
- 26.Sankaram MB, Marsh D, Gierasch LM, Thompson TE. Biophys J. 1994;66:1959–1968. doi: 10.1016/S0006-3495(94)80989-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones JD, Gierasch LM. Biophys J. 1994;67:1534–1545. doi: 10.1016/S0006-3495(94)80627-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones JD, Gierasch LM. Biophys J. 1994;67:1546–1561. doi: 10.1016/S0006-3495(94)80628-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L, Tai PC, Briggs MS, Gierasch LM. J Biol Chem. 1987;262:1427–1429. [PubMed] [Google Scholar]
- 30.Miller JD, Wilhelm H, Gierasch L, Gilmore R, Walter P. Nature. 1993;366:351–354. doi: 10.1038/366351a0. [DOI] [PubMed] [Google Scholar]
- 31.Swain JF, Gierasch LM. J Biol Chem. 2001;276:12222–12227. doi: 10.1074/jbc.M011128200. [DOI] [PubMed] [Google Scholar]
- 32.Luirink J, Sinning I. Biochim Biophys Acta. 2004;1694:17–35. doi: 10.1016/j.bbamcr.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Pohlschroder M, Hartmann E, Hand NJ, Dilks K, Haddad A. Annu Rev Microbiol. 2005;59:91–111. doi: 10.1146/annurev.micro.59.030804.121353. [DOI] [PubMed] [Google Scholar]
- 34.Connolly T, Gilmore R. Cell. 1989;57:599. doi: 10.1016/0092-8674(89)90129-3. [DOI] [PubMed] [Google Scholar]
- 35.Gilmore R, Walter P, Blobel G. J Cell Biol. 1982;95:470–477. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer DI, Krause E, Dobberstein B. Nature. 1982;297:647. doi: 10.1038/297647a0. [DOI] [PubMed] [Google Scholar]
- 37.Tajima S, Lauffer L, Rath VL, Walter P. J Cell Biol. 1986;103:1167–1178. doi: 10.1083/jcb.103.4.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter P, Blobel G. J Cell Biol. 1981;91:557–561. doi: 10.1083/jcb.91.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siu FW, Spanggord RJ, Doudna JA. RNA. 2007;13:240–250. doi: 10.1261/rna.135407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spanggord RJ, Siu F, Ke A, Doudna JA. Nat Struct Mol Biol. 2005;12:1116–1122. doi: 10.1038/nsmb1025. [DOI] [PubMed] [Google Scholar]
- 41.Bradshaw N, Walter P. Mol Biol Cell. 2007 doi: 10.1091/mbc.E07-02-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hainzl T, Huang S, Sauer-Eriksson AE. Proc Natl Acad Sci USA. 2007;104:14911–14916. doi: 10.1073/pnas.0702467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freymann DM, Keenan RJ, Stroud RM, Walter P. Nature. 1997;385:361–364. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- 44.Gu SQ, Peske F, Wieden HJ, Rodnina MV, Wintermeyer W. RNA. 2003;9:566–573. doi: 10.1261/rna.2196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramirez UD, Freymann DM. Acta Crsytallogr D Biol Crystallogr. 2006;62:1520–1534. doi: 10.1107/S0907444906040807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramirez UD, Minasov G, Focia PJ, Stroud RM, Walter P, Kuhn P, Freymann DM. J Mol Biol. 2002;320:783–799. doi: 10.1016/s0022-2836(02)00476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA. Science. 2000;287:1232–1239. doi: 10.1126/science.287.5456.1232. [DOI] [PubMed] [Google Scholar]
- 48.Zheng N, Gierasch LM. Mol Cell. 1997;1:79–87. doi: 10.1016/s1097-2765(00)80009-x. [DOI] [PubMed] [Google Scholar]
- 49.Diener JL, Wilson C. Biochemistry. 2000;39:12862–12874. doi: 10.1021/bi001180s. [DOI] [PubMed] [Google Scholar]
- 50.Lentzen G, Dobberstein B, Wintermeyer W. FEBS Lett. 1994;348:233–238. doi: 10.1016/0014-5793(94)00599-0. [DOI] [PubMed] [Google Scholar]
- 51.Rosendal KR, Wild K, Montoya G, Sinning I. Proc Natl Acad Sci USA. 2003;100:14701–14706. doi: 10.1073/pnas.2436132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuelsson T, Olsson M. Nucl Acids Res. 1993;21:847–853. doi: 10.1093/nar/21.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keenan RJ, Freymann DM, Walter P, Stroud RM. Cell. 1998;94:181–191. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- 54.Cleverley RM, Zheng N, Gierasch LM. J Biol Chem. 2001;276:19327–19331. doi: 10.1074/jbc.M011130200. [DOI] [PubMed] [Google Scholar]
- 55.Lütcke H. Eur J Biochem. 1995;228:531–550. doi: 10.1111/j.1432-1033.1995.tb20293.x. [DOI] [PubMed] [Google Scholar]
- 56.Zopf D, Bernstein HD, Walter P. J Cell Biol. 1993;120:1113–1121. doi: 10.1083/jcb.120.5.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halic M, Blau M, Becker T, Mielke T, Pool MR, Wild K, Sinning I, Beckmann R. Nature. 2006;444:507–511. doi: 10.1038/nature05326. [DOI] [PubMed] [Google Scholar]
- 58.Schaffitzel C, Oswald M, Berger I, Ishikawa T, Abrahams JP, Koerten HK, Koning RI, Ban N. Nature. 2006;444:503–506. doi: 10.1038/nature05182. [DOI] [PubMed] [Google Scholar]
- 59.Newitt JA, Bernstein HD. Eur J Biochem. 1997;245:720–729. doi: 10.1111/j.1432-1033.1997.00720.x. [DOI] [PubMed] [Google Scholar]
- 60.Cleverley RM, Gierasch LM. J Biol Chem. 2002;277:46763–46768. doi: 10.1074/jbc.M207427200. [DOI] [PubMed] [Google Scholar]
- 61.Dorman G, Prestwich GD. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 62.Fancy DA, Denison C, Kim K, Xie Y, Holdeman T, Amini F, Kodadek T. Chem Biol. 2000;7:697–708. doi: 10.1016/s1074-5521(00)00020-x. [DOI] [PubMed] [Google Scholar]
- 63.Fancy DA, Kodadek T. Proc Natl Acad Sci USA. 1999;96:6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valent QA, Kendall DA, High S, Kusters R, Oudega B, Luirink J. EMBO J. 1995;14:5494–5505. doi: 10.1002/j.1460-2075.1995.tb00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lutcke H, High S, Romisch K, Ashford AJ, Dobberstein B. EMBO J. 1992;11:1543–1551. doi: 10.1002/j.1460-2075.1992.tb05199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. Nature. 1989;340:482–486. doi: 10.1038/340482a0. [DOI] [PubMed] [Google Scholar]
- 67.Vrontou E, Economou A. Biochim Biophys Acta. 2004;1694:67–80. doi: 10.1016/j.bbamcr.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 68.Driessen AJ, Manting EH, van der Does C. Nat Struct Biol. 2001;8:492–498. doi: 10.1038/88549. [DOI] [PubMed] [Google Scholar]
- 69.Baud C, Karamanou S, Sianidis G, Vrontou E, Politou AS, Economou A. J Biol Chem. 2002;277:13724–13731. doi: 10.1074/jbc.M200047200. [DOI] [PubMed] [Google Scholar]
- 70.Vrontou E, Karamanou S, Baud C, Sianidis G, Economou A. J Biol Chem. 2004;279:22490–22497. doi: 10.1074/jbc.M401008200. [DOI] [PubMed] [Google Scholar]
- 71.Keramisanou D, Biris N, Gelis I, Sianidis G, Karamanou S, Economou A, Kalodimos CG. Nat Struct Mol Biol. 2006;13:594–602. doi: 10.1038/nsmb1108. [DOI] [PubMed] [Google Scholar]
- 72.Triplett TL, Sgrignoli AR, Gao FB, Yang YB, Tai PC, Gierasch LM. J Biol Chem. 2001;276:19648–19655. doi: 10.1074/jbc.M100098200. [DOI] [PubMed] [Google Scholar]
- 73.Eichler J, Wickner W. J Bacteriol. 1998;180:5776–5779. doi: 10.1128/jb.180.21.5776-5779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eichler J, Brunner J, Wickner W. EMBO J. 1997;16:2188–2196. doi: 10.1093/emboj/16.9.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Price A, Economou A, Duong F, Wickner W. J Biol Chem. 1996;271:31580–31584. doi: 10.1074/jbc.271.49.31580. [DOI] [PubMed] [Google Scholar]
- 76.Ulbrandt ND, London E, Oliver DB. J Biol Chem. 1992;267:15184–15192. [PubMed] [Google Scholar]
- 77.Joly JC, Leonard MR, Wickner WT. Proc Natl Acad Sci USA. 1994;91:4703–4707. doi: 10.1073/pnas.91.11.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jilaveanu LB, Oliver DB. J Biol Chem. 2007;282:4661–4668. doi: 10.1074/jbc.M610828200. [DOI] [PubMed] [Google Scholar]
- 79.Ding H, Mukerji I, Oliver D. Biochemistry. 2001;40:1835–1843. doi: 10.1021/bi002058w. [DOI] [PubMed] [Google Scholar]
- 80.Ramamurthy V, Oliver D. J Biol Chem. 1997;272:23239–23246. doi: 10.1074/jbc.272.37.23239. [DOI] [PubMed] [Google Scholar]
- 81.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 82.Rusch SL, Kendall DA. Biochim Biophys Acta. 2007;1768:5–12. doi: 10.1016/j.bbamem.2006.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caruthers JM, McKay DB. Curr Opin Struct Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 84.Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, Jacobs WR, Jr, Sacchettini JC. Proc Natl Acad Sci USA. 2003;100:2243–2248. doi: 10.1073/pnas.0538077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vassylyev DG, Mori H, Vassylyeva MN, Tsukazaki T, Kimura Y, Tahirov TH, Ito K. J Mol Biol. 2006;364:248–258. doi: 10.1016/j.jmb.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 86.Zimmer J, Li W, Rapoport TA. J Mol Biol. 2006;364:259–265. doi: 10.1016/j.jmb.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 87.Papanikolau Y, Papadovasilaki M, Ravelli RB, McCarthy AA, Cusack S, Economou A, Petratos K. J Mol Biol. 2007;366:1545–1557. doi: 10.1016/j.jmb.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 88.Song M, Kim H. J Biochem (Tokyo) 1997;122:1010–1018. doi: 10.1093/oxfordjournals.jbchem.a021840. [DOI] [PubMed] [Google Scholar]
- 89.Papanikou E, Karamanou S, Baud C, Frank M, Sianidis G, Keramisanou D, Kalodimos CG, Kuhn A, Economou A. J Biol Chem. 2005;280:43209–43217. doi: 10.1074/jbc.M509990200. [DOI] [PubMed] [Google Scholar]
- 90.Cavanaugh LF, Palmer AG, III, Gierasch LM, Hunt JF. Nat Struct Mol Biol. 2006;13:566–569. doi: 10.1038/nsmb0706-566. [DOI] [PubMed] [Google Scholar]
- 91.Osborne AR, Clemons WM, Jr, Rapoport TA. Proc Natl Acad Sci USA. 2004;101:10937–10942. doi: 10.1073/pnas.0401742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chou YT, Gierasch LM. J Biol Chem. 2005;280:32753–32760. doi: 10.1074/jbc.M507532200. [DOI] [PubMed] [Google Scholar]
- 93.Musial-Siwek M, Rusch SL, Kendall DA. J Mol Biol. 2007;365:637–648. doi: 10.1016/j.jmb.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kimura E, Akita M, Matsuyama S, Mizushima S. J Biol Chem. 1991;266:6600–6606. [PubMed] [Google Scholar]
- 95.Musial-Siwek M, Rusch SL, Kendall DA. Biochemistry. 2005;44:13987–13996. doi: 10.1021/bi050882k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ahn T, Kin H. J Biol Chem. 1996;271:12372–12379. doi: 10.1074/jbc.271.21.12372. [DOI] [PubMed] [Google Scholar]
- 97.Wang L, Miller A, Kendall DA. J Biol Chem. 2000;275:10154–10159. doi: 10.1074/jbc.275.14.10154. [DOI] [PubMed] [Google Scholar]
- 98.Chan KM, Delfert D, Junger KD. Anal Biochem. 1986;157:375–380. doi: 10.1016/0003-2697(86)90640-8. [DOI] [PubMed] [Google Scholar]