

Abstract

A general solution for the synthesis of various oxetan-3-ones is developed. This reaction uses readily available propargylic alcohols as substrates and proceeds without the exclusion of moisture or air (‘open flask’). Notably, oxetan-3-one, a highly valuable substrate for drug discovery, can be prepared in one-step from propargyl alcohol in a fairly good yield. The facile formation of the strained oxetane ring provides strong support for the intermediacy of α-oxo gold carbenes. This safe and efficient generation of gold carbenes via intermolecular alkyne oxidation offers a potentially general entry into α-oxo metal carbene chemistry without using hazardous diazo ketones.
Oxetan-3-ones1 contain a strained four-membered ring and possess considerable synthetic/medicinal utility. They have been incorporated into steroid skeletons,2 used to prepare oxetanocin derivatives,3 and converted into 3-aminooxetanes4 including 3-aminooxetane-3-carboxylic acid.4a Moreover, Rogers-Evans, Carreira and co-workers have shown that the oxetane ring can serve as a surrogate of a gem-dimethyl group,5a resemble a carbonyl group,5b and offer alternatives to 1,3-heteroatom-substituted cyclohexanes in spirocyclic structures5c in drug discovery; in these studies, the parent oxetan-3-one serves as an essential starting material for introducing the oxetane ring. However, synthesis of oxetan-3-ones typically demand multiple synthetic steps and/or highly functionalized substrates.1 For example, oxetan-3-one itself was prepared in 45a or 54 steps with 23% or 13% overall yield, respectively, highlighting the challenge of constructing this strained skeleton and the lack of efficient preparative methods.
α-Diazo ketones have been used to prepare oxetan-3-ones (Scheme 1),6 but those diazo substrates are in general hazardous and their preparations are non-trivial. We have recently developed a practical and efficient synthesis of dihydrofuran-3-ones via gold-catalyzed intermolecular oxidation of terminal alkynes,7 where a C-C triple bond is converted into a reactive α-oxo gold carbene intermediate in the proposed catalytic cycle. This strategy would render alkynes equivalent to α-diazo ketones in accessing α-oxo metal carbene chemistry,8 which could offer significant synthetic and economic benefits. We speculated that this equivalency could substitute the α-diazo ketone moiety in oxetan-3-one synthesis6 with a simple C-C triple bond (Scheme 1); as a result, readily available propargylic alcohols could serve as direct substrates. Herein, we report a successful implementation of the design and the development of a practical solution to oxetan-3-one synthesis; moreover, the ease of forming this strained ring provides convincing support for the formation of α-oxo gold carbene intermediates via intermolecular alkyne oxidation.
Scheme 1.

Synthesis of Oxetan-3-Ones from Propargylic Alcohols?
Propargylic alcohol 1 was chosen as substrate for our initial study. To our delight, the expected oxetan-3-one (i.e., compound 2) was indeeded formed under the optimal reaction conditions we previously developed7 (Table 1, entry 1) and moreover, in a fairly good NMR yield. A variety of other oxidants were examined, and some results are shown in entries 2-7. 3-Methoxycarbonyl-5-bromopyridine N-oxide (4) led to a slightly improved yield (entry 7) and was chosen to screen catalysts. Among the different gold catalysts examined (entries 8-11), (2-biphenyl)Cy2PAuNTf2 gave a better result than Ph3P (entry 9). Of note, without using any gold catalyst, no oxetan-3-one 2 was observed under the acidic reaction conditions, and PtCl2 was not effective in promoting this reaction (entry 12). One of the side products in these reactions was mesylate 3, likely due to the reaction of gold carbene A with MsOH. To our delight, the use of HNTf2 minimized this side reaction, leading to a further improved yield (entry 13), and oxetan-3-one 2 was isolated in 71% yield. Little product (<5%) was observed without using acid.
Table 1.
Reaction Conditions Optimization.a

| entry | L | R | acidb | conditions | yieldc |
|---|---|---|---|---|---|
| 1 | Ph3P | 3,5-Cl2 | MsOH | DCE, rt, 6 h | 58% |
| 2 | Ph3P | 3-Br | MsOH | DCE, rt, 6 h | 50% |
| 3 | Ph3P | 4-Et | MsOH | DCE, rt, 6 h | 22% |
| 4 | Ph3P | 4-Ac | MsOH | DCE, rt, 6 h | 44% |
| 5 | Ph3P | 2-Br | MsOH | DCE, rt, 6 h | 58% |
| 6 | Ph3P | 2,4-Cl2 | MsOH | DCE, rt, 6 h | 61% |
| 7 | Ph3P | R1d | MsOH | DCE, rt, 6 h | 62% |
| 8 | IPr | R1d | MsOH | DCE, rt, 6 h | 33% |
| 9 | Le | R1d | MsOH | DCE, rt, 6 h | 66% |
| 10 | (4-CF3Ph)3P | R1d | MsOH | DCE, rt, 6 h | 53% |
| 11 | Et3P | R1d | MsOH | DCE, rt, 6 h | 47% |
| 12 | PtCl2 | R1d | MsOH | toluene, 80 °C | <5% |
| 13 | L e | R 1 d | Tf2NH | DCE, rt, 3 h | 73% f |
[1] = 0.05 M; DCE: 1, 2-dichloroethane.
1.2 equivalent.
Estimated by 1H NMR using diethyl phthalate as internal reference.
R1 = 3-MeO2C-5-Br.
(2-Biphenyl)Cy2P.
71% isolated yield.
With the optimal reaction conditions in hand, we then probed the reaction scope using various secondary propargylic alcohols as substrates. As shown in Table 2, this reaction proceeded smoothly in the presence of various functional groups. For example, remote Ph (entry 2) and vinyl (entry 3) groups were tolerated, suggesting that their reaction with the gold carbene moiety were insignificant; moreover, a Ph group at the propargylic position (entry 4) did not interfere the carbene O-H insertion to a significant extent. A MOM-protected HO group (entry 5) was compatible with the acidic reaction conditions, confirming the buffering effect of N-oxide 4. However, some deprotection of N-Boc groups was observed at room temperature; a serviceable yield (60%, entry 6) was obtained when the reaction was run at −20 °C. In addition, substrates containing an azido (entry 7) and a bromo (entry 8) groups were suitable as well.
Table 2.
Reaction Scope with Secondary Propargyl Alcoholsa 








[5] = 0.05 M.
Isolated yields.
2-Bromopyridine N-oxide and MsOH were used instead.
Temperature: −20 °C; time: 16 h.
Expansion of this oxetanone formation chemistry to tertiary propargylic alcohols was initially met with low yields likely due to increased tendency of forming propargylic cations under the acidic reaction conditions. To overcome this problem, an electron-withdrawing carboxylate group was installed to the alkyne terminus. Expectedly, the gold catalysis proceeded much slower, and mild heating was required to complete the reaction within 24 h; moreover, the combination of 4-acetylpyridine N-oxide and IPrAuNTf2 worked better. As shown in Table 3, high to excellent yields were observed with various substrates. For asymmetric substrates, low diastereoselectivities were observed (entries 2 and 3).
Table 3.
Reaction Scope With Tertiary Propargyl Alcoholsa 

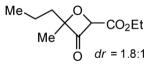
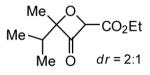





[7] = 0.1 M.
Isolated yields.
Reaction temperature: 40 °C.
Temperature: 60 °C.
The synthetic utility of this chemistry is underscored by a one-step preparation of oxetan-3-one. As discussed above, the synthesis of this highly useful O-heterocycle4,5 requires multiple steps and is low yielding. It is commercially available but expensive.9 To our delight, this volatile heterocycle was formed in 71% NMR yield directly from cheap propargyl alcohol following this chemistry, and the catalyst loading could be lowered to 1 mol % without much affecting the yield (Scheme 2); moreover, a 51% NMR yield was achievable with cheaper and less amounts of reagents and a higher substrate concentration. Coupled with a subsequent Wittig reaction5a upon simple workup or a Strecker reaction10 directly, this reaction led to oxetane derivatives 9 and 10 in 62% and 60% overall yields, respectively [5 mol % of (2-biphenyl)Cy2PAuNTf2 used].
Scheme 2.

A One-Step Synthesis of Oxetan-3-Ones and Its Synthetic Conversion without Purification.
With chiral propargylic alcohols readily available,11 this chemistry provides easy access to chiral oxetan-3-ones. For example, enantiomerically enriched alcohol 5b11c (80% e.e.) was converted into oxetan-3-one 6b (81% e.e.) with no apparent racemization detected.
In conclusion, we have developed a general solution for the synthesis of various oxetan-3-ones. This reaction uses readily available propargylic alcohols as substrates and proceeds without the exclusion of moisture or air (‘open flask’). Notably, oxetan-3-one, a highly valuable substrate for drug discovery, can be prepared in one step from propargyl alcohol in a fairly good yield. The facile formation of the strained oxetane ring provides strong support for the intermediacy of α-oxo gold carbenes. This efficient generation of gold carbenes via intermolecular alkyne oxidation would potentially offer a general entry into α-oxo metal carbene chemistry without using hazardous diazo ketones.
Supplementary Material
Acknowledgment
We thank NSF (CAREER award CHE-0969157), NIGMS (R01 GM084254) and UCSB for generous financial support.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).For examples, see: Dejaegher Y, Kuz’menok NM, Zvonok AM, De Kimpe N. Chem. Rev. 2002;102:29–60. doi: 10.1021/cr990134z.
- (2).For examples, see: Pons M, Simons SS., Jr. J. Org. Chem. 1981;46:3262–3264. Rowland AT, Bennett PJ, Shoupe TS. J. Org. Chem. 1968;33:2426–2436.
- (3).Kitagawa M, Hasegawa S, Saito S, Shimada N, Takita T. Tetrahedron Lett. 1991;32:3531–3534. [Google Scholar]
- (4)(a).Kozikowski AP, Fauq AH. Synlett. 1991;1991:783–784. [Google Scholar]; (b) Hamzik PJ, Brubaker JD. Org. Lett. 2010;12:1116–1119. doi: 10.1021/ol100119e. [DOI] [PubMed] [Google Scholar]
- (5)(a).Wuitschik G, Rogers-Evans M, Müller K, Fischer H, Wagner B, Schuler F, Polonchuk L, Carreira EM. Angew. Chem. Int. Ed. 2006;45:7736–7739. doi: 10.1002/anie.200602343. [DOI] [PubMed] [Google Scholar]; (b) Wuitschik G, Rogers-Evans M, Buckl A, Bernasconi M, Märki M, Godel T, Fischer H, Wagner B, Parrilla I, Schuler F, Schneider J, Alker A, Schweizer WB, Müller K, Carreira EM. Angew. Chem. Int. Ed. 2008;47:4512–4515. doi: 10.1002/anie.200800450. [DOI] [PubMed] [Google Scholar]; (c) Burkhard JA, Guérot C, Knust H, Rogers-Evans M, Carreira EM. Org. Lett. 2010;12:1944–1947. doi: 10.1021/ol1003302. [DOI] [PubMed] [Google Scholar]
- (6).For selected examples, see: Marshall JR, Walker J. J. Chem. Soc. 1952;467 Thijs L, Cillissen PJM, Zwanenburg B. Tetrahedron. 1992;48:9985–9990. Padwa A, Sa MM. Quimica Nova. 1999;22:815.
- (7).Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For monographs regarding metal carbene chemistry, see: Dörwald FZ. Metal Carbenes in Organic Synthesis. Wiley-VCH; Weinheim; Germany: 1999. Barluenga J, Rodrïguez F, Fañanàs FJ, Flòrez J. Metal Carbenes in Organic Synthesis. In: Dötz KH, editor. Topics in Organometallic Chemistry. Vol. 13. Springer; Berlin: 2004. pp. 59–122. Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds : From Cyclopropanes to Ylides. Wiley; New York: 1998.
- (9).$220/g from Synthonix.
- (10).Mai K, Patil G. Synth. Commun. 1985;15:157–163. [Google Scholar]
- (11).For selective reviews, see: Trost BM, Weiss AH. Adv. Synth. Catal. 2009;351:963–983. doi: 10.1002/adsc.200800776. Pu L. Tetrahedron. 2003;59:9873–9886. Frantz DE, Fässler R, Tomooka CS, Carreira EM. Acc. Chem. Res. 2000;33:373–381. doi: 10.1021/ar990078o.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.