

Summary
Innate and adaptive immunity are considered critical to protection against mucosal candidal infections. Among innate anti-Candida mechanisms, oral and vaginal epithelial cells have antifungal activity. The mechanism is fungistatic, acid-labile, and includes a requirement for cell contact by intact, but not necessarily live, epithelial cells. The purpose of this study was to use the acid-labile property to further characterize the effector moiety. Surface material extracted from PBS-, but not acid-treated epithelial cells, significantly inhibited the growth of Candida blastoconidia in a dose-dependent manner which was abrogated by prior heat and protease treatment. Proteins extracted from PBS-treated cells bound blastoconidia and hyphae more intensely than those from acid-treated cells. Proteins from PBS-treated cells eluted from Candida revealed two unique bands of approximately 33 and 45 kDa compared to acid-treated cells. Mass spectrometry identified these proteins as Annexin-A1 and actin, respectively. Oral epithelial cells stained positive for Annexin-A1, but not actin. Western blots showed reduced Annexin-A1 in proteins from acid-treated epithelial cells compared to those from PBS-treated epithelial cells. Lastly, it was demonstrated that immunoprecipitation of Annexin-A1 from proteins extracted from PBS-treated oral epithelial cells results in abrogation of inhibitory activity. Taken together, these results indicate that Annexin-A1 is a strong candidate for the epithelial cell anti-Candida effector protein.
Keywords: Candida albicans, pithelial cells, Innate immunity, Oral mucosa
Introduction
Candida albicans, the causative agent in the majority of cases of mucosal candidiasis, is a fungal organism that is both a commensal of the gastrointestinal and reproductive tracts, and an opportunistic pathogen of the same mucosal tissues (Sobel, 1992; Sobel, 2002). As a commensal, C. albicans asymptomatically colonizes both oral and vaginal epithelial surfaces. Clinically, oropharyngeal candidiasis (OPC) is a significant problem in immunocompromized individuals and is extremely common during human immunodeficiency virus (HIV) infection, especially when CD4+ T cells are reduced (Klein et al., 1984; Macher, 1988). In contrast, vulvovaginal candidiasis (VVC) is common in immunocompetent, otherwise healthy women (Sobel, 1992; Sobel, 2002).
To date, host defense mechanisms against mucosal candidiasis remain poorly understood. While cell-mediated immunity by Th1-type CD4+ T cells is considered a critical host defense mechanism against mucosal C. albicans infections, innate mechanisms are considered to have protective roles as well. One example involves epithelial cells. Our laboratory has shown that epithelial cells from the oral and vaginal mucosa of humans, vaginal mucosa of nonhuman primates, and vaginal mucosa of mice inhibit the growth of C. albicans in vitro at relatively low effector to target (E:T) ratios (Steele et al., Aug. 1999; Steele et al., Sept. 1999; Fidel et al., 2000; Steele et al., 2000). Additionally, HIV-infected persons with OPC and women with recurrent VVC have been shown to have reduced oral and vaginal epithelial cell anti-Candida activity (Barousse et al., 2001; Steele et al., 2000), respectively, providing clinical evidence that epithelial cells represent a protective innate host immune defense mechanism against C. albicans infections at the oral and vaginal mucosa.
Studies on the properties of the epithelial cell anti-Candida activity demonstrated that both oral and vaginal epithelial cells have a strict requirement for cell contact with C. albicans with no demonstrable role for soluble factors (Steele et al., 1999; Steele et al., 2000). In addition, epithelial cell anti-Candida activity is sensitive to heat and detergents, but resistant to fixation and irradiation, is not mediated by phagocytosis, oxidative or nonoxidative mechanisms such as defensins or calprotectins, and is fungistatic, not fungicidal (Nomanbhoy et al., 2002). Studies to identify the effector moiety demonstrated that the activity was sensitive to treatment with periodic acid as part of an acid-labile mechanism (Yano et al., 2005). The purpose of this study was to use the acid-labile property to further characterize the effector moiety and identify possible candidates for the antifungal activity.
Materials and Methods
Human subjects
Oral epithelial cells were obtained exclusively from healthy volunteers. Informed consent was obtained from each participant, and all procedures were conducted in accordance with the guidelines of the Institutional Review Board at Louisiana State University Health Sciences Center.
Oral epithelial cell isolation
Oral epithelial cells were isolated as previously described (Steele et al., Aug. 1999; Steele et al., 2001). While gently scraping the epithelium by teething action, 10–15 ml of unstimulated saliva from each participant was expectorated into a polypropylene centrifuge tube and centrifuged at 3000 rpm for 5 min. The cell pellet was washed and resuspended with sterile Hanks’ Balanced Salt Solution (HBSS) (Life Techonologies, Carlsband, CA), and passed over a 20μm nylon membrane (Small parts Inc., Miami Lakes, FL). The epithelial cell-enriched population collected from the membrane was washed, resuspended in cryopreservative solution (50% FBS, 25% RPMI 1640 tissue culture medium, 15% dimethyl sulfoxide), and stored at − 70°C until use. At the time of use, the cells were thawed, washed twice in PBS, and enumerated by Trypan blue dye exclusion. Viability was consistently 60–85% before and after thawing.
Target cells
C. albicans 3153A from the National Collection of Pathogenic Fungi (London, UK) was grown on Sabouraud dextrose agar (Becton Dickinson, Sparks, MD) at 34°C. One colony was used to incubate 10 ml of phytone-peptone (PP) broth (Becton Dickinson) supplemented with 0.1% glucose for 18 h at 25°C in a shaking water bath. The blastoconidia were collected, washed with PBS, and enumerated on a hemacytometer using Trypan blue dye exclusion. Hyphae were produced as previously described (Steele et al., 2000). Briefly, the blastoconidia were resuspended in PP broth containing 10% FBS and 1% penicillin, incubated for 3 h at 37°C in 5% CO2 and washed with PBS.
Growth inhibition
[3H]-glucose uptake
The growth inhibition assay was performed as previously described (Steele et al., Aug. 1999; Steele et al., 2000; Steele et al., 2001). Briefly, stationary-phase blastoconidia were added to individual wells of a 96-well microtitier plate (Costar, Cambridge, MA) at 105 cells/ml in a volume of 100 μl of PP broth supplemented with 10% FBS and 1% Penicillin-streptomycin. Epithelial cells were added to triplicate wells in a volume of 100 μl of PP broth at an effector to target (E:T) ratio of 5:1, which was found to be optimal for this assay (Steele et al., 2000). Controls included effector cells and target cells cultured alone. The cultures were incubated for 9 h at 37°C in 5% CO2 in the presence of 1 μCi [3H]-glucose (ICN, Costa Mesa, CA) during which Candida transformed largely into hyphal forms (Steele et al., 2000). Following the incubation, 100 μl of sodium hypochlorite solution (bleach) was added to all wells and left for 5 min, and the cell extracts were harvested onto glass fiber filters using a PHD cell harvester (Cambridge technologies, Watertown, MA). The incorporated [3H]-glucose was measured by liquid scintillation. The incorporation of glucose by Candida blastoconidia/hyphae and epithelial cells during the 9 h incubation was generally 15,000–30,000 cpm and 500–2,000 cpm, respectively. The percent growth inhibition was calculated as follows: % growth inhibition = 1 [(mean experimental cpm mean effector cpm)/mean Candida cpm] × 100.
For experiments requiring restriction of the yeast form of Candida, C. albicans 3153A that spontaneously mutated and lacks the ability to form hyphae (3153Ab) was used. The yeast was maintained on Sabouraud Dextrose agar, and the blastoconidia were prepared in the same manner as 3153A. The standard [3H]-glucose uptake assay using 3153A and 3153Ab was conducted in parallel, and similar levels of [3H]-glucose uptake were confirmed. Cocultures were examined under a microscope to confirm the blastoconidia morphology before harvesting. Hyphal forms of Candida were prepared as described above, by incubating blastoconidia at 1×105 cells/ml (concentration used in the co-culture assay) in PP broth with 10% FBS and 1% penicillin and streptomycin for 3 h at 37 °C prior to the coculture with epithelial cells. Following incubation and confirmation of the hyphal form by microscopy, hyphae were harvested, washed, and equal volumes used in the standard [3H]-glucose uptake assay as described above.
Quantitative plate count
Experiments involving periodic acid-treated epithelial cells require a quantitative plate count method to measure the growth inhibition of C. albicans due to high constitutive [3H]-glucose uptake by acid-treated epithelial cells (Steele et al., Aug. 1999; Steele et al., 2000; Steele et al., 2001). Briefly, effector and target cell cultures were prepared as described above in the absence of [3H]-glucose. Following the 9 h incubation, 100 μl of 0.3% Triton X-100 (Sigma) was added to each well, and adherent Candida cells were scraped from the bottom surface of the wells with a pipette tip. The contents of each well were serially diluted (1:10) and plated on Sabouraud dextrose agar. CFU counts were determined after 48 h incubation at 34°C. Wells containing C. albicans alone were included as controls. The percent growth inhibition was calculated as follows: % growth inhibition = (1 experimental CFU/Candida CFU) × 100.
Soluble Protein Analyses
Epithelial cell treatment
Epithelial cells were pretreated with periodic acid (5 mM in PBS) (Fisher Scientific, Fair Lawn, NJ) for 15 min at 37°C. Controls included epithelial cells incubated in PBS alone. Following incubation, the cells were washed with PBS and cell viability was assessed by Trypan blue dye exclusion. Viability was consistently 60–85% following treatment. Viable cells (5×105 cells/ml) were used in the quantitative plate count assay to measure growth inhibition activity.
Extraction of protein moieties on epithelial cells
PBS- or acid-treated epithelial cells (107 cells/ml) were transferred into 2 ml microcentrifuge tubes, and the surface proteins were biotinylated by incubating the cells with EZ-Link®Sulfo- NHS-LC-Biotin (1 mg/ml in PBS supplemented with 1 mM magnesium chloride [MgCl2] and 0.1 mM calcium chloride [CaCl2]) (Pierce, Rockford, IL) for 30 min at 4°C on a nutator. Following the incubation, the cells were washed three times with PBS supplemented with 100 mM glycine, and resuspended in 500 μl of sterile lysis buffer [50 mM trimethylol aminomethane (Tris; Bio-Rad, Hercules, CA), 5 mM EGTA tetrasodium (Sigma), 150 mM sodium chloride (NaCl), 1% Triton X (Sigma), 1 μg/ml protease inhibitors cocktail (Sigma), 5 μl/ml phenylmethanesulfonyl fluoride (Sigma)] supplemented with 60 mM octyl β-D-glucopyranoside (OBG) (Sigma). The cells were vigorously shaken by a pellet pestle motor for 30 seconds, incubated on ice for 10 min, and then centrifuged at 14000 rpm for 15 min. The supernatants containing cell surface proteins were removed from the cell pellets and collected into 5 ml Falcon tubes. This procedure was repeated two times, and the resulting supernatants were pooled. The proteins from PBS- and acid-treated epithelial cells were dialyzed against PBS (Slide-A-lyzer Dialysis cassette, 3500 molecular weight cutoff; Pierce) overnight at 4°C, and the total protein concentrations were measured. Briefly, BCA protein assay reagents (Pierce) were added to undiluted and diluted proteins and standards (2 mg/ml bovine serum albumin serially diluted 1:2) and incubated for 30 min at 65°C. The absorbance values were determined at 595 nm by a Multiskan Ascent photometric plate reader (Labsystems, Franklin, MA).
Growth inhibition by protein moieties
Blastoconidia (105 cells/ml) were incubated with dialyzed proteins from epithelial cells treated with PBS or acid as described above. Candida was directly added to the wells containing 100 μl of extracted proteins at ~250 μg/ml in the standard [3H]-glucose uptake assay. In addition, proteins were either denatured by heat (incubated for 1 h at 95°C) or digested with proteases (50 units/ml, incubated 3 h at 37°C and dialyzed against PBS overnight), and the growth inhibition assay was performed in parallel. Controls included Candida incubated with vehicles (PBS or PP broth) and fresh epithelial cells.
Binding assay
Blastoconidia and hyphae were incubated with 100 μg/ml of biotinylated proteins from PBS-or acid-treated epithelial cells for 30 min at 37°C on a nutator. Candida was washed three times with PBS and cytospun on glass slides coated with Vectabond Reagent (Vector, Burlingame, CA). The slides were fixed in chilled acetone for 5 min, washed with PBS, and incubated with R.T.U. Horseradish Peroxidase Streptavidin (Vector) for 30 min at room temperature. The slides were washed three times with PBS and incubated with 3-amino-9-ethylcarbazole (AEC Chromogen) supplied in HRP-AEC Cell and Tissue Staining Kit (R&D Systems) for 3 min or until the proper intensity of staining emerged. The slides were washed with distilled water for 5 min, dried and mounted with Aqueous Mounting Medium (R&D Systems). Controls included Candida incubated with dialyzed lysis buffer alone.
Slides were examined under a light microscope (Nikon Instruments, Melville, NY) for morphometric analysis to measure staining intensity. Five areas at 10x magnification (~37,000 μm2/each) per slide were identified in areas of positive staining. These areas were gated, and the stained areas were marked (“painted”) using Metavue Software (Universal Imaging Corp., Downington, PA). For each slide, staining intensity was calculated as a percent threshold per multiple units of area.
Western blots
Proteins extracted from PBS- or acid-treated epithelial cells were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blots were conducted. Briefly, the proteins and molecular weight standards were first prepared for SDS-PAGE with Laemmli sample buffer (Bio-Rad) supplemented with β-mercaptoethanol. Following heating for 5 min at 95°C, proteins and the standards were loaded onto 10% polyacrylamide gels (Bio-Rad) in Tris/glycine/SDS buffer (Bio-Rad) under constant current of 20 mA. Following electrophoresis, the proteins and the standards from the SDS-PAGE were transferred to a nitrocellulose membrane at 100 V for 1 h in tris-glycine transfer buffer with 20% methanol. The membrane was washed two times with PBS with 0.1% Tween. The signal was amplified using the Western Blot Amplification Module as per manufacturer’s instructions (Bio-Rad). Finally, the blotted biotinylated proteins were visualized by Opti-4CN substrate (Bio-Rad). The images of protein expression were obtained using a MultiImage Light Cabinet (Alpha Innotech, San Leandro, CA).
In addition, western blots were performed on proteins eluted from Candida following the binding assay. For this, proteins extracted from PBS- or acid-treated epithelial cells were incubated with blastoconidia or hyphae for 30 min at 37°C on a nutator as described above. After washing, both forms of Candida were resuspended with elution buffer (7 M urea, 2 M thiourea, 4% CHAPS, 2% OBG and 20% glycerol) for 2 h at room temperature with frequent agitation. Following incubation, Candida was centrifuged for 15 min at 14000 rpm. Eluted proteins were collected. Controls included Candida incubated with buffer alone and subjected to the same elution protocol.
Mass spectrometry
Proteins extracted from PBS- or acid-treated epithelial cells were separated by SDS-PAGE and visualized by Coomassie blue (Bio-Rad). Areas on the gel where protein bands of interest were observed (unique to PBS-treatment and not present with acid treatment) were excised and prepared for matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) analysis. The 5 most abundant peaks were used to query all entries of the NCBI-nr protein database using the Mascot search program (Matrix Sciences, London, United Kingdom). The Mascot uses a Probability Based Mowse (molecular weight search) score. The protein integrity score is represented as (−10) × Log(P), where P is the probability that the observed match is a random event. A match is considered a significant, non-random event if the protein integrity score is higher than 75 (P<0.05) and the percent confidence interval is higher than 99.7.
Following mass spectrometry, western blots were performed on proteins from PBS- and acid-treated cells to confirm the presence/absence of Annexin-A1. Recombinant human Annexin-A1 protein (10 ng; R&D Systems) was used as a positive control. Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane as described above. The membrane was washed with PBS with 0.1% Tween and blocked in 3% nonfat milk for 1 h on an orbital shaker. After additional washes, the membrane was incubated overnight at 4°C on an orbital shaker with polyclonal goat anti-human Annexin-A1 antibody (0.2 μg/ml; R&D Systems) in 2% nonfat milk. The membrane was washed and incubated with biotinylated anti-goat immunoglobulin G (IgG; 0.05 μg/ml; R&D Systems) in PBS with 0.1% Tween and 1% BSA for 1 h at 25°C on an orbital shaker. After washing, signal was amplified with Western Blot Amplification Module and visualized by Opti-4CN substrate (Bio-Rad). The images of protein expression were obtained using a MultiImage Light Cabinet (Alpha Innotech).
Immunohistochemical Staining
Oral epithelial cells were cytospun onto glass slides fixed with Vectabond Reagent (Vector). Slides were fixed in chilled acetone for 5 min and rehydrated (PBS) for 5 min. The endogenous peroxidase activity was blocked by incubating the sections in 3% hydrogen peroxide (peroxidase blocking reagent; R&D Systems, Minneapolis, MN.) for 5 min. After washing with PBS, nonspecific protein-binding sites in the tissue were blocked by incubating the tissue in normal rabbit serum (R&D Systems) for 15 min. This was followed by the addition of two additional blocking reagents, avidin-0.1% sodium azide (avidin blocking reagent) (R&D Systems) and biotin-0.1% sodium azide (biotin blocking reagent) (R&D Systems) (each for 15 minutes) with a PBS wash in between. Following blocking, the sections were washed with PBS and incubated for 1 h at 37°C with monoclonal rabbit anti-human Annexin-A1 or actin antibodies (10 μg/ml; Abcam, Cambridge, MA). Negative controls consisted of cells incubated with isotype-specific, purified rabbit immunoglobulin (Dako Corp., Cambridge, MA). After incubation with primary antibodies, the slides were washed in PBS and incubated with a biotinylated anti-rabbit immunoglobulin G (IgG; 10 μg/ml; R&D Systems) for 30 min. Next, the sections were washed and incubated for 30 min with high-sensitivity streptavidin-horseradish peroxidase conjugate (R&D Systems). To washed sections, the substrate 3-amino-9-ethylcarbazole chromogen (AEC) (R&D Systems) was added for 5 to 10 min. Slides were examined under a light microscope (Nikon Instruments).
Immunoprecipitation
Annexin-A1 was immunoprecipitated from protein extracts of PBS-treated cells using the Immunoprecipitation Kit-Dynabeads® Protein G (Invitrogen, Carlsbad, CA). To conjugate the antibody to the beads, polyclonal goat anti-human Annexin-A1 antibody (10 μg; R&D Systems, Minneapolis, MN) was incubated for 10 minutes at 25 °C on a nutator with 1.5 mg (50 μl) of magnetic beads in 200 μl of buffer provided in the kit. Following conjugation, the complex was resuspended in buffer and washed by removing the supernatant after the tube was exposed to the MPC®-E Magnetic Particle Concentrator magnet (Dynal, Oslo, Norway). For immunoprecipitation, 500 ul of proteins extracted from PBS-treated cells were added to the tube containing the antibody conjugated beads and incubated for 10 min at 25 °C on a nutator. The supernatant was removed after exposure to the magnet and stored at 4 °C for further testing. The bead-antibody-protein complex was washed three times in buffer as described above and transferred to a fresh tube. The antibody-protein complex was eluted from the beads by the addition of 20 ul of elution buffer as per manufacturer’s instructions. The suspension was incubated for 2 min at 25 °C on a nutator to dissociate the complex. After incubation, the supernatant containing eluted antibody and protein was removed while exposing the suspension to the magnet. The supernatant was stored at 4 °C prior to use.
Western blots were conducted on protein extracts before and after immunoprecipitation as described above using polyclonal goat anti-human Annexin-A1 antibody (0.2 μg/ml; R&D Systems) and recombinant human Annexin-A1 (10 ng; R&D Systems) as a positive control. The eluted protein-antibody complex was also included to confirm immunoprecipitation of Annexin-A1. Lastly, protein extracts were tested before and after immunoprecipitation in the standard growth inhibition assay.
Statistical analysis
The unpaired Student’s t-test was used to analyze data. Significant differences were defined at a confidence level where the P-value was <0.05. All statistics were evaluated using Prism Software (Graph Pad, San Diego, CA).
Results
Effect of oral epithelial cell surface proteins on anti-Candida activity
Based on the facts that epithelial cells mediate their inhibitory activity against Candida through an acid-labile property of the cell surface moiety and that the activity does not require live epithelial cells, we first sought to determine whether the extracted soluble form of the cell surface protein moiety could inhibit C. albicans growth. Results in Fig. 1A show that cell surface proteins extracted from PBS-treated epithelial cells exhibited nearly equivalent level of antifungal activity to intact epithelial cells (P>0.05), while the activity of the same amount of proteins extracted from acid-treated cells was significantly abrogated (P=0.0019). Fresh epithelial cells confirmed the acid-labile property (68.0 ± 12.4 % for PBS treatment vs. 8.8 ± 4.2 % inhibition for acid treatment) (data not shown). In addition, the surface proteins extracted from PBS-treated cells demonstrated antifungal activity in a dose dependent manner, whereas the surface proteins from acid-treated cells showed negligible antifungal activity at all concentrations (Fig. 1B).
Figure 1. Anti-Candida activity of oral epithelial cell surface proteins.
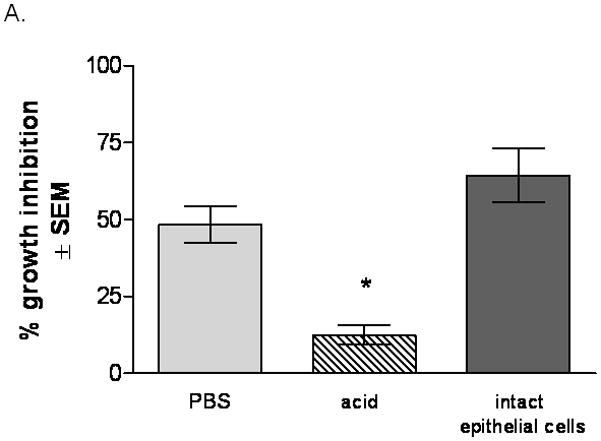
(A) Cell surface proteins from PBS- and acid-treated cells were extracted and placed with Candida in the growth inhibition assay. The standard epithelial cell-Candida coculture was included as a positive control Results are cumulative of 4 experiments (B) Proteins extracted from PBS- and acid-treated cells were placed in a dose response manner with Candida based on total protein. Figure represents cumulative results for 4 repeats. Results are expressed as % growth inhibition of Candida by oral epithelial cell proteins ± SEM, standard error of the mean.
Confirmation of the anti-Candida activity by a protein moiety
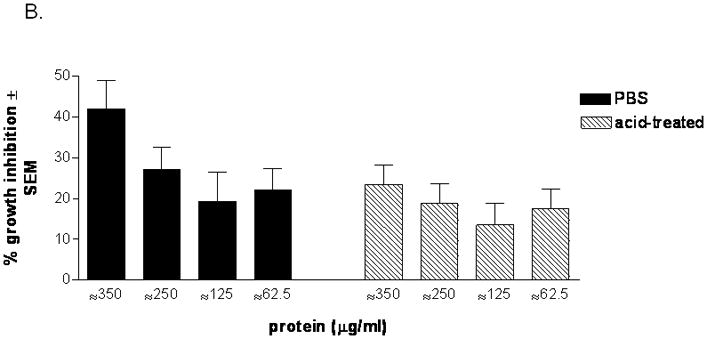
In order to confirm whether the effector moiety is of protein origin, extracted proteins from PBS-treated cells were denatured by heat or digested with proteases prior to the growth inhibition assay. Untreated proteins were included as a positive control. The results in Fig. 2 show that the anti-Candida activity was significantly abrogated by protease treatment (P=0.047). Heat treatment also reduced the activity, but statistical significance was not attained (P=0.13).
Figure 2. Confirmation of the effector moiety as a protein.
Extracted proteins from PBS-treated cells were denatured by heat or digested with proteases prior to the growth inhibition assay. Untreated proteins were included as a positive control. Results are expressed as % growth inhibition of Candida by oral epithelial cell proteins ± SEM, standard error of the mean, for 5 experiments. An asterisk represents a significant reduction in anti-Candida activity (P<0.05) compared to activity prior to treatment.
Epithelial cell anti-Candida activity against blastoconidia and hyphae
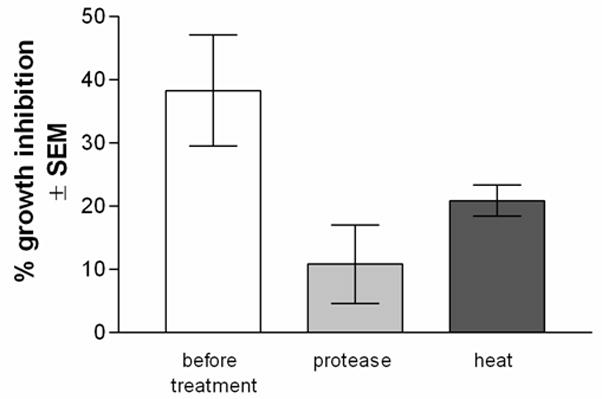
In order to conduct future binding studies of extracted proteins with Candida yeast and hyphae, it was first important to confirm that the abrogation of the anti-Candida activity by acid-treated epithelial cells could occur with both morphological forms of Candida. The results in Fig. 3 show that, compared to buffer-treated cells, the growth inhibition activity of acid-treated epithelial cells was reduced against both blastoconidia and hyphal forms of Candida when initiated and held in each respective state for the duration of the assay (P=0.002 and P=0.004, respectively). These levels of activity were similar to that for the standard assay where blastoconidia transform into hyphae during incubation.
Figure 3. Epithelial cell anti-Candida activity against blastoconidia and hyphae.
Growth inhibition of both morphological forms of Candida by PBS- and acid-treated epithelial cells was evaluated. Results are expressed as % growth inhibition of Candida by oral epithelial cell proteins ± SEM, standard error of the mean, for 5 experiments.
Binding of cell surface proteins on Candida blastoconidia and hyphae
To address the physical interaction between the cell surface proteins and Candida, biotin was added to PBS- and acid-treated epithelial cells prior to extraction. Equal amounts of biotinylated extracted proteins were incubated with Candida blastoconidia and hyphae. Proteins bound to Candida were then visualized by the addition of streptavidin-horseradish peroxidase and chromogen substrate. Controls included Candida incubated with dialyzed lysis buffer alone and stained in parallel. The results in Fig. 4A show that the surface proteins extracted from PBS-treated epithelial cells bound both Candida blastoconidia and hyphae more intensely than those from acid-treated cells.
Figure 4. Binding of oral epithelial cell surface proteins on Candida blastocondia and hyphae.
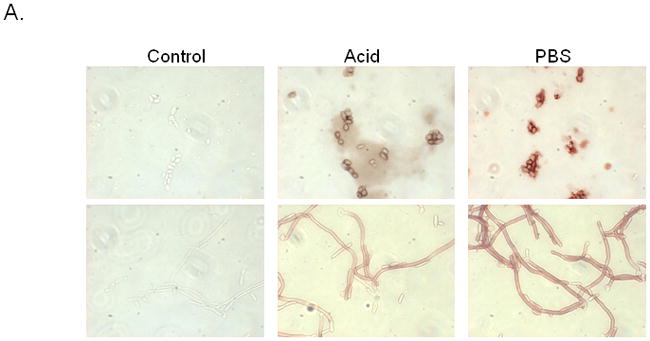
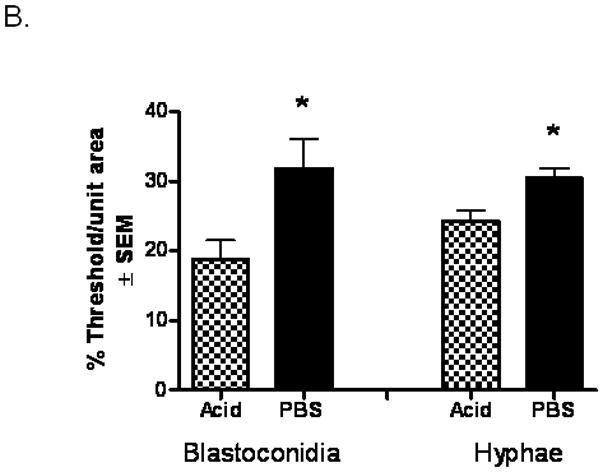
(A) PBS- and acid-treated proteins were biotinylated, extracted, and incubated with Candida blastoconidia and hyphae. Bound proteins were visualized by the addition of streptavidin-horseradish peroxidase and chromogen substrate. Controls included Candida incubated with dialyzed lysis buffer alone. (B) Morphometric analysis. A percent threshold of positive staining of bound proteins on Candida blastoconidia and hyphae per multiple units of area was quantified (37,328 μm2 ) for each image., standard error of the mean. Asterisks indicate significance. Figures shown are representative of 5 repeats.
To quantify the bound protein, morphometric analysis was performed which showed a significant decrease in staining intensity (bound protein) on blastoconidia (P=0.03) and hyphae (P=0.018) incubated with surface proteins from acid-treated epithelial cells compared to proteins from buffer-treated epithelial cells (Fig. 4B).
Visualization of anti-Candida effector protein candidates
To further evaluate the interaction between epithelial cell surface proteins and Candida, extracted biotinylated surface proteins from PBS- or acid-treated epithelial cells were bound to and eluted from Candida blastoconidia and hyphae and eluted proteins visualized by western blots. The results in Fig. 5 show that bands of approximately 45 and 33 kDa were observed in samples from PBS- but not acid-treated epithelial cells.
Figure 5. Visualization of anti-Candida effector protein candidates.
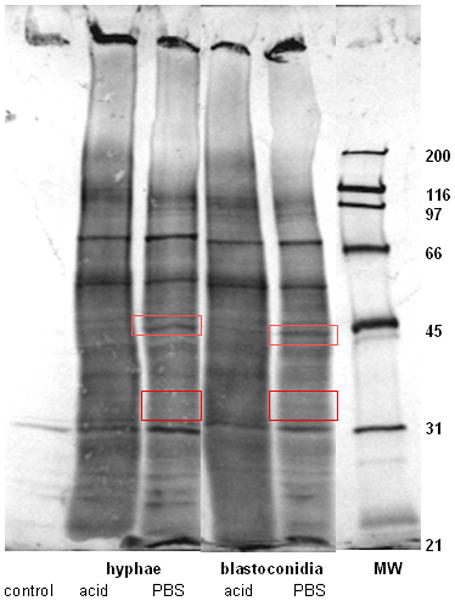
Extracted biotinylated surface proteins from PBS- and acid-treated epithelial cells were bound to and eluted from Candida blastoconidia and hyphae. Eluted proteins were visualized by western blot. Bands of interest at 33 and 45 kDa in PBS-treated proteins were boxed in this representative image of 5 repeats.
Identification of the candidate effector proteins
The two bands observed in the Western blot results were excised from SDS-PAGE preparations and analyzed by mass spectrometry. The 45 kDa protein was identified to be actin (Table 1) with a reproducible protein integrity score (78.0 ± 10) (integrity score >75 considered accurate match). Similarly, the 33 kDa protein was identified to be Annexin-A1 with the reproducible significant protein integrity scores of 97 ± 23.81. Similar regions excised from lanes containing acid-treated proteins showed no protein candidates with significant integrity scores (24 ± 2.5 and 24, respectively).
Table 1.
Protein integrity scores of excised bands from PBS-treated proteins and comparable areas in PA-treated proteins at 45 and 33 kDa.
| PBS | Acid | |||
|---|---|---|---|---|
| Protein | Integrity score | Protein | Integrity score | |
| 45 kDa | Actin, cytoplasmic 2 | 88, 68 | Protein UNQ/PRO 1567 | 27, 22 |
| Actin, alpha skeletal muscle | 51, 39 | C3orf14 | 22, 19 | |
| Coiled-coil somain-containing protein 26 | 38 | Succinyl-CoA ligase | 18 | |
| Actin, aortic smooth muscle | 35 | Transmembrane protein 126A | 16 | |
| Mast cell-expressed membrane protein-1 | 29 | V-set and transmembrane domain- containing protein 2 precursor | 14 | |
| 33 kDa | Annexin-A1 | 142, 88, 61 | Peroxisomal membrane protein 2 | 24 |
| Protein SSX3 | 22 | IL-1 receptor type I precursor | 20 | |
| Coiled-coil domain containing protein72 | 20 | Electron transfer flavoprotein subunit beta | 19 | |
| Mps one binder kinase activator-like 1A | 20 | AMSH-like protease | 19 | |
| C12orf60 | 19 | Mitochondrial proteolipid | 17 |
Annexin-A1 and Actin surface expression on oral epithelial cells
To determine whether Annexin-A1 and/or actin were present on the epithelial cell surface, oral epithelial cells were isolated from saliva as described above, cytospun onto slides, and immunohistochemically stained for Annexin-A1 or actin. Negative controls consisted of cells incubated with isotype-specific, purified rabbit immunoglobulin. Results in Fig. 6 show surface expression of Annexin-A1 on oral epithelial cells, whereas Actin surface expression was absent.
Figure 6. Annexin-A1 and Actin expression on oral epithelial cells.
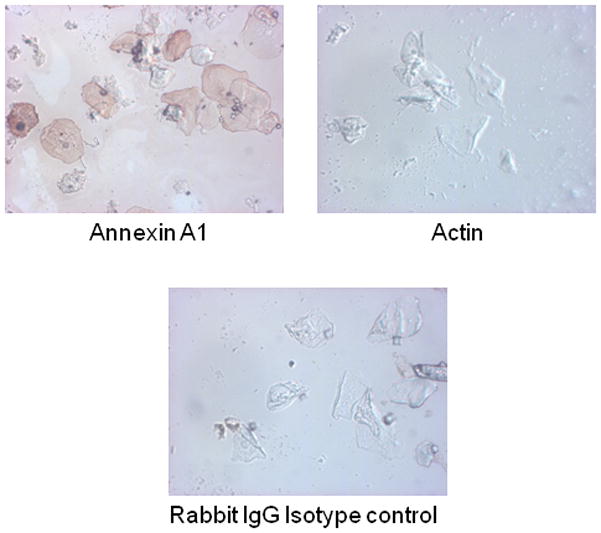
Oral epithelial cells were isolated from saliva and cytospun onto glass slides. Cells were incubated with Annexin-A1 or actin primary antibody and biotinylated secondary antibody then visualized by the addition of streptavidin-horseradish peroxidase and chromogen substrate. Controls included a Rabbit IgG isotype antibody. Magnification 10x. Figure shows representative images of 3 repeats.
Acid treatment reduces Annexin-A1 on oral epithelial cells
To confirm that acid treatment denatured/removed Annexin-A1 from the surface of oral epithelial cells, western blots were performed using proteins extracted from PBS- and acid-treated cells. Results in Figure 7 show significantly less Annexin-A1 in proteins from acid-treated cells compared to those from PBS-treated cells.
Figure 7. Reduction of Annexin-A1 in extracted proteins from acid-treated oral epithelial cells.
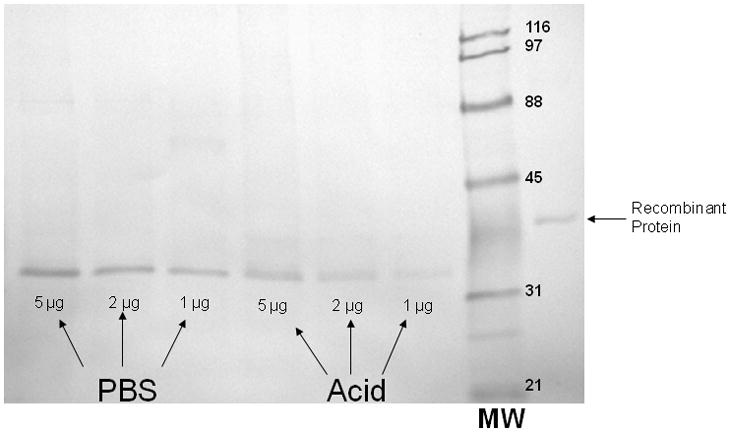
Western blots were performed using proteins extracted from PBS- and acid-treated cells in a dose response manner based on total protein. Recombinant human Annexin-A1 protein was used as a control. Figure shows a representative image of 3 repeats.
Loss of Anti-Candida activity after immunoprecipitation of Annexin-A1
Immunoprecipitation was used to examine the functional activity of Annexin-A1 in cellular protein extracts. For this, anti-human Annexin-A1 antibody-conjugated magnetic beads were employed followed by elution of the antibody-protein complex from the beads. Western blots were conducted before and after immunoprecipitation to confirm reduction/removal of Annexin-A1. Western blot results in Figure 8A show that a significant amount of Annexin-A1 was removed by immunoprecipitation. The eluted antibody-protein complex shows the presence of precipitated Annexin-A1 as well as the heavy (~45 kDa) and light (~26 kDa) chains of the goat anti-human Annexin-A1 antibody. To confirm the additional bands present were the antibody chains, an additional western blot was performed using only biotinylated anti-goat immunoglobulin G (detection) antibody. This revealed the presence of the antibody chains only (data not shown).
Figure 8. Abrogation of Anti-Candida activity following immunoprecipitation of Annexin-A1.
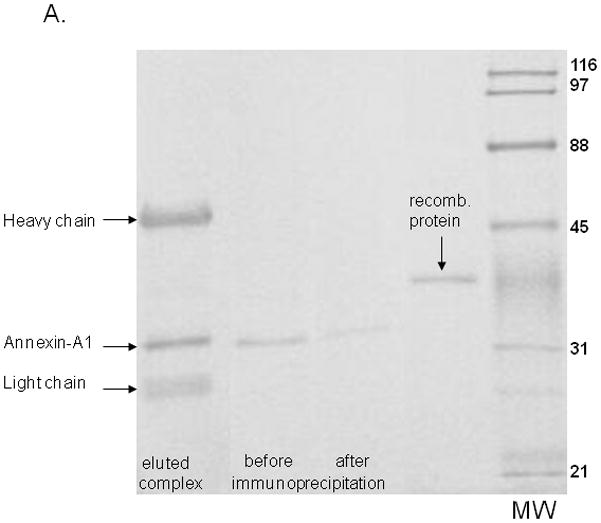
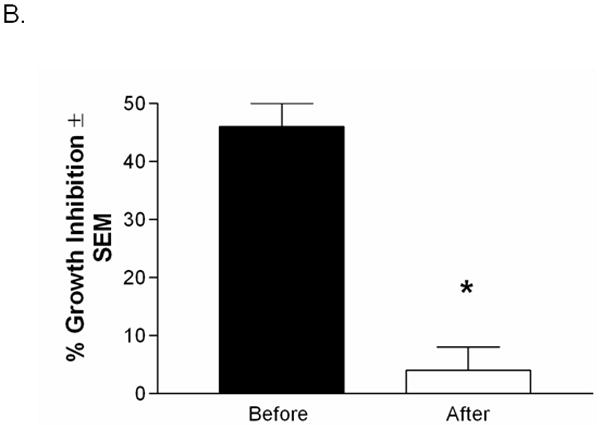
(A) Western blots were performed using protein extracts before and after immunoprecipitation. Figure also shows eluted protein and the heavy and light chains of the Annexin-A1 antibody used for immunoprecipitation. Recombinant human Annexin-A1 protein was used as a control. Image is representative of 3 repeats. (B) Protein extracts were used in the standard growth inhibition assay before and after immunoprecipitation. Controls included Candida incubated with vehicles (PBS or PP broth) and fresh epithelial cells alone. Results are expressed as % growth inhibition of Candida by oral epithelial cell proteins ± SEM, standard error of the mean, for 3 experiments.
To examine anti-Candida activity, protein extracts (~250 μg protein/ml) were used in the standard growth inhibition assay before and after immunoprecipitation. Results in Figure 8B show significant abrogation of anti-Candida activity by supernatants subjected to immunoprecipitation of Annexin-A1 compared to before immunoprecipitation (P=0.018).
Discussion
To date, studies show that oral and vaginal epithelial cell anti-Candida activity has a strict requirement for cell contact via an acid-labile effector moiety. However, the effector function of the moiety does not require metabolic activity of the epithelial cell (Nomanbhoy et al., 2002). Hence, it was possible to remove the moiety, evaluate the activity in soluble form, characterize it further, and begin to determine its identity. The sensitivity of the moiety to acid treatment provided the tool by which to conduct the studies.
The first set of results showed that the cell surface proteins extracted from PBS-, but not acid-treated, epithelial cells were able to inhibit the growth of Candida similar to intact cells. In addition, proteins from PBS-treated cells demonstrated the activity in a dose dependent manner. The trend toward somewhat lower activity by the soluble proteins compared to intact epithelial cells may be due to the soluble form of the effector protein having a lower affinity when not attached to the epithelial cells. Alternatively, the concentration of the effector moiety may have been different in the two forms although higher activity could not be achieved by further concentration of the soluble form (data not shown). Nonetheless, these results provide evidence that the effector moiety can act independent of epithelial cells. A second set of studies confirmed that the effector moiety was a protein; the anti-Candida activity was significantly abrogated when the extracted surface proteins were denatured with proteases. A similar reduction occurred with heat although statistical significance was not achieved, presumably due to incomplete denaturation.
Taking into account the action by the soluble form of the protein moiety, the next set of experiments sought to evaluate the physical characteristics of the protein moiety interaction with Candida. For this, we first confirmed that the cellular activity could occur with both morphological forms of Candida initiated and held in each respective state throughout the assay, together with the acid labile sensitivity against both morphological forms. Then we utilized biotinylation of the epithelial cells prior to protein extraction to visibly evaluate the interaction of the proteins with both morphological forms of Candida via streptavidin enzyme/substrate reaction. Results revealed that proteins extracted from acid-treated epithelial cells had significantly reduced binding capacity to both forms of Candida compared to PBS-treated epithelial cells. Thus, acid-treatment appears to either remove or damage/inactivate/denature much of the effector protein moiety. Indeed, the amounts of protein collected from acid-treated epithelial cells were consistently lower than those from the same number of PBS-treated cells (data not shown). Of particular note in these studies, biotinylation of the epithelial cells prior to extraction restricted the pool of proteins evaluated to predominantly cell surface proteins which were of primary interest, despite the potential presence of both surface and cytoplasmic proteins following extraction.
Based on the differential binding of proteins from PBS- and acid-treated epithelial cells to Candida, we began studies to identify candidates for the effector protein moiety. For this, biotinylated surface proteins from PBS- and acid-treated epithelial cells were extracted, bound to and eluted from Candida, and the resulting proteins analyzed by western blots, a technique which again allowed us to exclusively visualize epithelial cell surface proteins specific to Candida. Results revealed bands of 45 and 33 kDa unique to PBS-treated epithelial cell derived proteins and not seen in the acid-treated protein pool. These results suggested two viable candidates for the effector moiety.
Proteomic analyses were conducted in order to identify the two candidate proteins. Several initial approaches failed. The first was to label the biotinylated surface proteins from PBS- and acid-treated epithelial cells with two distinct fluorophores and to perform two-dimensional gel electrophoresis (2D-DIGE). However, this approach proved limiting because the noncovalent bonding between biotin and fluorophore-conjugated horseradish peroxidase dissociated during the elution process. A second attempt at 2D-DIGE using two CyDye DIGE fluor minimal dyes (Mayrhofer et al., 2006), which covalently bind lysine residues of proteins, was equally limiting; the labeled proteins could not be detected on 2D-gels due to the low levels of eluted proteins. A second approach was to visualize the bands seen in the western blot results on Coomassie-stained 1D SDS-PAGE gels. However, because the high sensitivity of western blots achieved by enzymatic amplification could not be duplicated under Coomassie staining, none of the bands in the eluted proteins could be detected. Thus, we were restricted to using Coomassie-stained 1D SDS-PAGE gels of extracted proteins (not bound to and eluted from Candida). Conveniently, the same 45 and 33 kDa bands of interest were present and absent, respectively, for PBS- and acid-treated extracted epithelial cell proteins. Hence, although not as optimal as Candida-eluted, we felt confident that the same proteins were being visualized. Accordingly, following excision of the matching bands and analyses by mass spectrometry, the candidate effector proteins of 45 and 33 kDa were identified to be actin and Annexin-A1, respectively. We recognize, however, that this evidence remains indirect being that the proteins were not confirmed to be Candida-specific.
To this end, actin would not appear to be involved in anti-Candida activity because the activity does not involve metabolic reactions with the epithelial cells and are not involved in inhibitory activities. Yet the acid treatment obviously appears to have affected actin-associated activities of the epithelial cells. In contrast, Annexin-A1 is a known mediator of anti-inflammatory actions (Croxtall et al., 2003) and an inhibitor of signal-transduction pathways that lead to cell proliferation (Liu et al., 2007). These properties are consistent with the mechanism of the anti-Candida activity and as such represent a viable effector moiety candidate.
Immunohistochemical staining supports this concept as well since Annexin-A1, but not actin, was shown to be present on the epithelial cell surface. Thus, although the biotinylation process obviously interacted with some cytoplasmic proteins as well, the surface expression of Annexin-A1 indeed provided compelling evidence that Annexin-A1 represented a strong candidate for the antifungal effector moiety.
With the identification of Annexin-A1 as a strong candidate, we used western blots to confirm its acid-labile property. Indeed, a significant reduction of Annexin-A1 was evident in protein extracts from acid-treated cells when compared to those taken from PBS-treated cells. The lack of complete elimination of Annexin-A1 from the cells is consistent with initial experiments showing a significant reduction, but not complete elimination of inhibitory activity. Nevertheless, these results continue to support a strong candidacy of Annexin-A1 as the effector moiety. Related to these experiments, we also observed a difference in size of the Annexin-A1 proteins extracted from the cells and the recombinant protein (33 kDa vs. 39 kDa, respectively). This is likely due to Annexin-A1 existing as both cytoplasmic and membrane-bound protein. Hence, the process of removing the surface proteins most likely cleaved the protein, resulting in the smaller size. Irrespective, proteomic analysis recognized the peptide sequence accurately enough to reveal a strong match.
A final series of experiments were to confirm that Annexin-A1 was responsible for the anti-Candida activity. To this end, the abrogation of activity following immunoprecipitation of Annexin-A1 from the protein extracts provides strong support for Annexin-A1 as the effector moiety. In fact, the abrogation was more dramatic than that demonstrated by acid treatment although each procedure showed a small amount of Annexin-A1 remaining via western blot. These results suggest that a threshold amount of Annexin-A1 is likely required for anti-Candida activity. Irrespective, this is the first report suggesting Annexin-A1 has antifungal activity and the first report to identify the elusive effector protein moiety. We recognize, however, that as much as Annexin-A1 appears to represent the effector moiety exclusively, contributions by other moieties are possible for complete activity. We hypothesize that Annexin-A1 on epithelial cells interacts with Candida and signals Candida to retard or halt its growth as part of a sophisticated symbiotic relationship Annexin-A1, as a non-inflammatory, static inhibitor of Candida growth, may also aid Candida as a commensal by holding its numbers at levels that would not initiate inflammatory responses that could ultimately kill Candida. Studies are ongoing to further investigate its mechanism of action.
Acknowledgments
This work was supported by National Institute of Health Public Health Service grants DE 12178 (Fidel) from the National Institute of Dental and Craniofacial Research.
This work was also supported in part by the Louisiana Vaccine Center and South Louisiana Institute for Infectious Diseases Research sponsored by the Louisiana Board of Regents.
References
- Barousse MM, Steele C, Dunlap K, et al. Growth Inhibition of Candida albicans by human vaginal epithelial cells. J Infect Dis. 2001;184:1489–1493. doi: 10.1086/324532. [DOI] [PubMed] [Google Scholar]
- Croxtall JD, Gilroy DW, Solito E, et al. Attenuation of glucocorticoid functions in an Anx-A1−/− cell line. Biochem J. 2003;371:927–935. doi: 10.1042/BJ20021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidel PL, Jr, Cutright JL, Steele C. Effects of Reproductive hormones on experimental vaginal candidiasis. Infect Immun. 2000;68:651–657. doi: 10.1128/iai.68.2.651-657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Harris CA, Small CB, Moll B, Lesser M, Friedland GH. Oral candidiasis in high risk patients as the initial manifestation of the acquired immunodeficiency syndrome. N Engl J Med. 1984;311:354–357. doi: 10.1056/NEJM198408093110602. [DOI] [PubMed] [Google Scholar]
- Liu N, Zhang YP, Han S, et al. Annexin A1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase A2 activation in adult rats following spinal cord injury. J Neuropathol Exp Neurol. 2007;66:932–943. doi: 10.1097/nen.0b013e3181567d59. [DOI] [PubMed] [Google Scholar]
- Macher AM. The pathology of AIDS. Public Health Report. 1988;103:246–254. [PMC free article] [PubMed] [Google Scholar]
- Mayrhofer C, Krieger S, Allmaier G, Kerjaschki D. DIGE compatible labelling of surface proteins on vital cells in vitro and in vivo. Proteomics. 2006;6:579–585. doi: 10.1002/pmic.200500104. [DOI] [PubMed] [Google Scholar]
- Nomanbhoy F, Steele C, Yano J, Fidel PL., Jr Vaginal and oral epithelial cell anti-Candida activity. Infect Immun. 2002;70:7081–7088. doi: 10.1128/IAI.70.12.7081-7088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel JD. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clin Infect Dis. 1992;4:S148–S153. doi: 10.1093/clinids/14.supplement_1.s148. [DOI] [PubMed] [Google Scholar]
- Sobel JD. Pathogenesis of recurrent vulvovaginal candidiasis. Curr Infect Dis Rep. 2002;4:514–519. doi: 10.1007/s11908-002-0038-7. [DOI] [PubMed] [Google Scholar]
- Steele C, Leigh JE, Swoboda RK, Fidel PL., Jr Growth inhibition of Candida by human oral epithelial cells. J Infect Dis. 2000;182:1479–1485. doi: 10.1086/315872. [DOI] [PubMed] [Google Scholar]
- Steele C, Leigh JE, Swoboda RK, Ozenci H, Fidel PL., Jr Potential role for a carbohydrate moiety in anti-Candida activity of human oral epithelial cells. Infect Immun. 2001;69:7091–7099. doi: 10.1128/IAI.69.11.7091-7099.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C, Ozenci H, Luo W, Scott M, Fidel PL., Jr Growth inhibition of Candida albicans by vaginal cells from naive mice. Med Mycol. 1999;37:251–260. [PubMed] [Google Scholar]
- Steele C, Ratterree M, Fidel PL., Jr Differential susceptibility to experimental vaginal candidiasis in macaques. J Infect Dis. 1999;180:802–810. doi: 10.1086/314964. [DOI] [PubMed] [Google Scholar]
- Yano J, Lilly E, Steele C, Fortenberry D, Fidel PL., Jr Oral and vaginal epithelial cell anti-Candida activity is acid-labile and does not require live epithelial cells. Oral Microbiol Immunol. 2005;20:199–205. doi: 10.1111/j.1399-302X.2005.00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]