

In the race between DNA sequencing throughput and computer speed, sequencing is winning by a mile. Sequencing throughput has recently been improving at a rate of about 5-fold per year1, while computer performance generally follows “Moore's Law,” doubling only every 18 or 24 months2. As this gap widens, the question of how to design higher-throughput analysis pipelines becomes critical. If analysis throughput fails to turn the corner, research projects will continually stall until analyses catch up.
How do we close the gap? One option is to invent algorithms that make better use of a fixed amount of computing power. Unfortunately, algorithmic breakthroughs of this kind, like scientific breakthroughs, are difficult to plan or foresee. A more practical option is to concentrate on developing methods that make better use of multiple computers and processers. When many computer processors work together in parallel, a software program can often finish in significantly less time.
While parallel computing has existed for decades in various forms3–5, a recent manifestation called “cloud computing” holds particular promise. Cloud computing is a model whereby users access compute resources from a vendor over the Internet1, such as from the commercial Amazon Elastic Compute Cloud6, or the academic DOE Magellan Cloud7. The user can then apply the computers to any task, such as serving web sites, or even running computationally intensive parallel bioinformatics pipelines. Vendors benefit from vast economies of scale8, allowing them to set fees that are competitive with what users would otherwise have spent building an equivalent facility, and potentially saving all the ongoing costs incurred by a facility that consumes space, electricity, cooling, and staff support. Finally, because the pool of resources available “in the cloud” is so large, customers have substantial leeway to “elastically” grow and shrink their allocations.
Cloud computing is not a panacea: it poses problems for developers and users of cloud software, requires large data transfers over precious low-bandwidth Internet uplinks, raises new privacy and security issues, and is an inefficient solution for some types of problems. On balance, though, cloud computing is an increasingly valuable tool for processing large datasets, and it is already used by the US federal government9, pharmaceutical10 and Internet companies11, as well as scientific labs12 and bioinformatics services13, 14. Furthermore, several bioinformatics applications and resources have been developed to specifically address the challenges of working with the very large volumes of data generated by second-generation sequencing technology (Table 1).
Table 1.
Bioinformatics Cloud Resources
| Applications | |
|---|---|
| CloudBLAST34 | Scalable BLAST in the Clouds http://www.acis.ufl.edu/~ammatsun/mediawiki-1.4.5/index.php/CloudBLAST_Project |
| CloudBurst19 | Highly Sensitive Short Read Mapping http://cloudburst-bio.sf.net |
| Cloud RSD26 | Reciprocal Smalest Distance Ortholog Detection http://roundup.hms.harvard.edu |
| Contrail27 | De novo assembly of large genomes http://contrail-bio.sf.net |
| Crossbow22 | Alignment and SNP Genotyping http://bowtie-bio.sf.net/crossbow/ |
| Myrna25 | Differential expression analysis of mRNA-seq http://bowtie-bio.sf.net/myrna/ |
| Quake35 | Quality guided correction of short reads http://github.com/davek44/error_correction/ |
| Analysis Environments & Datasets | |
|---|---|
| AWS Public Data | Cloud copies of Ensembl, GenBank, 1000 Genomes Data, etc… http://aws.amazon.com/publicdatasets/ |
| CLoVR | Genome and metagenome annotation and analysis http://clover.igs.umaryland.edu |
| Cloud BioLinux | Genome Assembly and Alignment http://www.cloudbiolinux.com/ |
| Galaxy29 | Platform for interactive large-scale genome analysis http://galaxy.psu.edu |
MapReduce and Genomics
Parallel programs run atop a parallel “framework” to enable efficient, fault-tolerant parallel computation without making the developer's job too difficult. The Message Passing Interface (MPI) framework3, for example, gives the programmer ample power to craft parallel programs, but requires relatively complicated software development. Batch processing systems such as Condor4, are very effective for running many independent computations in parallel, but are not expressive enough for more complicated parallel algorithms. In between, the MapReduce framework15 is efficient for many (although not all) programs, and makes the programmer's job simpler by automatically handling duties such as job scheduling, fault tolerance, and distributed aggregation.
MapReduce was originally developed at Google to streamline analyses of very large collections of webpages. Google's implementation is proprietary, but Hadoop16 is a popular open source alternative maintained by the Apache Software Foundation. Hadoop/MapReduce programs comprise a series of parallel computational steps (Map and Reduce), interspersed with aggregation steps (Shuffle). Despite its simplicity, Hadoop/MapReduce has been successfully applied to many large-scale analyses within and outside of DNA sequence analysis17–21.
In a genomics context, Hadoop/MapReduce is particularly well suited for common “Map-Shuffle-Scan” pipelines (Figure 1) that use the following paradigm:
Map: many reads are mapped to the reference genome in parallel on multiple machines.
Shuffle: the alignments are aggregated so that all alignments on the same chromosome or locus are grouped together and sorted by position.
Scan: the sorted alignments are scanned to identify biological events such as polymorphisms or differential expression within each region.
Figure 1. Map-Shuffle-Scan framework used by Crossbow.
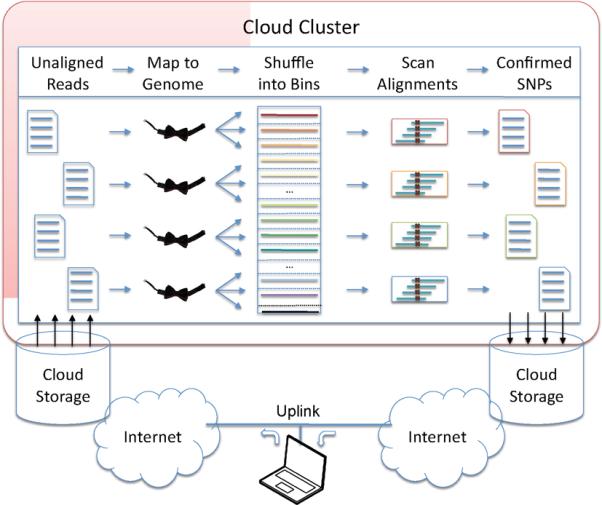
Users begin by uploading the sequencing reads into the cloud storage. Hadoop, running on a cluster of virtual machines in the cloud, then maps the unaligned reads to the reference genome using many parallel instances of Bowtie. Hadoop then automatically shuffles the alignments into sorted bins determined by chromosome region. Finally, many parallel instances of SOAPsnp scan the sorted alignments in each bin. The final output is a stream of SNP calls stored within the cloud that can be downloaded back to the user's local computer.
For example, the Crossbow22 genotyping program leverages Hadoop/MapReduce to launch many copies of the short read aligner Bowtie23 in parallel. After Bowtie has aligned the reads (which may number in the billions for a human re-sequencing project) to the reference genome, Hadoop automatically sorts and aggregates the alignments by chromosomal region. It then launches many parallel instances of the Bayesian SNP caller SOAPsnp24 to accurately call SNPs from the alignments. In our benchmark test on the Amazon cloud, Crossbow genotyped a human sample comprising 2.7 billion reads in ~4 hours, including the time required for uploading the raw data, for a total cost of $85 USD22.
Programs with abundant parallelism tend to scale well to larger clusters; i.e., increasing the number of processors proportionally decreases the running time, less any additional overhead or non-parallel components. Several comparative genomics pipelines have been shown to scale well using Hadoop19, 22, 25, 26, but not all genomics software is likely to follow suit. Hadoop, and cloud computing in general, tends to reward “loosely coupled” programs where processors work independently for long periods and rarely coordinate with each other. But some algorithms are inherently “tightly coupled,” requiring substantial coordination and making them less amenable to cloud computing. That being said, PageRank20 (Google's algorithm for ranking web pages) and Contrail27 (a large-scale genome assembler) are examples of relatively tightly coupled algorithms that have been successfully adapted to MapReduce in the cloud.
Cloud computing obstacles
To run a cloud program over a large dataset, the input must first be deposited in a cloud resource. Depending on data size and network speed, transfers to and from the cloud can pose a significant barrier. Some institutions and repositories connect to the Internet via high-speed backbones such as Internet2 and JANET, but each potential user should assess whether their data generation schedule is compatible with transfer speeds achievable in practice. A reasonable alternative is to physically ship hard drives to the cloud vendor28.
Another obstacle is usability. The rental process is complicated by technical questions of geographic zones, instance types, and which software image the user plans to run. Fortunately, efforts such as the Galaxy project29 and Amazon's Elastic MapReduce service30 enhance usability by allowing customers to launch and manage resources and analyses through a point-and-click web interface.
Data security and privacy are also concerns. Whether storing and processing data in the cloud is more or less secure than doing so locally is a complicated question, depending as much on local policy as on cloud policy. That said, regulators and Institutional Review Boards are still adapting to this trend, and local computation is still the safer choice when privacy mandates apply. An important exception is HIPAA; several HIPAA-compliant companies already operate cloud-based services31.
Finally, cloud computing often requires re-designing applications for parallel frameworks like Hadoop. This takes expertise and time. A mitigating factor is that Hadoop's “streaming mode” allows existing non-parallel tools to be used as computational steps. For instance, Crossbow uses the non-cloud programs Bowtie and SOAPsnp, albeit with some small changes to format intermediate data for the Hadoop framework. New parallel programming frameworks, such as DryadLINQ32 and Pregel33 can also help in some cases by providing richer programming abstractions. But for problems where the underlying parallelism is sufficiently complex, researchers may have to develop sophisticated new algorithms.
Recommendations
With biological datasets accumulating at ever faster rates, it is better to prepare for distributed and multi-core computing sooner rather than later. The cloud provides a vast, flexible source of computing power at a competitive cost, potentially allowing researchers to analyze ever-growing sequencing databases while relieving them of the burden of maintaining large computing facilities. On the other hand, the cloud requires large, possibly network-clogging data transfers, it can be challenging to use, and it isn't suitable for all types of analysis tasks. For any research group considering the use of cloud computing for large-scale DNA sequence analysis, we recommend a few concrete steps:
Verify that your DNA sequence data will not overwhelm your network connection, taking into account expected upgrades for any sequencing instruments.
Determine whether cloud computing is compatible with any privacy or security requirements associated with your research.
Determine whether necessary software tools exist and can run efficiently in a cloud context. Is new software needed, or can existing software be adapted to a parallel framework? Consider the time and expertise required.
Consider cost: what is the total cost of each alternative?
Consider the alternative: is it justified to build and maintain, or otherwise gain access to a sufficiently powerful non-cloud computing resource?
If these prerequisites are met, then computing “in the cloud” can be a viable option to keep pace with the enormous data streams produced by the newest DNA sequencing.
Acknowledgements
The authors were supported in part by NSF grant IIS-0844494 and by NIH grant R01-LM006845.
References
- 1.Stein LD. The case for cloud computing in genome informatics. Genome Biol. 2010;11:207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore GE. Cramming more components onto integrated circuits. Electronics. 1965;38:4–1965. [Google Scholar]
- 3.Dongarra JJ, Otto SW, Snir M, Walker D. A message passing standard for MPP and workstations. Commun. ACM. 1996;39:84–1996. [Google Scholar]
- 4.Litzkow M, Livny M, Mutka M. Condor: A Hunter of Idle Workstations. 8th International Conference of Distributed Computing Systems.1988. [Google Scholar]
- 5.Dagum L, Menon R. OpenMP: An Industry-Standard API for Shared-Memory Programming. IEEE Comput. Sci. Eng. 1998;5:46–1998. [Google Scholar]
- 6.Amazon Elastic Compute Cloud. http://aws.amazon.com/ec2/
- 7.DOE Magellan Cloud. http://magellan.alcf.anl.gov/
- 8.Markoff J, Hansell S. Hiding in Plain Sight, Google Seeks More Power. The New York Times; 2006. http://www.nytimes.com/2006/06/14/technology/14search.html. [Google Scholar]
- 9.Apps.gov. https://apps.gov/
- 10.Eli Lilly On What's Next In Cloud Computing. http://www.informationweek.com/cloud-computing/blog/archives/2009/01/whats_next_in_t.html.
- 11.Netflix Selects Amazon Web Services to Power Mission-Critical Technology Infrastructure. http://phx.corporate-ir.net/phoenix.zhtml?c=176060&p=irol-newsArticle&ID=1423977&highlight=
- 12.AWS Case Study: Harvard Medical School. http://aws.amazon.com/solutions/case-studies/harvard/
- 13.DNAnexus. http://dnanexus.com/
- 14.Spiral Genetics. http://www.spiralgenetics.com/
- 15.Jeffrey D, Sanjay G. MapReduce: simplified data processing on large clusters. Commun. ACM. 2008;51:107–2008. [Google Scholar]
- 16.Hadoop. http://hadoop.apache.org/
- 17.Lin J, Dyer C. Data-Intensive Text Processing with MapReduce. Synthesis Lectures on Human Language Technologies. 2010;3:1–2010. [Google Scholar]
- 18.Chu C-T, et al. Map-Reduce for Machine Learning on Multicore. Advances in Neural Information Processing Systems. 2007;19:281–2007. [Google Scholar]
- 19.Schatz MC. CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics. 2009;25:1363–2009. doi: 10.1093/bioinformatics/btp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brin S, Page L. The Anatomy of a Large-Scale Hypertextual Web Search Engine. Seventh International World-Wide Web Conference.1998. [Google Scholar]
- 21.Matthews SJ, Williams TL. MrsRF: an efficient MapReduce algorithm for analyzing large collections of evolutionary trees. BMC Bioinformatics. 2010;11(Suppl 1):S15. doi: 10.1186/1471-2105-11-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–2009. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Hansen K, Leek J. Cloud-scale RNA-seq differential expression analysis. Submitted for Publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wall D, et al. Cloud computing for comparative genomics. BMC Bioinformatics. 2010;11:259. doi: 10.1186/1471-2105-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schatz MC, Sommer DD, Kelley DR, Pop M. De Novo Assembly of Large Genomes using Cloud Computing. In Preparation. [Google Scholar]
- 28.AWS Import/Export. http://aws.amazon.com/importexport/
- 29.Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–2005. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amazon Elastic MapReduce. http://aws.amazon.com/elasticmapreduce/
- 31.Creating HIPAA-Compliant Medical Data Applications With AWS. http://aws.amazon.com/about-aws/whats-new/2009/04/06/whitepaper-hipaa/
- 32.Yu Y, et al. DryadLINQ: A System for General-Purpose Distributed Data-Parallel Computing Using a High-Level Language. Symposium on Operating System Design and Implementation (OSDI).2008. [Google Scholar]
- 33.Malewicz G, et al. Pregel: a system for large-scale graph processing. PODC '09: Proceedings of the 28th ACM symposium on Principles of distributed computing.2009. [Google Scholar]
- 34.Matsunaga A, Tsugawa M, Fortes J. CloudBLAST: Combining MapReduce and Virtualization on Distributed Resources for Bioinformatics Applications. IEEE Fourth International Conference on eSciene.2008. [Google Scholar]
- 35.Kelley DR, Schatz MC, Salzberg SL. Quality guided correction and filtration of errors in short reads. Manuscript in preparation. [Google Scholar]