

Abstract
Fragile X syndrome is an X-linked disorder caused by the inactivation of the FMR-1 gene with symptoms ranging from impaired cognitive functions to seizures, anxiety, sensory abnormalities, and hyperactivity. Males are more severely affected than heterozygote (H) females, who, as carriers, have a 50% chance of transmitting the mutated allele in each pregnancy. fmr-1 knockout (KO) mice reproduce fragile X symptoms, including hyperactivity, seizures, and abnormal sensory processing. In contrast to the expectation that wild-type (WT) males born to H (fmr-1+/−) mothers (H> WT) are behaviorally normal and indistinguishable from WT males born to WT mothers (WT> WT); here, we show that H> WT offspring are more active than WT> WT offspring and that their hyperactivity is similar to male KO mice born to H or KO (fmr-1−/−) mothers (H> KO/KO> KO). H> WT mice, however, do not exhibit seizures or abnormal sensory processing. Consistent with their hyperactivity, the effect of the D2 agonist quinpirole is reduced in H> WT as well as in H> KO and KO> KO mice compared to WT> WT offspring, suggesting a diminished feedback inhibition of dopamine release. Our data indicate that some aspects of hyperactivity and associated dopaminergic changes in ‘fragile X’ mice are a maternal fmr-1 genotype rather than an offspring fmr-1 genotype effect.
Keywords: fragile X, environmental, maternal, dopamine, FMRP, locomotor activity
INTRODUCTION
In studying knockout (KO) phenotypes, it is a common practice to compare the behavior of KO and wild-type (WT) littermates born to heterozygote (H) parents. Many older studies with KO animals, however, used non-littermate WT controls as KO and WT lines were separately maintained. There are advantages and disadvantages to both approaches. A behavioral difference between KO and WT littermates is usually interpreted as genetic. Yet, the phenotype of the KO littermates may not be purely genetic but rather the result of an interaction between genetic and environmental, or more specifically parental effects because the parents’ heterozygosity could lead to non-genetic effects. Additionally, it is rarely realized that WT littermate controls (born to H parents) may also have abnormalities due to parental effects. In contrast, WT offspring born to WT parents in a non-littermate breeding strategy have neither genetic nor parental effects and, therefore, genuinely represent the baseline ‘normal’ behavior. However, the phenotype of KO offspring of a non-littermate breeding (born to KO parents) can be difficult to interpret because it could be the result of both genetic and parental effects. These questions have practical implications because in human conditions and diseases with Mendelian inheritance the parents’ heterozygosity is not normally considered as a risk factor in genetically unaffected progeny. To separate the contribution of genetic and nongenetic factors to a phenotype, we used a novel strategy of breeding and analyzing littermate and non-littermate groups of animals in parallel (Figure 1a). We tested this idea by using fmr-1 KO mice representing an animal model of the fragile X syndrome (Consortium, 1994).
Figure 1.

Locomotor activity is modulated by the mother’s fmr genotype. (a) Experiments involved two WT and two KO male offspring groups born to WT (solid circle), H (gray circle), and KO (open circle) mothers as shown. (b) Total activity during a 2-h test. Two cohorts of animals were tested and since they were not statistically different from each other the data from the two experiments were combined. Main effect ANOVA showed both a maternal and offspring genotype effect (see in the Results and Discussion section). Univariate ANOVA with the four groups showed significant group differences (F(3, 87) = 8.75, p = 0.00003; LSD post hoc test *p < 0.05; ***p < 0.0005; N = 20–26). WT and KO male offspring are indicated by solid and open columns, respectively (mean±SE). Littermates are highlighted by a gray background. (c) In the first 20 min of the test, there was an offspring but not a mother fmr genotype effect (see main effect ANOVA in the Results and Discussion section). A follow-up univariate ANOVA with the four groups showed significant differences between the WT and KO offspring (F(3, 88) = 11.72, p = 0.00002; LSD post hoc test *p < 0.05; ***p < 0.0005). (d and e) The offspring fmr genotype effect was still apparent between 20 and 60 min but disappeared later (60–120 min) in the test. In contrast, a mother fmr genotype effect became evident during these periods (see main effects ANOVA in the Results and Discussion section). A follow-up univariate ANOVA with the four groups showed significant group differences (F(3, 88) = 13.04, p < 0.00001 in d and F(3, 88) = 5.77, p = 0.001 in e; LSD post hoc test *p < 0.05; ***p < 0.0005). Data in (b–e) are normalized for 1 h to allow a direct comparison of activity through the trial but the scale of the graphs is different.
Fragile X syndrome is an X-linked neuropsychiatric disorder caused, in most cases, by a triplet expansion in the FMR-1 gene leading to the inactivation of the gene (Hagerman, 1996). Although most fragile X individuals inherit a premutated allele that undergoes triplet expansion resulting in a full mutation, some inherit a fully mutated allele from H carrier mothers. Males with the full mutation always have some degree of mental retardation while H females with a full mutation are not, or are only moderately affected (Toledano-Alhadef et al, 2001). Besides mental retardation, fragile X individuals can have seizures, attention deficit, hyperactivity, anxiety, and symptoms associated with autism (Chen and Toth, 2001; Consortium, 1994; D’Hooge et al, 1997; Musumeci et al, 2000; Nielsen et al, 2002; Peier et al, 2000; Yun et al, 2006).
Here we report that in the mouse model of fragile X hyperactivity is maternally transmitted, at least partly, while seizures, reduced startle and increased prepulse inhibition are strictly Mendelian traits. Since hyperactivity has been linked to low tonic stimulation of inhibitory dopamine (DA) D2 autoreceptors and consecutively to high phasic DA activity in the striatum (Grace, 2001; Koob et al, 1981; Koob and Swerdlow, 1988; Szczypka et al, 2001), we tested the function of these receptors by quinpirole, a D2 receptor preferring drug (Cory-Slechta et al, 1996; Starke et al, 1989; Usiello et al, 2000). These experiments indicated that the D2 receptor or its coupling/signaling may be the molecular target of the maternal effect.
MATERIALS AND METHODS
Animals
fmr1-KO mice (Consortium, 1994) on the FVB (FVB/NJ-Fmr1tm1Cgr) background as well as FVB WT mice (FVB/NJ) were purchased from The Jackson Laboratory, Bar Harbor, ME. KO mice were backcrossed more than 10 times and can be considered congenic with genetic differences only at and possibly around the targeted fmr-1 gene. For our experiments, animals were generated as illustrated in Figure 1a using breeding cages with two females and one male. Experiments were conducted using male offspring aged 8–12 weeks. All animals were housed in groups of up to five per cage (in most cases 3–4 animals) with 12 h light/dark cycle with lights on at 0600 hours. Food and water were available ad libitum. Animal experiments were carried out in accordance with the Weill Cornell Medical College of Cornell University IACUC guidelines.
Drugs
d-Amphetamine sulfate, an indirect DA agonist, (±)-Quinpirole dihydrochloride, a D2/3R agonist with preferential activity at the presynaptic D2 receptor at low concentrations ( < 0.2 mg/kg), (±)-6-chloro-PB hydrobromide (SKF-81297), a D1R agonist and chloro-APB hydrobromide (SKF 82958), another D1R agonist were obtained from Sigma (St Louis, MO). (+)-PD 128907 hydrochloride (a preferential D3R agonist) was purchased from Tokris Bioscience (Ellisville, MO). All drugs were dissolved in sterile saline and administered intraperitoneally (i.p.) at 0.1 ml/10 g animal weight.
Locomotor Activity
Locomotor behavior was recorded using infrared beamequipped activity chambers (27 × 27 cm, Med Associates Inc., St Albans, VT). The animals were allowed to habituate to the behavioral testing room for at least 45 min prior to the onset of any experiments. During 2 h recordings, activity was measured in 20 min intervals to assess group differences and habituation during the 2 h trial. This experiment was repeated with a second batch of animals and these animals were retested on the second, third, and fourth consecutive days to assess long-term habituation.
In experiments that investigated the effect of dopaminergic drugs, additional animals were used. These animals were first allowed to explore the locomotor chamber for 20 min, then they were briefly removed for the administration of the appropriate drug and immediately returned to the same testing chamber for a 40-min observational period. Activity was recorded from min 5 to 20 and from 20 to 40 min of the postinjection period. To minimize the number of animals required, a crossover study design was used to test the locomotor effect of dopaminergic drugs but animals were not used after the 4 mg/kg amphetamine dose. Behavioral tests were repeated in 7-day intervals not more than four times. Prior studies in our laboratory showed no habituation to the locomotor boxes when tests were repeated in 7-day intervals up to four times (Supplementary Figure S1).
Audiogenic Seizures
Mice were placed into a sound-attenuating chamber equipped with a plexiglass door for observation. After a 5-min habituation period, animals were subjected to a 115-dB noise (5–20 kHz) until seizure onset (approximately 20–25 s) but not longer than 60 s. Latency to seizure and seizure incidence were recorded. Subjects were tested for seizure susceptibility only once and were not reused in other behavioral tests.
Acoustic Startle Response and PPI
Mice were tested in an SR-pilot startle reflex apparatus (San Diego Instruments, San Diego, CA) (Chen and Toth, 2001). Background noise was 65 dB. After a 5-min habituation period, subjects were presented three consecutive startle stimuli (65–115 dB, 5–20 kHz, 40 ms), with 30 s intervals. Then, in a pseudorandom order, six prepulse (75 dB) followed in 100 ms by startle stimuli (100–115 dB) and six startle stimuli alone (115 dB) were presented. The interval between the stimuli varied from 15 to 30 s. Due to the short duration of the startle stimulus (40 ms) and because seizure is not elicited within 20–25 s of exposure to sound, seizures could not have confounded the startle response. PPI of startle response was calculated in percent by the following formula: 100− ((mean value of startle response of prepulse trials/mean value of startle response of startle alone trials) × 100). The effect of prepulse on startle was not tested at low startle intensities (65–90 dB). Response to the various startle intensities were measured in a crossover study at a test frequency of one per day.
Data Analysis
One-way, two-way, and main effects ANOVAs were used in various experiments as described in the text followed by various post hoc test. Fisher’s exact test was used in the seizure experiments. Statistical significance was assumed at p < 0.05.
RESULTS AND DISCUSSION
Wild-Type Offspring of Carrier Mothers Exhibit Hyperactivity
Two WT and two fmr-1 KO groups were generated representing littermates and non-littermates as shown in Figure 1a. While one of the WT groups had WT parents (WT> WT) the other group (H> WT) had H mothers (and WT fathers). Two KO groups were generated and tested in most experiments (H> KO and KO> KO) even if the medical relevance of the KO> KO group is limited because homozygote carriers, even for one full and one premutated or two premutated alleles, are extremely rare (Mazzocco and Holden, 1996; Mila et al, 1996). Therefore, the most relevant groups to compare in our experiments were the WT> WT representing individuals with neither maternal nor offspring genotype effects, the H> WT representing individuals with only maternal genotype effect and H> KO individuals with both maternal and offspring genotype effects. First, we analyzed the locomotor activity of these groups during the 2-h trial and were surprised to see that the total activity of WT> WT and H> WT offspring was different. As the combined data from two cohorts of animals show, analyzed by one-way ANOVA (Figure 1b), H> WT animals, compared to WT> WT mice, had increased locomotor activity that was indistinguishable (NS) from the activity measured in the KO> KO group. These data indicate that a partial deficiency in fmr-1 expression in the mother (in a mosaic pattern) is sufficient to elicit a maternal effect on WT offspring locomotor activity. However, activity in the H> KO group was still higher than activity in the H> WT group suggesting that a combination of maternal and offspring genotype effects may be responsible for the high activity in this group (Figure 1b). To analyze further the maternal and offspring genotype effect, main effect ANOVA with maternal genotype (WT and M (mutant) that included KO and H) and offspring genotype (WT and KO) as factors was performed. This analysis showed both maternal and offspring genotype effects (maternal: F(1, 88) = 4.92, p = 0.03, LSD post hoc test WT vs M, p = 0.00003; offspring: F(1, 88) = 4.26, p = 0.04, LSD post hoc test WT vs KO, p = 0.00003).
Since animals displayed high activity initially that was gradually reduced during the 2-h trial (Supplementary Figure S2), we also analyzed the maternal and offspring genotype effects at different stages of locomotor habituation. Activity measured in the first 20 min (0–20 min) was reduced by one-third during the 20–40 min period that was further reduced by approximately one-third by the 60–80 min period with activity levelling off afterwards specifying three time windows (0–20, 20–60, and 60–120 min). The increased length of these periods is consistent with the initially fast and then slower locomotor habituation. While main effects ANOVA showed only an offspring genotype effect during the first 20 min (F(1, 88) = 16.14, p = 0.0001, LSD p < 0.00001) (Figure 1c), both maternal and offspring genotype effects were seen in the 20–60 min period (maternal: F(1, 88) = 5.64, p = 0.02, LSD WT vs M p < 0.00001; offspring: F(1, 88) = 8.79, p = 0.004, LSD WT vs KO p < 0.00001) (Figure 1d). It is possible that the effect of the maternal fmr genotype is present all along but is not detectable at the beginning of the test because of the overall high activity. Then, during the 60–120 min period, while the maternal effect was still present (F(1, 88) = 3.76, p = 0.05, LSD p < 0.001), the offspring genotype effect disappeared (F(1, 88) = 1.33, p = 0.25) (Figure 1e). Follow-up univariate ANOVA (see legend of Figure 1) also consistently showed increased activity of H> WT mice as compared to WT> WT mice and no difference between H> WT and KO> KO mice when maternal effect was seen by main effect ANOVA. Interestingly, activity of H> KO mice was higher than the activity of H> WT animals in all periods suggesting that the H maternal and offspring genotype effects interact (in H> KO mice). Surprisingly, the activity of H> KO mice was also higher than the activity of KO> KO mice during the 60–120 min period. Further studies will be necessary to resolve the apparent paradox of why offspring hyperactivity is less severe with mothers with homozygous deletion of the fmr-1 gene.
Next, the second cohort of animals was used to test if the maternal effect was still present on re-exposure to the locomotor chambers in 24 h. The first 20 min of the second test was essentially the continuation of the end of the first trial showing only maternal effect (main effects ANOVA; maternal: F(1, 39) = 4.00, p = 0.05, LSD WT vs M p = 0.005; offspring: F(1, 39) = 0.002, p = 0.96, NS) and indeed the difference in activity between WT> WT and H> WT mice was still apparent (Supplementary Figure S3A). This demonstrates that the maternal genotype effect on activity is not limited to within test habituation but also applies to repeated test habituation. However, the maternal effect later disappeared (20–120 min on day 2) and indeed the difference between WT> WT and H> WT mice was no longer detectable (Supplementary Figure S3B and C). In parallel, the offspring genotype effect became once again apparent (Supplementary Figure S3B and C for the 20–60 and 60–120 min periods). Further re-exposure on days 3 and 4 resulted in only offspring genotype effect (data not shown). These data show that while offspring genotype dependent hyperactivity is constitutive (detectable through the four consecutive trials except between 60 and 120 min on day 1 and between 0 and 20 min on day 2), the maternal effect on activity is present initially but disappears later. We speculate that this almost reciprocal relationship between the maternal and offspring genotype-related hyperactivity reflects a time-dependent predominance of the maternal effect during day 1 and at the beginning of day 2 making the offspring effect too small to be detectable. Although we do not currently know why the maternal effect disappears during the second day as the environment becomes more familiar on repeated testing, we speculate that the novelty and stress content of the environment is necessary to elicit the maternal genotype dependent component of the hyperactivity.
Next, we analyzed if the fmr genotype of the mother alters the litter size and the gender and/or genotype distribution of offspring. The average litter size of H and KO mothers (7.44±0.32 (SE) and 6.10±0.37; no. of litters (N) = 94 and 73, respectively) was similar to that of WT mothers (6.50±0.35, N = 81), although a significant difference in litter size was observed between KO and H mothers (mother’s genotype effect by one-way ANOVA: F(2, 245) = 4.019, p = 0.019; Bonferroni’s post hoc analysis of H vs KO mothers: p = 0.021). The percent of males per litter in the three groups was not statistically different (51.91, 48.2, and 48.53% for offspring of H, KO, and WT mothers). Finally, the ratio of WT and KO littermates born to H mothers was 1.00 : 1.01 indicating no bias toward WT or KO offspring. These data show that the mother’s genotype (WT vs H/KO) has no appreciable effect on the number and on the expected gender- and genotype-distribution of the offspring.
Reduced Startle Response, Increased Prepulse Inhibition (PPI) and Seizure Susceptibility of ‘Fragile X’ Offspring is not Maternally Transmitted
We have previously reported that KO> KO males, compared to WT> WT males, have reduced auditory startle response at 115 dB startle intensity (by approximately 14%) (Chen and Toth, 2001). Here we determined if this difference is due to a genotype or maternal effect. As we have reported the KO> KO data and because the three other genotypes are sufficient to determine the maternal and offspring genotype contribution, here we compared the startle response of WT> WT, H> WT, and H> KO mice. While the startle response of WT> WT and H> WT mice was similar, the response of H> KO mice was reduced by 24% (compared to WT> WT) and by 25% (compared to H> WT) mice, indicating a genotype effect (Figure 2a). This genotype dependent difference in startle was also seen at reduced startle intensity (100 and 110 dB, Figure 2a). No maternal effect was seen at any of these intensities (100–115 dB). Startle amplitude was dramatically reduced below 100 dB and the genotype effect was no longer apparent at these startle intensities. Startle was not detectable below 85 dB (Figure 2a).
Figure 2.

Changes in startle and PPI responses are offspring genotype-dependent. (a) Startle is reduced at 100, 110, and 115 dB in H> KO mice but not in H> WT mice as compared to WT> WT mice indicating that this abnormality is dependent on the offspring’s fmr genotype (two-way genotype × stimulus intensity ANOVA; genotype: F(2, 205) = 12.29, p = 0.000009; LSD post hoc test, *p < 0.05 and §p < 0.05, H> KO vs H> WT and WT> WT, respectively; N = 11–16 per group). Startle is elicited at or above 85 dB (stimulus intensity: F(8, 205) = 170.43, p < 0.000001; LSD post hoc test #p < 0.05 vs 65 dB in all groups). (b) PPI is increased at 75 dB prepulse and at 100, 110, and 115 dB startle intensity in H> KO mice as compared to WT> WT and H> WT mice indicating an offspring genotype effect (two-way genotype × stimulus intensity ANOVA; genotype: F(2, 89) = 14.92, p = 0.000003, LSD post hoc test, *p < 0.05, dotted line represents trend only; startle intensity: F(2, 89) = 23.86, p < 0.000001; N = 11–16 per group).
We have also reported that KO> KO males, compared to WT> WT males, have increased PPI (75 vs 50% PPI at 75/115 dB prepulse/startle intensity) (Chen and Toth, 2001), and here we compared WT> WT, H> WT, and H> KO mice. PPI was significantly higher in H> KO mice (64%) than in WT> WT (36%) and H> WT (46%) mice (Figure 2b). The difference in PPI was also seen at lower (100 and 110 dB) startle intensities (Figure 2b). These data indicate that modulation of PPI is also offspring-genotype dependent. Finally, we also reported the occurrence of audiogenic seizures in KO> KO but not in WT> WT mice that started with ‘wild running’ followed by clonic (jumping) and tonic convulsion frequently leading to death (Chen and Toth, 2001). Seizures were again observed in H> KO, as in KO> KO mice, while seizures never occurred in H> WT and WT> WT mice (Table 1). Taken together, tests showed hyperactivity as the only behavioral characteristic of the offspring that correlates with the mother’s fmr genotype while the other traits (reduced startle reactivity, increased PPI, and seizure susceptibility) correlate with the offspring’s fmr genotype. An offspring genotype effect was also seen on locomotor activity indicating that KO offspring of H mothers acquire both the maternal and offspring genotype effects while the WT offspring of H mothers acquire only the maternal effect.
Table 1.
Occurrence of Audiogenic Seizures is an Offspring fmr Genotype-Dependent Behavior
| Mother | Offspring | Clonic-tonic seizure (N) | No seizure (N) | Total (N) |
|---|---|---|---|---|
| WT | WT | 0 | 13 | 13 |
| H | WT | 0 | 11 | 11 |
| H | KO | 3 | 8 | 11 |
| KO | KO | 5 | 13 | 18 |
Occurrence of seizures is significantly associated with the offspring’s genotype (χ2 = 7.798, p = 0.0052), but not with the mother’s fmr genotype (χ2 = 4.67, p = 0.099).
Wild-Type Offspring of H Mothers have Altered D2 Receptor Function
Lesion and restoration experiments have linked hyperactivity to low tonic stimulation of inhibitory DA D2 autoreceptors and successively to high phasic DA activity in the dorsal and/or ventral striatum (Grace, 2001; Koob et al, 1981; Koob and Swerdlow, 1988; Szczypka et al, 2001). Since the D2/3 receptor agonist quinpirole, at concentrations of 0.05–0.2 mg/kg, inhibits locomotor activity primarily via D2 autoreceptor-mediated inhibition of DA release in rodents (Cory-Slechta et al, 1996; Starke et al, 1989; Usiello et al, 2000), we could directly probe D2 autoreceptor function in H> WT mice. Since data displayed in Figure 1 showed that the maternal effect becomes apparent during the 20–60 min period in the locomotor test, drug was administered at 20 min and behavior was recorded between 40 and 60 min in these experiments (Figure 3a). Data in Figure 3b, normalized to saline controls (as well as the raw locomotor data in Supplementary Figure S4) show that quinpirole (0.05–0.1 mg/kg) significantly reduced activity (main effect ANOVA: treatment F(2, 145) = 17.55, p < 0.000001). Although there was no difference between the groups in drug-induced inhibition at the lower 0.05 mg/kg dose (Figure 3b, left panel), there was a maternal effect at 0.1 mg/kg as quinpirole resulted in less suppression of locomotor activity in offspring born to M than to WT mothers (main effects ANOVA, maternal genotype: F(1, 31) = 4.79, p = 0.035; LSD post hoc test, WT vs M mothers, p = 0.036) (Figure 3b, right panel). One may argue that higher behavioral activity in the hyperactive groups needs more quinpirole to be effective, but the lower dose of the drug (0.05 mg/kg) was equally effective in reducing activity in WT> WT and hyperactive offspring (by approximately 30%, Figure 3b, left panel). Since only the higher dose, that is presumably less selective for the presynaptic D2 receptor, revealed the behavioral difference, it is possible that the hyperactive mice have an increased D2 postsynaptic receptor function leading to increased activity. However, administration of even higher than 1 mg/kg quinpirole was shown not to cause hyperactivity in mice making the postsynaptic D2 receptor as an unlikely target for quinpirole at the 0.1 mg/kg dose in our experiments (Wang et al, 2000). Interestingly, the behavioral difference among the groups shown in Figure 3b was not clearly evident when activity was measured immediately following drug (0.1 mg/kg) administration (5–20 min after drug, Figure 3c). Indeed, all groups were initially (5–20 min after 0.1 mg/kg drug, Figure 3c) highly sensitive to quinpirole (60–80% reduction in activity) but the hyperactive groups of mice later (20–40 min after drug, Figure 3b) became less sensitive (40–50% reduction). Since its dose dependence, we speculate that the reduced drug effect in the hyperactive groups of mice could be due to enhanced D2 receptor desensitization. Among DA receptors, the D2 is particularly sensitive to agonist-induced phosphorylation and β-arrestin and/or GRK2/3-mediated sequestration (Cho et al, 2006). Drug disposition differences between WT> WT and hyperactive animals are also possible but less likely because the similar response of the different groups of animals to the low dose of quinpirole.
Figure 3.

Effects of various dopaminergic drugs on locomotor activity in WT and KO mice. WT and KO male offspring are indicated by solid and open columns, respectively (mean + SE). Littermates are highlighted by a gray background. (a) The maternal effect becomes apparent from 20 min in the test is shown. Also illustrated is the dosing and testing timeline for the locomotor assays. (b) Suppression of locomotor activity by quinpirole is impaired in the hyperactive groups of mice (H> WT, H> KO, and KO> KO) at the higher 0.1 mg/kg dose during the 40–60 min period (main effects ANOVA, maternal genotype: F(1, 31) = 4.79, p = 0.035; LSD post hoc test, WT vs mutant (H and KO) mothers, p = 0.036; direct comparison of the groups with one-way ANOVA with LSD post hoc test: *p < 0.05; dashed line represents trend only; no. of offspring (N) = 8–12 per group). (c) Quinpirole dose dependently suppresses locomotor activity in all groups of mice 5–20 min after drug administration (neither maternal nor genotype effect is detected) (main effects ANOVA, maternal genotype: F(1, 32) = 0.41, p = 0.52; offspring genotype: F(1, 32) = 1.18, p = 0.28; no. of offspring (N) = 8–12). (d) D3 receptor function is not altered in hyperactive mice. PD128907 reduced habituated locomotor activity as recorded between 40 and 60 min of the locomotor test equally among the various, including WT> WT and H> WT, groups (no. of offspring (N) = 9–12 per group); thus no maternal effect is seen. (e) Amphetamine resulted in increased locomotor activity at 4 mg/kg in all groups (see text) but was less effective in the H> KO group (*p < 0.05) (no. of offspring (N) = 7–12 per group). There was no difference between WT> WT and H> WT mice indicating the absence of maternal effect.
Since quinpirole is a D2/D3 agonist, it is possible that the blunted drug response of hyperactive mice involves the D3 rather than the D2 receptor. To test this possibility, locomotor activity was measured following the administration of the preferential D3 agonist PD128907 (Figure 3d). The drug, administered at 0.1 and 0.3 mg/kg, reduced locomotor activity (main effects ANOVA, treatment: F(2, 88) = 8.17, p = 0.00056) equally in all groups (no maternal or genotype effect present; maternal genotype effect: F(1, 27) = 1.91, p = 0.17 and F(1, 29) = 0.17, p = 0.68 for 0.1 and 0.3 mg/kg; offspring genotype effect: F(1, 27) = 1.89, p = 0.18 and F(1, 29) = 0.14, p = 0.70 for 0.1 and 0.3 mg/kg, respectively). Taken together, these data suggest that the D2 rather than the D3 receptor is responsible for the differential behavioral effect of quinpirole in the WT> WT vs hyperactive mice. These data are also consistent with the findings that deletion of the D2 receptor abolishes the locomotor suppressant effect of quinpirole (Usiello et al, 2000; Wang et al, 2000).
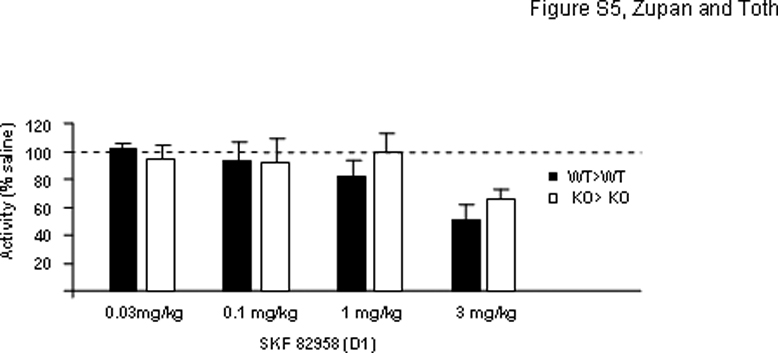
Next, we studied possible postsynaptic DA receptor alterations in hyperactive mice. Most but not all D1-like agonists result in increased locomotor activity at low to moderate doses and some of them can even inhibit activity at higher doses (Desai et al, 2005). The difficulty to correlate the pharmacology (activation of adenylate cyclase) of these drugs with their behavioral effect is likely due to the limited selectivity and intrinsic activity of D1-like agonists. The D1-like agonist SKF 82958 resulted in no increase in activity up to 1 mg/kg while the higher 3 mg/kg dose caused similar reductions in activity in WT> WT and KO> KO mice (Supplementary Figure S5). SKF 81297, another D1 receptor agonist, at doses which have been reported to increase activity in mice (Kim et al, 2000) also failed to increase activity in our mice (data not shown). Finally, the indirect DA receptor agonist amphetamine was tested to further probe the dopaminergic system. Although amphetamine was without effect at the 2 mg/kg dose, the 4 mg/kg dose significantly increased locomotor activity in all groups (between 40 and 60 min; main effects ANOVA, treatment: F(2, 83) = 252.89, p < 0.000001; LSD post hoc test: saline vs 4 mg/kg, p < 0.000001; Figure 3e). There seemed to be, however, an offspring genotype effect (F(2, 83) = 4.12, p = 0.02) indicated by the blunted response of H> KO mice to amphetamine as compared to that of the WT> WT and H> WT mice (LSD post hoc test, WT vs KO: p < 0.05). A similar difference in amphetamine response between WT and KO mice was also reported by Ventura et al (2004). These data suggest an offspring genotype effect (hyperactivity in H> KO) but not a maternal effect (H> WT mice are normal) in the amphetamine response; thus, amphetamine is not informative as a probe to decipher the mechanism of maternal effect.
CONCLUSION
Here we show that by generating two littermate (H> WT and H> KO) and two non-littermate (WT> WT and KO> KO) offspring of heterozygote and homozygote fmr-1 KO and WT mothers in parallel, it is possible to identify and separate behavioral abnormalities that are caused by genetic and maternal effects. Although all of these four combinations were used in most of our experiments, three groups (WT> WT, H> WT, and H> KO) are sufficient to determine the genotype and maternal effects.
We demonstrate that hyperactivity of offspring can be maternally transmitted (both WT and KO pups are hyperactive as long as the mother is H or KO) while reduced startle, increased PPI and audiogenic seizures were strictly dependent on the offspring’s genotype. Importantly, both H and KO females exhibit hyperactivity (Musumeci et al, 2000; Qin et al, 2005) suggesting that this behavior may be transmitted epigenetically from mother to offspring. Since the D2 receptor is fundamentally important in regulating the DA system and behavioral activity, and because the response of hyperactive offspring to a D2 agonist is reduced, the receptor or its coupling/signalling is a candidate target for the maternal effect.
The maternal genotype effect in the fragile X mouse model is consistent with data in a recent human study that analyzed maternal–child interactions in dyads affected by fragile X syndrome. Mothers of children with fragile X (all carriers but unknown full or permutation status) use primarily ‘maintaining behavior’ (maintaining the attention of a child) in interaction with their children and less ‘directive behaviors’ (structural requests from the mother) and these mothers have clinically significant levels of stress, depression and anxiety at a prevalence rate higher than that of the general public (Wheeler et al, 2007). Also, child behavior problems in these families contributed to maternal stress, and mothers with higher stress engaged in fewer interactions with their children. These raise the possibility that the environment provided by mothers with fragile X children may have influences on the development of their children.
Maternal genotype effects involving CNS-specific genes have previously been reported in rodents but none of these studies investigated the maternal effect in a disease context. For example, mutation of the circadian clock gene (Clock−/−) or the gene encoding the cyclic AMP response element-binding protein (CREB-αΔ−/−) disrupts maternal nurturing behavior and leads to suboptimal pup care resulting in stunted pup growth or neonate death (Hoshino et al, 2006; Jin et al, 2005). Also, the maternal tau−/− genotype has a strong negative effect on offspring weight regardless of the pup genotype in Syrian hamsters (Oklejewicz et al, 2001). Similarly, genetic inactivation of the tryptophan hydroxylase1 gene resulting in a deficiency in peripheral serotonin synthesis in the mother but not in the offspring leads to small and abnormally developed pups (Cote et al, 2007). Some other studies reported maternal genotype effects on offspring behavior. Lack of several proteins and hormones, including adrenomedullin, have been shown to alter maternal physiology and consequently offspring behavior (Li et al, 2006). In another report, variability in maternal care (Francis and Meaney, 1999) was demonstrated to alter the level of anxiety in the offspring via an epigenetic regulation of the glucocorticoid receptor (Weaver et al, 2004). To the best of our knowledge, maternal transmission of a phenotype specific for a disease-associated gene has not yet been reported, thus making our findings the first indication that some psychiatric disease-related phenotypes may be maternal genotype dependent.
We believe that our finding has also applicability to the general rules of breeding genetically modified and matched control mice. It is now a routine practice to use heterozygote parents to generate mutant mice and their WT littermate controls. The behavioral differences between such groups reflect mostly genetic effects. However, it has become more and more obvious that human disease is linked to both genetic and environmental such as parental effects. By adding WT> WT mice as a second control group, it would be possible to identify and dissociate parentally and genetically transmitted behavioral phenotypes in the offspring. The combination and interaction of these effects would better reflect the full spectrum of phenotypes linked to disease-associated mutations.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the FRAXA Foundation.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
No conflict of interest for Bojana Zupan and Miklos Toth.
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp).
References
- Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- Cho DI, Beom S, Van Tol HH, Caron MG, Kim KM. Characterization of the desensitization properties of five dopamine receptor subtypes and alternatively spliced variants of dopamine D2 and D4 receptors. Biochem Biophys Res Commun. 2006;350:634–640. doi: 10.1016/j.bbrc.2006.09.090. [DOI] [PubMed] [Google Scholar]
- Consortium TD–BFX. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Cory-Slechta DA, Zuch CL, Fox RA. Comparison of the stimulus properties of a pre- vs. a putative postsynaptic dose of quinpirole. Pharmacol Biochem Behav. 1996;55:423–432. doi: 10.1016/s0091-3057(96)00113-x. [DOI] [PubMed] [Google Scholar]
- Cote F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J, et al. Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci USA. 2007;104:329–334. doi: 10.1073/pnas.0606722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai RI, Terry P, Katz JL. A comparison of the locomotor stimulant effects of D1-like receptor agonists in mice. Pharmacol Biochem Behav. 2005;81:843–848. doi: 10.1016/j.pbb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- D’Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, et al. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience. 1997;76:367–376. doi: 10.1016/s0306-4522(96)00224-2. [DOI] [PubMed] [Google Scholar]
- Francis DD, Meaney MJ. Maternal care and the development of stress responses. Curr Opin Neurobiol. 1999;9:128–134. doi: 10.1016/s0959-4388(99)80016-6. [DOI] [PubMed] [Google Scholar]
- Grace A. Psychostimulant actions on dopamine and limbic system function: relevance to the pathophysiology and treatment of ADHD. In: Solanto MV, Arnsten AFT, editors. Stimulant Drugs and ADHD: Basic and Clinical Neuroscience. Oxford University Press; New York: 2001. pp. 134–155. [Google Scholar]
- Hagerman RJ. Physical and behavioral phenotype. In: Cronister A, editor. Fragile X Syndrome: Diagnosis, Treatment and Research. The Johns Hopkins University Press; Baltimore: 1996. pp. 3–87. [Google Scholar]
- Hoshino K, Wakatsuki Y, Iigo M, Shibata S. Circadian Clock mutation in dams disrupts nursing behavior and growth of pups. Endocrinology. 2006;147:1916–1923. doi: 10.1210/en.2005-1343. [DOI] [PubMed] [Google Scholar]
- Jin SH, Blendy JA, Thomas SA. Cyclic AMP response element-binding protein is required for normal maternal nurturing behavior. Neuroscience. 2005;133:647–655. doi: 10.1016/j.neuroscience.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Kim DS, Szczypka MS, Palmiter RD. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci. 2000;20:4405–4413. doi: 10.1523/JNEUROSCI.20-12-04405.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Swerdlow NR. The functional output of the mesolimbic dopamine system. Ann N Y Acad Sci. 1988;537:216–227. doi: 10.1111/j.1749-6632.1988.tb42108.x. [DOI] [PubMed] [Google Scholar]
- Koob GF, Stinus L, Le Moal M. Hyperactivity and hypoactivity produced by lesions to the mesolimbic dopamine system. Behav Brain Res. 1981;3:341–359. doi: 10.1016/0166-4328(81)90004-8. [DOI] [PubMed] [Google Scholar]
- Li M, Yee D, Magnuson TR, Smithies O, Caron KM. Reduced maternal expression of adrenomedullin disrupts fertility, placentation, and fetal growth in mice. J Clin Invest. 2006;116:2653–2662. doi: 10.1172/JCI28462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzocco MM, Holden JJ. Neuropsychological profiles of three sisters homozygous for the fragile X premutation. Am J Med Genet. 1996;64:323–328. doi: 10.1002/(SICI)1096-8628(19960809)64:2<323::AID-AJMG18>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Mila M, Castellvi-Bel S, Gine R, Vazquez C, Badenas C, Sanchez A, et al. A female compound heterozygote (pre- and full mutation) for the CGG FMR1 expansion. Hum Genet. 1996;98:419–421. doi: 10.1007/s004390050232. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, et al. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Derber WJ, McClellan DA, Crnic LS. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 2002;927:8–17. doi: 10.1016/s0006-8993(01)03309-1. [DOI] [PubMed] [Google Scholar]
- Oklejewicz M, Pen I, Durieux GC, Daan S. Maternal and pup genotype contribution to growth in wild-type and tau mutant Syrian hamsters. Behav Genet. 2001;31:383–391. doi: 10.1023/a:1012274419184. [DOI] [PubMed] [Google Scholar]
- Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL. (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet. 2000;9:1145–1159. doi: 10.1093/hmg/9.8.1145. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB. A null mutation for Fmr1 in female mice: effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience. 2005;135:999–1009. doi: 10.1016/j.neuroscience.2005.06.081. [DOI] [PubMed] [Google Scholar]
- Starke K, Gothert M, Kilbinger H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, et al. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30:819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Toledano-Alhadef H, Basel-Vanagaite L, Magal N, Davidov B, Ehrlich S, Drasinover V, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351–360. doi: 10.1086/321974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav Pharmacol. 2004;15:433–442. doi: 10.1097/00008877-200409000-00018. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB. Dopamine D2 long receptor-deficient mice display alterations in striatum-dependent functions. J Neurosci. 2000;20:8305–8314. doi: 10.1523/JNEUROSCI.20-22-08305.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Wheeler A, Hatton D, Reichardt A, Bailey D. Correlates of maternal behaviours in mothers of children with fragile X syndrome. J Intellect Disabil Res. 2007;51:447–462. doi: 10.1111/j.1365-2788.2006.00896.x. [DOI] [PubMed] [Google Scholar]
- Yun SW, Platholi J, Flaherty MS, Fu W, Kottmann AH, Toth M. Fmrp is required for the establishment of the startle response during the critical period of auditory development. Brain Res. 2006;1110:159–165. doi: 10.1016/j.brainres.2006.06.086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.