

Abstract
Human Islet Amyloid Polypeptide (hIAPP) is a highly amyloidogenic protein found in islet cells of patients with type II diabetes. Because hIAPP is highly toxic to beta-cells under certain conditions, it has been proposed that hIAPP is linked to the loss of beta-cells and insulin secretion in type II diabetics. One of the interesting questions surrounding this peptide is how the toxic and aggregation prone hIAPP peptide can be maintained in a safe state at the high concentrations that are found in the secretory granule where it is stored. We show here zinc, which is found at millimolar concentrations in the secretory granule, significantly inhibits hIAPP amyloid fibrillogenesis at concentrations similar to those found in the extracellular environment. Zinc has a dual effect on hIAPP fibrillogenesis: it increases the lag-time for fiber formation and decreases the rate of addition of hIAPP to existing fibers at lower concentrations, while having the opposite effect at higher concentrations. Experiments at an acidic pH which partially neutralizes the change in charge upon zinc binding show inhibition is largely due to an electrostatic effect at His18. High-resolution structures of hIAPP determined from NMR experiments confirm zinc binding to His18 and indicate zinc induces localized disruption of the secondary structure of IAPP in the vicinity of His18 of a putative helical intermediate of IAPP. The inhibition of the formation of aggregated and toxic forms of hIAPP by zinc provides a possible mechanism between the recent discovery of linkage between deleterious mutations in the SLC30A8 zinc transporter, which transports zinc into the secretory granule, and type II diabetes.
INTRODUCTION
Human Islet Amyloid Polypeptide (hIAPP) is a polypeptide hormone secreted from pancreatic β-cells in response to glucose or other chemical signals. Under normal conditions, hIAPP is involved in glycemic control.1 However, in type II diabetics hIAPP aggregates in the pancreas to form dense, insoluble extracellular fibrillar deposits known as amyloid fibers. The amyloid deposits are composed of β-sheet aggregates with a characteristic structure similar to those found in Alzheimer’s, Parkinson’s, Huntington’s and a variety of other degenerative diseases.2 While hIAPP aggregates at nanomolar concentrations in vitro, forming both the amyloid fibers characteristic of type II and smaller oligomeric species that have been repeatedly linked to β-cell destruction,3–7 it is safely stored in the secretory granule at millimolar concentrations.8 The aggregation of hIAPP is highly sensitive to circumstances in which the experiment is run; both the kinetics and the morphology and yield of the final amyloid product of IAPP can be influenced by subtle variations in experimental conditions.9, 10 Given that hIAPP in isolation spontaneously aggregates at concentrations 2 to 3 orders of magnitude lower than those present in the secretory granule where it is stored, it is reasonable to look for other factors that act as chaperones to stabilize hIAPP in a non-toxic form in normal individuals. 11, 12
There is considerable evidence that the dysregulation of such amyloid chaperones acts as a trigger for the pathological aggregation of other amyloid proteins.11, 12 The dysregulation of metal ion homeostasis in particular has received attention due to the early discovery of a high prevalence of metal ions in amyloid deposits. High affinity metal binding sites have been identified in a high percentage of amyloid proteins including β2-microglubulin,13, 14 amyloid-beta (Aβ),15–20 α-synuclein,21–23 superoxide dismutase,24, ABri,25 NAC,26 and mammalian prion protein.27 In some cases, such as with Parkinson’s disease, clinical and epidemiological evidence directly links an increased heavy metal intake in environmental contaminated areas to an abnormally high incidence of neurodegenerative disease.28–30 In other cases, a pathological amyloid aggregation appears to be caused by a deficient metal transport process as shown by the significant reduction in amyloid plaques found in mice deficient for the zinc transporter ZnT3.31
The binding of metal ions can have profound effects on amyloid aggregation, frequently causing rapid precipitation, a dramatic increase in fibillogenesis, changes in the toxicity of the amyloid aggregates, and alterations in the morphology of the final aggregate product. Even the transient millisecond exposure to zinc upon the synaptic pulsing of neurons can rapidly stimulate Aβ aggregation.20 In contrast to other amyloid proteins for which high-affinity metal binding sites have been identified, the influence of metal binding on hIAPP has not been clearly established. There is evidence that copper can stimulate the production of H2O2 by hIAPP,32 possibly generating oxidative stress in a manner similar to Aβ.33 In addition to its possible role in generating β-cell oxidative stress, copper (II) has also been found to mediate the membrane-interaction of a fragment (17–29) of hIAPP.34 Similar modifications of membrane interactions have been found with calcium and full-length hIAPP.35 While evidence exists for the interaction of Cu(II) with hIAPP, the ability of other metals to affect hIAPP has been almost entirely unexplored. Zinc is of particular interest as the zinc content of pancreatic β-cells is among the highest in the body and various clinical and epidemiological studies suggest zinc deficiency is a common symptom of type II diabetes.36, 37 The recent discovery between the genetic linkage between the SLC30A8 gene,38–41 which transports zinc into the secretory granule where insulin and hIAPP are stored,42–44 and type II diabetes suggests zinc could have an impact on hIAPP cytotoxicity towards β-cells.
Materials and Methods
Peptide synthesis
Human-IAPP (sequence KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY) was synthesized with an amidated C-terminus and a disulfide bridge between residues 2 and 7 using t-Boc-protected amino acids on MBHA resin using a standard solid-phase synthesis protocol.45 The crude product was dissolved in 3.5M GuHCl and purified on a reverse-phase HPLC on a Waters semipreparative C18 column equilibrated in 0.1% trifluoricacetic acid and eluted with a linear gradient from 0% to 80% acetonitrile at a flow rate of 10 ml/min. After lyophilization, the semi-pure product was then dissolved in pure water to 2 mg/ml and oxidized with Thallium (III) trifluoroacetate according to the method of Page et al,46 using a 2 hour reaction time with stirring followed by additional purification by reverse-phase HPLC. The purity of the final product (>95%) was checked by analytical HPLC and MALDI-MS.
Kinetic studies using thioflavin fluorescence
The kinetics of hIAPP amyloid formation was measured using the increase in fluorescence intensity upon binding of the amyloid fiber to the amyloid specific dye Thioflavin T (ThT). Before the start of the experiment hIAPP was first solubilized in hexafluoroisopropanol at a concentration of 250 μM to break up any pre-existing aggregates. To start the aggregation experiment, the lyophilized peptide was first dissolved in deionized water at 4 ºC to a concentration of 250 μM, spun through a 0.22 μm filter, and then diluted to a concentration of either 5 μM or 10 μM with a buffer containing the appropriate amount of ZnCl2 (100 mM Tris with 100 mM NaCl and 25 μM ThT for experiments at pH 7.5, 100 mM sodium acetate with 100 mM NaCl and 25 μM ThT for experiments at pH 5.5). The pH of each sample was individually adjusted to the correct value due to the acidity of ZnCl2. All buffers were previously passed through a Chelex-100 column to remove trace amounts of endogenous zinc. Tris buffer was used due to its low affinity for zinc.
Experiments were performed in sealed Corning 96 well clear bottom half area, non-binding surface plates. Time traces were recorded with Biotek Synergy 2 plate reader using a 485 excitation filter and a 505 emission filter at a constant temperature of 25°C without shaking. The time-dependence of ThT fluorescence was fitted to a sigmoidal growth model that has empirically been found to reproduce most of the features of amyloid aggregation:
| Eq.1 |
where I0 and Imax are the initial and maximum fluorescence values, t1/2 is the time required to reach half intensity, and k is an apparent first order rate constant for the addition of hIAPP to existing fibers. 47 The lag-time t0, the time predicted by nucleation dependent polymerization theory before detectable amyloid formation occurs, is described by t0=t1/2-2/k.
Electron microscopy
Solutions of hIAPP were allowed to incubate for 2 days at pH 7.5 before loading in the presence of either 0,100 or 1000 μM ZnCl2. Samples were prepared identically to those used for ThT kinetic experiments except the concentration of hIAPP was 100 μM and ThT was not included in the sample. After incubation, 5μL aliquots were loaded onto Formvar-coated copper grids (Ernest F. Fullam, Inc., Latham, NY) for 2 minutes, washed twice with 10 μl deionized water, and then negatively stained for 90 seconds with 2% uranyl acetate. Samples were imaged with a Philips CM10 Transmission Electron Microscope at 6500x or 15000x magnification.
NMR sample preparation
Preformed aggregates of hIAPP were removed by dissolving the peptide in hexafluoroisopropanol (HFIP) and removing the solvent by lyophillization as described above. The lyophilized powder was then dissolved first in deuterated HFIP/trifluoroethanol (TFE) and then buffer so that the final solution was 1mM hIAPP in 22% HFIP, 8%TFE, 20 mM deuterated Tris at pH 7.5. The spectrum with zinc was obtained by adding zinc to a final concentration of 10 mM from a 100 mM stock solution.
NMR data collection and processing
All the NMR spectra of hIAPP1–37 were recorded at 30 ºC using a 900 MHz Bruker Avance NMR spectrometer equipped with a triple-resonance z-gradient cryogenic probe optimized for 1H-detection. Backbone and side-chain assignments were done with the help of homonuclear experiments such as 2D 1H-1H TOCSY (Total Correlation Spectroscopy) and 2D 1H-1H NOESY (Nuclear Overhauser Enhancement Spectroscopy) recorded at 80 and 300 ms mixing time, respectively. Complex data points were acquired for quadrature detection in both frequency dimensions for the 2D experiments and all the spectra were zero-filled in both dimensions to yield matrices of 2048 × 2048 points. Proton chemical shifts were referenced to the methyl signal of 2,2-dimethyl-2-silapentane-5-sulfonate (Cambridge Isotope Laboratories) as an external reference at 0 ppm. All 2D spectra were processed using TopSpin 2.1 software and analyzed using SPARKY.48 Initial resonance assignments were carried out using a standard approach reported elsewhere.49
Structure calculations
The final structure was calculated with the CYANA 2.1 program package using simulated annealing in combination with molecular dynamics in torsion angle space.50 Dihedral angle restraints were calculated directly from the from the NOE connectivities.51 During the first round of structure calculations, only unambiguous long-range NOE constraints were used to generate a low-resolution fold for the structure. Assignments of the remaining ambiguous NOE cross-peaks were made in an iterative fashion by applying a structure-aided filtering strategy in repeated rounds of structure calculations.52 After the complete assignment of resonances, a total of 100 conformers were calculated in 8000 annealing steps each and the best 10 low energy conformers were selected and visualized using MOLMOL.53
RESULTS
Zinc Reduces the Extent of hIAPP Amyloid Formation
In order to determine if zinc can directly affect amyloidogenesis by hIAPP, we monitored the rate of fibril formation of solutions of 5 μM and 10 μM hIAPP in the presence of micromolar to millimolar concentrations of ZnCl2 using the fluorescence of the dye Thioflavin T (Figure 1) as a marker. Thioflavin T’s fluorescence increases markedly upon binding to the stacked β-sheets of amyloid fibers making it a useful specific marker of amyloid formation as other prefibrillar aggregates which lack the characteristic cross β-sheet superstructure of the amyloid fibers do not increase its fluorescence substantially. Zinc significantly affects the formation of IAPP amyloid fibers, as shown by the sharp reduction in Thioflavin T fluorescence when zinc is added to the incubation solution (Figure 2). Amyloid formation is strongly affected by zinc in the micromolar concentration range found in the secretory granule, with the final Thioflavin T fluorescence value at the end of the incubation period decreasing to an intensity of about half the zinc free value at 250 μM ZnCl2 before apparent saturation is reached at approximately 750 μM ZnCl2 (Figure 2).
Figure 1. Zinc inhibits hIAPP amyloid fibril formation at pH 7.5.

Amyloid formation by hIAPP as a function of zinc concentration as followed by the amyloid-specific dye Thioflavin T. (A) 5μM and (B) 10 μM hIAPP with indicated concentrations of ZnCl2. Traces are averages for n=9 (5 μM hIAPP) and n =7 (10 μM hIAPP) runs. The kinetic analysis of these traces and the variation of the final THT fluorescence intensity as a function of zinc concentration (along with the associated error bars) are given in Figures 4 and 2 respectively.
Figure 2. Zinc decreases the equilibrium concentration of hIAPP fibers at pH 7.5.

Variation in the fluorescence intensity of the amyloid-bonding dye Thioflavin T as a function of ZnCl2 concentration after fibrillogenesis has completed as extracted from the experimental results presented in Figure 1. Values for 5 μM and 10 μM were determined after 2 and 1 day(s) respectively. The intensity values were normalized to hIAPP in the absence of zinc. Error bars indicate standard error of measurement (S.E.M) for n=9 (5 μM hIAPP) and n =7 (10 μM hIAPP). Inset shows the effect at lower ZnCl2 concentrations (0 - 1000 μM).
Zinc Reduces Amyloid Deposition But Does Not Significantly Alter the Final Fiber Morphology
The results from the Thioflavin T fluorescence assay suggest the stability of the amyloid fibers is negatively affected by zinc. However, some inhibitors are known to interfere with the binding of Thioflavin T to amyloid fibers, leading to a false positive result for inhibition.54, 55 Accordingly, we confirmed the results of the Thioflavin T assay by examining the ultrastructure of the amyloid fibers by electron microscopy. In the absence of zinc, 100 μM solutions of hIAPP massively aggregate to form the dense mats of the long branched fibers typical of amyloid proteins within 48 hours (Figure 3a). The addition of either 100 μM or 1mM ZnCl2 during incubation causes a significant reduction in the amount of amyloid deposition on the grid (compare Figures 3b and 3c to 3a), giving an independent confirmation of the results of the Thioflavin T assay.
Figure 3. Zinc reduces the density of hIAPP fibrils while maintaining a similar fibril morphology.

Top: Electron micrographs showing the effect of zinc on total amyloid deposition and fiber morphology. Aliquots from a 100 μM hIAPP solution incubated with (A) 0 μM ZnCl2, (B) 100 μM ZnCl2 and (C) 1000 μM ZnCl2 were deposited on copper grids after an incubation period of 2 days and imaged at 6500x magnification. Bottom: Electron micrographs of hIAPP incubated with (D) 0 μM ZnCl2, (E) 100 μM ZnCl2 and (F) 1000 μM ZnCl2 imaged at 15000x magnification.
The morphology of amyloid fibers is frequently sensitive to the conditions at which they assemble (18–20). Typically, amyloid fibers grown in the presence of an amyloid inhibitor are usually both thinner and shorter than those grown in its absence due to an enhancement of the fiber breakage rate and impairment of the association of the protofilament subunits that make up the amyloid fiber bundle.56–60 Interestingly, while the total amount of fibers is greatly reduced by zinc, the overall morphology of the individual amyloid fibers remains similar within the limits of the resolution of our instrument. Fibril diameters of ~10 nm were observed consistently in all samples in agreement with previous reports,61 indicating zinc does not lead to a decrease in filament width (Figure 3d-f). Noticeably shorter or significantly thinner fibers were not seen, indicating Zn+2 does not likely significantly enhance breakage of the fiber or greatly impair the lateral attachment of protofilaments to mature amyloid fibers. Similar results have been obtained with CuCl2.62 Furthermore, large amorphous non-fibrillar precipitates, which have frequently been observed with other metal-complexed amyloids,20, 25, 63 were not detected at either zinc concentration. The inability of the zinc to alter the final fiber morphology likely indicates that some of the IAPP forms fibers are relatively unaffected by zinc while another significant fraction remains soluble and not extensively aggregated.
Zinc Decreases the Rate of Amyloid Formation at Low Concentrations but Stimulates it at Higher Concentrations
Amyloid fibers are typically formed in a multistep process that is highly dependent on the formation of energetically unfavorable nuclei. In the initial stages of aggregation before a sufficient population of nuclei are formed (the lag-phase), amyloid formation proceeds very slowly and only trace amounts of amyloid fibers are formed. Once a critical concentration of nuclei is reached, aggregation proceeds exponentially by the addition of protein to the ends of the existing fibers until virtually all of the protein is converted to amyloid at equilibrium. Examination of the effect of an inhibitor on the kinetic profile is frequently helpful in distinguishing its mode of action. The length of the lag-time before detectable amounts of amyloid fibers are formed, for example, is an indication of the relative stability of early intermediates in the aggregation process. A compound that stabilizes this rare intermediate can be expected according to the nucleation polymerization model to reduce the lag-time, while one that destabilizes it can be expected to have the opposite effect unless it is excessively stabilized to form a stable intermediate. Similarly, the steepness of the polymerization reaction is a reflection of the rate of growth at fiber ends and secondary nucleation (creation of growth sites by fiber breakage or branching).
It has been empirically found that amyloid kinetics frequently follow a sigmoidal growth model (Eq. 1).47 Analysis of the aggregation kinetics by this equation reveals a complex dependence of the aggregation rate on the zinc concentration with differing effects on the rate of nucleation and the rate of fibril growth. In particular, the nucleation rate is affected in a multi-modal fashion as depicted in a plot of the lag-time before the onset of fiber growth (Figure 4). While low concentrations of zinc up to approximately 100 μM cause a modest increase in the lag-time of 5 μM hIAPP (Figure 4a inset), this effect is reversed as the zinc concentration is further increased with the lag-time decreasing nearly linearly before it plateaus at approximately half the zinc free value at 1000 μM ZnCl2 (Figure 4a). A similar effect can be seen when hIAPP is incubated at 10 μM concentration, although the concentration range in which the lag-time is decreased is comparatively much less (Figure 4b).
Figure 4. Zinc has a bimodal effect on hIAPP fibrillogenesis at pH 7.5.

Analysis of the kinetics of hIAPP at pH 7.5 according to a sigmoidal growth model. A variation in the lag-time (t1/2-2/k) before detectable fiber formation as a function of ZnCl2 at (A) 5 μM and (B) 10 μM hIAPP. Variations in the apparent first-order elongation rate constant (k) at (C) 5 μM and (D) 10 μM hIAPP. Error bars indicate standard error of measurement (S.E.M) for n=9 (5 μM hIAPP) and n =7 (10 μM hIAPP). Insets show the effect from 0–500 μM ZnCl2.
While zinc has a relatively modest effect on the nucleation rate at physiological concentrations, it has a stronger and relatively consistent effect on the fibril growth rate. Zinc has a strong inhibitory effect on the fibril growth rate of 5 μM hIAPP at all concentrations decreasing it monotonically at all zinc concentrations except the two highest (Figure 4c). However, at 10 μM hIAPP the multimodal effect of zinc becomes apparent for the elongation rate as well, decreasing the fiber growth rate sharply up to 250 μM (approximately 80% reduction) but then slowly increasing the rate at millimolar concentrations. Two competing mechanisms are apparently in simultaneous operation - one mechanism dominant at lower ZnCl2 concentrations leading to an increase in the lag-phase and retardation of fibril growth and another dominant at higher ZnCl2 concentrations leading to a both reduction in the lag-phase and acceleration of fibrillation but a reduction in the amount of the total amount of amyloid present at equilibrium.
Inhibition of Fiber Formation is Specific for Metals that are Good Ligands for Histidine
To determine if the effect of ZnCl2 on the aggregation of IAPP is specific to Zn or is mediated by a non-specific process, fibril formation of 7.5 μM IAPP was monitored in the presence of 100 μM NH4Cl, CaCl2, MgCl2, ,ZnCl2 or CuCl2. As shown in Figure 5, the strongly kosmotropic salt NH4Cl had little effect on the aggregation of IAPP in comparison to ZnCl2. The equilibrium THT fluorescence at 3000 minutes, lag-time, and elongation rate are similar to the control sample without NH4Cl. Similarly, the chaotropic salts CaCl2 and MgCl2 also had little effect on the aggregation kinetics. This is significant because, although Ca+2 and Mg+2 are divalent cations like Zn+2, they are poor ligands for imidazole which implies imidazole has a specific role in zinc-influenced IAPP aggregation. On the other hand CuCl2, which has a strong affinity for the imidazole group and is known to specifically bind to IAPP, had an even stronger effect than ZnCl2. This is an indication that ZnCl2 exerts its influence by specific binding to imidazole, rather than through a non-specific change in solvent structure, at least at lower cation concentrations.
Figure 5. Inhibition of aggregation is specific for cations that bind imidazole.

100 μM NaCl, NH4Cl,MgCl2, CaCl2, or ZnCl2 was added to 7.5 μM hIAPP in 100 mM Tris, 100 mM NaCl, pH 7.5 buffer and amyloid formation was followed by changes in the fluorescence of the amyloid specific dye Thioflavin T (25 μM). Samples were shaken at 60 Hz. Traces are averages for n= 8 experiments.
Fibrillogenesis is Acclerated by Zinc at Acidic pH
To partially isolate the electrostatic effect of zinc binding we performed experiments at a lower pH. In contrast to the complex effects zinc has on the kinetic profile at pH 7.5, zinc has a much simpler effect at pH 5.5 when His-18 is already protonated and the change in charge upon zinc binding is drastically less. At pH 5.5, zinc decreases the lag-time and increases the elongation rates in a monotonic and nearly linear fashion (Figure 6) in contrast to the more complex effects seen at pH 7.5. These results suggest that while the inhibitory effect of zinc is likely due to the unfavorable electrostatics of incorporating zinc into the amyloid fiber, an additional mechanism also exists that is not dominated by this effect and favors the creation of the amyloid fiber.
Figure 6. Zinc accelerates hIAPP amyloid fibril formation at pH 5.5.

Kinetics of hIAPP amyloid formation at pH 5.5. (A) Amyloid formation by 10 μM hIAPP as a function of zinc concentration. (B) Decrease in the lag-time (t1/2-2/k) before detectable fiber formation as a function of ZnCl2 concentration according to the sigmoidal growth model (Eq. 1). (C) Increase in the elongation rate as a function of ZnCl2 concentration according to the sigmoidal growth model.
Zinc Binding Causes Localized Disruption of the Secondary Structure Near His-18
To gain structural insight into this process, we solved the atomic-level resolution structure of human-IAPP in an organic solvent solution (22% HFIP 8%TFE) which strongly promotes the formation of α-helices in the presence and absence of a ten-fold excess of ZnCl2 (Figure 7) using NMR experiments. This solvent mixture was used to both minimize the rapid aggregation of IAPP at neutral pH and to capture some features of the helical intermediate proposed as a critical step in the progression of the largely unstructured hIAPP monomer to amyloid fibers. 64, 65 Helix-formation is frequently thermodynamically linked to aggregation,64, 65 possibly because of the smaller conformational space that must be searched to bring two aggregation-prone regions together for peptides that are initially partially helical as opposed to completely unstructured.8, 66, 67 In hIAPP this relationship has been shown by the dramatic decrease in the length of the lag-phase by the addition of cosolvents which favor the formation of the helical conformation,68–70 the rapid onset of aggregation after the conformational transition to the helical state that occurs upon binding to phospholipid membranes,5, 71 8, 70 and by the effect of mutations that are expected to alter the intermolecular interfaces likely to be present in helical oligomers but not in the β-sheet conformation of the amyloid fiber.72, 73
Figure 7.

The finger print region of 2D 1H-1H NOESY spectra of hIAPP in the absence (A) and presence (B) of 10 mM ZnCl2 showing the connectivities among Hα nuclei and resonance assignment.
The structure of hIAPP in the absence of zinc is defined by a kinked helix from R11 to T30 (Figure 8, statistics are reported in Table 1). The kink is most likely stabilized by a clustering of hydrophobic residues on one side of the helix. The N- and C-terminal regions beyond these residues are disordered, consistent with previously determined structures of hIAPP. 74–76 As expected, it strongly resembles the structure of hIAPP in 35% HFIP .74 Significantly, the structure is also a relatively close match to the crystal structure of the hIAPP dimer fused to maltose-binding protein (MBP) as it preserves the helix-kink-helix architecture that is the central motif in both structures.72 The similarities in this respect is likely significant, as previous studies have shown that the helix-kink-helix motif detected here is very likely an early on-pathway intermediate to aggregation.72 Mutations that destabilized the N-terminal helix or disrupted the packing interface between helices resulted in severely disrupted amyloid formation.72, 73 Conformational changes in the NMR structure after zinc binding can therefore reasonably be interpreted as a conformational change in early intermediates in hIAPP aggregation.
Figure 8. Zinc binding causes a local disruption of the secondary structure in the vicinity of His-18.
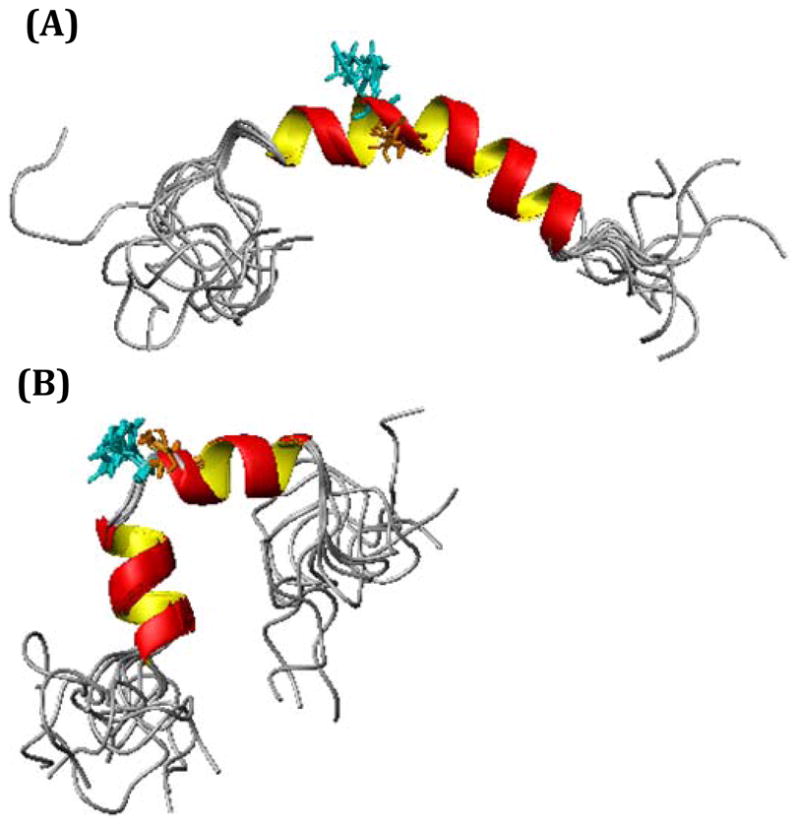
High-resolution NMR structures of hIAPP in the absence (A) and presence of 10 mM ZnCl2 (B).
Table 1.
Statistical information for the hIAPP and hIAPP-Zn+2 structural ensembles
| IAPP | IAPP+Zn2+ | ||
|---|---|---|---|
| Distance constraints | Total | 435 | 460 |
| Intraresidual | 156 | 171 | |
| Interresidual | 279 | 289 | |
| Sequential (i-j = 1) | 135 | 153 | |
| Medium (i-j = 2–4) | 144 | 136 | |
| Structural statistics | Violated distance constraints | 0 | 4 |
| Violated angle constraints | 0 | 0 | |
| RMSD of all backbone atoms (Å) | |||
| Q10 – L27 | 0.47 ± 0.19 | ||
| Q10 – T30 | 0.33 ± 0.12 | ||
| RMSD of all heavy atoms (Å) | |||
| Q10 – L27 | 1.15 ± 0.34 | ||
| Q10 – T30 | 1.21 ± 0.26 | ||
| Ramachandran plot | Residues in most favored region (%) | 72.1 | 64.5 |
| Residues in additionally allowed region (%) | 22.4 | 29.4 | |
| Residues in generously allowed region (%) | 5.5 | 6.1 |
The structure of IAPP in the presence of zinc has similarities but important differences from the zinc-free structure (Figure 8). The structural differences are localized around His18, and can be correlated with the chemical shift changes observed (a plot of the changes in Hα and HN chemical shifts is provided in Figure 9). A significant change in the chemical shift of the Hε and Hδ atoms on the imidazole side chain of His-18 by 0.208 ppm and 0.106 ppm respectively was observed. These values are similar in magnitude to values previously reported for the change in chemical shift upon Zn+2 binding to the imidazoles of Aβ.77 By contrast the Asn14 and Tyr37 residues of hIAPP, which are infrequent but possible ligands for zinc,78 did not show significant chemical shift differences when compared to the zinc-free spectra. The loss of the F15-H18 and L16-S19 dαHN(i,i+3) NOEs and the F15-S19 dαHN (i,i+4) (Figure 10) indicates zinc binding disrupts the backbone conformation in the immediate vicinity of His-18, causing an unwinding of the helix from residues V17-S19. The remainder of the peptide outside of this region is unaffected by the presence of zinc.
Figure 9.

Alpha proton chemical shift indexes (CSI) for hIAPP (A) and hIAPP in the presence of 10 mM zinc (B). The CSI was calculated by subtracting the appropriate random coil shifts reported in the literature. A CSI ≤ 0.1 is considered indicative of helical conformations. Changes in Hα (C) and HN (D) chemical shifts upon binding to zinc.
Figure 10.

Top: Histogram of NOEs versus the amino acid residues for hIAPP (A) without ZnCl2 and (B) with ZnCl2, showing the number of intra-residue, sequential (i – j = 1), and medium range (i – j = 2, 3, 4) NOEs. Bottom: Summary of the sequential and medium range NOE connectivities for hIAPP (C) without ZnCl2 and (D) with ZnCl2, The intensities of the observed NOEs are represented by the thickness of lines and are classified as strong, medium, and weak, corresponding to upper bound constraints of 2.9, 4.5, and 6 Å, respectively.
DISCUSSION
The most significant finding in this paper is the inhibition of hIAPP aggregation by zinc at concentrations typical for the extracellular space where amyloidosis occurs (10–25 μM).79 At pH 7.5, zinc was found to adversely affect the kinetics of the aggregation reaction by increasing the length of the lag phase and decreasing the cooperativity of the transition. An extracellular concentration of 25 μM ZnCl2 leads to an approximate 25% increase in the length of the lag phase and a 50% reduction in the elongation rate (Figure 4b and 4d). While the reduction at 25 μM is significant, it is important to remember that the distribution of zinc is neither spatially homogenous nor static in time. Zinc is concentrated near the plasma membrane which is both a catalyst for aggregation and the likely target for many of hIAPP’s toxic effects. The temporal dynamics of zinc distribution, particularly the spike in zinc concentration that occurs upon the exocytosis of the insulin/hIAPP granule, may also contribute to a magnified effect of zinc on hIAPP aggregation. When the secretory granule is exocytosed, the fusion of the secretory granule with the beta cell plasma membrane releases the entire contents of the secretory granule into the extracellular space (see a cartoon depiction in Figure 11). Since the concentration of zinc within the granule is much higher than in the extracellular environment (mM vs. μM),36, 79 emptying the granule therefore causes a brief spike in the zinc concentration near the site of exocytosis.80 This is important because the release of the granule contents does not occur simultaneously with the opening of the granule fusion pore, typically 1–2 seconds elapse before the granule contents are fully released but in some cases the process takes minutes to complete.81, 82 The acidic pH of the granule, on the other hand, is neutralized immediately upon the opening of the fusion pore and the exposure of the granule contents to the extracellular matrix. During this time hIAPP is both highly concentrated and at a pH very favorable for aggregation. It is also in close proximity to the plasma membrane which is both a target for attack and a dramatic facilitator of aggregation.5, 71, 83 The simultaneous release of zinc with hIAPP may allow hIAPP to safely diffuse away from the plasma membrane and be diluted in the bloodstream before it can aggregate on the cell surface.
Figure 11. Cartoon schematic of how zinc may influence IAPP aggregation and toxicity.

(A) In the secretory granule, IAPP is prevented from aggregation by the acidic environment and the inhibitory action of insulin. (B) Exocytosis of the secretory granule simultaneously releases IAPP and zinc into the extracellular space. (C) A high local concentration of IAPP near the plasma membrane is prevented from disrupting the β-cell by transient binding to zinc. (D) Zinc dissociates from IAPP at the low equilibrium concentrations found in the extracellular space, however, IAPP is now diluted past the critical concentration for aggregation.
In contrast to the micromolar inhibition of aggregation seen at neutral pH, zinc is not likely to directly contribute to the observed stability of hIAPP in the secretory granule. The acidic environment of the secretory granule considerably reduces the rate of aggregation (Figure 6 vs. Figure 4), similar to the results of previously reported groups.56, 84 We found that, contrary to its effects at neutral pH (7.5), zinc enhances rather than retards aggregation. However, it is unlikely that zinc contributes to pathological aggregation in the secretory granule in this manner, even at the zinc concentrations present in the secretory granule. First, the effect is diminished by the reduced affinity of the protonated His18 residue for zinc. Moreover, aggregation in the secretory granule is diminished to a much greater extent first by the acidic pH and further by the strong inhibitory action of insulin.12, 56, 85–88 The combined action of these two strong inhibiting effects is likely to suppress aggregation to a point beyond the lifetime of the secretory granule. It should be mentioned, however, that zinc may have a strong indirect effect on hIAPP fibrillogenesis by modulating the monomer to hexamer equilibrium of insulin,89 since the hexameric and monomeric forms of insulin likely display different affinities for hIAPP.72, 87
While the action of zinc on hIAPP fibrillogenesis at pH 5.5 is probably not directly physiologically relevant, it is useful in illuminating the mechanism of zinc inhibition of fibrillogenesis. Zinc exhibits both inhibitory and catalytic effects on the aggregation rate at pH 7.5, with the inhibitory effect being dominant at lower concentrations (Figure 4). The propensity for a protein to form amyloid is determined by a variety of factors similar to those that influence protein folding including a favorable potential for hydrogen bonding, favorable hydrophobic interactions, and favorable electrostatics. From an exclusively electrostatic point of view, the binding of zinc should strongly disfavor the amyloid state, as His18 is located in the hydrophobic interior of the fiber near a turn between strands 1 and 2.56, 90 Charged residues in amyloid fibers are intrinsically disfavored unless stabilized by salt bridges as the parallel stacking of the β-sheets brings the charges on each monomer unit close in space in the final structure.91, 92 This is particularly true of charged residues in the low dielectric environment of the interior of the fiber. At pH 5.5, this effect is mitigated as His18 already carries a positive charge. In contrast to pH 7.5, zinc enhances the rate of fibrillogenesis uniformly and nearly linearly with concentration at pH 5.5 indicating the inhibitory effect is indeed largely electrostatic in origin and related to destabilization of the amyloid fiber by the addition of a charge in an unfavorable position.
While the origin of zinc inhibition is relatively clear, the origin of its catalytic effect is less certain and cannot be concretely established from the data from this study. The pathway to amyloid formation is complex and involves a variety of on- and off-pathway intermediates. Stabilization of any on-pathway intermediate would be expected to reduce the length of the lag-phase and may also increase the fiber elongation rate through either direct addition of the intermediate to the fiber or interconversion to a placque-competent species. Alternatively, zinc may enhance both aspects of the fibrillogenesis rate by breaking existing amyloid fibers into smaller fragments, which then act as new sites for fiber growth.93 However, we did not see significant fragmentation of the amyloid fibers by electron microscopy, only a reduced amount of amyloid deposition was observed (Figure 3). It therefore seems more likely that stabilization of on-pathway intermediates, rather than increased secondary nucleation, is responsible for the partial compensation of zinc inhibition at high zinc concentrations.
While the exact binding constant of zinc to hIAPP was not determined in this study, zinc’s effects are clearly noticeable in the micromolar concentration range. Since this apparent binding constant is too high for hIAPP to be attached to only a single histidine,94 another residue is likely to act as a second ligand which must be either a non-histidine residue or another histidine on an adjacent molecule. The NMR structure places Ser19 in close proximity to the sidechain of His 18, opening the possibility that the zinc ion is chelated by the sidechains of both His18 and Ser19. While the hydroxyl group of serine is a hard ligand and is less than ideal for zinc binding in comparison to histidine, the favorable entropic caused by the chelating effect may overcome the less favorable enthalpy contribution to binding. The backbone oxygen of Ser19, another possible but enthalpically unfavorable ligand, is exposed in the zinc bound structure of IAPP and may be a possible binding site.
Another possibility is that zinc bridges multiple molecules of IAPP. There is some precedence for this type of bridging interaction as zinc catalyzes the formation of hexamers of insulin, which shares some sequence homology with hIAPP. Each insulin hexamer is held together by two zinc molecules, which each bind three insulin monomers by coordination to three histidines.95 A similar structure may be present in the hIAPP-Zn molecule, although the exact structure of the putative hIAPP-Zn oligomers (if they exist) remains to be determined.
Supplementary Material
Acknowledgments
This study was supported by research funds from NIH (DK078885 to A.R.) and Michigan Diabetes Research Training Center at the University of Michigan. We thank Professor Robert Kennedy of the University of Michigan for discussion on the role of zinc in beta-cell function. We also thank the 900 MHz NMR facility at the Michigan State University.
Footnotes
Complete references 38, 39, and 95. This material is available free of charge via the Internet at http://pubs.acs.org".
References
- 1.Scherbaum WA. Exp Clin Endocr Diab. 1998;106:97–102. doi: 10.1055/s-0029-1211958. [DOI] [PubMed] [Google Scholar]
- 2.Harrison RS, Sharpe PC, Singh Y, Fairlie DP. Rev Physiol Biochem Pharmacol. 2007;159:1–77. doi: 10.1007/112_2007_0701. [DOI] [PubMed] [Google Scholar]
- 3.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. J Am Chem Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knight JD, Miranker AD. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 6.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Proc Natl Acad Sci USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirzabekov TA, Lin MC, Kagan BL. J Biol Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 8.Knight JD, Hebda JA, Miranker AD. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 9.Goldsbury CS, Cooper GJ, Goldie KN, Muller SA, Saafi EL, Gruijters WT, Misur MP, Engel A, Aebi U, Kistler J. J Struct Biol. 1997;119:17–27. doi: 10.1006/jsbi.1997.3858. [DOI] [PubMed] [Google Scholar]
- 10.Radovan D, Smirnovas V, Winter R. Biochemistry. 2008;47:6352–60. doi: 10.1021/bi800503j. [DOI] [PubMed] [Google Scholar]
- 11.Alexandrescu AT. Protein Sci. 2005;14:1–12. doi: 10.1110/ps.04887005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larson JL, Miranker AD. J Mol Biol. 2004;335:221–231. doi: 10.1016/j.jmb.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 13.Calabrese MF, Eakin CM, Wang JM, Miranker AD. Nat Struct Mol Biol. 2008;15:965–71. doi: 10.1038/nsmb.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eakin CM, Knight JD, Morgan CJ, Gelfand MA, Miranker AD. Biochemistry. 2002;41:10646–56. doi: 10.1021/bi025944a. [DOI] [PubMed] [Google Scholar]
- 15.Talmard C, Yona RL, Faller P. J Biol Inorg Chem. 2009;14:449–455. doi: 10.1007/s00775-008-0461-9. [DOI] [PubMed] [Google Scholar]
- 16.Talmard C, Bouzan A, Faller P. Biochemistry. 2007;46:13658–13666. doi: 10.1021/bi701355j. [DOI] [PubMed] [Google Scholar]
- 17.Talmard C, Guilloreau L, Coppel Y, Mazarguil H, Faller P. Chembiochem. 2007;8:163–165. doi: 10.1002/cbic.200600319. [DOI] [PubMed] [Google Scholar]
- 18.Huang XD, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. J Biol Inorg Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 19.Goldstein L, Leopold M, Huang X, Bowden E, Tanzi R, Bush A. Invest Ophth Vis Sci. 1999;40:S530–S530. [Google Scholar]
- 20.Noy D, Solomonov I, Sinkevich O, Arad T, Kjaer K, Sagi I. J Am Chem Soc. 2008;130:1376–83. doi: 10.1021/ja076282l. [DOI] [PubMed] [Google Scholar]
- 21.Uversky VN, Li J, Fink AL. J Biol Chem. 2001;276:44284–96. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 22.Yamin G, Glaser CB, Uversky VN, Fink AL. J Biol Chem. 2003;278:27630–5. doi: 10.1074/jbc.M303302200. [DOI] [PubMed] [Google Scholar]
- 23.Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Biochemistry. 2005;44:9096–107. doi: 10.1021/bi048778a. [DOI] [PubMed] [Google Scholar]
- 24.Potter SZ, Zhu H, Shaw BF, Rodriguez JA, Doucette PA, Sohn SH, Durazo A, Faull KF, Gralla EB, Nersissian AM, Valentine JS. J Am Chem Soc. 2007;129:4575–83. doi: 10.1021/ja066690+. [DOI] [PubMed] [Google Scholar]
- 25.Khan A, Ashcroft AE, Korchazhkina OV, Exley C. J Inorg Biochem. 2004;98:2006–2010. doi: 10.1016/j.jinorgbio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Khan A, Ashcroft AE, Higenell V, Korchazhkina OV, Exley C. J Inorg Biochem. 2005;99:1920–1927. doi: 10.1016/j.jinorgbio.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Jobling MF, Huang XD, Stewart LR, Barnham KJ, Curtain C, Volitakis I, Perugini M, White AR, Cherny RA, Masters CL, Barrow CJ, Collins SJ, Bush AI, Cappai R. Biochemistry. 2001;40:8073–8084. doi: 10.1021/bi0029088. [DOI] [PubMed] [Google Scholar]
- 28.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Neurotoxicol. 1999;20:239–247. [PubMed] [Google Scholar]
- 29.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Neurol. 1997;48:650–658. doi: 10.1212/wnl.48.3.650. [DOI] [PubMed] [Google Scholar]
- 30.Rybicki BA, Johnson CC, Uman J, Gorell JM. Mov Disord. 1993;8:87–92. doi: 10.1002/mds.870080116. [DOI] [PubMed] [Google Scholar]
- 31.Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Proc Natl Acad Sci USA. 2002;99:7705–10. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masad A, Hayes L, Tabner BJ, Turnbull S, Cooper LJ, Fullwood NJ, German MJ, Kametani F, El-Agnaf OM, Allsop D. FEBS Lett. 2007;581:3489–93. doi: 10.1016/j.febslet.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 33.Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Proc Natl Acad Sci USA. 1995;92:1989–93. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pappalardo G, Milardi D, Magri A, Attanasio F, Impellizzeri G, La Rosa C, Grasso D, Rizzarelli E. Chem-Eur J. 2007;13:10204–15. doi: 10.1002/chem.200700576. [DOI] [PubMed] [Google Scholar]
- 35.Sciacca MFM, Pappalardo M, Milardi D, Grasso DM, La Rosa C. Arch Biochem Biophys. 2008;477:291–298. doi: 10.1016/j.abb.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 36.Foster MC, Leapman RD, Li MX, Atwater I. Biophys J. 1993;64:525–32. doi: 10.1016/S0006-3495(93)81397-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor CG. Biometals. 2005;18:305–312. doi: 10.1007/s10534-005-3686-x. [DOI] [PubMed] [Google Scholar]
- 38.Sladek R, et al. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 39.Zeggini E, et al. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirchhoff K, Machicao F, Haupt A, Schafer SA, Tschritter O, Staiger H, Stefan N, Haring HU, Fritsche A. Diabetologia. 2008;51:597–601. doi: 10.1007/s00125-008-0926-y. [DOI] [PubMed] [Google Scholar]
- 41.Staiger H, Machicao F, Stefan N, Tschritter O, Thamer C, Kantartzis K, Schafer SA, Kirchhoff K, Fritsche A, Haring HU. PloS one. 2007;2:e832. doi: 10.1371/journal.pone.0000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lemaire K, Ravier MA, Schraenen A, Creemers JWM, Van De Plas R, Granvik M, Van Lommel L, Waelkens E, Chimienti F, Rutter GA, Gilon P, in't Veld PA, Schuit FC. Proc Natl Acad Sci USA. 2009;106:14872–14877. doi: 10.1073/pnas.0906587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salazar G, Falcon-Perez JM, Harrison R, Faundez V. PloS one. 2009;4 doi: 10.1371/journal.pone.0005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruchat SM, Elks CE, Loos RJF, Vohl MC, Weisnagel SJ, Rankinen T, Bouchard C, Perusse L. Acta Diabet. 2009;46:217–226. doi: 10.1007/s00592-008-0080-5. [DOI] [PubMed] [Google Scholar]
- 45.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int J Pep Res. 2007;13:31–44. [Google Scholar]
- 46.Page K, Hood CA, Patel H, Fuentes G, Menakuru M, Park JH. J Pep Sci. 2007;13:833–838. doi: 10.1002/psc.909. [DOI] [PubMed] [Google Scholar]
- 47.Naiki H, Higuchi K, Nakakuki K, Takeda T. Lab Invest. 1991;65:104–10. [PubMed] [Google Scholar]
- 48.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 1999. [Google Scholar]
- 49.Wuthrich K. NMR of proteins and nucleic acids. John Wiley and Sons; New York: 1986. [Google Scholar]
- 50.Guntert P, Mumenthaler C, Wuthrich K. J Mol Biol. 1997;273:283–98. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 51.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 52.Herrmann T, Guntert P, Wuthrich K. J Mol Biol. 2002;319:209–27. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 53.Koradi R, Billeter M, Wuthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 54.Groenning M, Norrman M, Flink JM, van de Weert M, Bukrinsky JT, Schluckebier G, Frokjaer S. J Struct Biol. 2007;159:483–497. doi: 10.1016/j.jsb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Hudson SA, Ecroyd H, Kee TW, Carver JA. FEBS J. 2009;276:5960–5972. doi: 10.1111/j.1742-4658.2009.07307.x. [DOI] [PubMed] [Google Scholar]
- 56.Abedini A, Raleigh DP. Biochemistry. 2005;44:16284–16291. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- 57.Porat Y, Mazor Y, Efrat S, Gazit E. Biochemistry. 2004;43:14454–14462. doi: 10.1021/bi048582a. [DOI] [PubMed] [Google Scholar]
- 58.Janciauskiene S, Eriksson S, Carlemalm E, Ahren B. Biochem Biophy Res Comm. 1997;236:580–585. doi: 10.1006/bbrc.1997.7014. [DOI] [PubMed] [Google Scholar]
- 59.Tatarek-Nossol M, Yan LM, Schmauder A, Tenidis K, Westermark G, Kapurniotu A. Chem Biol. 2005;12:797–809. doi: 10.1016/j.chembiol.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Cao P, Meng F, Abedini A, Raleigh DP. Biochemistry. 2009 doi: 10.1021/bi901751b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldsbury CS, Cooper GJS, Goldie KN, Muller SA, Saafi EL, Gruijters WTM, Misur MP. J Struct Biol. 1997;119:17–27. doi: 10.1006/jsbi.1997.3858. [DOI] [PubMed] [Google Scholar]
- 62.Masad A, Hayes L, Tabner BJ, Turnbull S, Cooper LJ, Fullwood NJ, German MJ, Kametani F, El-Agnaf OMA, Allsop D. FEBS Lett. 2007;581:3489–3493. doi: 10.1016/j.febslet.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 63.Olofsson A, Lindhagen-Persson M, Vestling M, Sauer-Eriksson AE, Ohman A. FEBS J. 2009;276:4051–60. doi: 10.1111/j.1742-4658.2009.07113.x. [DOI] [PubMed] [Google Scholar]
- 64.Abedini A, Raleigh DP. Prot Eng Des Sel. 2009;22:453–9. doi: 10.1093/protein/gzp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abedini A, Raleigh DP. Phys Biol. 2009;6:15005. doi: 10.1088/1478-3975/6/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hall D, Hirota N, Dobson CM. J Mol Biol. 2005;351:195–205. doi: 10.1016/j.jmb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 67.Nanga RP, Brender JR, Vivekanandan S, Popovych N, Ramamoorthy A. J Am Chem Soc. 2009;131:17972–9. doi: 10.1021/ja908170s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Higham CE, Jaikaran E, Fraser PE, Gross M, Clark A. FEBS letters. 2000;470:55–60. doi: 10.1016/s0014-5793(00)01287-4. [DOI] [PubMed] [Google Scholar]
- 69.Williamson JA, Loria JP, Miranker AD. J Mol Biol. 2009;393:383–96. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saraogi I, Hebda JA, Becerril J, Estroff LA, Miranker AD, Hamilton AD. Angew Chem Int Ed. 2009 doi: 10.1002/anie.200901694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jayasinghe SA, Langen R. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 72.Wiltzius JJ, Sievers SA, Sawaya MR, Eisenberg D. Protein Sci. 2009;18:1521–30. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koo BW, Hebda JA, Miranker AD. Prot Eng Des Sel. 2008;21:147–54. doi: 10.1093/protein/gzm076. [DOI] [PubMed] [Google Scholar]
- 74.Cort JR, Liu Z, Lee GM, Huggins KN, Janes S, Prickett K, Andersen NH. Prot Eng Des Sel. 2009;22:497–513. doi: 10.1093/protein/gzp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patil SM, Xu S, Sheftic SR, Alexandrescu AT. J Biol Chem. 2009;284:11982–91. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nanga RP, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A. J Am Chem Soc. 2009;131:8252–61. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaggelli E, Janicka-Klos A, Jankowska E, Kozlowski H, Migliorini C, Molteni E, Valensin D, Valensin G, Wieczerzak E. J Phys Chem B. 2008;112:100–109. doi: 10.1021/jp075168m. [DOI] [PubMed] [Google Scholar]
- 78.Alberts IL, Nadassy K, Wodak SJ. Protein Sci. 1998;7:1700–16. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aspinwall CA, Brooks SA, Kennedy RT, Lakey JRT. J Biol Chem. 1997;272:31308–31314. doi: 10.1074/jbc.272.50.31308. [DOI] [PubMed] [Google Scholar]
- 80.Qian WJ, Gee KR, Kennedy RT. Anal Chem. 2003;75:3468–75. doi: 10.1021/ac0341057. [DOI] [PubMed] [Google Scholar]
- 81.Barg S, Olofsson CS, Schriever-Abeln J, Wendt A, Gebre-Medhin S, Renstrom E, Rorsman P. Neuron. 2002;33:287–99. doi: 10.1016/s0896-6273(02)00563-9. [DOI] [PubMed] [Google Scholar]
- 82.Michael DJ, Ritzel RA, Haataja L, Chow RH. Diabetes. 2006;55:600–7. doi: 10.2337/diabetes.55.03.06.db05-1054. [DOI] [PubMed] [Google Scholar]
- 83.Jha S, Sellin D, Seidel R, Winter R. J Mol Biol. 2009;389:907–20. doi: 10.1016/j.jmb.2009.04.077. [DOI] [PubMed] [Google Scholar]
- 84.Mishra R, Geyer M, Winter R. Chembiochem. 2009;10:1769–72. doi: 10.1002/cbic.200900237. [DOI] [PubMed] [Google Scholar]
- 85.Gilead S, Wolfenson H, Gazit E. Angew Chem Int Ed. 2006;45:6476–6480. doi: 10.1002/anie.200602034. [DOI] [PubMed] [Google Scholar]
- 86.Knight JD, Williamson JA, Miranker AD. Prot Sci. 2008;17:1850–6. doi: 10.1110/ps.036350.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jaikaran E, Nilsson MR, Clark A. Biochem J. 2004;377:709–716. doi: 10.1042/BJ20030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei L, Pervushin K, Mu Y. Biochemistry. 2009 doi: 10.1021/bi802097b. [DOI] [PubMed] [Google Scholar]
- 89.Lemaire K, Ravier MA, Schraenen A, Creemers JW, Van de Plas R, Granvik M, Van Lommel L, Waelkens E, Chimienti F, Rutter GA, Gilon P, in't Veld PA, Schuit FC. Proc Natl Acad Sci USA. 2009;106:14872–7. doi: 10.1073/pnas.0906587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luca S, Yau WM, Leapman R, Tycko R. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Richardson JS, Richardson DC. Proc Natl Acad Sci USA. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reumers J, Maurer-Stroh S, Schymkowitz J, Rousseau F. Hum Mutat. 2009;30:431–7. doi: 10.1002/humu.20905. [DOI] [PubMed] [Google Scholar]
- 93.Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. Science. 2009;326:1533–7. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 94.Aruga R. Transit Met Chem. 1983;8:56–58. [Google Scholar]
- 95.Baker, et al. Phil Trans Roy Soc Lond. 1988;319:369–456. doi: 10.1098/rstb.1988.0058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.