

Abstract
Defining the pathways through which tumors progress is critical to our understanding and treatment of cancer. We do not routinely sample patients at multiple time points during the progression of their disease, and thus our research is limited to inferring progression a posteriori from the examination of a single tumor sample. Despite this limitation, inferring progression is possible because the tumor genome contains a natural history of the mutations that occur during the formation of the tumor mass. There are two approaches to reconstructing a lineage of progression: (1) inter‐tumor comparisons, and (2) intra‐tumor comparisons. The inter‐tumor approach consists of taking single samples from large collections of tumors and comparing the complexity of the genomes to identify early and late mutations. The intra‐tumor approach involves taking multiple samples from individual heterogeneous tumors to compare divergent clones and reconstruct a phylogenetic lineage. Here we discuss how these approaches can be used to interpret the current models for tumor progression. We also compare data from primary and metastatic copy number profiles to shed light on the final steps of breast cancer progression. Finally, we discuss how recent technical advances in single cell genomics will herald a new era in understanding the fundamental basis of tumor heterogeneity and progression.
Keywords: Tumor progression, Tumor heterogeneity, Copy number aberrations, CGH microarrays, Cancer phylogenetics
1. Introduction
The process of cancer progression, from initiation in normal tissue to full‐blown tumor and eventual metastasis, is – despite intense study – still a mystery for most cancers. What is clear is that cancer is a genetic disease and that the dysregulation of cancer cells involves multiple levels of genetic control, including DNA point mutations, epigenetic alterations, chromosome copy number changes, inversions and translocations. As human tumors progress, these mutations accumulate in the genome and can be analyzed by cytogenetic and genomic techniques. Based on the assumption that mutational complexity increases with time, this ‘permanent record’ can be used to reconstruct the patterns of development after the tumor is excised from the patient. In this article we describe how we, and others, have used these approaches to study tumor progression using whole‐genome copy number analysis and how the observed patterns in breast cancer relate to the current models for tumor progression.
2. A brief history of tumor heterogeneity
In breast cancer the malignant cells often arise from ductal tissue and are constrained by the duct structure until they begin to invade surrounding stromal tissue. They exhibit regions of growth, regions of hypoxia and necrosis and regions of interaction with blood vessels and lymph ducts. It would be surprising if all cells in a tumor were identical. As early as the 1800s, Rudolf Virchow and other early pathologists observed the morphological heterogeneity of tumor cells using the first compound microscopes (Brown and Fee, 2006). The subsequent development of sophisticated staining methods allowed pathologists to visualize and categorize the morphology of tumor cells in detail, and to score various characteristics including nuclear size, mitotic index and differentiated structures. These characteristics are used to score the grade of a tumor which aids clinicians in determining how aggressively to treat a patient. However, many pathologists have noted that cells from different regions of a tumor differ in their morphological characteristics (Fitzgerald, 1986; Hirsch et al., 1983; Kruger et al., 2003; van der Poel et al., 1997). Taking into account this heterogeneity, pathologists will examine many tissue sections from several regions of the tumor, but generally report only the highest grade for clinical treatment (Ignatiadis and Sotiriou, 2008; Komaki et al., 2006).
In the early 1980s, a new arsenal of tools was developed by cytogeneticists to investigate tumor heterogeneity at the genome level: chromosome G‐banding, spectral karyotyping (SKY) and fluorescence in situ hybridization (FISH). A particularly large body of data concerning genetic heterogeneity comes from interphase FISH studies. Using specific DNA probes, FISH can reveal the copy number of a limited number of chromosomal loci across a large number of cells. By comparing the copy numbers of representative genomic loci using specific DNA probes across multiple tumor samples, various studies reported tumors as either ‘homogeneous’ (monoclonal) or ‘heterogeneous’ (polyclonal) (Farabegoli et al., 2001; Maley et al., 2006; Mora et al., 2001; Pantou et al., 2005; Roka et al., 1998; Sauter et al., 1995; Shipitsin et al., 2007; Teixeira et al., 1996; Zojer et al., 1998).
A more complete characterization of the tumor genome was obtained by visualizing metaphase chromosomes by Giemsa staining. The resulting G‐banding karyotypes provided chromosome specific landmarks and made it possible to accurately identify chromosome abnormalities in tumor genomes (Mitelman et al., 1997; Trent, 1985). As with FISH, it was observed that subpopulations of cells from the same tumor showed distinct sets of chromosomal rearrangements, indicating the presence of multiple clones (Coons et al., 1995, 1995, 1995, 1996). Using this technique, recurrent chromosome events began to be catalogued, providing the first notion that such events might be ordered in tumor development.
The heterogeneity of tumors has since been repeatedly validated using various molecular markers, including mRNA expression (Bachtiary et al., 2006; Cole et al., 1999); protein expression (Allred et al., 2008; Johann et al., 2009); and DNA sequencing (Khalique et al., 2007; Lips et al., 2008). The question then becomes one of understanding the role of heterogeneity in tumor progression. A number of studies have shown that despite the genetic diversity in heterogeneous tumor, neighboring clones often share many common mutations (Maley et al., 2006; Navin et al., 2010; Pantou et al., 2005; Shipitsin et al., 2007; Teixeira et al., 1996; Torres et al., 2007). Thus it seems unlikely that genetic heterogeneity is simply the result of random unselected variation. Instead, heterogeneous clones may represent discrete time points in the progression of the disease. By deconvoluting genomic heterogeneity we can therefore order a lineage of clones and identify mutations involved in the early or late stages of tumor progression.
With the advent of genomic techniques, such as microarrays and next‐generation sequencing, it has become possible to survey the entire genome to at much higher resolution than previously possible. Deep sequencing of heterogeneous tumors using next‐generation sequencing has shown that some tumors contain more alleles than would be expected in a single clones (Campbell et al., 2008; Shah et al., 2009). But it is difficult if not impossible to determine from sequence alone the number of clones present (and to which genomes the reads belong). As an alternative strategy, comparative genomic hybridization (CGH) microarrays can measure the precise location of chromosome breakpoints and the amplitude of copy number events that differ between divergent tumor subpopulations (Benetkiewicz et al., 2006; Navin et al., 2010; Shah et al., 2009; Shipitsin et al., 2007; Torres et al., 2007). This information can be used to track chromosome breakpoint markers as they are inherited and persist through successive subpopulations of clones that progress to form the tumor.
3. Modeling tumor progression
In recent years, several general models have been proposed to explain tumor progression. These models make different assumptions concerning the proliferative capacity of the major populations of tumor cells and thus lead to testable predictions concerning their lineage. The first model for tumor progression to gain widespread acceptance appeared in a landmark theoretical paper by Peter Nowell in 1976, where he combined two seemingly unrelated fields: evolutionary biology and tumor biology (Nowell, 1976). Nowell proposed that tumor cells obey the laws of natural selection, undergoing positive selection when advantageous mutations occur and negative selection when deleterious mutations arise. The two major schemes based on this fundamental tenet are collectively referred to as clonal evolution. They share the common assumption that the majority of tumor cells have the potential to undergo unlimited proliferation, but differ in the number of clonal subpopulations that they predict will form the mass of tumor.
The monoclonal evolution model states that solid tumors undergo a brief period of heterogeneity in the early stages of tumor progression, followed by a clonal expansion of a single population of cells, which forms the mass of the tumor (Figure 1a). It is assumed that a single clone undergoes positive selection and outcompetes all other subpopulations by the time the tumor is large enough to be detected. Evidence supporting the monoclonal evolution model originally came from methods that followed only a small number of traits, such as X‐inactivation in tumors, RFLP analysis of carcinomas, plasma cell immunoglobulin sequences and microsatellite markers (Endoh et al., 2001, 1974, 1965, 2004, 1992, 1994, 1994). Recent genomic data have also supported this model in a subset of breast cancers by showing that multiple samples within the same tumors contain highly similar copy number profiles by CGH microarrays (Navin et al., 2010).
Figure 1.
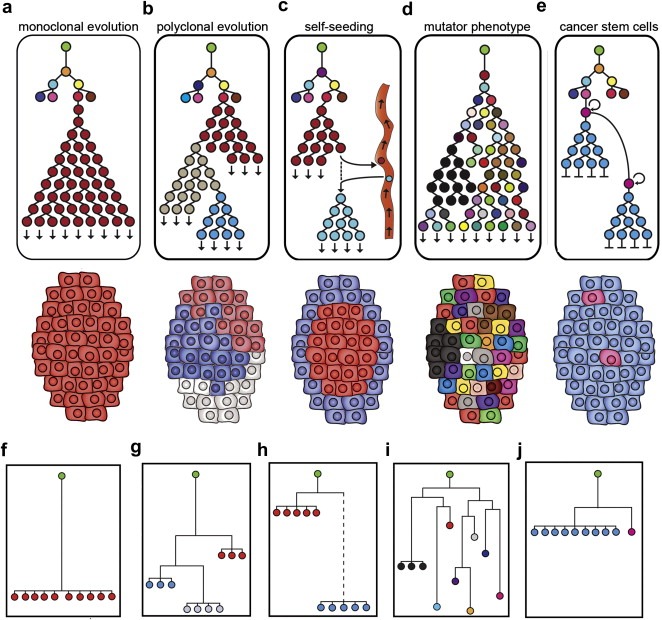
Tumor progression models and lineages. Green root nodes represent normal diploid cells, colored nodes are different tumor clones. (a–e) Models for tumor progression and phylogenetic lineages. (f–j) Hypothetical neighbor‐joining (NJ) trees constructed using 10 copy number profiles from a single tumor. (a) Monoclonal evolution forms a monogenomic tumor (b) Polyclonal evolution forms a polygenomic tumor (c) Self‐seeding results in a tumor with a divergent peripheral subpopulation (d) Mutator phenotype generates a tumor with many diverse clones (e) Cancer stem cell progression results in a tumor with a minority of pink cancer stem cells (f) NJ tree of a monogenomic tumor (g) NJ tree of a polygenomic tumor (h) NJ tree of a self‐seeded tumor with a dotted line representing a large phylogenetic distance (i) NJ of a mutator phenotype tumor (j) NJ tree of a cancer stem cell tumor.
In contrast, the polyclonal evolution model posits that solid tumors undergo an early period of heterogeneity followed by the expansion of multiple, divergent clones to form the mass of the tumor (Figure 1b). Empirical evidence supporting this model comes from a variety of studies including interphase FISH experiments, immunohistochemistry of tumor sections, gene expression studies and array CGH experiments (Aubele et al., 1999; Bachtiary et al., 2006). Recent experiments have supported the polyclonal evolution model by showing that genetic related clones with divergent genomes may cohabit the same tumor (Navin et al., 2010; Shipitsin et al., 2007).
This raises an interesting question about polyclonal evolution: do cohabiting clones within a single tumor suggest a cooperative relationship? In contrast to monoclonal evolution, in which a single dominant clone outcompetes all others, the polyclonal model implies an evolutionary advantage to cohabitation. In the tumor microenvironment resources – including oxygen, vasculature, stroma and growth factors – are scarce, so a selective advantage for having two or more clones seems highly plausible. The nature of their interaction may be mutualistic, commensal or perhaps even parasitic (See (Marusyk and Polyak, 2009) for an excellent review on these interactions). Clone interactions merit further study, as they may imply that targeting a single clone with therapy could lead to the rise or the demise of neighboring subpopulations.
In recent years, several variations of the polyclonal evolution model have been proposed, including the self‐seeding hypothesis and the mutator phenotype. The self‐seeding hypothesis posits that tumor clones leave the primary site, intravasate into the circulatory system, develop or subsist at a distant site for a period of time, then return to the primary tumor where they establish new subpopulations (Norton, 2008; Norton and Massague, 2006). This variation on the venerable ‘seed and soil’ theory (Paget, 1889) implies that circulating tumor cells have a homing mechanism that attracts them back to their site of origin. It also predicts that new clones will aggregate at the periphery of the tumor surface, or where vasculature leads into the tumor (Figure 1c) and that tumors are ‘built’ out of the sequential accretion of clones. Recently, homing behavior and self‐seeding was demonstrated in a mouse model using both human tumor cell lines and pleural effusion cells (Kim et al., 2009). These investigators showed that specific cytokine attractants (IL‐6 and IL‐8) and mediators of infiltration (MMP1 and fascin‐1) were integral factors in this self‐seeding process. Among the various implications of these results, the authors raise the counter‐intuitive notion that the presence of a primary tumor mass might act as a ‘sponge’ for circulating tumor cells, and by ‘soaking them up’ actually reduce the potential for distant metastases, the major cause of breast cancer mortality.
The mutator phenotype model, originally set forth by Lawrence Loeb (Loeb et al., 1974), is related to polyclonal evolution but differs by proposing that tumors consist of a large diversity of small clones rather than a few dominant clonal subpopulations (Figure 1d). In this model the rate of random mutations in tumor cells is thought to increase drastically perhaps by introduction of mutations into DNA polymerase itself (Bielas and Loeb, 2005; Loeb et al., 1974). Clonal expansions may occur, but a large diversity of tumor genomes are generated by random, non‐expanded mutations. Evidence for this model comes largely from a DNA capture sequencing approach, from which it was estimated that the mutation rate increased from more than 200‐fold in neoplastic tissues (Bielas et al., 2006). The mutator phenotype has also been extended to copy number changes, suggesting that tumor progression is driven by random, non‐expanded amplifications and deletions that generate genomic instability (Heng et al., 2006, 2006). While this model differs in predicting a larger diversity of genetic clones, it shares the primary assumption of clonal evolution: that the majority of tumor cells have to potential to proliferate indefinitely.
In the late 1990s an alternative model emerged that challenged the primary assumption of the previous models by assuming that only a minority of tumor cells could proliferate indefinitely. The cancer stem cell (CSC) hypothesis became widely accepted as the leading model for tumor progression. The CSC hypothesis posits that a rare population of stem cells within the solid tumor is the only subpopulation with the ability for unlimited proliferation (Figure 1e). The model assumes: (1) a rare population of cancer stem cells proliferate indefinitely, (2) the majority of tumor cells have limited proliferation, and (3) the rare cells continuously give rise to the major population. Cancer stem cells were originally believed to arise from normal stem cells, but it is now thought that any somatic cell may become a cancer stem cell (Clarke et al., 2006).
Evidence for the CSC hypothesis originally came from studying normal hematopoietic stem cells and the malignant stem cells that arise during leukemogenesis. The first empirical evidence came with the invention of fluorescence‐activated cell sorting (FACS) which allowed the isolation of human leukemic stem cells using surface markers (Lapidot et al., 1994). These human cancer stem cells were reimplanted into immunocompromised mice, in which they were fully capable of initiating leukemia, while other reimplanted cancer cells could not (Bonnet and Dick, 1997). The isolation and reimplantation assay has become the gold standard for identifying cancer stem cells and has been used to identify cancer stem cells in breast carcinomas (Al‐Hajj et al., 2003), brain tumors (Singh et al., 2004), colon cancers (O'Brien et al., 2007) and pancreatic tumors (Li et al., 2007). The CSC model is also attractive to clinicians, because it suggests that the entire tumor can be eradicated by targeting only the cancer stem cell population (Campbell and Polyak, 2007).
4. Models and their phylogenetic implications
Each progression model for tumorigenesis implies a different phylogenetic tree structure. In Figure 1a–e, we show phylogenetic trees as clones progress during the development of a solid tumor, and in Figure 1f–j, we show hypothetical trees that can be reconstructed from clones after the tumor develops and has been excised from the patient. In the latter case, we show a hypothetical tumor that was sampled 10 times and profiled by microarray CGH, from which neighbor‐joining trees were calculated by profile correlations (discussed in detail in the subsequent sections). In monoclonal evolution, the tree would show a period of heterogeneity followed by the expansion of a single dominant clone (Figure 1a). The expected neighbor‐joining tree of a monogenomic tumor would be flat with a single branch of highly similar nodes that have diverged an equal distance from the green root node (Figure 1f). In homogeneous tumors, it is by definition impossible to gain further information about lineage unless the apparently rare precursor cells can be identified and isolated. In heterogeneous tumors, however, lineage can be inferred by assuming that mutational complexity increases with time. Borrowing from evolutionary biology, we can use phylogenetic inference to determine the genetic distance between a set of observed genomes, and to estimate the common ancestors (Whelan, 2008).
In the polyclonal evolution model, a phylogenetic tree would consist of several highly similar subpopulations (red, gray and blue) that have diverged from each other and clonally expanded, each forming a significant mass of the tumor (Figure 1b). In the neighbor‐joining tree we would expect multiple branches of highly similar nodes (red, gray and blue), each represents a clonal subpopulation that has diverged an equal distance from the root node (Figure 1g). While each subpopulation is genetically distinct, they contain a set of common mutations that are inherited and persistent throughout their evolution.
In the self‐seeding tree, a brief period of heterogeneity would result in a series of diverse clones, followed by the expansion of a single red subpopulation (Figure 1d). As this subpopulation migrates into the vasculature and develops offsite, it may acquire a number of new mutations before reseeding the primary to form a new subpopulation. The neighbor‐joining tree for this model would have a large phylogenetic distance (dotted lines) between the original subpopulation (red) and the re‐seeded clones (blue), representing a gap in the evolution of the primary tumor (Figure 1h). In order to fully characterize the lineage of a self‐seeded tumor it would be necessary to detect and analyze clones in circulating tumor cells and metastatic tissues.
In stark contrast to the previous models, the mutator phenotype model predicts a large and diverse phylogenetic tree represented by many different node colors (Figure 1d). In the neighbor‐joining tree we would expect to see many different nodes, each diverging a unique distance from the green root node (Figure 1i). While some common mutations would be shared between the 10 copy number profiles, a large number of diverse non‐expanded mutations would generate a complex multi‐branched tree structure.
The CSC model has been subject to multiple interpretations (Adams and Strasser, 2008; Campbell and Polyak, 2007; Clarke et al., 2006; Fabian et al., 2009; Polyak, 2007b). For our purposes, we will focus on one version of the cancer stem cell hypothesis in which any normal somatic cell (not necessarily a normal stem cell) can undergo the generation of heterogeneity typical of cancer initiation, followed by dedifferentiation to a cancer stem cell. Thereafter, the cancer stem cell (pink) continually gives rise to the majority of tumor cells (blue) which terminate the lineage after limited number of divisions (Figure 1e). The resulting cancer stem cell tree is characterized by a flat structure with many terminal nodes. The neighbor‐joining tree would appear similar to the monogenomic tumors, in which the majority of cells occupy a single branch (blue); however if the cancer stem cells can be detected and have distinguishing genomic aberrations, then the nodes (pink) would be placed in separate branches, a short phylogenetic distance away from the major nodes (Figure 1j).
5. Inferring tumor progression
In theory, there are two ways to infer progression from primary tumor genomes: (1) comparing different tumors, and (2) comparing clones within single tumors. Until recently the most common approach has involved surveying single samples from archived tumor collections and cataloguing the order and frequency of genetic events. This approach has been widely applied to reconstruct progression in many different cancer types using large collection of tumors (Bilke et al., 2005, 2005, 2006, 2002, 2005, 2009, 2009, 2008). In most studies the underlying assumption is that mutations accumulate as the tumor progresses and only rarely are lost. Specific genetic lesions can thus be classified as early or late, relative to the total complexity of the tumor genome. The limitation to this approach is that while a few structural aberrations can be clearly classified as early, placement of high frequency events into ordered pathways has been problematic. Surgically resected tumors from archived collections represent relatively advanced cases with large numbers of genomic aberrations. With a few exceptions, the vast majority of mutational events occur at low frequency across tumor collections, indicating that each tumor travels down a unique mutational pathway.
Tumor evolution can also be observed by comparing multiple samples in an individual tumor. Many observations including our own (Navin et al., 2010) have demonstrated that most breast cancers show significant heterogeneity in their genomic profiles, making it possible to identify clones that represent various time points in the progression of the tumor. By isolating and comparing tumor clones from within a single tumor, we can then reconstruct a detailed lineage of how the tumor developed, assuming that mutations are persistent and inherited between clones. The lineage trees identified by intra‐tumor analysis can then be compared to the predicted trees from the general models to shed light on the tumor progression pathways in breast cancer.
6. Inferring progression from inter‐tumor comparisons
Early studies of tumor genome progression involved longitudinal comparisons of the karyotypes of tumors from large collections (Heim and Mittleman, 2009). The general theory was that tumor genomes with the fewest chromosome aberrations contained the earliest mutations in tumor progression. An extension to this approach involved separating non‐invasive precursors to breast cancer (DCIS) or low grade and comparing them to high grade tumors (Tsarouha et al., 1999). Most of these tumors were pseudo‐diploid and therefore easy to karyotype by G‐banding. In tumors with few chromosomal aberrations, the most frequent event involved the gain of the entire 1q chromosome arm and the loss of the 16q arm (Hoglund et al., 2002; Tsarouha et al., 1999). This combination of gain and loss seems to be the earliest event in some breast cancer and often occurs through pericentric recombination and the generation of an either 1q:16p translocation (followed by loss of the reciprocal product) or a 1q:1q isochromosome. These tumors were mostly hormone receptor‐ positive, and had the best prognoses. However, in these studies of tumor progression, the collections often consisted of a diverse mixture of subtypes, with each evolving down a different mutational pathway.
A milestone in understand the diversity of breast cancers came with advances in gene expression microarrays. In 2000, Sørlie and Perou et al. proposed that breast tumors could be classified into five different subtypes based on the expression of a few hundred mRNA transcripts. This stratification of breast cancer had important implications for studying tumor progression in that each subtype could be studied as an independent disease. The original five subtypes include: luminal A, dominated by the ER+ tumors with the best prognosis; luminal B, characterized as more advanced and often more genomically complex; ERBB2‐like, often amplified at the ERBB2 growth factor receptor locus; basal‐like, most often negative for ER, PR and ERBB2 (‘triple‐negative’); and normal‐like, with expression patterns most closely related to normal breast tissue (Perou et al., 2000; Sorlie et al., 2001). This classification scheme has been shown to be extremely robust and has further been refined using more advanced technology on larger cohorts (Calza et al., 2006; Carey et al., 2006; Hu et al., 2006; Sorlie et al., 2003).
More recently, high‐resolution CGH microarrays have been used to study the genome structure of these subtypes and shown that they progress by different genomic rearrangement patterns (Bergamaschi et al., 2006; Chin et al., 2006; Kwei et al., 2010). In the basal‐like tumors, Bergamaschi identified a higher numbers of gains and losses than luminal A, and the luminal B and ERBB2+ had more frequent high‐level amplifications (Bergamaschi et al., 2006). Our work has also shown that breast cancers can be classified into at least four distinct patterns of genomic rearrangements, suggesting different progression patterns (Hicks et al, 2006). The ‘Simplex’ pattern had broad segments of duplications and deletions. ‘Complex I’ had a “sawtooth” appearance with narrow segments of deletions and duplications affecting more or less all chromosomes. ‘Complex II’ resembled the ‘simplex’ but had at least one localized region of clustered peaks of amplifications called “firestorms.” The fourth pattern was called “flat” defined profiles with no clear gains or losses. A calculated index reflecting the complex rearrangements called firestorms, was found to be significantly associated with survival independent of other clinical parameters. Another aCGH study identified three subtypes (with overlapping characteristics to the Hicks et al. classes) that varied with respect to level of genomic instability (Chin et al., 2006). In summary, the expression subtypes of breast cancer were highly correlated to different genomic rearrangement patterns, suggesting that inter‐tumor comparison studies should be restricted to individual subtypes.
These microarray CGH studies corroborated many of the previously identified chromosome arm imbalances and translocations that were reported by Teixeria using G‐banding in metaphase cells (Teixeira et al., 1994, 1995, 1996). However, CGH microarrays also identified many additional focal aberrations (<1 mb) that could not be detected by G‐banding and allowed uncultured tumors to be analyzed. CGH data can also be mathematically segmented to detect numerous chromosome ‘breakpoints’ that characterize tumor genomes. When such methods are applied to tumor profiles it is possible to distinguish imbalances ranging from whole chromosomes and chromosome arms to events of ∼30 kb. For example in the segmented copy number pattern shown in Figure 2a (upper panel) we observe the gain at least one copy of the q arm of chromosome 1 and the loss of 1 copy of chromosome 16q. These two changes in copy number are the most frequent events observed in breast cancer and are also the most highly correlated with each other. That they are highly correlated is not surprising, because they are likely the result of a single event – a pericentromeric and apparently reciprocal translocation between chromosomes 1 and 16, followed by the loss of the hybrid containing 16q and 1p. A similar event often occurs between chromosome 16 and chromosome 8 leading to profiles such as that in Figure 2b; the 16q arm is lost along with the 8p arm, followed by the doubling of the 16p–8q hybrid. Interestingly, the breakpoints of these translocation and rearrangements do not pinpoint a specific location or gene important for the cancer process.
Figure 2.
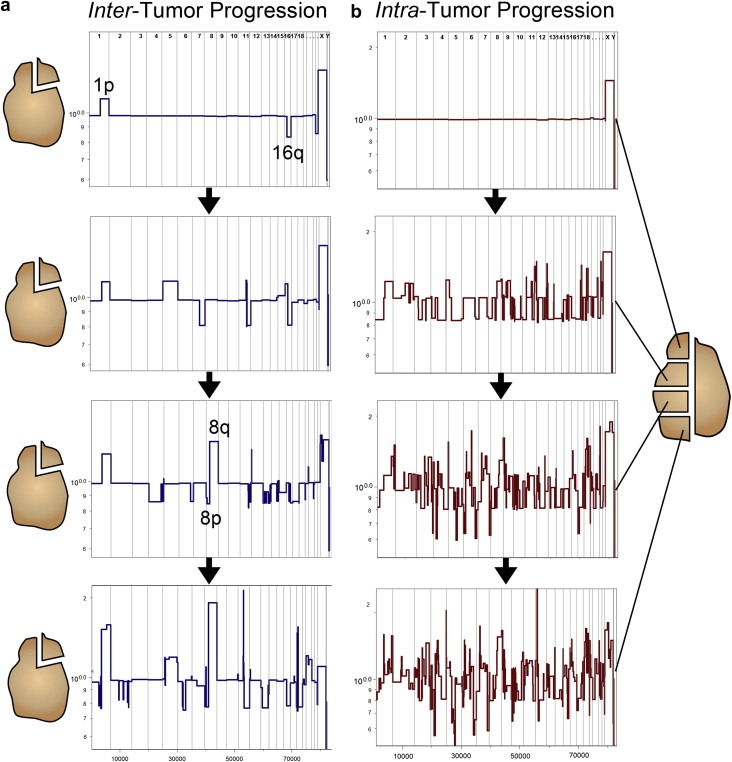
Inter and intra‐tumor comparisons of copy number profiles. (a) Inter‐tumor comparisons. A single sample was resected from four different luminal A breast tumors and CGH profiles were measured and segmented. The profiles shown were ordered based on increasing genomic complexity. (b) Intra‐tumor comparisons. Four samples were taken from a single heterogeneous basal‐like breast carcinoma. Nuclei were isolated from each quadrant and samples were flow‐sorted by ploidy, followed by microarray CGH profiling. The profiles are ordered based on increasing numbers of chromosome breakpoints.
In luminal A tumors, the earliest event that can be detected by inter‐tumor comparisons is the translocation of chromosomes 1p and 16q. By ordering tumor profiles based on increasing numbers of chromosome rearrangements, we can readily identify ‘early’ profiles that contain only a gain of 1q and loss of 16p (Figure 2a, upper panels); these profiles are simple in that they contain no other copy number changes. We can also detect ‘late’ profiles that often contain the 1q gain and/or 16p loss but have also acquired a numerous additional amplifications and deletions (Figure 2a, lower panels). This early 1p–16q event can also be seen in frequency plots of hundreds of luminal A tumors, which clearly show the gain of 1q and 16p in the progression of this subtype (Figure 3a). With the exception of the concurrent gain and loss of the 8q and 8p arms (often appearing simultaneously), loss of 11q and 22q is also apparently accomplished through arm swapping with multiple other chromosome partners (personal communication with Dr. Anders Zetterberg) the rest of the events appear to be distributed more or less evenly across multiple tumors. The genome profiles of luminal B tumors are in general more complex than luminal A profiles, usually characterized by the appearance of multiple amplified regions or ‘firestorms’ (Hicks et al., 2006). Their frequency plots, however, do not differ a great deal from luminal A, indicating that the additional events are distributed throughout the genome.
Figure 3.
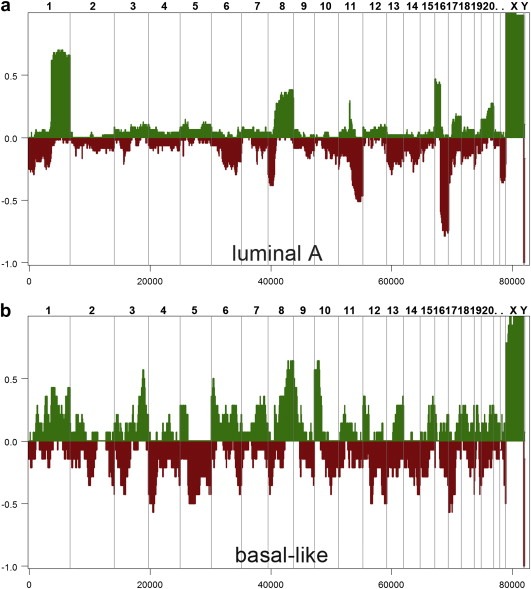
Frequency plots of luminal A and basal‐like breast tumors. Microarray CGH was used to generate copy number profiles from collections of luminal A and basal‐like breast tumors. The frequency plots were calculated from segmented copy number profiles. (a) Luminal A frequency plot was calculated from 45 tumor samples. (b) Basal‐like frequency plot was calculated from 23 tumor samples.
Conversely, the inter‐tumor comparisons of basal‐like tumors show a very different pattern of genome progression. As exemplified by the tumor profile in Figure 2b (which was measured from intra‐tumor comparisons), the basal‐like subtype most often presents a ‘sawtooth’ pattern characterized by multiple broad deletions rather than reciprocal gains and losses seen in the luminals. Also, the deletion breakpoints are not necessarily pericentromeric. Although the sawtooth pattern varies a greatly from tumor to tumor even in the early stages of its development, these tumors ultimately share a series of common markers distinct from the luminals. By calculating frequency plots from the segmented profiles, we can show that these genomes are characterized by frequent gains at the ends of 3q, 6q and 10p and losses at 4p, 5q and 17p (Figure 3b). In fact, the progression patterns of the luminal A and basal‐like subtypes are so drastically different, that the only events that they share is the loss of 8p and gain of 8q (Figure 3). Thus, the basal‐like and luminal A breast tumors show markedly different patterns of genome progression.
In summary, only a limited number of conclusions can be drawn from longitudinal surveys of breast tumor genomes. Although certain events are frequently observed in certain subtypes, it is difficult to draw a roadmap in which even a few of the observed events are precisely ordered. Furthermore, the roles that these genomic events play in the initiation or proliferation of cancer is still open to speculation. The broad distribution of breakpoints makes it unlikely that they act through gene fusion or disruption and these events represent at most twofold changes in gene dosage. It is also difficult to discern the biological impact of gene dosage effects during progression, when whole chromosome arms are deleted or amplified. In the case of 16q deletion, the copy number of six cadherin genes is decreased, perhaps decreasing cell–cell interaction, but the copy number of hundreds of other genes is also reduced. In the case of 8q arm amplification, there is a drastic increase in the gene dosage of CMYC, but also many other potential oncogenes. Inter‐tumor analysis is also confounded because most samples represent a single time point in the later stages of tumor progression, often containing numerous genetic aberrations. Thus it is difficult to understand the importance of any single amplification or deletion event during a specific stage of tumor progression.
7. Inferring progression from intra‐tumor comparisons
An alternative approach to studying large sets of tumors is to infer progression from multiple samples of individual heterogeneous tumors (Figure 2b). Genomic heterogeneity can serve as a permanent record of the mutations that occurred during tumorigenesis, allowing us to reconstruct progression, in a manner analogous to a forensic investigator. The tumor genome holds strong evidence, acquiring a multitude of stable mutations during tumor progression. If we assume that mutational complexity increases with time, then we can temporally order a set of genomes based on increasing numbers of mutations. However, in using genomic approaches to studying tumor heterogeneity we encounter a new problem: mixed populations of cells. Unlike many cytological techniques where individual tumor cells can be observed, most genomic techniques measure signal from a complex mixture of cell types, including multiple tumor clones and an amalgamation of normal cells collectively referred to as stroma (Hanahan and Weinberg, 2000).
In order to compare the genomes of heterogeneous clones, we need to first isolate the individual subpopulations and remove any normal cells to ‘purify’ the measured signal. One method for isolating subpopulations from within a single tumor involves using surface receptors that are displayed on different clones. For example, by isolating tumor subpopulations via FACS using CD44+ CD24− and CD44− CD24+ receptors and measuring copy number profiles, it was shown that these subpopulations were highly similar, but did differed by a few genomic aberrations (Shipitsin et al., 2007). This approach requires a priori knowledge of which receptors can distinguish clonal subpopulations. Another method for isolating tumor subpopulations involves sampling multiple distinct regions of a tumor by macro‐dissection or laser‐capture micro‐dissection. Using macro‐dissection it has been shown that different quadrants of single breast tumors show divergent copy number profiles, suggesting the presence of multiple, genetically related clones in the tumor (Teixeira et al., 1995, 1996, 2007). Similar results have been found, using laser‐capture micro‐dissection and copy number quantification to identify divergent clonal subpopulations (Aubele and Werner, 1999; Glockner et al., 2002). A caveat of this method is that tumor clones must be regionally segregated in the tumor in order to be detected. Furthermore, the mixing of normal cells may severely decrease the overall signal of the tumor subpopulations in different sectors.
Coupling macro‐dissection with flow‐sorting nuclei by DNA content provides an alternative method for isolating heterogeneous tumor subpopulations. Cytometrists have long been aware that many solid tumors contain multiple aneuploid distributions of cells with different mean DNA indices (Coons et al., 1995; Giaretti et al., 1996; Kallioniemi, 1988). However, until recently a genomic analysis of subpopulations that differ in ploidy had not been investigated (Corver et al., 2008). We developed a method to isolate clonal subpopulations in tumors called Sector‐Ploidy‐Profiling (SPP) which involves macro‐dissecting tumors into multiple sectors (6–12), isolating nuclei, staining with DAPI, flow‐sorting by total DNA content and using high‐resolution comparative genomic hybridization microarrays to measure genome‐wide copy number (Navin et al., 2010). This method has the advantage of isolating multiple tumor subpopulations that differ in ploidy, even when they are intermixed within single sectors of the tumor.
8. Reconstructing phylogenetic trees within tumors
We applied SPP to a collection of high grade ductal carcinomas and quantified 8–20 copy number profiles per tumor (Navin et al., 2010). We then clustered and compared profiles within tumors, which showed two classes of genomic structural variation: ‘monogenomic’ and ‘polygenomic’. Monogenomic tumors appear to contain a single major clonal subpopulation with a highly stable chromosome structure. Polygenomic tumors contain multiple clonal tumor subpopulations that occupied the same sectors or separate anatomic locations. Assuming that mutational complexity increases with time, we used Pearson correlations and neighbor‐joining to construct phylogenetic trees. In the monogenomic tumors we often observed a flat tree structure in which all nodes were highly correlated and diverged an equal distance from the root node. We show an empirically derived neighbor‐joining tree for a monogenomic tumor in Figure 4a. This tree structure is highly similar to the predicted tree (Figure 1f), with a small degree of variation in the correlation between nodes. In these homogenous tumors, a more detailed genetic lineage is difficult to infer, because no other intermediate subpopulations can be measured.
Figure 4.
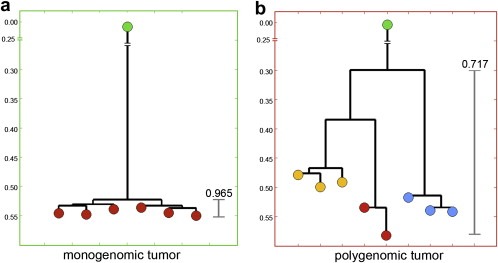
Empirical neighbor‐joining trees. Multiple copy number profiles were measured in two different breast tumors by Sector‐Ploid‐Profiling. Pearson correlations were calculated between all profiles and neighbor‐joining trees were constructed. (a) A monogenomic tumor showing a flat tree structure with a high correlation between all nodes (b) A polygenomic tree showing three groups of highly similar profiles (yellow, red, blue) that represent distinct subpopulations.
From polygenomic tumors we can infer a detailed phylogenetic lineage. In Figure 4b we show an empirically derived neighbor‐joining tree from a polygenomic tumor with three major subpopulations (red, yellow and blue nodes). In many polygenomic breast tumors, the inferred trees showed that copy number profiles were clearly related, and shared the majority of chromosome breakpoints, suggesting a common genetic lineage from a single precursor cell. Importantly, the copy number profiles in these tumors were always organized into highly similar groups, representing clonal subpopulations. Thus, genomic heterogeneity can be ascribed to a few major subpopulations rather than a series of gradual intermediates. From these data we conclude that the majority of chromosome breakpoints are inherited from previous subpopulations and persist through the evolution of more advanced subpopulations, as clones expand to form the mass of the tumor.
9. Genomic heterogeneity and progression in basal‐like tumors
As described in previous sections, breast cancer is a genomically heterogeneous disease both across cohorts and within individual tumors. Notwithstanding the genomic diversity, the expression‐based subtypes (luminal A, luminal B, Her2/erbb2, basal‐like and normal‐like, plus the recently added ‘claudin‐low’) provide useful a tool for grouping tumors of shared characteristics (Perou et al., 2000). The basal‐like breast cancers are a particularly aggressive subtype that has been the focus of intense study, and are often characterized by triple negative receptor status (ER, PR and Her2) (Rakha et al., 2008; and reviewed by Podo et al., 2010). Recently, the genome structure of basal‐like breast tumors has been revealed by array CGH showing a ‘sawtooth’ copy number pattern caused by the loss of many broad genomic regions (Bergamaschi et al., 2006; Chin et al., 2007; Hicks et al., 2006). In our study of genomic heterogeneity using SPP, the basal‐like tumors consisted of a number of heterogeneous but clearly related genome structures, providing evidence of clonal progression (Navin et al., 2010). In macro‐dissected sectors of each of two basal‐like breast tumors we identified three or four clonal subpopulations: Diploid (D), Hypodiploid (H), and Aneuploid (A1‐A2). Assuming that mutational complexity increases with time, we ordered the genomic profiles by increasing numbers of chromosome breakpoints (Figure 5, middle panel). We found that the basal‐like tumors progressed from diploid to hypodiploid, which correlated with a downward shift in total DNA content (as indicated by the FACS histogram) and loss of many chromosomes in the genome profiles (Figure 5b, lower panel). The hypodiploid subpopulation then diverged to form the A1 subpopulation, correlating with a drastic increase in total DNA content and multiple genome‐wide focal amplifications and deletions (Figure 5c). Finally the A1 subpopulation mutated by a massive amplification of the KRAS locus on chromosome 12p12.1 and a homozygous deletion of the EFNA5 tumor suppressor to form the A2 subpopulation, correlating which another upward shift in total DNA content by FACS (Figure 5d). From this, we infer a step‐wise pattern of progression in the basal‐like tumors in which much of the genome is first deleted, followed by endoreduplication to generate a highly aneuploid genome that continues to acquires many focal amplifications and deletions of cancer genes. The large degree of genomic heterogeneity in this subtype make them an ideal tool for studying tumor progression and may result in useful clinical diagnostic markers to determine how far along these tumors have progressed in breast cancer patients.
Figure 5.
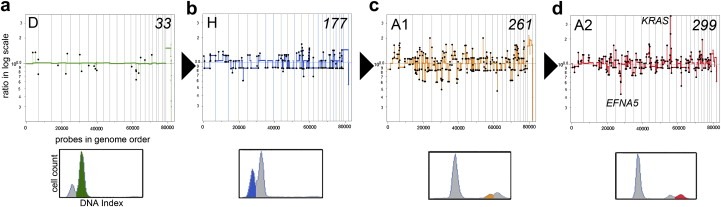
Genomic progression in a basal‐like breast tumor. Sector‐Ploidy‐Profiling was used to measure and compare twenty copy number profiles from 6 different sectors of a basal‐like tumor. (Upper Panels) Copy number profiles were segmented, clustered and coalesced to generate representative profiles from each major subpopulation: diploid (D), hypodiploid (H), aneuploid 1 (A1) and aneuploid 2 (A2). The profiles were ordered based on increasing numbers of chromosome breakpoints from 33 to 299. (Lower Panels) FACS Histograms of ploidy. (a) Diploid subpopulation with a 2N total DNA content by FACS (b) Hypodiploid subpopulation with a downward shift in ploidy (c) Aneuploid 1 subpopulation shows an upwards shift in ploidy (d) Aneuploid 2 subpopulation shows the highest total DNA content.
10. Metastatic progression of the genome
In many cases, tumor lineages can be traced all the way to the final step of progression: metastasis. While seemingly an obvious extension of the studies on primary tumors, metastatic studies are few because the material is rare. Metastases are seldom excised or biopsied in late stage patients unless part of a dedicated study, and recurrence – sometimes years after the surgery – is often treated by different physicians at different institutions. Therefore matching the correct primary and metastatic tumor samples from the same patient is often formidable. Distant metastasis, however, is nearly always the direct cause of patient mortality, and understanding its relationship to the primary tumor is of paramount importance.
A major question revolves around determining which cells are capable of initiating metastasis and how they can be identified. Do most cells in an invasive carcinoma have the ability to metastasize, or only a subset of highly malignant clones? Do tumor clones from the primary and metastatic tumors share the majority of chromosome aberrations or do they acquire new mutations that confer metastatic potential? One way to investigate these questions involves tracking tumor cells in the primary tumor, circulating tumor cells and metastasis by CGH profiling. By comparing a large number of patients with only primary tumors to those with primary tumors and metastasis it may also be possible to identify high frequency mutations that are associated with metastatic potential. Such biomarkers would be clinically useful in predicting whether a patient with a primary tumor may form a metastasis.
Using high‐resolution CGH microarrays, our lab is currently engaged in a study with several clinical collaborators to investigate the relationship of genome structure in a large collection of metastatic breast cancers. To date, we have found a very high correlation (>0.90) between the primary and metastatic copy number profiles in our analysis of 20 metastatic patients. In Figure 6 we present our segmented CGH results from a single patient with a primary breast carcinoma and a liver metastasis. We calculated a Pearson's Correlation between the segmented profiles, showing a very high value (PCC = 0.96) (Figure 5c). Even very complex rearrangements at specific loci, such as chromosome 10p (Figure 5c, lower panel), where our high‐resolution microarrays detected 8 amplifications at 10 kb resolution, we observed nearly identical breakpoints in the primary and metastatic tumors. Based on the comparison of the hundreds of genomic profiles in our database, we believe that is highly unlikely that these complex multifocal events could arise independently with identical patterns from an otherwise undifferentiated progenitor cell. Similar results were recently reported using high‐resolution CGH microarrays to study metastatic prostate cancer patients (Liu et al., 2009b). In their comparative analysis of copy number profiles, they too showed that primary and metastatic genomes were highly similar, diverging by only a few (if any) specific genetic events. Other studies using lower resolution methods have likewise reported that metastatic profiles are highly similar to primaries and diverge by only a few, if any, genetic events (Bockmuhl et al., 2004; Hovey et al., 1998; Israeli et al., 2004; Jiang et al., 2005). Thus, the genomic data suggest that metastatic tumors are often seeded by clones prominent in the primary tumor and do not undergo much further evolution in the metastasis. However, to truly track the progression of tumor cells as they leave the primary tumor, migrate through the circulatory system and seed the metastasis, even finer resolution is required, capable of following single cell lineages.
Figure 6.
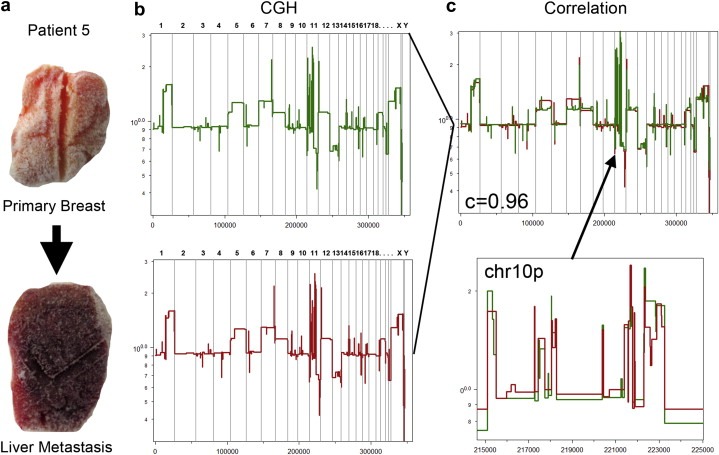
Genomic progression in a metastatic breast cancer patient. (a) A frozen primary breast tumor and metastatic liver tumor resected from a single patient. (b) CGH profiles measured by Representational Oligonucleotide Microarray Analysis (ROMA). The segmented breast tumor profile is plotted in green and liver profile is plotted in red. (c) (Upper Panel) Segmented breast and liver profiles are plotted and a high Pearson's correlation was calculated (c = 0.96) between the copy number events. (Lower Panel) An enlarged region of Chromosome 10p is plotted showing the high similarity between complex rearrangements in the primary and metastatic tumors.
11. Single cell genomics
A major problem that confounds the analysis of tumor progression is the presence of mixed cell populations. In inter‐tumor comparisons, single samples from heterogeneous tumors may reflect a mixture of tumor clones at various stages of progression, thus diluting the detection of high frequency chromosome mutations. As we discussed, in intra‐tumor comparison, clones must first be stratified and isolated based on differences in region, surface markers or ploidy to conduct genome‐wide comparisons. However, this requirement is effectively eliminated by single cell analysis – the ultimate level of genome stratification. By analyzing the whole genomes of single tumor cells we can address questions such as: Are the major clonal subpopulation profiles represented in the genomes of single cells? Do monogenomic tumors really contain identical genomes in every tumor cell? Using current genomic techniques, the minor subpopulations would almost certainly be masked by the overwhelming signal from the major tumor subpopulations in a mixture. Single cell copy number profiles are particularly useful, as they would allow detailed phylogenetic trees to be reconstructed, potentially illuminating the models for tumor progression.
Recent advances in whole genome amplification (WGA) methods now allow DNA from a single cell to be amplified to microgram quantities (Sigma‐Aldrich GenomePlex©, Rubicon Genomics PicoPlex©). However, the amplified DNA is not a perfect copy of the genome, but rather a representative library of random fragments covering less than 10% of the human genome (unpublished). Efforts to quantify whole‐genome copy number from WGA DNA have shown that it is indeed possible, albeit at low resolution (Le Caignec et al., 2006). However, major issues exist with the overall signal:noise ratio, standard deviation and dynamic range which permits only large (>10 megabase) chromosome aberrations to be detected in single tumor cells (Fuhrmann et al., 2008; Imle et al., 2009; Klein et al., 1999). One study did achieve a higher resolution (>3 mb) by applying tiling oligonucleotide microarrays to single cell WGA fragments (Geigl et al., 2009). However, such resolution is not a big improvement over traditional single cell cytological techniques such as G‐banding. In principle, array CGH is problematic for measuring copy number from WGA samples, since probes target predefined sequences only a fraction of the genomic DNA is amplified. Thus, the majority of microarray probes in any experiment will not hybridize to their respective targets at all.
To address this problem, our lab developed a technique using next‐generation sequencing to randomly sequence WGA amplified libraries from a single cell and measure copy number by read depth within fixed intervals in the human genome. Recent studies have shown that read depth from next‐generation sequencing can be used to accurately measure genomic copy number in DNA from millions of cells (Alkan et al., 2009; Chiang et al., 2009). In single cells, next‐generation Illumina sequencing has the advantage of being agnostic to the genomic position of the random DNA fragments, and thus the random fragments would not be ‘missed’ as with CGH microarrays. We call our method Single Nucleus Sequencing (SNS). Briefly, it involves sorting single nuclei, amplifying DNA by WGA, constructing sequencing libraries, and sequencing the DNA at 36 cycles using next‐generation illumina GA2 analyzers. We then align the massive number of sequencing reads to the human genome and measure read depth in 50 kb fixed intervals across the genome, resulting in a copy number profile. We then divide the experimental sample by a reference sample that was deep‐sequenced to generate a genomic copy number profile and segment the profiles for comparative analysis.
We applied SNS to an SK‐BR‐3 breast cancer cell line and showed that it can accurately generate a high‐quality copy number profile with a small standard deviation and large dynamic range (Figure 7a). We then compared the single cell profile to an array CGH profile performed on millions of SK‐BR‐3 cells. All of the major amplifications (RD2, MYC, ERBB2) and deletions (DCC) detected in the CGH experiment using millions of cells were also detected in the single cell sequencing experiments (Compare Figure 7a to b). Interestingly, the single cell profiles are highly quantile, clearly showing the four major tetraploid states in the SK‐BR‐3 genome. We applied SNS to 14 single SK‐BR‐3 cells and observed highly similar copy number profiles, suggesting that the SK‐BR‐3 cell culture was highly clonal. In summary, SNS permits accurate copy number profiling of single cells at 50 kb resolution on a single Illumina flowcell lane. Currently, we are applying this method to 100 cells from a frozen basal‐like breast tumor to see if the pattern of genomic loss followed by amplification can be observed at the single cell level. Single cell copy number data is particularly useful for reconstructing phylogenetic lineages to see if we can find evidence for clonal evolution.
Figure 7.
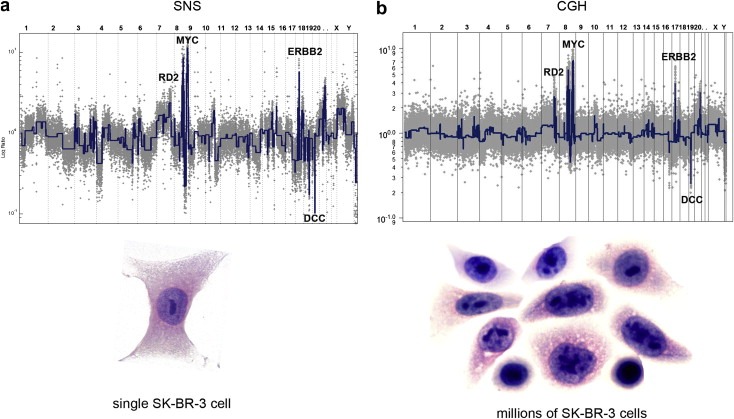
Single cell vs. million cell copy number profiles. Single Nucleus Sequencing (SNS) was used to profile copy number in a single SK‐BR‐3 cell and microarray CGH was used to profile copy number in approximately one million SK‐BR‐3 cells. (a) Next‐generation sequencing was used to measure genome‐wide read depth in a single SK‐BR‐3 cell on one Illumina flowcell lane. Reads were counted in 50 kb intervals across the human genome to generate a copy number profile (gray) from which segments were calculated (blue). (b) CGH profile of one million SK‐BR‐3 cells. The copy number ratio data are plotted in gray and the segmented profile in blue.
12. Final remarks
Models for tumor progression are useful only to the degree that they provoke experimental tests of their predictions or provide insight into cancer therapy. The clonal evolution models predict that all of the tumor cells need to be targeted by therapy and eliminated in order to cure the disease. In contrast, certain aspects of the self‐seeding model would predict that complete removal of the primary tumor might actually increase the number of potentially metastatic cells in circulation by removing their preferred homing target. Further, the cancer stem cell model predicts that targeting a small subpopulation of cells (CD44+/CD24−) would effectively treat the disease, irrespective of the rest of the tumor cells. This prediction has lead to an intense study of eradicating cancer stem cells with chemical inhibitors.
The cancer stem cell model has been built mainly on evidence that a relatively rare population of CD44+/24− cells can form tumors in immunocompromised (NOD/SCID) mice more efficiently than the bulk of tumor cells. But many have questioned the xenotransplantation model, because of the artificiality of injecting a subpopulation of human tumor cells into a foreign mouse microenvironment. Particularly strong evidence in melanoma showed that 27% of unselected cancer cells could give rise to tumors when the mouse model was more severely immunocompromised (NOD/SCID/IL‐2rg) (Quintana et al., 2008). In recent years, many reviewers have challenged the cancer stem cell hypothesis (Adams and Strasser, 2008; Campbell and Polyak, 2007; Fabian et al., 2009; Polyak, 2007a; Shipitsin and Polyak, 2008). What has mainly been questioned is whether the majority of tumor cells have limited proliferation potential, or ‐ as the clonal evolution models state – can continue proliferating indefinitely. These experiments suggest that the ability of a minority of enriched cancer stem cells to grow well in NOD/SCID mice (while the vast majority of tumor cells could not) may be the result of their ability to evade the mouse immune system (Shackleton et al., 2009), or simply a subpopulation that can recruit mouse stroma well (Adams and Strasser, 2008). Thus, the value of the cancer stem cell hypothesis for human patient treatment depends on (1) whether targeting cancer stem cells can effectively cure the disease and (2) if cancer stem cells can seed metastatic tumors.
In breast cancer, the clonal evolution models (monoclonal, polyclonal and self‐seeding) are strongly supported by genomic evidence from intra‐tumor, inter‐tumor and primary metastasis comparisons. Moreover, these results often contradict the assumptions of the cancer stem cell model and mutator phenotype. The major distinction between the clonal evolution and the cancer stem cell models is the potential for unlimited replication in the majority of tumor cells. However, in heterogeneous breast tumors, when we compare the copy number profiles from isolated subpopulations, we see evidence that clones diverge, but share the majority of chromosome breakpoints, suggesting a sequential evolution (Navin et al., 2010; Shipitsin et al., 2007; Torres et al., 2007). This strongly suggests that the majority of tumor cells continue to proliferate to form new subpopulations, rather than being regenerated from cancer stem cells. When an advantageous genotype is achieved, the clone will undergo clonal expansion and continue this process to form the mass of the tumor.
The continued proliferation of tumor cells is also evident in inter‐tumor comparisons in which we and others observe common mutational pathways that can be inferred by comparing simple CGH profiles to complex aneuploid profiles (Hicks et al., 2006, 2002, 2005, 1999). For example, in the luminal A subtype, we found a few simple profiles that contain only a gain of chromosome 1p and loss of 16q, while numerous complex aneuploid profiles include these events in addition to many other lesions. Thus, we can infer that 1p gain and 16q loss is an early event in the evolution of luminal A breast cancers, after which they continue to proliferate and acquire later events. By comparing the genome profiles of primary and metastatic tumors, we also see evidence that the majority of tumor clones arise by clonal expansions and continued proliferation. If cancer stem cells seeded the metastatic tumor, then it would be highly improbable that they would generate aneuploid profiles with nearly identical breakpoints to the primary tumor. Thus, we see strong evidence for monoclonal and polyclonal evolution (or Self‐seeding) in breast cancer.
The mutator phenotype model for progression by the random accumulation of non‐expanded mutations is highly unlikely as a cause for breast cancer. This model would predict a large diversity of unrelated clones in heterogeneous tumors. However, many experiments have shown that when multiple samples are taken from single heterogeneous tumors, and compared, they share the majority of genetic mutations (Aubele et al., 1999, 2007, 1995, 1996, 2007). We reached a similar conclusion using SPP to analyze 6–20 samples from individual heterogeneous tumors (Navin et al., 2010). Our analysis showed that profiles from the same tumor often clustered into a few highly similar groups, rather than a series of gradual intermediates or unrelated profiles (Figure 4b). These data suggests that when an advantageous genotype is achieved, a clone will undergo clonal expansion to form a mass of tumor cells. Furthermore, our data and others have shown that the copy number profiles in the primary and metastatic tumors show a high degree of similarity in many cancer types (Bockmuhl et al., 2004; Hovey et al., 1998; Israeli et al., 2004; Jiang et al., 2005; Liu et al., 2009b). This contradicts the mutator phenotype, because it predicts that a metastatic tumor would be seeded by a new series of random clones.
Biological models are by definition built upon incomplete information. At best, these explicit models for tumor progression provide guideposts for further exploration. As technology continues to evolve, the analysis of cancer samples of complex mixtures will give way to methods aimed at the individual cell. Such methods will enable single cancer cells to be tracked as they progress to form the primary tumor and traced as they migrate through the body to seed the metastasis. In the near future the cost of deep sequencing a mammalian genome, whether from a tumor sample or a few disseminated cells will be approximately equivalent to the current price of a microarray experiment. Single cell genomes are also ideal for constructing detailed lineages of tumor progression, because individual mutations in a genome can be traced as they are inherited and expanded in subpopulations. As we bring the magnifying glass closer, we may also be able to track the genetic stepping stones for tumor growth, or follow the genetic changes in circulating tumor cells as they progress from the primary to metastasis. Perhaps, we will find evidence that individual circulating tumor cells return to the primary tumor after developing offsite as the self‐seeding model suggests. It is then that these predictive genetic models will have realized their full value.
Acknowledgements
We would like to thank Michael Ronemus for reviewing this manuscript, and members of the Wigler and Hicks laboratories for experimental work and discussions. We would also like to thank Michael Wigler, Anders Zetterberg and Anne‐Lise Børresen‐Dale for useful discussions. This work was supported by a T32 fellowship to N.N. from NCI, and Grants from the Department of the Army (W81XWH04‐1‐0477) and Breast Cancer Foundation to provided support to J.H.
Navin Nicholas E., Hicks James, (2010), Tracing the tumor lineage, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.04.010.
References
- Adams, J.M. , Strasser, A. , 2008. Is tumor growth sustained by rare cancer stem cells or dominant clones?. Cancer Res. 68, 4018–4021. [DOI] [PubMed] [Google Scholar]
- Al-Hajj, M. , Wicha, M.S. , Benito-Hernandez, A. , Morrison, S.J. , Clarke, M.F. , 2003. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U S A. 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan, C. , Kidd, J.M. , Marques-Bonet, T. , Aksay, G. , Antonacci, F. , Hormozdiari, F. , Kitzman, J.O. , Baker, C. , Malig, M. , Mutlu, O. , 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred, D.C. , Wu, Y. , Mao, S. , Nagtegaal, I.D. , Lee, S. , Perou, C.M. , Mohsin, S.K. , O'Connell, P. , Tsimelzon, A. , Medina, D. , 2008. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin. Cancer Res. 14, 370–378. [DOI] [PubMed] [Google Scholar]
- Aubele, M. , Mattis, A. , Zitzelsberger, H. , Walch, A. , Kremer, M. , Hutzler, P. , Höfler, H. , Werner, M. , 1999. Intratumoral heterogeneity in breast carcinoma revealed by laser-microdissection and comparative genomic hybridization. Cancer Genet. Cytogenet. 110, 94–102. [DOI] [PubMed] [Google Scholar]
- Aubele, M. , Werner, M. , 1999. Heterogeneity in breast cancer and the problem of relevance of findings. Anal. Cell Pathol. 19, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtiary, B. , Boutros, P.C. , Pintilie, M. , Shi, W. , Bastianutto, C. , Li, J.H. , Schwock, J. , Zhang, W. , Penn, L.Z. , Jurisica, I. , 2006. Gene expression profiling in cervical cancer: an exploration of intratumor heterogeneity. Clin. Cancer Res. 12, 5632–5640. [DOI] [PubMed] [Google Scholar]
- Benetkiewicz, M. , Piotrowski, A. , Diaz De Stahl, T. , Jankowski, M. , Bala, D. , Hoffman, J. , Srutek, E. , Laskowski, R. , Zegarski, W. , Dumanski, J.P. , 2006. Chromosome 22 array-CGH profiling of breast cancer delimited minimal common regions of genomic imbalances and revealed frequent intra-tumoral genetic heterogeneity. Int. J. Oncol. 29, 935–945. [PubMed] [Google Scholar]
- Bergamaschi, A. , Kim, Y.H. , Wang, P. , Sorlie, T. , Hernandez-Boussard, T. , Lonning, P.E. , Tibshirani, R. , Borresen-Dale, A.L. , Pollack, J.R. , 2006. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 45, 1033–1040. [DOI] [PubMed] [Google Scholar]
- Bielas, J.H. , Loeb, K.R. , Rubin, B.P. , True, L.D. , Loeb, L.A. , 2006. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. U S A. 103, 18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas, J.H. , Loeb, L.A. , 2005. Mutator phenotype in cancer: timing and perspectives. Environ. Mol. Mutagen. 45, 206–213. [DOI] [PubMed] [Google Scholar]
- Bilke, S. , Chen, Q.R. , Westerman, F. , Schwab, M. , Catchpoole, D. , Khan, J. , 2005. Inferring a tumor progression model for neuroblastoma from genomic data. J. Clin. Oncol. 23, 7322–7331. [DOI] [PubMed] [Google Scholar]
- Bockmuhl, U. , You, X. , Pacyna-Gengelbach, M. , Arps, H. , Draf, W. , Petersen, I. , 2004. CGH pattern of esthesioneuroblastoma and their metastases. Brain Pathol. 14, 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet, D. , Dick, J.E. , 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737. [DOI] [PubMed] [Google Scholar]
- Brown, T.M. , Fee, E. , 2006. Rudolf Carl Virchow: medical scientist, social reformer, role model. Am. J. Public Health. 96, 2104–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calza, S. , Hall, P. , Auer, G. , Bjohle, J. , Klaar, S. , Kronenwett, U. , Liu, E.T. , Miller, L. , Ploner, A. , Smeds, J. , 2006. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 8, R34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, P.J. , Pleasance, E.D. , Stephens, P.J. , Dicks, E. , Rance, R. , Goodhead, I. , Follows, G.A. , Green, A.R. , Futreal, P.A. , Stratton, M.R. , 2008. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. U S A. 105, 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, L.L. , Polyak, K. , 2007. Breast tumor heterogeneity: cancer stem cells or clonal evolution?. Cell Cycle. 6, 2332–2338. [DOI] [PubMed] [Google Scholar]
- Carey, L.A. , Perou, C.M. , Livasy, C.A. , Dressler, L.G. , Cowan, D. , Conway, K. , Karaca, G. , Troester, M.A. , Tse, C.K. , Edmiston, S. , 2006. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 295, 2492–2502. [DOI] [PubMed] [Google Scholar]
- Chiang, D.Y. , Getz, G. , Jaffe, D.B. , O'Kelly, M.J. , Zhao, X. , Carter, S.L. , Russ, C. , Nusbaum, C. , Meyerson, M. , Lander, E.S. , 2009. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 6, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, K. , DeVries, S. , Fridlyand, J. , Spellman, P.T. , Roydasgupta, R. , Kuo, W.L. , Lapuk, A. , Neve, R.M. , Qian, Z. , Ryder, T. , 2006. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 10, 529–541. [DOI] [PubMed] [Google Scholar]
- Chin, S.F. , Wang, Y. , Thorne, N.P. , Teschendorff, A.E. , Pinder, S.E. , Vias, M. , Naderi, A. , Roberts, I. , Barbosa-Morais, N.L. , Garcia, M.J. , 2007. Using array-comparative genomic hybridization to define molecular portraits of primary breast cancers. Oncogene. 26, 1959–1970. [DOI] [PubMed] [Google Scholar]
- Clarke, M.F. , Dick, J.E. , Dirks, P.B. , Eaves, C.J. , Jamieson, C.H. , Jones, D.L. , Visvader, J. , Weissman, I.L. , Wahl, G.M. , 2006. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 66, 9339–9344. [DOI] [PubMed] [Google Scholar]
- Cole, K.A. , Krizman, D.B. , Emmert-Buck, M.R. , 1999. The genetics of cancer–a 3D model. Nat. Genet. 21, 38–41. [DOI] [PubMed] [Google Scholar]
- Coons, S.W. , Johnson, P.C. , Shapiro, J.R. , 1995. Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade human glioma. Cancer Res. 55, 1569–1577. [PubMed] [Google Scholar]
- Corver, W.E. , Middeldorp, A. , ter Haar, N.T. , Jordanova, E.S. , van Puijenbroek, M. , van Eijk, R. , Cornelisse, C.J. , Fleuren, G.J. , Morreau, H. , Oosting, J. , 2008. Genome-wide allelic state analysis on flow-sorted tumor fractions provides an accurate measure of chromosomal aberrations. Cancer Res. 68, 10333–10340. [DOI] [PubMed] [Google Scholar]
- Endoh, Y. , Tamura, G. , Kato, N. , Motoyama, T. , 2001. Apocrine adenosis of the breast: clonal evidence of neoplasia. Histopathology. 38, 221–224. [DOI] [PubMed] [Google Scholar]
- Fabian, A. , Barok, M. , Vereb, G. , Szollosi, J. , 2009. Die hard: are cancer stem cells the Bruce Willises of tumor biology?. Cytometry A. 75, 67–74. [DOI] [PubMed] [Google Scholar]
- Farabegoli, F. , Santini, D. , Ceccarelli, C. , Taffurelli, M. , Marrano, D. , Baldini, N. , 2001. Clone heterogeneity in diploid and aneuploid breast carcinomas as detected by FISH. Cytometry. 46, 50–56. [DOI] [PubMed] [Google Scholar]
- Fialkow, P.J. , 1974. The origin and development of human tumors studied with cell markers. N. Engl. J. Med. 291, 26–35. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, P.J. , 1986. Homogeneity and heterogeneity in pancreas cancer: presence of predominant and minor morphological types and implications. Int. J. Pancreatol. 1, 91–94. [DOI] [PubMed] [Google Scholar]
- Fuhrmann, C. , Schmidt-Kittler, O. , Stoecklein, N.H. , Petat-Dutter, K. , Vay, C. , Bockler, K. , Reinhardt, R. , Ragg, T. , Klein, C.A. , 2008. High-resolution array comparative genomic hybridization of single micrometastatic tumor cells. Nucleic Acids Res. 36, e39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigl, J.B. , Obenauf, A.C. , Waldispuehl-Geigl, J. , Hoffmann, E.M. , Auer, M. , Hormann, M. , Fischer, M. , Trajanoski, Z. , Schenk, M.A. , Baumbusch, L.O. , 2009. Identification of small gains and losses in single cells after whole genome amplification on tiling oligo arrays. Nucleic Acids Res. 37, e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaretti, W. , Monaco, R. , Pujic, N. , Rapallo, A. , Nigro, S. , Geido, E. , 1996. Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: association with degree of DNA aneuploidy. Am. J. Pathol. 149, 237–245. [PMC free article] [PubMed] [Google Scholar]
- Glockner, S. , Buurman, H. , Kleeberger, W. , Lehmann, U. , Kreipe, H. , 2002. Marked intratumoral heterogeneity of c-myc and cyclinD1 but not of c-erbB2 amplification in breast cancer. Lab. Invest. 82, 1419–1426. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- In Heim S., Mittleman F.(Eds.), Cancer Cytogenetics. third ed. John Wiley and Sons; ISBN 10:0-470-18179-6 [Google Scholar]
- Heng, H.H. , Bremer, S.W. , Stevens, J. , Ye, K.J. , Miller, F. , Liu, G. , Ye, C.J. , 2006. Cancer progression by non-clonal chromosome aberrations. J. Cell Biochem. 98, 1424–1435. [DOI] [PubMed] [Google Scholar]
- Heng, H.H. , Stevens, J.B. , Liu, G. , Bremer, S.W. , Ye, K.J. , Reddy, P.V. , Wu, G.S. , Wang, Y.A. , Tainsky, M.A. , Ye, C.J. , 2006. Stochastic cancer progression driven by non-clonal chromosome aberrations. J. Cell Physiol. 208, 461–472. [DOI] [PubMed] [Google Scholar]
- Hicks, J. , Muthuswamy, L. , Krasnitz, A. , Navin, N. , Riggs, M. , Grubor, V. , Esposito, D. , Alexander, J. , Troge, J. , Wigler, M. , 2005. High-resolution ROMA CGH and FISH analysis of aneuploid and diploid breast tumors. Cold Spring Harb. Symp. Quant. Biol. 70, 51–63. [DOI] [PubMed] [Google Scholar]
- Hicks, J. , Krasnitz, A. , Lakshmi, B. , Navin, N.E. , Riggs, M. , Leibu, E. , Esposito, D. , Alexander, J. , Troge, J. , Grubor, V. , 2006. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 16, 1465–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, F.R. , Ottesen, G. , Podenphant, J. , Olsen, J. , 1983. Tumor heterogeneity in lung cancer based on light microscopic features. A retrospective study of a consecutive series of 200 patients, treated surgically. Virchows Arch. A Pathol. Anat. Histopathol. 402, 147–153. [DOI] [PubMed] [Google Scholar]
- Hoglund, M. , Gisselsson, D. , Hansen, G.B. , Sall, T. , Mitelman, F. , 2002. Multivariate analysis of chromosomal imbalances in breast cancer delineates cytogenetic pathways and reveals complex relationships among imbalances. Cancer Res. 62, 2675–2680. [PubMed] [Google Scholar]
- Hoglund, M. , Frigyesi, A. , Sall, T. , Gisselsson, D. , Mitelman, F. , 2005. Statistical behavior of complex cancer karyotypes. Genes Chromosomes Cancer. 42, 327–341. [DOI] [PubMed] [Google Scholar]
- Hovey, R.M. , Chu, L. , Balazs, M. , DeVries, S. , Moore, D. , Sauter, G. , Carroll, P.R. , Waldman, F.M. , 1998. Genetic alterations in primary bladder cancers and their metastases. Cancer Res. 58, 3555–3560. [PubMed] [Google Scholar]
- Hu, Z. , Fan, C. , Oh, D.S. , Marron, J.S. , He, X. , Qaqish, B.F. , Livasy, C. , Carey, L.A. , Reynolds, E. , Dressler, L. , 2006. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 7, 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis, M. , Sotiriou, C. , 2008. Understanding the molecular basis of histologic grade. Pathobiology. 75, 104–111. [DOI] [PubMed] [Google Scholar]
- Imle, A. , Polzer, B. , Alexander, S. , Klein, C.A. , Friedl, P. , 2009. Genomic instability of micronucleated cells revealed by single-cell comparative genomic hybridization. Cytometry A. 75, 562–568. [DOI] [PubMed] [Google Scholar]
- Israeli, O. , Gotlieb, W.H. , Friedman, E. , Korach, J. , Goldman, B. , Zeltser, A. , Ben-Baruch, G. , Rienstein, S. , Aviram-Goldring, A. , 2004. Genomic analyses of primary and metastatic serous epithelial ovarian cancer. Cancer Genet. Cytogenet. 154, 16–21. [DOI] [PubMed] [Google Scholar]
- Jiang, J.K. , Chen, Y.J. , Lin, C.H. , Yu, I.T. , Lin, J.K. , 2005. Genetic changes and clonality relationship between primary colorectal cancers and their pulmonary metastases–an analysis by comparative genomic hybridization. Genes Chromosomes Cancer. 43, 25–36. [DOI] [PubMed] [Google Scholar]
- Johann, D.J. , Rodriguez-Canales, J. , Mukherjee, S. , Prieto, D.A. , Hanson, J.C. , Emmert-Buck, M. , Blonder, J. , 2009. Approaching solid tumor heterogeneity on a cellular basis by tissue proteomics using laser capture microdissection and biological mass spectrometry. J. Proteome Res. 8, 2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallioniemi, O.P. , 1988. Comparison of fresh and paraffin-embedded tissue as starting material for DNA flow cytometry and evaluation of intratumor heterogeneity. Cytometry. 9, 164–169. [DOI] [PubMed] [Google Scholar]
- Khalique, L. , Ayhan, A. , Weale, M.E. , Jacobs, I.J. , Ramus, S.J. , Gayther, S.A. , 2007. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J. Pathol. 211, 286–295. [DOI] [PubMed] [Google Scholar]
- Kim, M.Y. , Oskarsson, T. , Acharyya, S. , Nguyen, D.X. , Zhang, X.H. , Norton, L. , Massague, J. , 2009. Tumor self-seeding by circulating cancer cells. Cell. 139, 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, C.A. , Schmidt-Kittler, O. , Schardt, J.A. , Pantel, K. , Speicher, M.R. , Riethmüller, G. , 1999. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc. Natl. Acad. Sci. U S A. 96, 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki, K. , Sano, N. , Tangoku, A. , 2006. Problems in histological grading of malignancy and its clinical significance in patients with operable breast cancer. Breast Cancer. 13, 249–253. [DOI] [PubMed] [Google Scholar]
- Kruger, S. , Thorns, C. , Bohle, A. , Feller, A.C. , 2003. Prognostic significance of a grading system considering tumor heterogeneity in muscle-invasive urothelial carcinoma of the urinary bladder. Int. Urol. Nephrol. 35, 169–173. [DOI] [PubMed] [Google Scholar]
- Kwei, K.A. , Kung, Y. , Salari, K. , Holcomb, I.N. , Pollack, J.R. , 2010. Genomic instability in breast cancer: Pathogenesis and clinical implications. Mol. Oncol. 4, (3) 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot, T. , Sirard, C. , Vormoor, J. , Murdoch, B. , Hoang, T. , Caceres-Cortes, J. , Minden, M. , Paterson, B. , Caligiuri, M.A. , Dick, J.E. , 1994. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 367, 645–648. [DOI] [PubMed] [Google Scholar]
- Le Caignec, C. , Spits, C. , Sermon, K. , De Rycke, M. , Thienpont, B. , Debrock, S. , Staessen, C. , Moreau, Y. , Fryns, J.P. , Van Steirteghem, A. , 2006. Single-cell chromosomal imbalances detection by array CGH. Nucleic Acids Res. 34, e68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Heidt, D.G. , Dalerba, P. , Burant, C.F. , Zhang, L. , Adsay, V. , Wicha, M. , Clarke, M.F. , Simeone, D.M. , 2007. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Linder, D. , Gartler, S.M. , 1965. Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas. Science. 150, 67–69. [DOI] [PubMed] [Google Scholar]
- Lips, E.H. , van Eijk, R. , de Graaf, E.J. , Doornebosch, P.G. , de Miranda, N.F. , Oosting, J. , Karsten, T. , Eilers, P.H. , Tollenaar, R.A. , van Wezel, T. , 2008. Progression and tumor heterogeneity analysis in early rectal cancer. Clin. Cancer Res. 14, 772–781. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Bandyopadhyay, N. , Ranka, S. , Baudis, M. , Kahveci, T. , 2009. Inferring progression models for CGH data. Bioinformatics. 25, 2208–2215. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Laitinen, S. , Khan, S. , Vihinen, M. , Kowalski, J. , Yu, G. , Chen, L. , Ewing, C.M. , Eisenberger, M.A. , Carducci, M.A. , 2009. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 15, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb, L.A. , Springgate, C.F. , Battula, N. , 1974. Errors in DNA replication as a basis of malignant changes. Cancer Res. 34, 2311–2321. [PubMed] [Google Scholar]
- Maley, C.C. , Galipeau, P.C. , Finley, J.C. , Wongsurawat, V.J. , Li, X. , Sanchez, C.A. , Paulson, T.G. , Blount, P.L. , Risques, R.A. , Rabinovitch, P.S. , 2006. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 38, 468–473. [DOI] [PubMed] [Google Scholar]
- Marusyk, A. , Polyak, K. , 2009. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, T. , Fujii, H. , Arakawa, A. , Yamasaki, S. , Sonoue, H. , Hattori, K. , Kajiyama, Y. , Hirose, S. , Tsurumaru, M. , 2004. Loss of heterozygosity analysis shows monoclonal evolution with frequent genetic progression and divergence in esophageal carcinosarcoma. Hum. Pathol. 35, 322–327. [DOI] [PubMed] [Google Scholar]
- Mitelman, F. , Johansson, B. , Mandahl, N. , Mertens, F. , 1997. Clinical significance of cytogenetic findings in solid tumors. Cancer Genet. Cytogenet. 95, 1–8. [DOI] [PubMed] [Google Scholar]
- Mora, J. , Cheung, N.K. , Gerald, W.L. , 2001. Genetic heterogeneity and clonal evolution in neuroblastoma. Br. J. Cancer. 85, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin, N. , Krasnitz, A. , Rodgers, L. , Cook, K. , Meth, J. , Kendall, J. , Riggs, M. , Eberling, Y. , Troge, J. , Grubor, V. , 2010. Inferring tumor progression from genomic heterogeneity. Genome Res. 20, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, S. , Motomura, K. , Inaji, H. , Imaoka, S. , Koyama, H. , 1992. Clonal analysis of human breast cancer by means of the polymerase chain reaction. Cancer Res. 52, 6594–6597. [PubMed] [Google Scholar]
- Noguchi, S. , Motomura, K. , Inaji, H. , Imaoka, S. , Koyama, H. , 1994. Clonal analysis of predominantly intraductal carcinoma and precancerous lesions of the breast by means of polymerase chain reaction. Cancer Res. 54, 1849–1853. [PubMed] [Google Scholar]
- Norton, L. , 2008. Cancer stem cells, self-seeding, and decremented exponential growth: theoretical and clinical implications. Breast Dis. 29, 27–36. [DOI] [PubMed] [Google Scholar]
- Norton, L. , Massague, J. , 2006. Is cancer a disease of self-seeding?. Nat. Med. 12, 875–878. [DOI] [PubMed] [Google Scholar]
- Nowell, P.C. , 1976. The clonal evolution of tumor cell populations. Science. 194, 23–28. [DOI] [PubMed] [Google Scholar]
- O'Brien, C.A. , Pollett, A. , Gallinger, S. , Dick, J.E. , 2007. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 445, 106–110. [DOI] [PubMed] [Google Scholar]
- Paget, S. , 1889. The distribution of secondary growths in cancer of the breast. Lancet. 133, 571–573. [PubMed] [Google Scholar]
- Pandis, N. , Jin, Y. , Gorunova, L. , Petersson, C. , Bardi, G. , Idvall, I. , Johansson, B. , Ingvar, C. , Mandahl, N. , Mitelman, F. , 1995. Chromosome analysis of 97 primary breast carcinomas: identification of eight karyotypic subgroups. Genes Chromosomes Cancer. 12, 173–185. [DOI] [PubMed] [Google Scholar]
- Pantou, D. , Rizou, H. , Tsarouha, H. , Pouli, A. , Papanastasiou, K. , Stamatellou, M. , Trangas, T. , Pandis, N. , Bardi, G. , 2005. Cytogenetic manifestations of multiple myeloma heterogeneity. Genes Chromosomes Cancer. 42, 44–57. [DOI] [PubMed] [Google Scholar]
- Pathare, S. , Schaffer, A.A. , Beerenwinkel, N. , Mahimkar, M. , 2009. Construction of oncogenetic tree models reveals multiple pathways of oral cancer progression. Int. J. Cancer. 124, 2864–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Podo, F. , Buydens, L.M.C. , Degani, H. , Hillhorst, R. , Klipp, E. , Gribbestad, I.S. , van Huffel, S. , van Laarhooven, H.W.M. , Luts, J. , Monleon, D. , 2010. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 4, (3) 209–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak, K. , 2007. Breast cancer stem cells: a case of mistaken identity?. Stem Cell Rev. 3, 107–109. [DOI] [PubMed] [Google Scholar]
- Polyak, K. , 2007. Breast cancer: origins and evolution. J. Clin. Invest. 117, 3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana, E. , Shackleton, M. , Sabel, M.S. , Fullen, D.R. , Johnson, T.M. , Morrison, S.J. , 2008. Efficient tumour formation by single human melanoma cells. Nature. 456, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakha, E.A. , Reis-Filho, J.S. , Ellis, I.O. , 2008. Basal-like breast cancer: a critical review. J. Clin. Oncol. 26, 2568–2581. [DOI] [PubMed] [Google Scholar]
- Roka, S. , Fiegl, M. , Zojer, N. , Filipits, M. , Schuster, R. , Steiner, B. , Jakesz, R. , Huber, H. , Drach, J. , 1998. Aneuploidy of chromosome 8 as detected by interphase fluorescence in situ hybridization is a recurrent finding in primary and metastatic breast cancer. Breast Cancer Res. Treat. 48, 125–133. [DOI] [PubMed] [Google Scholar]
- Sauter, G. , Moch, H. , Gasser, T.C. , Mihatsch, M.J. , Waldman, F.M. , 1995. Heterogeneity of chromosome 17 and erbB-2 gene copy number in primary and metastatic bladder cancer. Cytometry. 21, 40–46. [DOI] [PubMed] [Google Scholar]
- Sawada, M. , Azuma, C. , Hashimoto, K. , Noguchi, S. , Ozaki, M. , Saji, F. , Tanizawa, O. , 1994. Clonal analysis of human gynecologic cancers by means of the polymerase chain reaction. Int. J. Cancer. 58, 492–496. [DOI] [PubMed] [Google Scholar]
- Selvarajah, S. , Yoshimoto, M. , Ludkovski, O. , Park, P.C. , Bayani, J. , Thorner, P. , Maire, G. , Squire, J.A. , Zielenska, M. , 2008. Genomic signatures of chromosomal instability and osteosarcoma progression detected by high resolution array CGH and interphase FISH. Cytogenet. Genome Res. 122, 5–15. [DOI] [PubMed] [Google Scholar]
- Shackleton, M. , Quintana, E. , Fearon, E.R. , Morrison, S.J. , 2009. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 138, 822–829. [DOI] [PubMed] [Google Scholar]
- Shah, S.P. , Morin, R.D. , Khattra, J. , Prentice, L. , Pugh, T. , Burleigh, A. , Delaney, A. , Gelmon, K. , Guliany, R. , Senz, J. , 2009. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 461, 809–813. [DOI] [PubMed] [Google Scholar]
- Shipitsin, M. , Campbell, L.L. , Argani, P. , Weremowicz, S. , Bloushtain-Qimron, N. , Yao, J. , Nikolskaya, T. , Serebryiskaya, T. , Beroukhim, R. , Hu, M. , 2007. Molecular definition of breast tumor heterogeneity. Cancer Cell. 11, 259–273. [DOI] [PubMed] [Google Scholar]
- Shipitsin, M. , Polyak, K. , 2008. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab. Invest. 88, 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S.K. , Hawkins, C. , Clarke, I.D. , Squire, J.A. , Bayani, J. , Hide, T. , Henkelman, R.M. , Cusimano, M.D. , Dirks, P.B. , 2004. Identification of human brain tumour initiating cells. Nature. 432, 396–401. [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.D. , van de Rijn, M. , Jeffrey, S.S. , 2001. Gene expression patterns of carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U S A. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Tibshirani, R. , Parker, J. , Hastie, T. , Marron, J.S. , Nobel, A. , Deng, S. , Johnsen, H. , Pesich, R. , Geisler, S. , 2003. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. U S A. 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, M.R. , Pandis, N. , Bardi, G. , Andersen, J.A. , Mandahl, N. , Mitelman, F. , Heim, S. , 1994. Cytogenetic analysis of multifocal breast carcinomas: detection of karyotypically unrelated clones as well as clonal similarities between tumour foci. Br. J. Cancer. 70, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, M.R. , Pandis, N. , Bardi, G. , Andersen, J.A. , Mitelman, F. , Heim, S. , 1995. Clonal heterogeneity in breast cancer: karyotypic comparisons of multiple intra- and extra-tumorous samples from 3 patients. Int. J. Cancer. 63, 63–68. [DOI] [PubMed] [Google Scholar]
- Teixeira, M.R. , Pandis, N. , Bardi, G. , Andersen, J.A. , Heim, S. , 1996. Karyotypic comparisons of multiple tumorous and macroscopically normal surrounding tissue samples from patients with breast cancer. Cancer Res. 56, 855–859. [PubMed] [Google Scholar]
- Torres, L. , Ribeiro, F.R. , Pandis, N. , Andersen, J.A. , Heim, S. , Teixeira, M.R. , 2007. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res. Treat. 102, 143–155. [DOI] [PubMed] [Google Scholar]
- Trent, J.M. , 1985. Cytogenetic and molecular biologic alterations in human breast cancer: a review. Breast Cancer Res. Treat. 5, 221–229. [DOI] [PubMed] [Google Scholar]
- Tsarouha, H. , Pandis, N. , Bardi, G. , Teixeira, M.R. , Andersen, J.A. , Heim, S. , 1999. Karyotypic evolution in breast carcinomas with i(1)(q10) and der(1;16)(q10;p10) as the primary chromosome abnormality. Cancer Genet. Cytogenet. 113, 156–161. [DOI] [PubMed] [Google Scholar]
- van der Poel, H.G. , Oosterhof, G.O. , Schaafsma, H.E. , Debruyne, F.M. , Schalken, J.A. , 1997. Intratumoral nuclear morphologic heterogeneity in prostate cancer. Urology. 49, 652–657. [DOI] [PubMed] [Google Scholar]
- Whelan, S. , 2008. Inferring trees. Methods Mol. Biol. 452, 287–309. [DOI] [PubMed] [Google Scholar]
- Zojer, N. , Fiegl, M. , Mullauer, L. , Chott, A. , Roka, S. , Ackermann, J. , Raderer, M. , Kaufmann, H. , Reiner, A. , Huber, H. , 1998. Chromosomal imbalances in primary and metastatic pancreatic carcinoma as detected by interphase cytogenetics: basic findings and clinical aspects. Br. J. Cancer. 77, 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]