

Abstract
Breast cancer is a heterogeneous disease, appreciable by molecular markers, gene‐expression profiles, and most recently, patterns of genomic alteration. In particular, genomic profiling has revealed three distinct patterns of DNA copy‐number alteration: a “simple” type with few gains or losses of whole chromosome arms, an “amplifier” type with focal high‐level DNA amplifications, and a “complex” type marked by numerous low‐amplitude changes and copy‐number transitions. The three patterns are associated with distinct gene‐expression subtypes, and preferentially target different loci in the genome (implicating distinct cancer genes). Moreover, the different patterns of alteration imply distinct underlying mechanisms of genomic instability. The amplifier pattern may arise from transient telomere dysfunction, although new data suggest ongoing “amplifier” instability. The complex pattern shows similarity to breast cancers with germline BRCA1 mutation, which also exhibit “basal‐like” expression profiles and complex‐pattern genomes, implicating a possible defect in BRCA1‐associated repair of DNA double‐strand breaks. As such, targeting presumptive DNA repair defects represents a promising area of clinical investigation. Future studies should clarify the pathogenesis of breast cancers with amplifier and complex‐pattern genomes, and will likely identify new therapeutic opportunities.
Keywords: Breast cancer, Genomic instability, DNA amplification, DNA repair defect, Basal-like, <i>BRCA1</i>
Abbreviations
- BER
base excision repair
- BFB
breakage-fusion-bridge
- CGH
comparative genomic hybridization
- CIN
chromosome instability
- CNA
copy-number alteration
- DCIS
ductal carcinoma in situ
- DM
double minute
- DSB
double-strand break
- ER
estrogen receptor
- FISH
fluorescence in situ hybridization
- HR
homologous recombination
- HSR
homogeneously staining region
- LOH
loss of heterozygosity
- MSI
microsatellite instability
- NER
nucleotide excision repair
- NHEJ
non-homologous end joining
- PR
progesterone receptor
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphism
1. Breast cancer molecular heterogeneity
Breast cancers exhibit a range of biologic and clinical behaviors. This diversity is reflected in the underlying morphologic and molecular variation, with a spectrum of histologic features and molecular pathologic markers that are useful in predicting clinical outcome and selecting appropriate therapy (Subramaniam and Isaacs, 2005). In particular, estrogen receptor (ER)/progesterone receptor (PR) positivity defines a tumor subset targetable by hormone modulation therapy (e.g. tamoxifen and aromatase inhibitors). Likewise, ERBB2 (HER2) amplification or overexpression identifies a distinct subset targetable with HER2 antagonists (e.g. trastuzumab). Tumors that are negative for ER, PR and HER2 (so‐called “triple negative”) are not amenable to these targeted therapies, and are often associated with poor prognosis (Reis‐Filho and Tutt, 2008; Podo et al., 2010).
More recently, genome‐scale analysis, and in particular microarray‐based gene‐expression profiling, has provided a refined molecular classification of breast cancer diversity. Expression patterns across “intrinsic” genes (more variant between than within tumors) have defined at least six molecular subtypes (Perou et al., 2000; Sorlie et al., 2001; Hennessy et al., 2009). Luminal subtypes A and B both express markers of the luminal epithelial layer of normal breast ducts (e.g. keratins 8/18) and are ER‐positive, though luminal B tumors are more highly proliferative and have less favorable prognosis. An ERBB2 subtype is associated with amplification and overexpression of ERBB2 (HER2) and neighboring genes at 17q12‐q21. A basal‐like subtype displays markers of the basal epithelial layer of normal breast ducts (e.g. keratins 5/6), is generally triple negative for ER, PR and HER2, and carries an unfavorable prognosis. Other triple‐negative classes include “normal‐like”, and the recently described “claudin‐low” subtype (Hennessy et al., 2009), underscoring that basal‐like and triple‐negative are highly overlapping but not equivalent classes (Reis‐Filho and Tutt, 2008).
There has been much speculation on the origins of the different molecular subtypes (Polyak, 2007; Stingl and Caldas, 2007). One possibility is that they arise from distinct breast epithelia progenitor cell types. For example, claudin‐low tumors have been noted to share markers (e.g. CD44+/CD24−) thought to characterize breast epithelial stem cells (Reis‐Filho and Tutt, 2008). Alternatively, the different subtypes might reflect distinct somatic DNA alterations (perhaps even arising through distinct mechanisms of genomic instability), ultimately influencing gene expression and cell phenotype. While these possibilities are not mutually exclusive, much interest has focused on characterizing breast cancer genomes and underlying genome instability.
2. Genomic instability
Genomic instability most accurately refers to an increased rate of genomic alteration, although some use the term to describe the state of the altered cancer genome. Genomic instability (also called “mutator phenotype”) is believed to be necessary for cells to accumulate the multiple mutations required for cancer to develop (Loeb, 2001), and is thought to result from dysfunction of genome “caretaker” genes (Kinzler and Vogelstein, 1997) early on during carcinogenesis.
Features of genomic instability were first well described in colorectal cancers (Lengauer et al., 1997, 1998), which exhibit either of two major types. Microsatellite instability (MSI) results from defects in DNA mismatch repair, leading to sequence mutations (including frameshifts) at simple repeats called microsatellites. More commonly, colorectal cancers exhibit chromosome instability (CIN), which leads to aneuploidy (chromosome number imbalances). The mechanism(s) underlying CIN are less well understood, but likely reflect dysfunction of chromosome duplication or segregation in mitosis. Virtually all colorectal cancers exhibit MSI or CIN, underscoring the profound relationship between genomic instability and tumorigenesis. Nevertheless, genomic rearrangements in breast cancer are typically quite complex and are likely to result from distinct mechanisms than those observed in colorectal cancer.
More recently, telomere dysfunction has been proposed as a mechanism underlying genomic instability (Artandi and DePinho, 2000). Telomeres function to cap and protect chromosome ends. Hyperproliferation of cells (hyperplasia) during early cancer progression can lead (in the normal absence of telomerase expression) to telomere attrition, driving chromosome fusion and breakage‐fusion‐bridge (BFB) cycles (discussed more later) resulting in complex, unbalanced chromosome rearrangements. In situ analysis of breast cancer progression supports a role of “telomere crisis” in the transition from ductal hyperplasia to ductal carcinoma in situ (DCIS; a precursor lesion) (Chin et al., 2004). Telomere crisis is presumed transient, with subsequent telomerase re‐expression stabilizing the rearranged genome.
The relative contribution of telomere crisis (vs. other mechanisms of genomic instability) to the etiology of breast cancer remains unclear. Also uncertain is whether there exists ongoing genomic instability in breast cancers. Hereditary breast cancers resulting from germline mutations in BRCA1, BRCA2, TP53 and CHEK2 also implicate possible underlying defects in DNA repair and cell‐cycle checkpoints (Ralhan et al., 2007), which will be discussed more later.
3. Genomic aberrations and instability in breast cancer
An established literature on breast cancer genomics has drawn from analysis by cytogenetics (of cultured cells and lines), fluorescence in situ hybridization (FISH), and conventional (chromosome‐based) comparative genomic hybridization (CGH) (Gray et al., 1994). This literature describes numerous gains and losses, including for example recurrent DNA amplifications (with presumptive oncogene driver) at 8p12 (FGFR1), 8q24 (MYC), 11q13 (CCND1), and 17q12 (ERBB2), among others. Some of such alterations have been noted to occur together (Courjal et al., 1997), suggesting cooperating events and implying molecular subgroups.
A marked advance in the field arrived with the development of array‐based CGH protocols (Pinkel et al., 1998; Pollack et al., 1999; Lucito et al., 2000). In array CGH, genomic DNA from tumor and from normal reference is comparatively hybridized to microarrays containing DNA sequence probes of known genome position (e.g. bacterial artificial chromosomes, cDNAs, or more recently, oligonucleotides). The resultant tumor/normal ratios provide a high‐resolution profile of DNA copy‐number alterations (CNAs) across the cancer genome (referenced to normal genome position). Current commercial CGH arrays and single nucleotide polymorphism (SNP) arrays (used to determine gene dosage and/or loss of heterozygosity (LOH)) contain hundreds of thousands to millions of oligonucleotide probes, yielding ultra‐high resolution genomic profiles.
Applying this technology, several contemporaneous studies defined the fine structure of breast cancer genomes (Loo et al., 2004; Wang et al., 2004; Fridlyand et al., 2006; Bergamaschi et al., 2006; Hicks et al., 2006; Chin et al., 2006). Together, these studies have provided a higher resolution catalog of recurrent alterations, pinpointing novel candidate cancer genes. A more provocative finding, though, was the discovery of distinct patterns of CNA among breast tumors (Fig. 1), since validated by others (Nordgard et al., 2008; Haverty et al., 2008). The consensus finding was three distinct patterns (or subtypes) of genomic alteration.
Figure 1.

Genomic profiling studies have identified distinct patterns of CNA among different breast cancers. Exemplary genomic profiles are shown here for individual breast tumors representing (A) “Simple” (“1q/16”) pattern, characteristic of luminal A tumors; (B) “Amplifier” (“firestorm”) pattern, typical of luminal B tumors. Note, in this tumor highest amplifications are seen within 1p, 2q and 6q (potentially pinpointing novel oncogenes), though more commonly occur at sites like 8p (FGFR), 11q (CCND1), and 20q (ZNF217); (C) “Amplifier” pattern, also typical for ERBB2 tumors. Note highest peak at 17q12 (ERBB2); (D) “Complex” (“sawtooth”) pattern, characteristic of basal‐like tumors. For each of the above graphs, the red trace represents the segmented CGH profile (tumor/normal fluorescence log2 ratio vs. genome map position), obtained using cghFLasso (Tibshirani and Wang, 2008). cDNA microarray CGH data are from Bergamaschi et al. (2006) and Bergamaschi et al. (2008). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The first type exhibits few CNAs, which tend to be gains or losses of whole chromosome arms, most characteristically gain of 1q and 16p, with loss of 16q (Fig. 1A). This pattern, dubbed “simple”, “simplex”, or “1q/16”, is associated mainly with ER‐positive, moderate to highly differentiated tumors, with luminal A gene‐expression patterns. While CDH1 (E‐cadherin) has been noted to reside on 16q, there is yet no evidence implicating it as a relevant target of 16q loss.
A second pattern of genomic alteration shows focal high‐level DNA amplifications, clustered on one or more chromosome arm, found on a background of simple to moderately complex gains/losses. This pattern, called “amplifier”, or “firestorm”, is characteristic of luminal B (Fig. 1B) and ERBB2 (Fig. 1C) subtype tumors. Commonly amplified sites include 8p12 (FGFR1), 8q24 (MYC), 11q13 (CCND1), 12q15 (MDM2), 17q12 (ERBB2), and 20q13 (ZNF217), as well as other less well‐characterized loci. Amplified genes often function in signaling, cell‐cycle regulation, and nucleic acid metabolism (Chin et al., 2006). Notably, tumor amplicons typically span several genes, where multiple “driver” oncogenes might confer selective growth advantage (Kao and Pollack, 2006).
A third type of genomic alteration is marked by a complex pattern of numerous low‐amplitude gains and losses, typically spanning short chromosome regions, and resulting in a “segmented” profile with many copy‐number transitions (Fig. 1D). This pattern, dubbed “complex, or “sawtooth”, is most common in triple‐negative, basal‐like tumors, and is also associated with TP53 mutation. Despite the complex patterns, preferential gain is seen at 10p, and loss at 3p, 4p, 4q, 5q, 14q, 15q, and in some studies 17q.
As noted, there is a good (if imperfect) correspondence between genomic pattern and gene‐expression subtype (Fig. 2A). Because the different genomic patterns target distinct loci, one implication is that the expression subtypes arise via alteration of distinct cancer genes. Even more intriguing, and a key conclusion of these genomic studies, is that the expression subtypes appear to originate via distinct underlying mechanisms of genomic instability.
Figure 2.
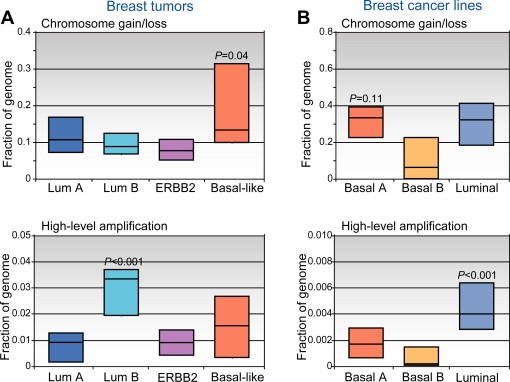
Molecular (gene‐expression) subtypes of breast cancer have been associated with distinct patterns of CNA. (A) Breast tumors classified by expression pattern (luminal A, luminal B, ERBB2, and basal‐like) display varying levels of low‐amplitude chromosome segment gain/loss (above), and of high‐level DNA amplification (below). Box plots display the 25th, 50th (median), and 75th percentiles of genome (cytoband) fraction altered among tumors classified (by expression profiles) to each subtype. Note that basal‐like tumors exhibit higher levels of segmental gain/loss, while luminal B tumors show more high‐level DNA amplifications (P‐values represent comparisons of the one group vs. all others). (B) Breast cancer cell lines, which can be classified by expression patterns to luminal or to one of either of two basal‐like groups (Neve et al., 2006; Kao et al., 2009), display analogous CNA patterns. Fraction of genome (genes) altered is displayed, as above. “Basal A” lines, which are most similar to basal‐like tumors (Kao et al., 2009), exhibit high levels of segmental gain/loss (P‐value compared to basal‐B group), while luminal lines show more high‐level DNA amplifications (compared to basal lines). Breast lines therefore provide a relevant model system to study the underlying genomic instabilities. Data shown are from Bergamaschi et al. (2006) and Kao et al. (2009).
Recently, “next‐generation” DNA sequencing has provided yet an additional view of the structural alterations in cancer genomes. Based on earlier paired‐end sequencing strategies (Volik et al., 2003), Futreal, Stratton and colleagues sequenced the paired ends of short genomic DNA fragments, yielding information both on copy number (by counting sequence reads) and on rearrangements (when paired ends mapped to discordant sites in the reference genome) (Campbell et al., 2008). Using this approach, they could classify rearrangements as occurring within amplicons, or if not then as being interchromosomal or intrachromosomal, with the latter further stratified as deletions, tandem duplication or inversions.
Despite the small number of specimens profiled, applying this approach to breast cancers (Stephens et al., 2009) recovered rearrangement patterns largely recapitulating the major array CGH types (Fig. 3). Luminal A subtype tumors showed few chromosome rearrangements. Luminal B tumors displayed many more rearrangements, which occurred mainly within amplicons. Triple‐negative basal‐like tumors exhibited numerous rearrangements, interestingly occurring mainly as small tandem duplications in the kilobase to megabase size range, largely unappreciated in prior array CGH profiles. The DNA sequences at rearrangement junctions were also informative (as discussed later).
Figure 3.
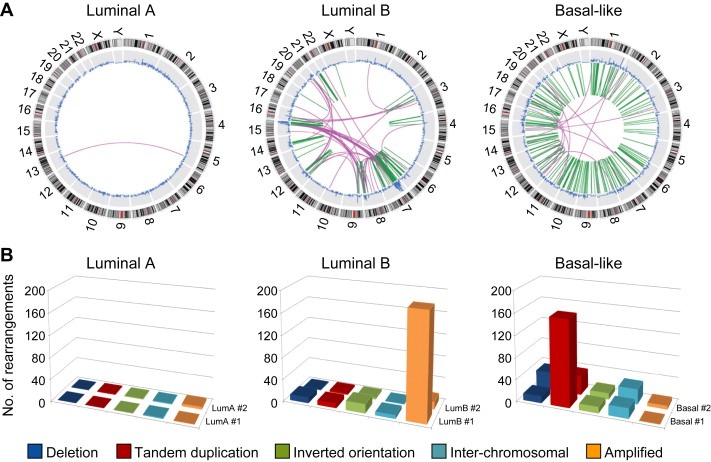
“Next‐Gen” DNA sequencing has revealed distinct patterns of genomic rearrangements among different breast cancers. (A) Exemplary patterns are shown for tumors classified by expression pattern as luminal A (left), luminal B (center), and basal‐like (right). In these “Circos” plots, the outer ring indicates genome position, the blue trace represents DNA copy number determined by counting sequencing reads (gains point to outside, losses to inside), and within the inner ring the green and purple lines represent intrachromosomal and interchromosomal rearrangements, respectively, identified by genome alignment of paired‐end sequence reads from small genomic fragments. Reprinted by permission from Macmillan Publishers Ltd: (Stephens et al., 2009). (B) Rearrangement architectures of luminal A (left), luminal B (center), and basal‐like (right) tumors, shown for the two tumors profiled from each group (the “front‐most” samples correspond to the Circos plots above). Bars indicate deletion (dark blue), tandem duplication (red), inverted orientation (green), interchromosomal rearrangements (light blue), and rearrangements within amplified regions (orange). Note, though the sample size is small, increased rearrangements within amplified regions are apparent in luminal B tumors, while intrachromosomal tandem duplications are prevalent in sporadic basal‐like tumors (as well as in BRCA1‐associated tumors, not shown). Data graphed here are from Stephens et al. (2009). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Of note, the genomic aberration patterns of breast tumors are largely recognizable in established breast cancer cell lines (Stephens et al., 2009; Neve et al., 2006; Kao et al., 2009) (Fig. 2B). Breast cancer lines with luminal expression patterns exhibit frequent DNA amplification, which typifies luminal B and ERBB2 subtype tumors. Likewise, cell lines with basal‐like expression patterns (“basal A” subgroup; Fig. 2B) show high numbers of low‐amplitude gain/loss, reminiscent of basal‐like breast tumors. By sequencing analysis, basal‐like breast lines also exhibit numerous intrachromosomal rearrangements of the tandem duplication variety (Stephens et al., 2009). Conspicuously absent among available breast lines is the simple genomic alteration pattern characteristic of luminal A tumors. Nonetheless, breast cancer cell lines should provide a useful model not only to discover and characterize novel breast cancer genes, but also to study the mechanisms underlying genomic instability.
These intriguing patterns of genomic alteration are likely the result of very distinct mechanisms. Below, we consider the possible causes for the amplifier and complex rearrangement phenotypes (the simple pattern being most consistent with a more subtle or absent instability).
4. Amplifier genomes and breast cancer
The amplifier pattern is characterized by focal high‐level DNA amplifications, and is associated mainly with luminal B subtype tumors. The ERBB2 subtype could be considered a special case, where the prominent amplification occurs at 17q12 (ERBB2).
Amplified DNA in cancer cells was first observed by cytogenetic analysis, where it occurred either within chromosomes as repeated units called homogeneously staining regions (HSRs), or as extrachromosomal copies called double minutes (DMs); in some experimental settings the two forms can interconvert (Hamlin et al., 1991). The structural organization of DNA amplicons has been studied in tumor cells, and in cultured mammalian cells following selection for drug resistance, e.g. DHFR amplification conferring methotrexate resistance. From such studies, proposed mechanisms of DNA amplification have been inferred (reviewed in Stark et al., 1989; Windle and Wahl, 1992; Albertson, 2006). In general, DNA amplification is thought to initiate with a chromosome break, often then followed by inappropriate cell‐cycle progression in the presence of this damaged DNA. One proposed mechanism of DNA amplification is based on episome formation (possibly resulting from a collapsed replication bubble), with subsequent extrachromosomal replication of DNA sometimes followed by reintegration into a chromosome. Such a mechanism is consistent with the head‐to‐tail tandem repeat structure of amplified MYCN observed often in neuroblastomas (Savelyeva and Schwab, 2001).
Another mechanism of DNA amplification is based on breakage‐fusion‐bridge (BFB) cycles, first described by McClintock (1941) in maize. BFB cycles are initiated when broken chromosomes (or sister chromatids) fuse, leading to a dicentric chromosome that then breaks apart during anaphase when the two centromeres are pulled to opposite spindle poles (Fig. 4). Depending on the site of the break, the result is a chromosome with an inverted duplication at the terminus. Further, because this chromosome has a broken end, the amplification process can be repeated in subsequent cell divisions. The initial break can occur at chromosome fragile sites (Coquelle et al., 1997; Hellman et al., 2002), or alternatively telomere attrition can provide the functional equivalent (O'Hagan et al., 2002). Anaphase bridges, a histologic marker of dicentric chromosomes (being pulled to opposite spindle poles), have been observed in early breast cancer progression (Chin et al., 2004). Furthermore, amplifier‐type breast tumors have been reported to show decreased telomere length and altered expression of telomere‐related genes, consistent with telomere attrition as a possible initiating mechanism (Fridlyand et al., 2006).
Figure 4.

DNA amplification arising by breakage‐fusion‐bridge (BFB) cycles. Cycle 1 initiates with chromosome breakage (A), occurring for example at a fragile site, or functionally emulated by critically shortened telomere sequences. Following chromosome replication in S‐phase to form sister chromatids (B), the break can be repaired (via NHEJ) by sister chromatid fusion (C). The resultant dicentric chromosome forms an anaphase bridge (D) when the two centromeres are pulled to opposite spindle poles, ultimately resolved by random chromosome breakage (E). The resultant broken chromosome provides a starting point for a subsequent BFB cycle. The blue arrowhead denotes a DNA segment residing proximal to the breakpoint, which becomes duplicated (note inverted structure) following the BFB cycle. Each subsequent BFB cycle can lead to additional duplication (cycle 2 product shown in (F)), resulting in an array of amplified DNA with characteristic inverted repeat architecture. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In a recent study, Bignell et al. (2007) analyzed amplicon‐associated rearrangements at the DNA sequence level. In a single ERBB2‐amplified breast cancer line, they identified at 17q12‐q21 (ERBB2) the precise inverted duplication architecture predicted by a sister chromatid BFB process. Nonetheless, amplicons elsewhere in the genome included head‐to‐tail direct repeats, indicating that different mechanisms of amplification are not mutually exclusive, and can even occur within the same cancer cell.
In complementary studies, Tlsty and colleagues explored the genetics of DNA amplification, using cultured cells exposed to the anti‐metabolite N‐(phosphonoacetyl)‐l‐aspartate (PALA), where resistance is conferred by amplification of CAD (encoding a multifunctional enzyme). Tumor cell lines representing diverse cancer types, but not normal fibroblasts, were found competent to amplify DNA (Tlsty, 1990; Tlsty et al., 1989). Cell fusions between tumor and normal cells defined amplification competence as a recessive phenotype (Tlsty et al., 1992). Furthermore, fusions between different cancer cells revealed at least two different complementation groups, which were distinct from TP53 mutation (Hall et al., 1997). The presumed caretaker genes were not identified.
Following up on Tlsty's studies, we have begun investigating CAD amplification (conferring PALA resistance) in breast cancer cells. In preliminary findings, a breast cancer line (MDA‐MB‐468) harboring focal DNA amplification at 7p11 (EGFR) exhibited the capacity (when exposed to PALA) to amplify DNA at the CAD locus (2p23) (Fig. 5). The amplification frequency was more than 1000‐fold higher than observed for a near‐diploid MSI breast cancer line (CAL51). This finding suggests the possibility that some breast cancers, possibly those already harboring amplicons, might retain a capacity (or permissivity) to amplify DNA de novo at other loci, i.e. an “amplifier” phenotype, with implications for cancer progression and therapy resistance.
Figure 5.

A possible “DNA amplifier” phenotype in breast cancer. (A) Segmented cDNA array CGH (log2 ratio) profile of two breast cancer cell lines, CAL51 (above), a diploid line with microsatellite instability (Seitz et al., 2003), and MDA‐MB‐468, a basal‐like line that also harbors focal high‐level DNA amplification at 7p11.12 (EGFR). To evaluate DNA amplification frequency, cells were plated and exposed to PALA at a concentration corresponding to nine times the lethal dose (LD50) for approximately four weeks, and the frequency of PALA resistant (PALAR) colonies (arising by CAD gene amplification at 2p23.3 (Tlsty et al., 1989)) determined. Despite comparably high cell plating efficiencies, MDA‐MB‐468 exhibited a more than 1000‐fold higher amplification frequency (PALAR colony frequencies indicated). This finding suggests the possibility that some breast cancers, possibly those already harboring amplicons, retain a competency to amplify DNA at other loci, i.e. an “amplifier” phenotype. (B) Oligonucleotide array CGH log2 ratios (blue dots) and segmented profile (red line) for chr2 in parent MDA‐MB‐468 line (above), and for three independent PALAR colonies (below), confirming focal DNA amplification at CAD (arrow). Note, the low‐level segmental gain of distal 2p seen in the parent line may provide an architecture contributing to the particularly high amplification rates (10−2) observed in MDA‐MB‐468; study of additional lines is warranted. Rare PALAR colonies of CAL51 exhibited whole chr2 gain (not shown). (C) Fluorescence in situ hybridization (FISH) confirming DNA amplification of CAD in a PALAR clone (below), compared to the parental line (above). Amplification of CAD in the PALAR clone is evident by the increased numbers of green (CAD locus) compared to red signals (control chr2 centromere probe). Note, the clustering of green signals observed in the interphase nucleus (filled arrow) and metaphase spread (open arrow) is indicative of in situ DNA amplification of CAD on chr2. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5. Complex patterns, basal‐like breast cancer, and BRCA1
The complex (sawtooth) pattern exhibits numerous low‐amplitude gains/losses and copy‐number transitions, and is characteristic of basal‐like breast cancers. Although the underlying mechanism is not yet known, the pattern is indicative of chromosome breakage throughout the genome, suggesting a more generalized process, e.g. a defect in DNA damage repair. Indeed, an intriguing hypothesis on the underlying mechanism is suggested by noted similarities between sporadic basal‐like breast cancers and hereditary breast cancers arising from germline BRCA1 mutation (Turner and Reis‐Filho, 2006). Sporadic basal‐like and BRCA1‐associated tumors exhibit similar histology, with high mitotic counts, medullary features, lymphocytic infiltrates, and “pushing” margins (Honrado et al., 2006). The vast majority of BRCA1‐associated breast tumors are also triple‐negative, and express basal markers and gene‐expression patterns (Sorlie et al., 2003; Foulkes et al., 2003). Notably, BRCA1 tumors also exhibit complex, low‐amplitude segmented profiles, with similar loci of preferential deletion (e.g. 4p, 4q, 5q) (Fridlyand et al., 2006; Tirkkonen et al., 1997; Jonsson et al., 2005). Finally, both sporadic basal‐like and BRCA1‐associated breast tumors appear to lack markers of a normal inactive X chromosome (Richardson et al., 2006). BRCA1 has diverse functions, but its tumor suppressive activity is most often connected to its role in DNA double‐strand break (DSB) repair (Jasin, 2002).
Multiple pathways exist to repair DNA damage, including for example mismatch repair, base excision repair (BER), nucleotide excision repair (NER), and DSB repair (Jackson and Bartek, 2009). DSBs, caused for example by ionizing radiation, reactive oxygen species (ROS) and collapsed DNA replication forks, are perhaps the most deleterious lesions. The two major pathways for DSB repair are homologous recombination (HR) and non‐homologous end joining (NHEJ) (Pardo et al., 2009). HR is an error‐free process that uses the sister chromatid (or homologous chromosome) to align and repair ends, and is the preferred mode of DSB repair in S/G2 phases of cell cycle (when sister chromatids are present). By contrast, NHEJ comprises error‐prone processes that join together two broken ends.
BRCA1 plays a key role in DSB repair, and in particular, in channeling repair towards error‐free HR (Scully et al., 2004). In the context of BRCA1 mutation, dysfunction of HR repair would lead to increased error‐prone repair, with resultant chromosome rearrangements and copy‐number transitions. Intriguingly, BRCA1 also appears to function in breast epithelial differentiation, regulating the differentiation of ER‐negative breast epithelial stem cells into ER‐positive luminal progenitors (Liu et al., 2008). Therefore, a unifying hypothesis states that sporadic basal‐like breast tumors harbor a dysfunction of BRCA1 (or BRCA1‐associated pathways), leading to both a complex pattern of genomic alteration, and an ER‐negative, basal‐like phenotype.
Some evidence supports a BRCA1 deficiency in sporadic basal‐like breast cancers. While BRCA1 mutations are rare in sporadic breast cancer (Futreal et al., 1994), BRCA1 (17q21) deletion/LOH has been reported (though does not appear to be specific for ER‐negative (including basal‐like) tumors) (Staff et al., 2003). Decreased BRCA1 transcript levels (Turner et al., 2007) and nuclear protein expression (Rakha et al., 2008) have also been observed in some basal‐like tumors. In addition, BRCA1 promoter hypermethylation has been reported in metaplastic breast cancers (a rare type of basal‐like tumor), as has overexpression of ID4, a negative regular of BRCA1 expression, in basal‐like cancers (Turner et al., 2007). Finally, paired‐end DNA sequencing analysis of rearrangements, including the tandem duplications prevalent in basal‐like breast cancers, has identified short (1–4 bp) regions of overlapping microhomology (i.e. identical sequences present in the contributing DNA segments, and once at the rearrangement junction), a finding less common of amplicon‐associated rearrangements in amplifier tumors (Stephens et al., 2009). Such microhomology, guiding the alignment of broken ends, has been considered a hallmark of the NHEJ process (Hefferin and Tomkinson, 2005), and possibly implies a defect in error‐free HR repair.
However, despite the intriguing hypothesis connecting complex genome patterns, basal‐like phenotype and BRCA1 dysfunction, at least one study supports the presence of functional BRCA1 in basal‐like tumors, evidenced by the common appearance of nuclear BRCA1 foci (Richardson et al., 2006). Furthermore, to our knowledge, there is as yet no direct evidence of a HR defect in sporadic basal‐like breast cancer.
Perhaps most notable, though, are the characteristics of hereditary breast cancers arising from germline mutation of BRCA2, which also has a key function in HR repair. BRCA2‐associated tumors are typically ER‐positive (not basal‐like), and exhibit neither the same preferential sites of loss (e.g. 4p, 4q, 5q), nor the complex pattern of genomic alteration (Stephens et al., 2009; Jonsson et al., 2005; van Beers et al., 2005; Waddell et al., 2009) (though at least one study did report finding complex‐pattern changes (Stefansson et al., 2009)). Of note, the main function of BRCA2 appears to be in regulating RAD51 activity in HR. In contrast, BRCA1 functions more as a signal integrator of the DNA damage response, acting not only in HR but also in other DNA repair pathways (e.g. NER, BER), as well as cell‐cycle (G2/M and spindle assembly) checkpoints, and more broadly in transcriptional regulation and chromatin remodeling (Venkitaraman, 2002; Narod and Foulkes, 2004). Therefore, it is possible that dysfunction of one or more of its other activities (in addition to or instead of HR) connects BRCA1 to the complex genome patterns in basal‐like tumors. In this regard, Alli et al. (2009) recently reported defective BER of oxidative DNA damage (a pathway also associated with BRCA1 function) in basal‐like cancer cell lines.
Alternatively, a different shared property of BRCA1‐associated and sporadic basal‐like breast tumors might underlie the observed complex‐pattern genome rearrangements. For example, both tumor types show frequent mutation of TP53, a master caretaker that controls cell fate in response to DNA damage, but also has a specific role in blocking HR between divergent sequences (which might otherwise lead to rearrangement) (Ralhan et al., 2007). In addition, BRCA1 and sporadic basal‐like tumors both show frequent deletions at 5q, a region harboring several genes functioning in DNA damage repair, including MSH3, RAD17, RAD50 and XRCC4 (Bergamaschi et al., 2006). Another shared feature appears to be dysfunction of the PTEN tumor suppressor (Saal et al., 2008). Loss of PTEN expression has been observed in sporadic basal‐like tumors, and BRCA1‐deficient tumors exhibit gross mutations (inversions, deletions) at the PTEN locus. Intriguingly, PTEN loss also results in defective HR (Mendes‐Pereira et al., 2009), though that dysfunction would seem superfluous in BRCA1‐deficient tumors (where ostensibly the PTEN gross mutations are themselves a likely consequence of a BRCA1‐associated HR defect).
Increased DNA breakage might also contribute to the etiology of complex‐pattern genomes. In this regard, another shared feature of hereditary BRCA1‐associated and sporadic basal‐like breast tumors is their high rates of cell proliferation. Indeed, analyzing gene‐expression patterns of complex pattern breast cancers, Fridlyand et al. (2006) identified enrichment of gene sets relating to cell proliferation (e.g. mitosis, cell cycle, DNA replication and repair), and of E2F1 target genes. Mechanistically, oncogene‐driven cell proliferation can induce DNA replication stress, where the stall and collapse of replication forks create DSBs (Halazonetis et al., 2008). Of note, collapsed replication forks will create single‐ended DSBs, most likely repaired by microhomology mediated break‐induced replication (MMBIR) (Hastings et al., 2009), and providing an alternative explanation for the observed microhomologies at rearrangement junctions (Stephens et al., 2009). However, we note that high proliferation rates also characterize many of the amplifier (luminal B) subtype breast tumors (Langerod et al., 2007).
Finally, the resultant constellation of CNAs might itself perpetuate or drive genomic instability. Analyzing the numerous gains in complex‐pattern genomes, Chin et al. (2006) noted enrichment of genes associated with RNA and cell metabolism, which might increase basal metabolism and possibly proliferation rates. In addition, widespread CNA, through the altered expression of many genes, might disrupt critical stoichiometric relationships within protein complexes (e.g. the mitotic spindle), possibly further promoting genomic instability (Pollack et al., 2002).
6. Clinical implications
Genomic instability and the resultant aberrations have important clinical implications. Most directly, the identified patterns of genomic alteration might provide a basis for improved breast cancer classification and prognostication. For example, amplifier tumors are associated with reduced survival (Hicks et al., 2006; Chin et al., 2006), with a quantitative relation between the number of amplifications and overall survival (Hicks et al., 2006). While several prior studies have identified specific amplified loci associated with unfavorable outcome, a general amplifier phenotype might instead underlie this association (Chin et al., 2006).
Also important are the therapeutic implications of genomic instability in breast cancer. Here, the amplification of specific oncogenes can provide therapeutic targets. A classic example is ERBB2 (HER2), an early success of molecularly targeted therapy (with trastuzumab) (Pegram et al., 2000). More generally, amplified oncogenes are promising targets based on likely oncogene addiction/dependency (Weinstein, 2002), and because the elevated expression (compared to normal tissues) should provide a wide therapeutic window. Genomic instability and the associated rearrangements might also lead to oncogenic fusion genes, which as novel creations could provide highly specific diagnostic markers and therapeutic targets.
A particularly exciting therapeutic area is the potential for targeting genomic instability itself. While genomic instability can be beneficial to tumors, by providing advantageous alterations, it can also create exploitable vulnerabilities. That is, by inducing gene or pathway dependencies not present in normal cells, genomic instability can generate “synthetic lethal” interactions (Kaelin, 2005) specifically in tumor cells. A striking example is that deficiency of BRCA1 or BRCA2 leads to marked sensitivity to inhibitors of poly(ADP‐ribose) polymerase (PARP) (Farmer et al., 2005; Bryant et al., 2005). PARP functions in the repair of single‐strand breaks, and presumably its inhibition leads to collapse of replication forks and DSBs that depend on HR for repair. Notably, because in BRCA mutation carriers only the tumors (and not normal tissues) are fully deficient in BRCA function, PARP inhibitors are likely to be highly tumor specific. A recent clinical trial supports the efficacy of PARP inhibition in tumors arising in BRCA mutation carriers (Fong et al., 2009), and trials evaluating sporadic triple‐negative breast tumors (with possible BRCA1 pathway defects) are ongoing. More generally, presumptive DNA repair defect(s) in complex pattern, basal‐like tumors might result in selective sensitivity to certain classes of DNA damaging agents, e.g. DNA crosslinking agents (Helleday et al., 2008).
7. Future directions
The study of breast cancer genomes and genomic instability is advancing rapidly, but much remains to be learned. The basis for the amplifier pattern remains uncertain, though continued sequence‐level analysis of amplicon architecture is likely to provide insight. Also unclear is whether amplification is largely a transient event (i.e. occurring at telomere crisis), or alternatively is an ongoing process shaping tumor genomes.
For the complex‐pattern breast tumors, future studies should clarify the nature of a possible defect in DSB repair (and, more broadly, in the DNA damage response), as well as the functionality of BRCA1 and BRCA1‐pathway components. Finally, for both the amplifiers and complex‐pattern tumors, future efforts (including unbiased screens) should identify selective vulnerabilities created by underlying genomic instability. Such studies will not only inform the mechanisms of genomic instability, but are likely to be clinically translatable, ultimately leading to new treatments for patients with breast cancer.
Acknowledgments
We wish to thank the Pollack lab for helpful discussion. This work was supported in part by grants from the NIH, R01 CA97139 (J.R.P.), T32 CA09151 (K.A.K and I.N.H.); and the California Breast Cancer Research Program, 8KB‐0135 (J.R.P). K.S. was supported by a Paul & Daisy Soros Fellowship, and the Medical Scientist Training Program.
Kwei Kevin A., Kung Yvonne, Salari Keyan, Holcomb Ilona N., Pollack Jonathan R., (2010), Genomic instability in breast cancer: Pathogenesis and clinical implications, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.04.001.
References
- Albertson, D.G. , 2006. Gene amplification in cancer. Trends Genet. 22, 447–455. [DOI] [PubMed] [Google Scholar]
- Alli, E. , Sharma, V.B. , Sunderesakumar, P. , Ford, J.M. , 2009. Defective repair of oxidative DNA damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res. 69, 3589–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi, S.E. , DePinho, R.A. , 2000. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr. Opin. Genet. Dev. 10, 39–46. [DOI] [PubMed] [Google Scholar]
- Bergamaschi, A. , Kim, Y.H. , Wang, P. , Sorlie, T. , Hernandez-Boussard, T. , Lonning, P.E. , Tibshirani, R. , Borresen-Dale, A.L. , Pollack, J.R. , 2006. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 45, 1033–1040. [DOI] [PubMed] [Google Scholar]
- van Beers, E.H. , van Welsem, T. , Wessels, L.F. , Li, Y. , Oldenburg, R.A. , Devilee, P. , Cornelisse, C.J. , Verhoef, S. , Hogervorst, F.B. , van't Veer, L.J. , Nederlof, P.M. , 2005. Comparative genomic hybridization profiles in human BRCA1 and BRCA2 breast tumors highlight differential sets of genomic aberrations. Cancer Res. 65, 822–827. [PubMed] [Google Scholar]
- Bergamaschi, A. , Kim, Y.H. , Kwei, K.A. , Choi, Y.-L. , Bocanegra, M. , Langerød, A. , Han, W. , Noh, D.-Y. , Huntsman, D.G. , Jeffrey, S.S. , Børresen-Dale, A.L. , Pollack, J.R. , 2008. CAMK1D amplification implicated in epithelial–mesenchymal transition in basal-like breast cancer. Mol. Oncol. 2, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell, G.R. , Santarius, T. , Pole, J.C. , Butler, A.P. , Perry, J. , Pleasance, E. , Greenman, C. , Menzies, A. , Taylor, S. , Edkins, S. , Campbell, P. , Quail, M. , Plumb, B. , Matthews, L. , McLay, K. , Edwards, P.A. , Rogers, J. , Wooster, R. , Futreal, P.A. , Stratton, M.R. , 2007. Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome Res. 17, 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, H.E. , Schultz, N. , Thomas, H.D. , Parker, K.M. , Flower, D. , Lopez, E. , Kyle, S. , Meuth, M. , Curtin, N.J. , Helleday, T. , 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Campbell, P.J. , Stephens, P.J. , Pleasance, E.D. , O'Meara, S. , Li, H. , Santarius, T. , Stebbings, L.A. , Leroy, C. , Edkins, S. , Hardy, C. , Teague, J.W. , Menzies, A. , Goodhead, I. , Turner, D.J. , Clee, C.M. , Quail, M.A. , Cox, A. , Brown, C. , Durbin, R. , Hurles, M.E. , Edwards, P.A. , Bignell, G.R. , Stratton, M.R. , Futreal, P.A. , 2008. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 40, 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, K. , de Solorzano, C.O. , Knowles, D. , Jones, A. , Chou, W. , Rodriguez, E.G. , Kuo, W.L. , Ljung, B.M. , Chew, K. , Myambo, K. , Miranda, M. , Krig, S. , Garbe, J. , Stampfer, M. , Yaswen, P. , Gray, J.W. , Lockett, S.J. , 2004. In situ analyses of genome instability in breast cancer. Nat. Genet. 36, 984–988. [DOI] [PubMed] [Google Scholar]
- Chin, K. , DeVries, S. , Fridlyand, J. , Spellman, P.T. , Roydasgupta, R. , Kuo, W.L. , Lapuk, A. , Neve, R.M. , Qian, Z. , Ryder, T. , Chen, F. , Feiler, H. , Tokuyasu, T. , Kingsley, C. , Dairkee, S. , Meng, Z. , Chew, K. , Pinkel, D. , Jain, A. , Ljung, B.M. , Esserman, L. , Albertson, D.G. , Waldman, F.M. , Gray, J.W. , 2006. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 10, 529–541. [DOI] [PubMed] [Google Scholar]
- Coquelle, A. , Pipiras, E. , Toledo, F. , Buttin, G. , Debatisse, M. , 1997. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 89, 215–225. [DOI] [PubMed] [Google Scholar]
- Courjal, F. , Cuny, M. , Simony-Lafontaine, J. , Louason, G. , Speiser, P. , Zeillinger, R. , Rodriguez, C. , Theillet, C. , 1997. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res. 57, 4360–4367. [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , Santarosa, M. , Dillon, K.J. , Hickson, I. , Knights, C. , Martin, N.M. , Jackson, S.P. , Smith, G.C. , Ashworth, A. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fong, P.C. , Boss, D.S. , Yap, T.A. , Tutt, A. , Wu, P. , Mergui-Roelvink, M. , Mortimer, P. , Swaisland, H. , Lau, A. , O'Connor, M.J. , Ashworth, A. , Carmichael, J. , Kaye, S.B. , Schellens, J.H. , de Bono, J.S. , 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Foulkes, W.D. , Stefansson, I.M. , Chappuis, P.O. , Begin, L.R. , Goffin, J.R. , Wong, N. , Trudel, M. , Akslen, L.A. , 2003. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 95, 1482–1485. [DOI] [PubMed] [Google Scholar]
- Fridlyand, J. , Snijders, A.M. , Ylstra, B. , Li, H. , Olshen, A. , Segraves, R. , Dairkee, S. , Tokuyasu, T. , Ljung, B.M. , Jain, A.N. , McLennan, J. , Ziegler, J. , Chin, K. , Devries, S. , Feiler, H. , Gray, J.W. , Waldman, F. , Pinkel, D. , Albertson, D.G. , 2006. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 6, 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal, P.A. , Liu, Q. , Shattuck-Eidens, D. , Cochran, C. , Harshman, K. , Tavtigian, S. , Bennett, L.M. , Haugen-Strano, A. , Swensen, J. , Miki, Y. , 1994. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 266, 120–122. [DOI] [PubMed] [Google Scholar]
- Gray, J.W. , Collins, C. , Henderson, I.C. , Isola, J. , Kallioniemi, A. , Kallioniemi, O.P. , Nakamura, H. , Pinkel, D. , Stokke, T. , Tanner, M. , 1994. Molecular cytogenetics of human breast cancer. Cold Spring Harb. Symp. Quant. Biol. 59, 645–652. [DOI] [PubMed] [Google Scholar]
- Hall, I.J. , Gioeli, D. , Weissman, B.E. , Tlsty, T.D. , 1997. Identification of additional complementation groups that regulate genomic instability. Genes Chromosomes Cancer. 20, 103–112. [PubMed] [Google Scholar]
- Hamlin, J.L. , Leu, T.H. , Vaughn, J.P. , Ma, C. , Dijkwel, P.A. , 1991. Amplification of DNA sequences in mammalian cells. Prog. Nucleic Acid Res. Mol. Biol. 41, 203–239. [DOI] [PubMed] [Google Scholar]
- Hastings, P.J. , Lupski, J.R. , Rosenberg, S.M. , Ira, G. , 2009. Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy, B.T. , Gonzalez-Angulo, A.M. , Stemke-Hale, K. , Gilcrease, M.Z. , Krishnamurthy, S. , Lee, J.S. , Fridlyand, J. , Sahin, A. , Agarwal, R. , Joy, C. , Liu, W. , Stivers, D. , Baggerly, K. , Carey, M. , Lluch, A. , Monteagudo, C. , He, X. , Weigman, V. , Fan, C. , Palazzo, J. , Hortobagyi, G.N. , Nolden, L.K. , Wang, N.J. , Valero, V. , Gray, J.W. , Perou, C.M. , Mills, G.B. , 2009. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 69, 4116–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, J. , Krasnitz, A. , Lakshmi, B. , Navin, N.E. , Riggs, M. , Leibu, E. , Esposito, D. , Alexander, J. , Troge, J. , Grubor, V. , Yoon, S. , Wigler, M. , Ye, K. , Borresen-Dale, A.L. , Naume, B. , Schlicting, E. , Norton, L. , Hagerstrom, T. , Skoog, L. , Auer, G. , Maner, S. , Lundin, P. , Zetterberg, A. , 2006. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 16, 1465–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis, T.D. , Gorgoulis, V.G. , Bartek, J. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Haverty, P.M. , Fridlyand, J. , Li, L. , Getz, G. , Beroukhim, R. , Lohr, S. , Wu, T.D. , Cavet, G. , Zhang, Z. , Chant, J. , 2008. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Genes Chromosomes Cancer. 47, 530–542. [DOI] [PubMed] [Google Scholar]
- Hefferin, M.L. , Tomkinson, A.E. , 2005. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst.). 4, 639–648. [DOI] [PubMed] [Google Scholar]
- Helleday, T. , Petermann, E. , Lundin, C. , Hodgson, B. , Sharma, R.A. , 2008. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer. 8, 193–204. [DOI] [PubMed] [Google Scholar]
- Hellman, A. , Zlotorynski, E. , Scherer, S.W. , Cheung, J. , Vincent, J.B. , Smith, D.I. , Trakhtenbrot, L. , Kerem, B. , 2002. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 1, 89–97. [DOI] [PubMed] [Google Scholar]
- Honrado, E. , Benitez, J. , Palacios, J. , 2006. Histopathology of BRCA1- and BRCA2-associated breast cancer. Crit. Rev. Oncol. Hematol. 59, 27–39. [DOI] [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA-damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin, M. , 2002. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 21, 8981–8993. [DOI] [PubMed] [Google Scholar]
- Jonsson, G. , Naylor, T.L. , Vallon-Christersson, J. , Staaf, J. , Huang, J. , Ward, M.R. , Greshock, J.D. , Luts, L. , Olsson, H. , Rahman, N. , Stratton, M. , Ringner, M. , Borg, A. , Weber, B.L. , 2005. Distinct genomic profiles in hereditary breast tumors identified by array-based comparative genomic hybridization. Cancer Res. 65, 7612–7621. [DOI] [PubMed] [Google Scholar]
- Kao, J. , Salari, K. , Bocanegra, M. , Choi, Y.L. , Girard, L. , Gandhi, J. , Kwei, K.A. , Hernandez-Boussard, T. , Wang, P. , Gazdar, A.F. , Minna, J.D. , Pollack, J.R. , 2009. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE. 4, e6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, J. , Pollack, J.R. , 2006. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer. 45, 761–769. [DOI] [PubMed] [Google Scholar]
- Kaelin, W.G. , 2005. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 5, 689–698. [DOI] [PubMed] [Google Scholar]
- Kinzler, K.W. , Vogelstein, B. , 1997. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 386, 761–763. [DOI] [PubMed] [Google Scholar]
- Langerod, A. , Zhao, H. , Borgan, O. , Nesland, J.M. , Bukholm, I.R. , Ikdahl, T. , Karesen, R. , Borresen-Dale, A.L. , Jeffrey, S.S. , 2007. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 9, R30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer, C. , Kinzler, K.W. , Vogelstein, B. , 1997. Genetic instability in colorectal cancers. Nature. 386, 623–627. [DOI] [PubMed] [Google Scholar]
- Lengauer, C. , Kinzler, K.W. , Vogelstein, B. , 1998. Genetic instabilities in human cancers. Nature. 396, 643–649. [DOI] [PubMed] [Google Scholar]
- Loeb, L.A. , 2001. A mutator phenotype in cancer. Cancer Res. 61, 3230–3239. [PubMed] [Google Scholar]
- Liu, S. , Ginestier, C. , Charafe-Jauffret, E. , Foco, H. , Kleer, C.G. , Merajver, S.D. , Dontu, G. , Wicha, M.S. , 2008. BRCA1 regulates human mammary stem/progenitor cell fate. Proc. Natl. Acad. Sci. U S A. 105, 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, L.W. , Grove, D.I. , Williams, E.M. , Neal, C.L. , Cousens, L.A. , Schubert, E.L. , Holcomb, I.N. , Massa, H.F. , Glogovac, J. , Li, C.I. , Malone, K.E. , Daling, J.R. , Delrow, J.J. , Trask, B.J. , Hsu, L. , Porter, P.L. , 2004. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 64, 8541–8549. [DOI] [PubMed] [Google Scholar]
- Lucito, R. , West, J. , Reiner, A. , Alexander, J. , Esposito, D. , Mishra, B. , Powers, S. , Norton, L. , Wigler, M. , 2000. Detecting gene copy number fluctuations in tumor cells by microarray analysis of genomic representations. Genome Res. 10, 1726–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B. , 1941. The stability of broken ends of chromosomes in Zea mays . Genetics. 26, 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Pereira, A.M. , Martin, S.A. , Brough, R. , McCarthy, A. , Taylor, J.R. , Kim, J.S. , Waldman, T. , Lord, C.J. , Ashworth, A. , 2009. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narod, S.A. , Foulkes, W.D. , 2004. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer. 4, 665–676. [DOI] [PubMed] [Google Scholar]
- Neve, R.M. , Chin, K. , Fridlyand, J. , Yeh, J. , Baehner, F.L. , Fevr, T. , Clark, L. , Bayani, N. , Coppe, J.P. , Tong, F. , Speed, T. , Spellman, P.T. , DeVries, S. , Lapuk, A. , Wang, N.J. , Kuo, W.L. , Stilwell, J.L. , Pinkel, D. , Albertson, D.G. , Waldman, F.M. , McCormick, F. , Dickson, R.B. , Johnson, M.D. , Lippman, M. , Ethier, S. , Gazdar, A. , Gray, J.W. , 2006. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgard, S.H. , Johansen, F.E. , Alnaes, G.I. , Bucher, E. , Syvanen, A.C. , Naume, B. , Borresen-Dale, A.L. , Kristensen, V.N. , 2008. Genome-wide analysis identifies 16q deletion associated with survival, molecular subtypes, mRNA expression, and germline haplotypes in breast cancer patients. Genes Chromosomes Cancer. 47, 680–696. [DOI] [PubMed] [Google Scholar]
- O'Hagan, R.C. , Chang, S. , Maser, R.S. , Mohan, R. , Artandi, S.E. , Chin, L. , DePinho, R.A. , 2002. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell. 2, 149–155. [DOI] [PubMed] [Google Scholar]
- Pardo, B. , Gomez-Gonzalez, B. , Aguilera, A. , 2009. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol. Life. Sci. 66, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram, M.D. , Konecny, G. , Slamon, D.J. , 2000. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat. Res. 103, 57–75. [DOI] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , Fluge, O. , Pergamenschikov, A. , Williams, C. , Zhu, S.X. , Lonning, P.E. , Borresen-Dale, A.L. , Brown, P.O. , Botstein, D. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Pinkel, D. , Segraves, R. , Sudar, D. , Clark, S. , Poole, I. , Kowbel, D. , Collins, C. , Kuo, W.L. , Chen, C. , Zhai, Y. , Dairkee, S.H. , Ljung, B.M. , Gray, J.W. , Albertson, D.G. , 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20, 207–211. [DOI] [PubMed] [Google Scholar]
- Podo, F. , Buydens, L.M. , Degani, H. , Hilhorst, R. , Klipp, E. , Gribbestad, I.S. , van Huffel, S. , van Laarhoven, H.W. , Luts, J. , Monleon, D. , Postma, J.G. , Schneiderhan-Marra, N. , Santoro, F. , Wouters, H. , Russnes, H.G. , Sørlie, T. , Tagliabue, E. , Børresen-Dale, A.L. , 2010. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 4, (3) 209–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak, K. , 2007. Breast cancer: origins and evolution. J. Clin. Invest. 117, 3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack, J.R. , Perou, C.M. , Alizadeh, A.A. , Eisen, M.B. , Pergamenschikov, A. , Williams, C.F. , Jeffrey, S.S. , Botstein, D. , Brown, P.O. , 1999. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 23, 41–46. [DOI] [PubMed] [Google Scholar]
- Pollack, J.R. , Sorlie, T. , Perou, C.M. , Rees, C.A. , Jeffrey, S.S. , Lonning, P.E. , Tibshirani, R. , Botstein, D. , Borresen-Dale, A.L. , Brown, P.O. , 2002. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. U S A. 99, 12963–12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakha, E.A. , El-Sheikh, S.E. , Kandil, M.A. , El-Sayed, M.E. , Green, A.R. , Ellis, I.O. , 2008. Expression of BRCA1 protein in breast cancer and its prognostic significance. Hum. Pathol. 39, 857–865. [DOI] [PubMed] [Google Scholar]
- Ralhan, R. , Kaur, J. , Kreienberg, R. , Wiesmuller, L. , 2007. Links between DNA double strand break repair and breast cancer: accumulating evidence from both familial and nonfamilial cases. Cancer Lett. 248, 1–17. [DOI] [PubMed] [Google Scholar]
- Reis-Filho, J.S. , Tutt, A.N. , 2008. Triple negative tumours: a critical review. Histopathology. 52, 108–118. [DOI] [PubMed] [Google Scholar]
- Richardson, A.L. , Wang, Z.C. , De Nicolo, A. , Lu, X. , Brown, M. , Miron, A. , Liao, X. , Iglehart, J.D. , Livingston, D.M. , Ganesan, S. , 2006. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 9, 121–132. [DOI] [PubMed] [Google Scholar]
- Scully, R. , Xie, A. , Nagaraju, G. , 2004. Molecular functions of BRCA1 in the DNA damage response. Cancer Biol. Ther. 3, 521–527. [DOI] [PubMed] [Google Scholar]
- Seitz, S. , Wassmuth, P. , Plaschke, J. , Schackert, H.K. , Karsten, U. , Santibanez-Koref, M.F. , Schlag, P.M. , Scherneck, S. , 2003. Identification of microsatellite instability and mismatch repair gene mutations in breast cancer cell lines. Genes Chromosomes Cancer. 37, 29–35. [DOI] [PubMed] [Google Scholar]
- Saal, L.H. , Gruvberger-Saal, S.K. , Persson, C. , Lovgren, K. , Jumppanen, M. , Staaf, J. , Jonsson, G. , Pires, M.M. , Maurer, M. , Holm, K. , Koujak, S. , Subramaniyam, S. , Vallon-Christersson, J. , Olsson, H. , Su, T. , Memeo, L. , Ludwig, T. , Ethier, S.P. , Krogh, M. , Szabolcs, M. , Murty, V.V. , Isola, J. , Hibshoosh, H. , Parsons, R. , Borg, A. , 2008. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 40, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savelyeva, L. , Schwab, M. , 2001. Amplification of oncogenes revisited: from expression profiling to clinical application. Cancer Lett. 167, 115–123. [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Thorsen, T. , Quist, H. , Matese, J.C. , Brown, P.O. , Botstein, D. , Eystein Lonning, P. , Borresen-Dale, A.L. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U S A. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Tibshirani, R. , Parker, J. , Hastie, T. , Marron, J.S. , Nobel, A. , Deng, S. , Johnsen, H. , Pesich, R. , Geisler, S. , Demeter, J. , Perou, C.M. , Lonning, P.E. , Brown, P.O. , Borresen-Dale, A.L. , Botstein, D. , 2003. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. U S A. 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff, S. , Isola, J. , Tanner, M. , 2003. Haplo-insufficiency of BRCA1 in sporadic breast cancer. Cancer Res. 63, 4978–4983. [PubMed] [Google Scholar]
- Stark, G.R. , Debatisse, M. , Giulotto, E. , Wahl, G.M. , 1989. Recent progress in understanding mechanisms of mammalian DNA amplification. Cell. 57, 901–908. [DOI] [PubMed] [Google Scholar]
- Stephens, P.J. , McBride, D.J. , Lin, M.L. , Varela, I. , Pleasance, E.D. , Simpson, J.T. , Stebbings, L.A. , Leroy, C. , Edkins, S. , Mudie, L.J. , Greenman, C.D. , Jia, M. , Latimer, C. , Teague, J.W. , Lau, K.W. , Burton, J. , Quail, M.A. , Swerdlow, H. , Churcher, C. , Natrajan, R. , Sieuwerts, A.M. , Martens, J.W. , Silver, D.P. , Langerod, A. , Russnes, H.E. , Foekens, J.A. , Reis-Filho, J.S. , van't Veer, L. , Richardson, A.L. , Borresen-Dale, A.L. , Campbell, P.J. , Futreal, P.A. , Stratton, M.R. , 2009. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 462, 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson, O.A. , Jonasson, J.G. , Johannsson, O.T. , Olafsdottir, K. , Steinarsdottir, M. , Valgeirsdottir, S. , Eyfjord, J.E. , 2009. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res. 11, R47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl, J. , Caldas, C. , 2007. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat. Rev. Cancer. 7, 791–799. [DOI] [PubMed] [Google Scholar]
- Subramaniam, D.S. , Isaacs, C. , 2005. Utilizing prognostic and predictive factors in breast cancer. Curr. Treat. Options. Oncol. 6, 147–159. [DOI] [PubMed] [Google Scholar]
- Tibshirani, R. , Wang, P. , 2008. Spatial smoothing and hot spot detection for CGH data using the fused lasso. Biostatistics. 9, 18–29. [DOI] [PubMed] [Google Scholar]
- Tirkkonen, M. , Johannsson, O. , Agnarsson, B.A. , Olsson, H. , Ingvarsson, S. , Karhu, R. , Tanner, M. , Isola, J. , Barkardottir, R.B. , Borg, A. , Kallioniemi, O.P. , 1997. Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res. 57, 1222–1227. [PubMed] [Google Scholar]
- Tlsty, T.D. , Margolin, B.H. , Lum, K. , 1989. Differences in the rates of gene amplification in nontumorigenic and tumorigenic cell lines as measured by Luria–Delbruck fluctuation analysis. Proc. Natl. Acad. Sci. U S A. 86, 9441–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty, T.D. , White, A. , Sanchez, J. , 1992. Suppression of gene amplification in human cell hybrids. Science. 255, 1425–1427. [DOI] [PubMed] [Google Scholar]
- Tlsty, T.D. , 1990. Normal diploid human and rodent cells lack a detectable frequency of gene amplification. Proc. Natl. Acad. Sci. U S A. 87, 3132–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, N.C. , Reis-Filho, J.S. , Russell, A.M. , Springall, R.J. , Ryder, K. , Steele, D. , Savage, K. , Gillett, C.E. , Schmitt, F.C. , Ashworth, A. , Tutt, A.N. , 2007. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 26, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Turner, N.C. , Reis-Filho, J.S. , 2006. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 25, 5846–5853. [DOI] [PubMed] [Google Scholar]
- Venkitaraman, A.R. , 2002. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 108, 171–182. [DOI] [PubMed] [Google Scholar]
- Volik, S. , Zhao, S. , Chin, K. , Brebner, J.H. , Herndon, D.R. , Tao, Q. , Kowbel, D. , Huang, G. , Lapuk, A. , Kuo, W.L. , Magrane, G. , De Jong, P. , Gray, J.W. , Collins, C. , 2003. End-sequence profiling: sequence-based analysis of aberrant genomes. Proc. Natl. Acad. Sci. U S A. 100, 7696–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell, N. , Arnold, J. , Cocciardi, S. , da Silva, L. , Marsh, A. , Riley, J. , Johnstone, C.N. , Orloff, M. , Assie, G. , Eng, C. , Reid, L. , Keith, P. , Yan, M. , Fox, S. , Devilee, P. , Godwin, A.K. , Hogervorst, F.B. , Couch, F. , Grimmond, S. , Flanagan, J.M. , Khanna, K. , Simpson, P.T. , Lakhani, S.R. , Chenevix-Trench, G. , 2009. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res. Treat. [DOI] [PubMed] [Google Scholar]
- Wang, Z.C. , Lin, M. , Wei, L.J. , Li, C. , Miron, A. , Lodeiro, G. , Harris, L. , Ramaswamy, S. , Tanenbaum, D.M. , Meyerson, M. , Iglehart, J.D. , Richardson, A. , 2004. Loss of heterozygosity and its correlation with expression profiles in subclasses of invasive breast cancers. Cancer Res. 64, 64–71. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Windle, B.E. , Wahl, G.M. , 1992. Molecular dissection of mammalian gene amplification: new mechanistic insights revealed by analyses of very early events. Mutat Res. 276, 199–224. [DOI] [PubMed] [Google Scholar]