

Abstract
The structure of the ring-opened product from direct oxidation of meso-tetraarylporphyrins has been controversial for three decades. Herein we show that bilitrienones 2 are obtained from oxidation of metal-free dodecasubstituted porphyrins 1 in the presence of sodium nitrite, trifluoroacetic acid and air oxygen. The presence of the para-nonyl groups in 1b stabilized the corresponding bilitrienone 2b, which was characterized by X-ray crystallography. In the absence of the para-nonyl groups bilitrienone 2a undergoes a rapid hydration reaction, giving biladienone 3a as the major isolated product. The molecular structures of 2b and 3a, and. the photochemical isomerization of 3a are discussed.
1. Introduction
The meso-oxidation of metallo-porphyrins and -chlorins has been investigated for several decades in order to gather insight into the important biological processes of heme catabolism,1 chlorophyll degradation2 and formation of algal biliproteins.3 The open-chain oxygenated tetrapyrroles, often designated generically as bile pigments, perform important biological functions, for example as the chromophores in biliproteins,4–6 and have been proposed for various applications, including as antiviral and antioxidant agents.7–9 In living organisms, bile pigments (e. g. biliverdins) are formed from the highly specific enzymatic oxidation of heme, catalyzed by heme oxygenase.10,11 On the other hand, the chemical syntheses of bile pigments can be achieved from pyrromethenone12–14 or dipyrromethene synthons,15 or from chemical oxidation of porphyrins and metalloporphyrins using O2 in the presence of ascorbic acid or hydrazine (so-called coupled oxidation of iron porphyrins),16,17 thallium(III) or cerium(IV) trifluoroacetate salts,18–20 N2O421 or sodium nitrate and trifluoroacetic acid,22 hydrogen peroxide23 or meta-chloroperoxybenzoic acid (mCPBA) in pyridine,24 by reactions of metalloporphyrin π-cation radicals with nucleophiles,25 or from photo-oxygenation.26–28 Whereas the structures of the oxidation products from b-octasubstituted porphyrins (e.g. octaethylporphyrin and protoporphyrin-IX) are well documented and were shown to involve the formation of formyl-bilitrienones, those from the related meso-substituted porphyrins, such as meso-tetraphenylporphyrin (TPP), have been subject of controversy for over three decades. The first spectral evidence for the formation of a bilitrienone from TPP was reported in 1980 by one of us.26
This compound was presumably unstable and rapidly underwent reversible addition of water, methanol or ethanol at the a-methine carbon bridge, forming the corresponding biladienones.28 Indeed, the involvement of a bilitrienone intermediate was controversial, since no NMR data were available for this product or its metal complexes. More recently, during our investigation of the oxidative ring-opening reaction of dodeca-substituted porphyrin 1, we isolated and characterized the air- and solvent-stable Ni(II), Zn(II) and Cu(II) complexes of its corresponding bilitrienone.22,24
2. Results and Discussion
The chemical oxidation of dodecasubstituted porphyrin 1a using 6 equiv of NaNO2/TFA in the presence of air gave the corresponding benzoylbiliverdin 2a, which was too unstable to isolate in pure form, due to its readily hydration to produce 3a.22,23 Two distinct isomers of 3a, purple and pink in color, were isolated in about 3:1 ratio after work-up of the oxidation reaction and purification by column chromatography. Both isomers showed broad absorption bands in the UV-Vis, indicating significant conformational flexibility. Mizutani and coworkers have also isolated the purple and pink fractions upon chemical oxidation of iron TPP and reported that the pink benzoylbiliverdin could be photoisomerized to the purple benzoylbiliverdin, while the reverse isomerization did not proceed.25 Heating the mixture of isomers 3a in the presence of a metal salt results in dehydration and metalation with formation of the corresponding stable metal complexes of 2a.23 Under the same conditions, porphyrin 1b was also oxidized, but in this case benzoylbiliverdin 2b was more resistant to spontaneous hydration than 2a, and was therefore isolated successfully in 70% yield, along with 15% yield of the corresponding hydrated form 3b. Separate studies by Smith,17–19 Fuhrhop,20 and Cavaleiro16 have shown that 5,10,15,20-tetraphenylporphyrin (TPP) is oxidized to give the corresponding biliverdin which rapidly undergoes nucleophilic addition of water to produce the corresponding hydrated biliverdin.
The 300 MHz 1H-NMR spectra of benzoylbiliverdins 3a24 showed the asymmetry of both tetrapyrroles, i.e. three N-H signals and one O-H signal that appeared at 11.5, 11.2, 9.0 and 6.0 ppm, respectively; all of these 1H-NMR absorptions disappeared upon performing a deuterium oxide shake on the tetrapyrrole that was purple in color. The 13C spectrum of 3a showed a benzoyl carbonyl peak at 187 ppm, the lactam carbonyl at 174 ppm and a quaternary carbon peak at 75 ppm. The tetrapyrrole that was pink in color, on the other hand, gave a 1H NMR spectrum with two N-H peaks appearing at 12.5 and 9.46, two distinct singlets, one of them corresponding to the hydroxyl group, appeared at 4.1 and 3.7 ppm. A deuterium oxide shake resulted to the disappearance of the two N-H protons and one of the singlet peaks that had previously appeared at 4.1 ppm. The 13C spectrum of the pink tetrapyrrole24 differed only slightly from that of the purple tetrapyrrole with very similar carbonyl frequencies appearing at 187 and 172.4 ppm, along with a quaternary carbon peak at 77.7 ppm. Characteristic benzoylbiliverdin fragments were obtained by high resolution EI-MS for both chromophores; these had M+Na ion peaks at m/z 903.4249 and 903.4224 for the pink and purple tetrapyrroles, respectively. Oxidations were also carried out using anhydrous methanol, ethanol, and methylamine to quench the reactions, and while methylamine gave no addition product at all, the methanol and ethanol adducts were formed in almost negligible amounts as inferred from the appearance of near-baseline m/z 913 and 927 peaks. Both steric and electronic (such as solvents being less nucleophilic than H2O) effects may be invoked to account for the above observations. Unfortunately, the amounts of the above products obtained were too little for any further analyses to be performed.

Biliverdins usually have the helical all-syn conformation due to an efficient intrachromophoric hydrogen bond system based on the N-H….N-H network. Studies carried out on biliverdin IXα esters have shown the ε(vis)/ε(UV) ratio to be a function of the verdin molecular extension. An increase in ratio indicates a change in the porphyrin-like helically coiled form (5Z, 10Z, 15Z) to a more extended form, (e.g., 5Z, 10E, 15Z or 5E, 10Z, 15Z), where the ratio is similar to that of a polyene.26–28 In our study, the ε(vis)/ε(UV) of the purple and pink biliverdin conformers was 0.47 and 0.64 respectively indicating a more stretched conformation for the pink conformer than the purple one. All free biliverdins normally tend to adopt the ZZZ helicoidal conformation with an ε(vis)/ε(UV) ratio of 0.25 in agreement with molecular orbital calculations.29–33 Helicoidal (ZZZ)–biliverdins could be photoisomerized to their extended EZZ or EZE conformers but the latter reconverted back to the helical forms.34 This explains the ~ 3:1 isolation of the violet biliverdin 3a versus the much more polar pink conformer of 3a.
When porphyrin 1b was oxidized under similar conditions to those for 1a, only bilatrienone 2b and biladienone 3b were obtained, with no traces of the pink conformer of 3b. This could be a result of the bulky alkyl substituent on the phenyl ring which do not favor the EZ isomerization of the C4–C5 bond; similar results were reported for etiobiliverdin by Mizutani and coworkers who reported that the energy difference between the ZZZ and EZZ isomer is ascribed to the steric repulsion between the β-alkyl groups and the pyrrole ring.25 We believe that the sole product of the chemical oxidation of porphyrin 1b is bilatrienone 2b along with some traces of starting material. However upon workup and chromatographic purification, biladienone 3b is isolated after addition of a water molecule across the methine carbon bridge. This could be further argued with the fact that upon subjecting 3b to higher temperatures e.g., 40 °C, compound 2b was isolated. This was confirmed by thin layer chromatography and UV-vis and 1H NMR spectroscopy.
A final confirmation of the molecular structure of biliverdin 2b and the purple benzoylbiliverdin 3a was provided by X-ray crystallography. Figure 1 shows the X-ray structure of benzoylbiliverdin 2b, with arbitrarily small C atoms for the n-nonyl groups. The central part of the molecule has a helical shape. Internal N-H…N hydrogen bonds allow each C(pyrrole)2 group to be reasonably planar, with mean deviations of 0.06 and 0.11 Å for these 11-atom fragments. However, these two planes form a dihedral angle of 29.3(2)° due to the helical twist of the acyclic molecule, which also brings phenyl groups of (C9H19)Ph substituents at opposite ends of the molecule into edge-to-face contact. Parameters of this interaction are C…centroid distance 3.456 Å and C–H…centroid angle 147°. Two of the nonyl groups are extended, while the other two are quite tortuous, as shown schematically in Fig. 1.
Figure 1.
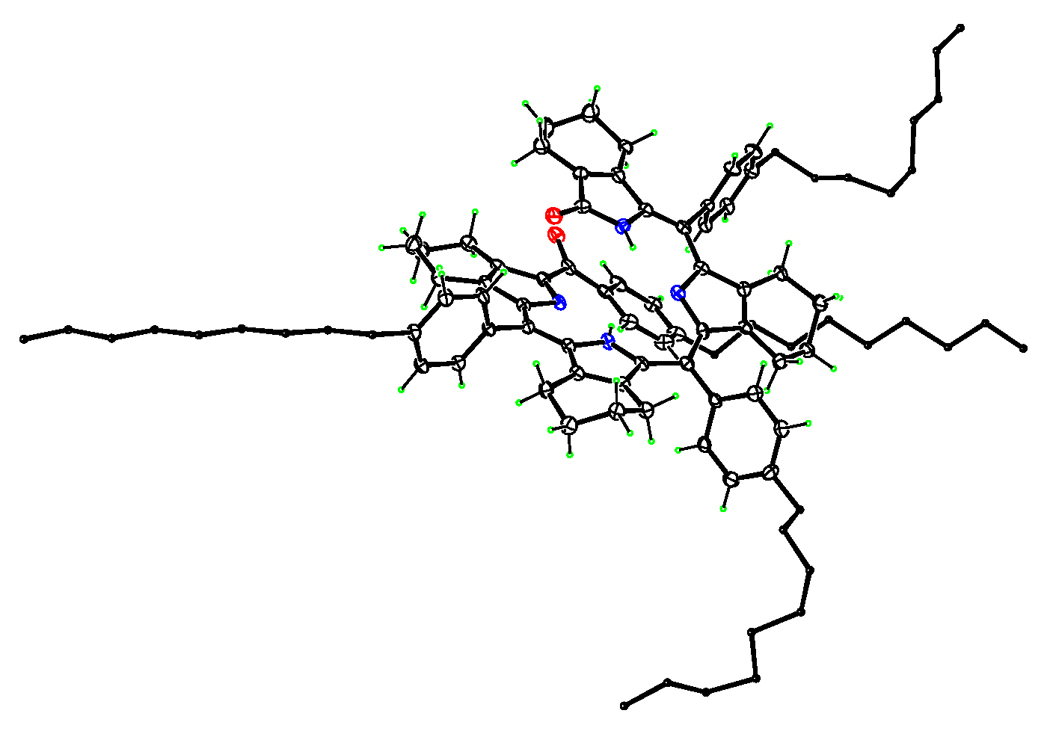
ORTEP diagram showing the molecular structure of 2b.
An examination of the X-ray structure of biliverdin 3a (obtained by slow diffusion of hexane into a solution of chloroform) shows the compound to be a biladienone, with the third meso-carbon (C15) a tetra-substituted one and bearing a hydroxyl group (see Figure 2). The existence of the −OH group is thus unequivocally established and corroborates the structural conclusions reached from 13C-NMR data, which showed the presence of a quaternary meso- carbon at δ 75 ppm. Within a single crystal, two molecules of the biliverdin associate with each other to form intermolecularly hydrogen-bonded pairs (Figure 3). Triads of hydrogen bonds exist about an inversion center, with the participating groups being the lactam unit on one molecule and the benzoyl carbonyl, the pyrrole −NH, and most importantly, the meso-OH on the other. The meso-OH•••O (1) bond, though slightly extended, lies at a nearly perfect hydrogen-bonding angle of ~179 thus allowing for moderately strong hydrogen bonds; the remaining hydrogen bonds lie within 20% of the optimum angle of 180°. Stabilization offered by the intermolecular association may be the raison d’etre for the hydroxyl group and may explain the readiness of the initially formed bilatrienone 2a to add a molecule of water across one of its double bonds to give the above biliverdin. The same, along with steric factors, may also explain why oxidations performed in methanol, ethanol, or methylamine media almost completely failed to give the corresponding addition products or gave so only in negligible quantities.
Figure 2.
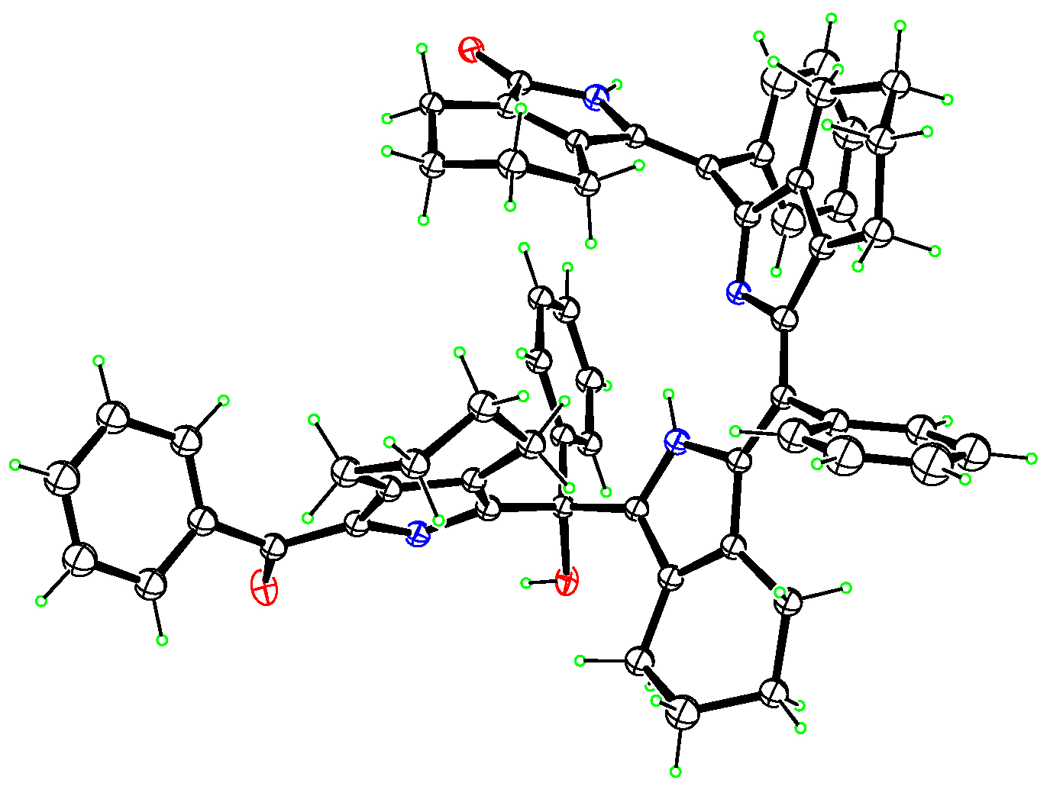
ORTEP diagram showing the molecular structure of 3a.
Figure 3.
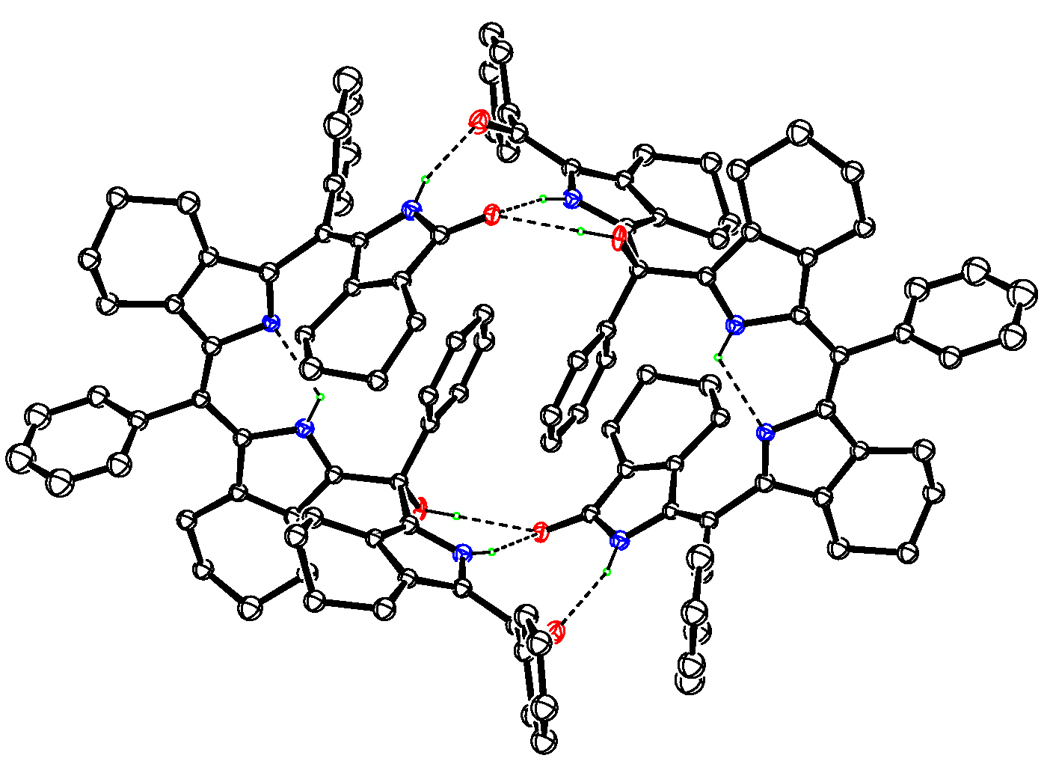
ORTEP diagram of 3a pair showing hydrogen bonding.
One consequence of the hydrogen bonding is that it predisposes the C (4)=C (5) double bond into an E-configuration, making the (EZ)-biladienone arrangement the most stable one for this biliverdin. This, however, does not discount the possibility of various other arrangements that may arise out of bond-rotation about the above double bond. Our theoretical calculations (B3LYP/6-31G*) do in fact predict the existence of various stereoisomers of the title biliverdin, which differ in energies by 6–10 Kcal/mol at room temperature, and may explain why the novel verdin is obtained as two different colored forms - namely the violet and the pink. In fact, similar isomeric biladienones were obtained by Mizutani et al., although from a β-unsubstituted porphyrin (TPP), and under oxidation conditions different than those reported herein.
3. Experimental
Silica gel 60 (70–230 and 230–400 mesh, Merck) or neutral alumina (Merck; usually Brockmann Grade III) were used for column chromatography. Analytical thin layer chromatography (TLC) was performed using Merck 60 F254 silica gel (pre-coated sheets, 0.2 mm thick). Reactions were monitored by TLC and spectrophotometry. 1H-NMR spectra were obtained in deuterochloroform or acetone-d6 solution, using a Brucker 250 or 400 MHz spectrometer; chemical shifts are expressed in ppm relative to residual chloroform (7.26 ppm) or internal TMS (0 ppm). Unless otherwise stated, electronic absorption spectra were measured in dichloromethane solution using a Hewlett-Packard 8450A spectrophotometer. Mass spectra were obtained at the Mass Spectrometry Facility at Louisiana State University. Benzaldehyde, BF3•OEt2, dichloro-dicyanobenzoquinone (DDQ) were used as purchased. All solvents were dried and purified according to literature procedures.
5,10,15,20-Tetra(4-nonanylphenyl)-2:3,7:8,12:13,17:18-tetrabutanoporphyrin (1b)
was prepared from the condensation of 3:4-butanopyrrole (0.25 g, 2.14 mmol) with 4-nonanyl-benzaldehyde, in freshly distilled and dry dichloromethane (214 mL) and in the presence of BF3•OEt2 (0.04 mL, 0.214 mmol), under argon. The reaction mixture was stirred for 1 h at room temperature, after which DDQ (0.7 g, 3.21 mmol) was added and the final mixture was refluxed under argon for 1 h to give a dark green solution. The solvent was reduced to dryness under vacuum and the resulting residue purified by alumina column chromatography using 2% methanol in dichloromethane for elution. Recrystallization from hot methanol gave purple crystals of the title porphyrin (0.5 g, 70% yield), m.p. >300 °C. 1H-NMR (CDCl3, drop of d-TFA), δ (ppm), 8.10 (d, 8H, J= 7.7 Hz) 7.91 (d, 8H, J= 7.8 Hz) 2.97-2.92, (t, 8H, J= 7.6 Hz) 2.33 (br s, 8H), 1.95-1.29 (m, 80H), 0.94-0.92 (t, 12H, J= 7.2 Hz). 13C-NMR (CDCl3) 144.1, 140.7, 136.2, 129.1, 118.5, 37.4, 33.5, 33.1, 31.29, 31.24, 30.97, 30.64, 27.3, 25.1, 24.2, 15.7. UV-Vis (CH2Cl2) λmax 446 nm (ε 66,100), 542 nm (5100), 615 nm (3700), 679 nm (55700), MS (MALDI) m/z 1335.30 (M+). Anal. Calcd for C96H126N4: C 84.03, H 9.56, N 4.09. Found: C 84.23, H 9.35, N 4.24. The 4-nonanylbenzaldehyde was obtained from the reaction of 1-octylbromide (50 mM, 10.8 g) in diethyl ether (50 mL) and under argon with magnesium turnings (10 g) at room temperature. After stirring for 30 min the reaction mixture was cooled to 0°C and bromobenzaldehyde (40 mM, 6.75 g) was slowly added. The final mixture was allowed to stir for 1 h and then quenched and washed with water (2 × 50 mL) and dried over Na2SO4. The organic solvents were evaporated under vacuum and the resulting yellow oil was taken up in hexane and purified by silica gel chromatography, using 3:1 hexane and ethyl acetate solution for elution. 1-(4-Bromophenyl)-1-hydroxynonane was obtained in 14.6 g (97 % yield); 1H-NMR (CDCl3,), δ (ppm), 7.45 (d, 2H, J= 8.4 Hz), 7.20 (d, 2H, J= 7.8 Hz), 4.61 (s, broad, 1H), 1.98 (s, 1H) 1.9-1.2 (m, 14H), 0.85 (t, 3H, J= 6.8 Hz). MS (FAB) m/z 299.17 (M+). To a solution of 1-(4-bromophenyl)-1-hydroxynonane (50 mM, 14.9 g) in 150 mL of acetonitrile, (300 mM, 27 mL) of trimethylsilylchloride (TMSC) and (300 mM, 45 g) of sodium iodide were added and the final mixture was stirred under argon for 12 h at room temperature. The reaction mixture was washed with an aqueous solution of Na2S2O3, dried over Na2SO4 and the organic solvent evaporated under reduced pressure affording an orange oil. The orange oil was diluted with 150 mL of DMSO and (750 mM, 2.85 g) of NaBH4 was slowly added. The resulting solution was stirred under argon for 12 h and then extracted into hexane and purified by silica gel chromatography using hexane for elution. The first band collected was 4-bromophenylnonane (10.7 g, 76.3%). 1H-NMR (CDCl3,), δ (ppm), 7.42 (d, 2H, J= 8.4 Hz), 7.08 (d, 2H, J= 7.8 Hz), 2.60 (t, 2H, J= 7.6 Hz) 1.58-1.29 (m, 14H), 0.91 (t, 3H, J= 7.2 Hz). MS (FAB) m/z 282.16 (M+). 4-Bromophenylnonane (15 mM, 4.25 g) was dissolved in freshly distilled THF (50 mL) and the reaction mixture cooled to −78°C. n-BuLi (46 mM, 26.5 mL) was then added dropwise and the mixture was stirred at −78°C for 1 h. Dry DMF (46 mM, 3.0 mL) was then added dropwise and the final mixture stirred under argon for 1 h. The reaction mixture was allowed to warm up to room temperature and then quenched with 100 mL of 1M HCl. The mixture was extracted into hexanes (3 × 100 mL), washed with water (2 × 100 mL), dried over Na2SO4 and the solvent removed under reduced pressure to give a yellow oil. 4-Nonanylbenzaldehyde was purified by silica gel chromatography using a 3:1 solution of hexane/ethyl acetate for elution to give an oil (2.3 g, 65% yield). 1H-NMR (CDCl3,), δ (ppm), 10.0 (s, 1H) 7.82 (d, 2H, J= 8.4 Hz), 7.36 (d, 2H, J=7.8 Hz), 2.74 (m, 2H) 1.69-1.29 (m, 14H), 0.93 (t, 3H, J= 7.2 Hz). MS (FAB) m/z 233.26 (M+).
Benzoylbiliverdins 2a and 3a
were prepared and characterized as we have previously described.24
Benzoylbiliverdins 2b and 3b
Porphyrin 1b (100 mg, 0.074 mmol) was dissolved in 2 mL of TFA and NaNO2 (30 mg, 0.45mM) was added to the solution while stirring under air at room temperature for 3 min. The reaction was quenched by pouring into 50 mL of water, following by extraction with dichloromethane (6 × 25 mL). The organic layers were washed with saturated aqueous NaHCO3 (2 × 100 mL), then with water (100 mL), and dried over anhydrous Na2SO4. The solvent was removed under vacuum and the resulting residue was taken up in chloroform and purified by alumina column chromatography using a gradient elution of chloroform to 1% methanol in chloroform. The title biliverdin 2b was the first green band eluted using pure chloroform (71 mg, 70%). The benzoylbiliverdin 3b (violet color) was eluted next using 1% methanol in chloroform solution (15 mg, 15%) yield. For the title biliverdin 2b: m.p. = 245-247 °C. UV-Vis (CH2Cl2) λmax 332 nm (ε 30600), 447 (31600), 683 (7800). MS (MALDI-TOF) m/z 1368.3 (M+). 1H NMR (CDCl3), δ (ppm), 12.80 (br s, 1H), 10.80 (br s, 1H), 8.22-6.75 (m, 16H), 2,76-1.33 (m, 96H), 0.94 (m, 12H): Anal. Calcd for C96H126N4O2: C 80.05, H 9.38, N 3.89. Found: C 80.07 H 9.07, N 3.83. For biliverdin 3b: m.p. = 185-190 °C; UV-Vis (CH2Cl2) λmax 345 nm (ε 47100), 576 nm (22700). MS (MALDI-TOF) m/z 1386.4 (M+). 1H NMR (CDCl3), δ (ppm), 11.52, 11.45 (2 br s, 2H), 8.86 (s, 1H), 7.56 (d, J= 8.0 Hz, 2H), 7.40 (d, J= 8.0 Hz, 2H), 7.3-7.1 (m, 12H), 6.10 (s, 1H), 2.7-1.24 (m, 96H), 0.94 (br s, 12H). 13C-NMR (CDCl3), δ (ppm), 172.2, 163.5, 146.6, 145.3, 144.3, 143.5, 143.2, 139.6, 138.8, 137.7, 136.6, 135.6, 135.1, 134.2, 133.5, 131.1, 129.8, 128.9, 128.5, 128.3, 127.2, 125.3, 120.6, 119.4, 74.1, 36.1, 35.8, 35.7, 32.1, 31.8, 31.6, 29.8, 29.7, 29.6, 29.5, 29.1, 24.9, 24.1, 23.4, 23.0, 22.9. Anal. Calcd for C96H128N4O3: C 83.19, H 9.31, N 4.04. Found: C 82.98, H 9.24, N 4.12.
Crystal Molecular structures
Bilatrienone 2b, C96H126N4O2·H2O·MeOH, Mr = 1418.1, triclinic, space group P-1, a = 14.222(5), b = 16.640(6), c = 19.510(10) Å, α = 92.097(14), β = 103.467(15), γ = 107.381(19)°, V = 4257(3) Å3, Z = 2, Mo-Kα radiation (λ = 0.71073 Å; = µ = 0.066 mm−1), T = 110 K, 22253 data by Nonius Kappa CCD, R = 0.122 (F2 > 2σ), for 11688 unique data having θmax = 23.0° and 906 refined parameters. Displacement parameters are large for three of the nonanyl groups, and anisotropic refinement of one of them was not possible. Solvent H atoms were not located. CCDC 745873, available from the Cambridge Crystallographic Data Centre.
Biladienone 3a, C60H56N4O3·CH2Cl2, Mr = 966.01, triclinic, space group P-1, a = 12.483(10), b = 13.993(12), c = 16.320(17) Å, α = 75.65(3), β = 75.83(3), γ = 67.91(6)°, V = 2522(4) Å3, Z = 2, Mo-Kα radiation (λ = 0.71073 Å; µ = 0.180 mm−1), T = 110 K, 26049 data by Nonius Kappa CCD, R = 0.151 (F2 > 2σ) for 6470 unique data having θmax=23.1° and 306 refined parameters. As a result of the small number (1771) of unique intensities having I>2 (I), only O and solvent Cl atoms were refined anisotropically. CCDC 745874, available from the Cambridge Crystallographic Data Centre.
4. Conclusions
It is shown that bilitrienones 2 are obtained from oxidation of metal-free dodecasubstituted porphyrins 1 in the presence of sodium nitrite, trifluoroacetic acid and air oxygen. Bilitrienone 2b is obtained from the 5,10,15,20-tetra(p-nonanylphenyl)porphyrin 1b, this being the first example of the unmodified metal-free open-chain tetrapyrrole to be reported; the structure of 2b is characterized by X-ray crystallography. In the absence of the para-nonanyl groups the initially-formed bilitrienone 2a undergoes a rapid and spontaneous hydration reaction to give biladienone 3a as the major isolated product. The molecular structure of 3a is also presented.
Acknowledgments
Support from the National Institutes of Health (CA 132861 to K.M.S.) and the National Science Foundation (CHE 0911629 to M.G.H.V.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ortiz de Montellano PR, Auclair K. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 12. Boston: Academic Press; 2003. pp. 183–210. [Google Scholar]
- 2.Kräutler BA. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 13. Boston: Academic Press; 2003. pp. 183–209. [Google Scholar]
- 3.(a) Frankenberg N, Lagarias JC. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 13. Boston: Academic Press; 2003. pp. 211–235. [Google Scholar]; (b) Gossauer A. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 13. Boston: Academic Press; 2003. pp. 237–274. [Google Scholar]
- 4.Braslavsky SE, Holzwarth AR, Schaffner K. Angew. Chem. Int. Ed. Engl. 1983;22:656–674. [Google Scholar]
- 5.Koerner R, Olmstead MM, Ozarowski A, Balch AL. Inorg. Chem. 1999;38:3262–3263. doi: 10.1021/ic9901113. [DOI] [PubMed] [Google Scholar]
- 6.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 7.Nakagami T, Taji S, Takahashi M, Yamanishi K. Microbiol. Immunol. 1992;36:381–390. doi: 10.1111/j.1348-0421.1992.tb02037.x. [DOI] [PubMed] [Google Scholar]
- 8.Bennett A, Siegelman HW. In: The Porphyrin. Dolphin D, editor. Vol. 6. New York: Academic Press; 1979. pp. 493–520. [Google Scholar]
- 9.Mori H, Otake T, Morimoto M, Ueba N, Kunita N, Nakagami T, Yamasaki N, Taji S. Jpn. J. Cancer Res. 1991;82:755–757. doi: 10.1111/j.1349-7006.1991.tb02698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmid R, McDonagh AF. Formation and metabolism of bile pigments in vivo. In: Dolphin D, editor. The Porphyrins. Part A. Vol 6. New York: Academic Press; 1979. pp. 257–292. [Google Scholar]
- 11.Frydman RB, Frydman B. Acc. Chem. Res. 1987;20:250–256. [Google Scholar]
- 12.Smith KM, Kishore D. Tetrahedron. 1983;39:1841–1847. [Google Scholar]
- 13.Cavaleiro JAS, Smith KM. J. Chem. Soc. Perkin Trans. 1. 1973:2149–2155. doi: 10.1039/p19730002149. [DOI] [PubMed] [Google Scholar]
- 14.(a) Boiadjiev EE, Lightner DA. Tetrahedron: Assymmetry. 2001;12:2551–2564. [Google Scholar]; (b) Kar AK, Lightner DA. Tetrahedron. 1998;54:12671–12690. [Google Scholar]
- 15.Smith KM. J. Chem. Soc., Perkin Trans 1. 1972:1471–1475. doi: 10.1039/p19720001471. [DOI] [PubMed] [Google Scholar]
- 16.(a) Bonnett R, McDonagh AF. J. Chem. Soc., Chem. Commun. 1970:237–238. [Google Scholar]; (b) Bonnett R, McDonagh AF. J. Chem. Soc., Perkin Trans. 1. 1973:881–888. doi: 10.1039/p19730000881. [DOI] [PubMed] [Google Scholar]
- 17.Crusats J, Suzuki A, Mizutani T, Ogoshi H. J. Org. Chem. 1998;63:602–607. doi: 10.1021/jo9714728. [DOI] [PubMed] [Google Scholar]
- 18.(a) Evans B, Smith KM, Cavaleiro JAS. Tetrahedron Lett. 1976:4863–4866. [Google Scholar]; (b) Evans B, Smith KM, Cavaleiro JAS. J. Chem. Soc., Perkin Trans. I. 1978:768–773. [Google Scholar]
- 19.Fuhrhop J-H, Mauzerall D. Photochem. Photobiol. 1971;13:453–458. [Google Scholar]
- 20.Wasser PKW, Fuhrhop J-H. Ann. N.Y. Acad. Sci. 1973;206:533–547. doi: 10.1111/j.1749-6632.1973.tb43235.x. [DOI] [PubMed] [Google Scholar]
- 21.Catalano MM, Crossley MJ, Harding MM, King LG. J. Chem. Soc., Chem. Commun. 1984:1535–1536. [Google Scholar]
- 22.Ongayi O, Fronczek F, Vicente MGH. Chem. Commun. 2003:2298–2299. doi: 10.1039/b306586c. [DOI] [PubMed] [Google Scholar]
- 23.Bonnett R, Dimsdale MJ. J. Chem. Soc., Perkin Trans. 1. 1972:2540–2548. doi: 10.1039/p19720002540. [DOI] [PubMed] [Google Scholar]
- 24.Ongayi O, Vicente MGH, Ou Z, Kadish KM, Kumar MR, Fronczek FR, Smith KM. Inorg. Chem. 2006;45:1463–1470. doi: 10.1021/ic050841c. [DOI] [PubMed] [Google Scholar]
- 25.Shine HJ, Padilla AG, Wu S-M. J. Org. Chem. 1979;44:4069–4075. [Google Scholar]
- 26.(a) Smith KM, Brown SB, Troxler RF, Lai J-J. Tetrahedron Lett. 1980;21:2763–2766. [Google Scholar]; (b) Smith KM, Brown SB, Troxler RF, Lai J-J. Photochem. Photobiol. 1982;36:147–152. doi: 10.1111/j.1751-1097.1982.tb04356.x. [DOI] [PubMed] [Google Scholar]
- 27.Matsuura T, Inoue K, Ranade AC, Saito I. Photochem. Photobiol. 1980;31:23–26. [Google Scholar]
- 28.(a) Cavaleiro JAS, Hewlins MJE, Jackson AH, Neves MGPMS. J. Chem. Soc., Chem. Commun. 1986:142–144. [Google Scholar]; (b) Cavaleiro JAS, Neves MGPMS, Hewlins MJE, Jackson AH. J. Chem. Soc., Perkin Trans. I. 1990:1937–11943. [Google Scholar]
- 29.Yamauchi T, Mizutani T, Wada K, Horii S, Furukawa H, Shigeyuki Masaoka, Chang H, Kitagawa S. Chem. Commun. 2005:2298–2299. doi: 10.1039/b414299c. [DOI] [PubMed] [Google Scholar]
- 30.Scheer H. Angew. Chem., Int. Ed. Eng. 1981;20:241–261. and references therein. [Google Scholar]
- 31.Niemevz F, Buldain YG. J. Porphyrins Pthalocyanines. 2004;8:989–995. [Google Scholar]
- 32.Iturraspe JB, Bari S, Frydman B. J. Am. Chem. Soc. 1989;111:1525–1527. [Google Scholar]
- 33.Blauer G, Wagniere G. J. Am. Chem. Soc. 1975;97:1949–1954. doi: 10.1021/ja00840a057. [DOI] [PubMed] [Google Scholar]
- 34.Pasternak R, Wagniere G. J. Am. Chem. Soc. 1979;101:1662–1667. [Google Scholar]
- 35.Burke MJ, Pratt DC, Moscowitz A. Biochemistry. 1972;11:4025–4031. doi: 10.1021/bi00772a003. [DOI] [PubMed] [Google Scholar]
- 36.Falk H, Hoellbacher G. Monatsh. Chem. 1978;109:1429–1449. [Google Scholar]
- 37.Wagniere G, Blauer G. J. Am. Chem. Soc. 1976;98:7806–7810. doi: 10.1021/ja00440a056. [DOI] [PubMed] [Google Scholar]
- 38.(a) Falk H, Muller N, Schlederer T. Monatsh.Chem. 1980;111:159–175. [Google Scholar]; (b) Falk H, Grubmayr K, Haslinger E, Schlederer T, Thirring K. Ibid. 1978;109:1451–1473. [Google Scholar]; (c) Falk H, Grubmayr K. Angew. Chem. 1977;89:487–488. [Google Scholar]