

Abstract
The process of megakaryopoiesis and platelet production is complex, with the potential for regulation at multiple stages. Megakaryocytes are derived from the hematopoietic stem cell through successive lineage commitment steps, and they undergo a unique maturation process that includes polyploidization, development of an extensive internal demarcation membrane system and finally formation of proplatelet processes. Platelets are shed from these processes into vascular sinusoids within the bone marrow. Megakaryocyte differentiation is regulated both positively and negatively by transcription factors and cytokine signaling. Thrombopoietin is the most important hematopoietic cytokine for platelet production. Clinically, acquired and inherited mutations affecting megakaryocytic transcription factors and thrombopoietin signaling have been identified in disorders of thrombocytopenia and thrombocytosis.
Transcriptional regulation of megakaryopoiesis
Megakaryocytes, like all blood cells, derive from the hematopoietic stem cell (HSC) (reviewed 1). During hematopoietic differentiation, the HSC gives rise to progressively committed progenitors, including the common myeloid progenitor (CMP) and the megakaryocyte-erythroid progenitor (MEP) (Figure 1, reviewed 2). MEPs are bipotential precursors that give rise to cells of both megakaryocytic and erythroid lineages. Multiple transcription factors including Runx1, Gata1, Fli1 and c-Myb form complex networks that regulate the differentiation of megakaryocytes both positively and negatively. For example, in addition to its requirement during embryogenesis for the formation of HSCs, Runx1 has an important role in megakaryocyte development, and inducible deletion of Runx1 in the adult mouse leads to thrombocytopenia and impaired megakaryopoiesis 3. Runx1 interacts with additional megakaryocytic factors including Gata1 and Fli1 4,5. Gata1 and its co-factor Friend of Gata1 (Fog1) are critical in promoting megakaryocyte-erythroid differentiaion, while at the same time inhibiting expression of Pu.1 and myeloid differentiation 6,7. Binding sites for Gata1 and Fli1 can be found in the enhancers of many megakaryocyte-specific genes 8, and Fli1 enhances the activity of Gata1 at megakaryocytic promoters and represses the activity of erythroid factors at erythroid promoters 9. Thus Fli1 expression may act to restrict the MEP to the megakaryocytic lineage. Accordingly, deletion of Fli1 is associated with loss of mature megakaryocytes in mouse models 10. In contrast, expression of the proto-oncogene c-Myb in the MEP favors erythropoiesis, and c-Myb expression is down-regulated during megakaryopoiesis 11. The miRNA miR-150, expressed during megakaryocyte maturation, acts post-transcriptionally to decrease c-Myb expression, and may influence whether the bipotential MEP adopts either the megakaryocyte or erythroid fate 12.
Figure 1.

Overview of megakaryopoiesis. Megakaryocytes are derived from the hematopoietic stem cell and proliferate and differentiate under the influence of TPO.
Endomitosis and proplatelet formation
As megakaryocytic cells become progressively differentiated they lose their proliferative ability and become polyploid through a variation of the cell cycle called endomitosis (reviewed 13). During differentiation, diploid promegakaryoblasts give rise to tetraploid megakaryoblasts and then successively larger and more polyploid promegakaryocytes and megakaryocytes. Mature megakaryocytes can be 150 μM or more in diameter and are the largest hematopoietic cells in the bone marrow. Although polyploidy is a defining feature of mature megakaryocytes, the purpose for acquiring multiple chromosome complements is unknown. It is theorized that polyploidization facilitates the massive protein and membrane synthesis required for later platelet production through functional gene amplification 14. In support of this notion, there is some correlation between the degree of megakaryocyte ploidy and platelet production; for example, neonatal megakaryocytes are typically of low ploidy and may have a reduced capacity to produce platelets relative to their adult counterparts 15. However, when mouse strains in which the modal megakaryocyte ploidy varies have been compared, there does not appear to be a direct relationship between ploidy and steady state platelet counts 16.
The mechanism of endomitosis is an enigma. Following G1 and S phases, endomitotic megakaryocytes enter mitosis and anaphase, separate their chromosomes, and begin the process of cleavage furrow formation 17,18. Subsequently, nuclei fail to completely separate and cleavage furrows regress before cytokinesis can be completed, resulting in the formation of a single cell with a multilobulated, polyploid nucleus 18 (Figure 2). Furrow regression may be due the failure to properly localize or activate RhoA at the contractile ring 19, but what regulates the switch from mitosis to endomitosis and how the polyploid cell avoids apoptotic triggers and continues to cycle remain important questions.
Figure 2.

Endomitosis. Diploid megakaryocyte progenitors progress through S phase and enter mitosis. Although anaphase is initiated, with separation of paired sister chromosomes and formation of the cleavage furrow, prior to completion of cytokinesis the cleavage furrow regresses, resulting in a tetraploid cell that re-enters G1.
Following nuclear polyploidization, megakaryocytes undergo cytoplasmic maturation involving the formation an extensive demarcation membrane system (DMS) as well as numerous alpha and dense granules (reviewed 16). The DMS is an elaborate membrane network that ultimately fills the megakaryocyte cytoplasm except for a narrow band at the periphery of the cell. The DMS is continuous with the plasma membrane of the cell and is in contact with the external environment 20, and it has been proposed to serve as a reservoir of membrane to support proplatelet formation 21. Alpha granules are formed in the Golgi complex and contain both endogenously produced proteins and proteins derived from the extracellular environment through receptor-mediated endocytosis and pinocytosis. These granules as well as other megakaryocyte organelles such as mitochondria and mRNAs will be transferred into the nascent platelets.
Two models have been proposed to explain platelet formation from mature megakaryocytes. In the fragmentation model, megakaryocytes travel from the bone marrow to the lung where they are broken up into platelets in the microvasculature 22. Alternatively, in the proplatelet model, megakaryocytes in the bone marrow develop multiple branching processes that extend into the marrow sinusoids to release platelets into the circulation 23. In support of the latter model, Italiano and colleagues used live cell microscopy to document the elaboration of a network of branching proplatelets by cultured megakaryocytes 24. Junt and colleagues later extended these results by imaging the bone marrow compartment in vivo, demonstrating that fluorescently-labeled megakaryocytes release platelet-like particles into marrow vascular sinusoids 25. These studies confirmed that the marrow is a site of platelet production, although a contribution from the lung cannot be excluded. The process and regulation of proplatelet formation is discussed in more detail elsewhere in this issue (see Thon and Italiano chapter).
TPO signaling in megakaryopoiesis
Multiple growth factors support megakaryopoesis, the most important of which is thrombopoietin (TPO). TPO belongs to the four-helix bundle family of cytokines, which includes erythropoietin, G-CSF, growth hormone and leukemia inhibitory factor among others. The TPO receptor c-Mpl was identified based on its homology to the oncogne v-Mpl, already known at the time as the transforming factor of the murine myeloproliferative leukemia virus 26. TPO and c-Mpl are critical for megakaryocyte growth and development, and in mouse models where one or the other is absent, platelets and megakaryocytes are reduced to approximately 10% of normal values 27,28. In addition to megakaryocytic cells, HSCs also express c-Mpl and depend on TPO signaling for their maintenance and expansion 29. Consequently, c-Mpl and Tpo-null mice are not only thrombocytopenic but have reduced HSCs and progenitors of all lineages 31.
The c-Mpl gene encodes a 635 amino acid protein consisting of a 25 amino acid signal peptide (1-25), a 465 amino acid extracellular domain (26-491), a 22 residue transmembrane domain (492-513) and an intracellular domain containing two conserved motifs termed box 1 (528-536) and box 2 (565-574). The extracellular domain is composed of two repeating modules; intriguingly, the membrane distal module appears to have an inhibitory effect on signaling, as its deletion results in constitutive activation of the receptor 32. c-Mpl does not have intrinsic kinase activity, but instead associates with the cytoplasmic tyrosine kinase Janus kinase 2 (Jak2) through its box 1 domain 33. Additional elements regulate receptor internalization and subsequent degradation following TPO binding. These include dileucine repeats located within box 2, Tyr591 and Tyr625 34,35.
TPO signaling depends on the activation of Jak2 (Figure 3). Jak2 associates with box 1 of c-Mpl through its FERM (band 4.1/ezrin/radixin/moesin) domain. Based on X-ray crystal studies of the erythropoietin receptor 36, it is believed that in the unliganded state c-Mpl exists as a homodimer, and that TPO binding results in a conformational change that brings the cytoplasmic tails of the receptor into closer proximity. Consequently, the Jak2 molecules associated with the receptor are brought close enough to each other to become activated through trans-autophosphorylation 37. Active Jak2 then phosphorylates itself on multiple residues and phosphorylates c-Mpl on at least Tyr625 and Tyr630 38. These phosphotyrosine residues provide docking sites for src homology 2 (SH2)-domain-containing signaling proteins that modulate receptor signaling.
Figure 3.
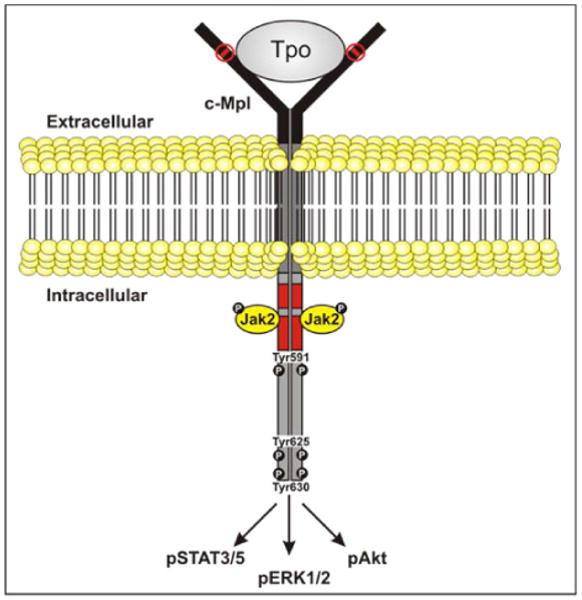
TPO signaling. c-Mpl is a homodimeric cytokine receptor that associates with the cytoplasmic tyrosine kinase Jak2. Following the binding of TPO, a conformation change occurs that leads to the activation of Jak2 and downstream signaling pathways including STAT, MAPK and PI3K.
Following the activation of Jak2, multiple signaling molecules are activated and mediate the cellular response to TPO. These include members of the signal transducer and activator of transcription (STAT), mitogen-activated protein kinase (MAPK) and phosphoinositol-3 kinase (PI3K) pathways (reviewed 39). Jak2 directly phosphorylates STAT family members including STAT1, 3, 5a and 5b 40. Once phosphorylated, these STAT proteins dimerize and translocate to the nucleus of the cell where they can bind to STAT-responsive transcriptional elements within genes such as p21 41, Bcl-xL 42 and cyclin D1 43. Constitutive activation of the Jak2/STAT pathway can lead to cytokine-independent growth and contribute to transformation, as demonstrated by the finding of mutant Jak2 in myeloproliferative disorders, translocations involving Jak2 in lymphoid leukemias, and constitutively active STAT5 in leukemic cell lines 44,45.
Jak2 also activates the small GTPase Ras and the MAPK cascade, culminating in the activation of extracellular signal-related kinase (ERK)1/2. Multiple studies have demonstrated the importance of TPO-induced MAPK signaling in megakaryocytic differentiation 46-48. The classical pathway by which TPO signaling is thought to activate Ras depends on the binding of the adaptor protein Shc to phosphorylated c-Mpl Tyr625 38,49 and the assembly of a complex containing the adaptor protein Grb2 and the guanine nucleotide exchange factor SOS 50,51. Ras then activates Raf-1, mitogen-induced extracellular kinase (MEK) and finally Erk 1/2 (reviewed 52). It is intriguing that although activation of MAPK is significantly reduced in the absence of c-Mpl Tyr625 and Tyr630, it is not eliminated 53, suggesting that activation of Erk1/2 can be mediated either through a Shc-independent mechanism, possibly through Grb2/Sos complexes recruited to Jak2 54. Alternatively, the small GTPase Rap1 can activate Erk1/2 via B-Raf independent of Ras 55.
The PI3K pathway is also essential for megakaryopoiesis 56. PI3K is composed of a kinase (p110) and a regulatory subunit (p85). TPO induces the formation of a complex between phosphorylated p85 and the adaptor Gab, although this complex has not been found to bind directly to c-Mpl 57; alternatively, PI3K may become activated indirectly through Ras 58. TPO-induced PI3K phosphorylates and activates the serine/threonine kinase Akt whose substrates include Forkhead, glycogen synthase kinase 3 beta (GSK-3β) and Bad 56,59,60, collectively promoting survival and proliferation of megakaryocytic cells. PI3K also activates mammalian target of rapamycin (mTOR), whose targets SK6 and 4E-BP1 increase proliferation and maturation of megakaryocytic progenitors 61,62. PI3K is itself negatively regulated by phosphatase and tensin homolog (PTEN), a tumor suppressor that promotes quiescence in hematopoietic stem cells (HSC)63. Although PTEN regulates the activity of Akt and mTOR, its role in TPO signaling and megakaryopoiesis has not yet been defined.
Negative regulation of TPO signaling and megakaryopoiesis
As with all proliferative stimuli, checks on TPO signaling and megakaryopoiesis are required to maintain homeostatic balance and prevent the development of thrombocythemia or leukemia. To ensure that signals are appropriately terminated, many positive regulators also induce their own inhibitors. For example, activation of Jak/STAT pathway induces the transcription of members of the suppressor of cytokine signaling (SOCS) family 64,65. This family includes at least 8 members that can inhibit Jak signaling in a variety of ways, including binding to the activation loop of Jak and targeting it for degradation, acting as a pseudosubstrate for Jak, or binding to phosphorylated tyrosines within the cytokine receptor itself (reviewed 66). Importantly, induction of a SOCS response from one receptor can negatively regulate another, thereby providing a mechanism for cytokine cross-talk; this is illustrated by the finding that treatment of megakaryocytes with interferon-α induces SOCS1, which then down-regulates TPO signaling through inhibition of Jak2 67. Some cancer cells may be able to avoid downregulation of Jak2 and this may be a strategy enabling them to grow; for example, the activity of mutant Jak2 V617F appears to be resistant to inhibition by SOCS3 68.
Jak2 has other binding partners that regulate its activity. For example, Lnk is an adaptor protein that inhibits growth in HSCs, erythroid and megakaryocytic cells 69,70. Lnk binds to phosphorylated tyrosines within Jak2 through its SH2-domain 71; however, the exact mechanism by which it inhibits TPO signaling is not yet understood. In addition to binding negative regulators, Jak2 may be phosphorylated within the FERM domain, inducing its dissociation from c-Mpl and thus providing another mechanism to ‘turn off’ signaling 72.
Clinical correlates
Congenital and familial thrombocytopenia syndromes are models for understanding the function of specific genes in megakaryopoiesis. For example, consistent with the importance of the transcription factors Runx1, Gata1 and Fli1 in megakaryocyte differentiation, mutations involving each of these factors have been found in association with an inherited thrombocytopenia syndrome. Runx1 mutations are associated with Familial Thrombocytopenia Disorder with Predisposition to AML, an autosomal dominant thrombocytopenia in which affected family members are prone to the development of myeloid leukemias 73,74. Reflecting the requirement for Gata1 in both megakaryopoiesis and erythropoiesis, mutations of Gata1 are associated with X-linked thrombocytopenia accompanied by anemia and red cell abnormalities such as thalassemia, dyserythropoiesis or porphyria (reviewed 75). Mutations typically are located within the N-terminal zinc finger of Gata1 and disrupt binding to its cofactor Fog1. Although specific Fli1 mutations have not been reported in the clinical setting, the expression of Fli1 is affected in patients with Jacobsen syndrome. This congenital disorder is characterized by thrombocytopenia, heart defects, mental retardation and hemizygous deletion of the terminal portion of chromosome 11q that includes Fli1 located at 11q23-24 76. Although other deleted genes might contribute to the platelet phenotype, over-expression of exogenous Fli1 in bone marrow cells from a patient with Jacobsen syndrome rescued defective megakaryopoiesis in vitro 77, supporting the conclusion that the thrombocytopenia in Jacobsen syndrome is due to the hemizygous loss of Fli1.
Altered TPO signaling contributes to both disorders of thrombocytopenia and thrombocytosis. The classic syndrome of deficient TPO signaling is seen in congenital amegakaryocytic thrombocytopenia (CAMT). CAMT is an autosomal recessive disorder in which c-Mpl is either absent or minimally functional 78,79. Circulating TPO levels are highly elevated due to the lack of receptor-mediated uptake, and without a functional receptor megakaryopoiesis is severely impaired. Children present at birth with severe thrombocytopenia and megakaryocytopenia, and because TPO is vital for maintenance of the HSC their disease nearly always progresses to aplastic anemia 80,81. Definitive treatment requires allogeneic HSC transplantation. Curiously, although in murine models the deletion of TPO phenocopies that of c-Mpl 31, no humans have been identified with an inherited deficiency of TPO.
Conversely, inherited mutations that result in increased TPO signaling have also been described in families with thrombocythemia and myeloproliferative disease. In familial thrombocythemia there is autosomal dominant transmission of high platelet counts. In some kindreds this is due to a mutation causing aberrant splicing of TPO mRNA resulting in a form that is translated more efficiently than the wild type transcript, leading to excessive TPO production 82,83. The clue to this diagnosis is that circulating TPO levels are elevated despite thrombocytosis. Additional families have been described in which mutations in c-Mpl result in constitutive activation of the receptor 84. Although these individuals also have thrombocytosis, circulating TPO levels are low because TPO is not being overproduced and is being taken up and degraded by the increased platelet mass.
Acquired mutations altering TPO signaling are characteristic of Philadelphia chromosome-negative myeloproliferative diseases, including polycythemia vera (PV), essential thrombocythemia (ET) and idiopathic myelofibrosis (IMF). Activating mutations of Jak2, most frequently the substitution Val617Phe 85-87 but rarely mutations within exon 12 88, are found in nearly all patients with PV and about half of those with ET and IMF. A minority of patients with ET and IMF carry activating mutations not of Jak2 but of c-Mpl 89. Complications of myeloproliferative disease include thrombosis, myelofibrosis and transformation to leukemia. Activated Jak2 seems to be an independent risk factor for thrombosis, as thrombosis in patients with Jak2 Val617Phe mutations can precede the development of overt overproduction of blood cells 90,91. In contrast, the development of myelofibrosis may be secondary to factors secreted by the increased numbers of megakaryocytes rather than a direct effect of c-Mpl and Jak2 signaling 92,93. Surprisingly, recent studies of patients with MPD who develop leukemia suggest that at least in some cases, the leukemic clone is derived from a cell that does not carry the Jak2 mutation 94. This, as well as studies of families with heritable MPD, provides evidence that Jak2 Val617Phe may not be the mutation that initiates this disease but may occur as a secondary mutation in a genetically predisposed cell 95,96.
Conclusions
Much has been learned about megakaryopoiesis since the discovery of TPO and its receptor. Furthermore, advances in genotyping have enabled researchers to identify the causative genes in several acquired and inherited disorders of platelet production. New tools, such as platforms that enable large scale studies of protein-protein and protein-DNA interactions, tissue-specific promoters that permit the deletion of megakaryocytic genes in mouse models, and developmental models using embryonic stem cells, will continue to expand the boundaries of our knowledge.
Acknowledgments
The author would like to thank Dr. Ian Hitchcock for his help in creating the figures.
Supported in part by NIH R01 DK049855
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. 2008;111:981–986. doi: 10.1182/blood-2007-05-088500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szalai G, LaRue AC, Watson DK. Molecular mechanisms of megakaryopoiesis. Cell Mol Life Sci. 2006;63:2460–2476. doi: 10.1007/s00018-006-6190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T, et al. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med. 2004;10:299–304. doi: 10.1038/nm997. [DOI] [PubMed] [Google Scholar]
- 4.Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003;101:4333–4341. doi: 10.1182/blood-2002-09-2708. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Yu M, Akie TE, Moran TB, Woo AJ, Tu N, et al. Differentiation-dependent interactions between RUNX-1 and FLI-1 during megakaryocyte development. Mol Cell Biol. 2009;29:4103–4115. doi: 10.1128/MCB.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nerlov C, Querfurth E, Kulessa H, Graf T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000;95:2543–2551. [PubMed] [Google Scholar]
- 7.Chou ST, Khandros E, Bailey LC, Nichols KE, Vakoc CR, Yao Y, et al. Graded repression of PU.1/Sfpi1 gene transcription by GATA factors regulates hematopoietic cell fate. Blood. 2009;114:983–994. doi: 10.1182/blood-2009-03-207944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisbacher M, Holmes ML, Newton A, Hogg PJ, Khachigian LM, Crossley M, et al. Protein-protein interaction between Fli-1 and GATA-1 mediates synergistic expression of megakaryocyte-specific genes through cooperative DNA binding. Mol Cell Biol. 2003;23:3427–3441. doi: 10.1128/MCB.23.10.3427-3441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starck J, Cohet N, Gonnet C, Sarrazin S, Doubeikovskaia Z, Doubeikovski A, et al. Functional cross-antagonism between transcription factors FLI-1 and EKLF. Mol Cell Biol. 2003;23:1390–1402. doi: 10.1128/MCB.23.4.1390-1402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metcalf D, Carpinelli MR, Hyland C, Mifsud S, Dirago L, Nicola NA, et al. Anomalous megakaryocytopoiesis in mice with mutations in the c-Myb gene. Blood. 2005;105:3480–3487. doi: 10.1182/blood-2004-12-4806. [DOI] [PubMed] [Google Scholar]
- 12.Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008;14:843–853. doi: 10.1016/j.devcel.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bluteau D, Lordier L, Di Stefano A, Chang Y, Raslova H, Debili N, et al. Regulation of megakaryocyte maturation and platelet formation. J Thromb Haemost. 2009;7 1:227–234. doi: 10.1111/j.1538-7836.2009.03398.x. [DOI] [PubMed] [Google Scholar]
- 14.Raslova H, Roy L, Vourc'h C, Le Couedic JP, Brison O, Metivier D, et al. Megakaryocyte polyploidization is associated with a functional gene amplification. Blood. 2003;101:541–544. doi: 10.1182/blood-2002-05-1553. [DOI] [PubMed] [Google Scholar]
- 15.Mattia G, Vulcano F, Milazzo L, Barca A, Macioce G, Giampaolo A, et al. Different ploidy levels of megakaryocytes generated from peripheral or cord blood CD34+ cells are correlated with different levels of platelet release. Blood. 2002;99:888–897. doi: 10.1182/blood.v99.3.888. [DOI] [PubMed] [Google Scholar]
- 16.Corash L, Levin J. The relationship between megakaryocyte ploidy and platelet volume in normal and thrombocytopenic C3H mice. Exp Hematol. 1990;18:985–989. [PubMed] [Google Scholar]
- 17.Vitrat N, Cohen-Solal K, Pique C, Le Couedic JP, Norol F, Larsen AK, et al. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood. 1998;91:3711–3723. [PubMed] [Google Scholar]
- 18.Geddis AE, Fox NE, Tkachenko E, Kaushansky K. Endomitotic megakaryocytes that form a bipolar spindle exhibit cleavage furrow ingression followed by furrow regression. Cell Cycle. 2007;6:455–460. doi: 10.4161/cc.6.4.3836. [DOI] [PubMed] [Google Scholar]
- 19.Lordier L, Jalil A, Aurade F, Larbret F, Larghero J, Debili N, et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood. 2008;112:3164–3174. doi: 10.1182/blood-2008-03-144956. [DOI] [PubMed] [Google Scholar]
- 20.Nakao K, Angrist AA. Membrane surface specialization of blood platelet and megakaryocyte. Nature. 1968;217:960–961. doi: 10.1038/217960a0. [DOI] [PubMed] [Google Scholar]
- 21.Schulze H, Korpal M, Hurov J, Kim SW, Zhang J, Cantley LC, et al. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood. 2006;107:3868–3875. doi: 10.1182/blood-2005-07-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharnoff JG, Scardino V. Pulmonary megakaryocytes in human fetuses and premature and full-term infants. Arch Pathol. 1960;69:139–141. [PubMed] [Google Scholar]
- 23.Wright JH. The Histogenesis of Blood Platelets. Journal of Morphology. 1910;21:263–277. [Google Scholar]
- 24.Italiano JE, Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147:1299–1312. doi: 10.1083/jcb.147.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junt T, Schulze H, Chen Z, Massberg S, Goerge T, Krueger A, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317:1767–1770. doi: 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- 26.Vigon I, Mornon JP, Cocault L, Mitjavila MT, Tambourin P, Gisselbrecht S, et al. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: identification of a member of the hematopoietic growth factor receptor superfamily. Proc Natl Acad Sci U S A. 1992;89:5640–5644. doi: 10.1073/pnas.89.12.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265:1445–1447. doi: 10.1126/science.8073287. [DOI] [PubMed] [Google Scholar]
- 28.Bunting S, Widmer R, Lipari T, Rangell L, Steinmetz H, Carver-Moore K, et al. Normal platelets and megakaryocytes are produced in vivo in the absence of thrombopoietin. Blood. 1997;90:3423–3429. [PubMed] [Google Scholar]
- 29.Fox N, Priestley G, Papayannopoulou T, Kaushansky K. Thrombopoietin expands hematopoietic stem cells after transplantation. J Clin Invest. 2002;110:389–394. doi: 10.1172/JCI15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murone M, Carpenter DA, de Sauvage FJ. Hematopoietic deficiencies in c-mpl and TPO knockout mice. Stem Cells. 1998;16:1–6. doi: 10.1002/stem.160001. [DOI] [PubMed] [Google Scholar]
- 32.Sabath DF, Kaushansky K, Broudy VC. Deletion of the extracellular membrane-distal cytokine receptor homology module of Mpl results in constitutive cell growth and loss of thrombopoietin binding. Blood. 1999;94:365–367. [PubMed] [Google Scholar]
- 33.Drachman JG, Kaushansky K. Structure and function of the cytokine receptor superfamily. Curr Opin Hematol. 1995;2:22–28. doi: 10.1097/00062752-199502010-00004. [DOI] [PubMed] [Google Scholar]
- 34.Dahlen DD, Broudy VC, Drachman JG. Internalization of the thrombopoietin receptor is regulated by 2 cytoplasmic motifs. Blood. 2003;102:102–108. doi: 10.1182/blood-2002-11-3468. [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock IS, Chen MM, King JR, Kaushansky K. YRRL motifs in the cytoplasmic domain of the thrombopoietin receptor regulate receptor internalization and degradation. Blood. 2008 doi: 10.1182/blood-2008-01-134049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- 37.Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 38.Drachman JG, Kaushansky K. Dissecting the thrombopoietin receptor: functional elements of the Mpl cytoplasmic domain. Proc Natl Acad Sci U S A. 1997;94:2350–2355. doi: 10.1073/pnas.94.6.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geddis AE, Linden HM, Kaushansky K. Thrombopoietin: a pan-hematopoietic cytokine. Cytokine Growth Factor Rev. 2002;13:61–73. doi: 10.1016/s1359-6101(01)00030-2. [DOI] [PubMed] [Google Scholar]
- 40.Schulze H, Ballmaier M, Welte K, Germeshausen M. Thrombopoietin induces the generation of distinct Stat1, Stat3, Stat5a and Stat5b homo- and heterodimeric complexes with different kinetics in human platelets. Exp Hematol. 2000;28:294–304. doi: 10.1016/s0301-472x(99)00154-x. [DOI] [PubMed] [Google Scholar]
- 41.Matsumura I, Ishikawa J, Nakajima K, Oritani K, Tomiyama Y, Miyagawa J, et al. Thrombopoietin-induced differentiation of a human megakaryoblastic leukemia cell line, CMK, involves transcriptional activation of p21(WAF1/Cip1) by STAT5. Mol Cell Biol. 1997;17:2933–2943. doi: 10.1128/mcb.17.5.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirito K, Watanabe T, Sawada K, Endo H, Ozawa K, Komatsu N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J Biol Chem. 2002;277:8329–8337. doi: 10.1074/jbc.M109824200. [DOI] [PubMed] [Google Scholar]
- 43.Magne S, Caron S, Charon M, Rouyez MC, Dusanter-Fourt I. STAT5 and Oct-1 form a stable complex that modulates cyclin D1 expression. Mol Cell Biol. 2003;23:8934–8945. doi: 10.1128/MCB.23.24.8934-8945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood. 2007;109:1678–1686. doi: 10.1182/blood-2006-01-029918. [DOI] [PubMed] [Google Scholar]
- 45.Najfeld V, Cozza A, Berkofsy-Fessler W, Prchal J, Scalise A. Numerical gain and structural rearrangements of JAK2, identified by FISH, characterize both JAK2617V>F-positive and -negative patients with Ph-negative MPD, myelodysplasia, and B-lymphoid neoplasms. Exp Hematol. 2007;35:1668–1676. doi: 10.1016/j.exphem.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 46.Rouyez MC, Boucheron C, Gisselbrecht S, Dusanter-Fourt I, Porteu F. Control of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17:4991–5000. doi: 10.1128/mcb.17.9.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojnuckarin P, Drachman JG, Kaushansky K. Thrombopoietin-induced activation of the mitogen-activated protein kinase (MAPK) pathway in normal megakaryocytes: role in endomitosis. Blood. 1999;94:1273–1282. [PubMed] [Google Scholar]
- 48.Fichelson S, Freyssinier JM, Picard F, Fontenay-Roupie M, Guesnu M, Cherai M, et al. Megakaryocyte growth and development factor-induced proliferation and differentiation are regulated by the mitogen-activated protein kinase pathway in primitive cord blood hematopoietic progenitors. Blood. 1999;94:1601–1613. [PubMed] [Google Scholar]
- 49.Miyakawa Y, Oda A, Druker BJ, Kato T, Miyazaki H, Handa M, et al. Recombinant thrombopoietin induces rapid protein tyrosine phosphorylation of Janus kinase 2 and Shc in human blood platelets. Blood. 1995;86:23–27. [PubMed] [Google Scholar]
- 50.Alexander WS, Maurer AB, Novak U, Harrison-Smith M. Tyrosine-599 of the c-Mpl receptor is required for Shc phosphorylation and the induction of cellular differentiation. Embo J. 1996;15:6531–6540. [PMC free article] [PubMed] [Google Scholar]
- 51.Skolnik EY, Batzer A, Li N, Lee CH, Lowenstein E, Mohammadi M, et al. The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science. 1993;260:1953–1955. doi: 10.1126/science.8316835. [DOI] [PubMed] [Google Scholar]
- 52.Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- 53.Luoh SM, Stefanich E, Solar G, Steinmetz H, Lipari T, Pestina TI, et al. Role of the distal half of the c-Mpl intracellular domain in control of platelet production by thrombopoietin in vivo. Mol Cell Biol. 2000;20:507–515. doi: 10.1128/mcb.20.2.507-515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brizzi MF, Dentelli P, Lanfrancone L, Rosso A, Pelicci PG, Pegoraro L. Discrete protein interactions with the Grb2/c-Cbl complex in SCF- and TPO-mediated myeloid cell proliferation. Oncogene. 1996;13:2067–2076. [PubMed] [Google Scholar]
- 55.Garcia J, de Gunzburg J, Eychene A, Gisselbrecht S, Porteu F. Thrombopoietin-mediated sustained activation of extracellular signal-regulated kinase in UT7-Mpl cells requires both Ras-Raf-1- and Rap1-B-Raf-dependent pathways. Mol Cell Biol. 2001;21:2659–2670. doi: 10.1128/MCB.21.8.2659-2670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geddis AE, Fox NE, Kaushansky K. Phosphatidylinositol 3-kinase is necessary but not sufficient for thrombopoietin-induced proliferation in engineered Mpl-bearing cell lines as well as in primary megakaryocytic progenitors. J Biol Chem. 2001;276:34473–34479. doi: 10.1074/jbc.M105178200. [DOI] [PubMed] [Google Scholar]
- 57.Miyakawa Y, Rojnuckarin P, Habib T, Kaushansky K. Thrombopoietin induces phosphoinositol 3-kinase activation through SHP2, Gab, and insulin receptor substrate proteins in BAF3 cells and primary murine megakaryocytes. J Biol Chem. 2001;276:2494–2502. doi: 10.1074/jbc.M002633200. [DOI] [PubMed] [Google Scholar]
- 58.Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker PJ. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4:798–806. doi: 10.1016/s0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 59.Nakao T, Geddis AE, Fox NE, Kaushansky K. PI3K/Akt/FOXO3a pathway contributes to thrombopoietin-induced proliferation of primary megakaryocytes in vitro and in vivo via modulation of p27(Kip1) Cell Cycle. 2007;7 doi: 10.4161/cc.7.2.5148. [DOI] [PubMed] [Google Scholar]
- 60.Soda M, Willert K, Kaushansky K, Geddis AE. Inhibition of GSK-3beta promotes survival and proliferation of megakaryocytic cells through a beta-catenin-independent pathway. Cell Signal. 2008;20:2317–2323. doi: 10.1016/j.cellsig.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raslova H, Baccini V, Loussaief L, Comba B, Larghero J, Debili N, et al. Mammalian target of rapamycin (mTOR) regulates both proliferation of megakaryocyte progenitors and late stages of megakaryocyte differentiation. Blood. 2006;107:2303–2310. doi: 10.1182/blood-2005-07-3005. [DOI] [PubMed] [Google Scholar]
- 62.Guerriero R, Parolini I, Testa U, Samoggia P, Petrucci E, Sargiacomo M, et al. Inhibition of TPO-induced MEK or mTOR activity induces opposite effects on the ploidy of human differentiating megakaryocytes. J Cell Sci. 2006;119:744–752. doi: 10.1242/jcs.02784. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 64.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 65.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 66.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 67.Wang Q, Miyakawa Y, Fox N, Kaushansky K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood. 2000;96:2093–2099. [PubMed] [Google Scholar]
- 68.Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W, et al. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood. 2007;109:4924–4929. doi: 10.1182/blood-2006-08-039735. [DOI] [PubMed] [Google Scholar]
- 69.Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200:569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, et al. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc Natl Acad Sci U S A. 2007;104:2349–2354. doi: 10.1073/pnas.0606238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest. 2008;118:2832–2844. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Funakoshi-Tago M, Pelletier S, Matsuda T, Parganas E, Ihle JN. Receptor specific downregulation of cytokine signaling by autophosphorylation in the FERM domain of Jak2. Embo J. 2006;25:4763–4772. doi: 10.1038/sj.emboj.7601365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 74.Owen CJ, Toze CL, Koochin A, Forrest DL, Smith CA, Stevens JM, et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. 2008;112:4639–4645. doi: 10.1182/blood-2008-05-156745. [DOI] [PubMed] [Google Scholar]
- 75.Ciovacco WA, Raskind WH, Kacena MA. Human phenotypes associated with GATA-1 mutations. Gene. 2008;427:1–6. doi: 10.1016/j.gene.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004;129A:51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- 77.Raslova H, Komura E, Le Couedic JP, Larbret F, Debili N, Feunteun J, et al. FLI1 monoallelic expression combined with its hemizygous loss underlies Paris-Trousseau/Jacobsen thrombopenia. J Clin Invest. 2004;114:77–84. doi: 10.1172/JCI21197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA, et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc Natl Acad Sci U S A. 1999;96:3132–3136. doi: 10.1073/pnas.96.6.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97:139–146. doi: 10.1182/blood.v97.1.139. [DOI] [PubMed] [Google Scholar]
- 80.Ballmaier M, Germeshausen M, Krukemeier S, Welte K. Thrombopoietin is essential for the maintenance of normal hematopoiesis in humans: development of aplastic anemia in patients with congenital amegakaryocytic thrombocytopenia. Ann N Y Acad Sci. 2003;996:17–25. doi: 10.1111/j.1749-6632.2003.tb03228.x. [DOI] [PubMed] [Google Scholar]
- 81.King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: a retrospective clinical analysis of 20 patients. Br J Haematol. 2005;131:636–644. doi: 10.1111/j.1365-2141.2005.05819.x. [DOI] [PubMed] [Google Scholar]
- 82.Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet. 1998;18:49–52. doi: 10.1038/ng0198-49. [DOI] [PubMed] [Google Scholar]
- 83.Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, et al. Familial essential thrombocythemia associated with one-base deletion in the 5′-untranslated region of the thrombopoietin gene. Blood. 1998;92:1091–1096. [PubMed] [Google Scholar]
- 84.Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103:4198–4200. doi: 10.1182/blood-2003-10-3471. [DOI] [PubMed] [Google Scholar]
- 85.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 86.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 87.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 88.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 90.Boissinot M, Lippert E, Girodon F, Dobo I, Fouassier M, Masliah C, et al. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood. 2006;108:3223–3224. doi: 10.1182/blood-2006-05-021527. [DOI] [PubMed] [Google Scholar]
- 91.De Stefano V, Fiorini A, Rossi E, Za T, Farina G, Chiusolo P, et al. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemost. 2007;5:708–714. doi: 10.1111/j.1538-7836.2007.02424.x. [DOI] [PubMed] [Google Scholar]
- 92.Yan XQ, Lacey D, Hill D, Chen Y, Fletcher F, Hawley RG, et al. A model of myelofibrosis and osteosclerosis in mice induced by overexpressing thrombopoietin (mpl ligand): reversal of disease by bone marrow transplantation. Blood. 1996;88:402–409. [PubMed] [Google Scholar]
- 93.Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood. 2002;100:3495–3503. doi: 10.1182/blood-2002-04-1133. [DOI] [PubMed] [Google Scholar]
- 94.Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, et al. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110:375–379. doi: 10.1182/blood-2006-12-062125. [DOI] [PubMed] [Google Scholar]
- 95.Kralovics R, Stockton DW, Prchal JT. Clonal hematopoiesis in familial polycythemia vera suggests the involvement of multiple mutational events in the early pathogenesis of the disease. Blood. 2003;102:3793–3796. doi: 10.1182/blood-2003-03-0885. [DOI] [PubMed] [Google Scholar]
- 96.Skoda R, Prchal JT. Lessons from familial myeloproliferative disorders. Semin Hematol. 2005;42:266–273. doi: 10.1053/j.seminhematol.2005.08.002. [DOI] [PubMed] [Google Scholar]