

Abstract
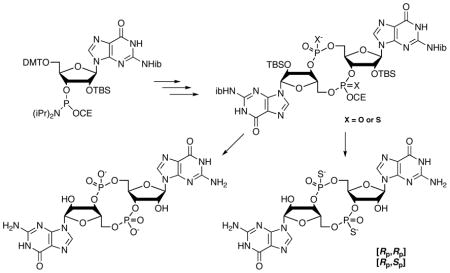
An integrated set of reactions and conditions that allow an eight step one-flask synthesis of the protected derivatives of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogs is reported. Deprotection is also carried out as a one-flask procedure, with the final products isolated by crystallization from the reaction mixture. Chromatography is only used for separation of the thiophosphate diastereomers.
The bacterial signaling molecule, cyclic diguanosine monophosphate (c-di-GMP, 7) is increasingly recognized as having widespread consequences for human health through its multiple roles, which include regulation of biofilm formation as well as virulence.1–4 Further, c-di-GMP has now been identified as a specific and high affinity ligand for the GEMM class of bacterial riboswitches that regulate gene expression.5 Recent crystal structures of the bound riboswitch aptamer6,7 suggest a molecular mechanism for the c-di-GMP signaling pathway through formation of a unique helix. These structures demonstrate asymmetrical binding of c-di-GMP, with the guanines deeply buried, but with the ribose phosphate ring partially exposed.
Better synthetic methods for c-di-GMP, as well as analogs and derivatives, are essential to fully explore and define its pathways and molecular mechanisms. Our first goal in revisiting the synthesis of c-di-GMP and its thiophosphate analogs was to develop a fast and convenient route to gram scale quantities from low-cost commercially available starting materials. Six laboratories, including ours, have reported routes to synthesis of c-di-GMP and a few analogs.8–15 These approaches use a range of chemistries, with two using phosphate triester chemistry for both the initial coupling and the cyclization,8,12 while the others begin with an amidite9–11,15 or H-phosphonate coupling,14 and then use a triester9,11,15 or H-phosphonate cyclization.10,14 A common feature of all of these approaches is the small amount of c-di-GMP prepared, with none reporting amounts over 200 mg, and most much less. All rely on reversed-phase HPLC for final purification, and all but the solid-phase route15 require several silica chromatography steps during the synthesis. The small amount of c-di-GMP obtained limits the opportunity for the preparation of derivatives. Further, as a phosphate triester cyclization does not lend itself to the preparation of thiophosphates, prior to our recent work16 only the analog with a single thiophosphate had been reported.8,13 We have recently prepared and characterized all of the thiophosphate diastereomers of c-di-GMP containing one, two, or three sulfur atoms,16 but in such modest yields that opportunities for further study are limited, beyond confirming that they showed similar polymorphism to that we had found for c-di-GMP itself.17 Thiophosphate analogs of defined stereochemistry are of particular interest as probes of phosphate/metal interaction, and because of the resistance of thiophosphates to enzymatic hydrolysis.
The method for preparation of c-di-GMP and c-di-(guanosine-monothiophosphate) we now report uses the standard commercial N-isobutyryl-2′-O-TBS protected guanosine phosphoramidite, 1, as the common starting material for both of the guanosines incorporated, and was designed such that the reagents used in each step do not interfere with subsequent steps, allowing the syntheses to be carried out as one-flask procedures. Two particular examples of the degree of integration of the reagents are that the dichloroacetic acid used for the first detritylation provides in the next step the pyridinium ion that is the promoter of the amidite coupling reaction, and that the dichloroacetate does not interfere with the H-phosphonate cyclization reaction. Thus, the eight steps to get to the protected cyclic derivatives 5a–c, which are isolated by extraction, are carried out by sequential addition of reagents without isolation of intermediates. We have also developed a two step, one-flask deprotection procedure in which the final products are isolated by crystallization, without the need for a fluoride scavenger,18 ion-exchange, or reversed-phase HPLC. There are no chromatographic steps in preparation of c-di-GMP, 7. The separation of the protected thiophosphate diastereomers 5b and 5c, on a silica column, is the only chromatographic step.
As shown in Scheme 1, the first step is hydrolysis of the first portion of the guanosine phosphoramidite, 1, by treatment with pyridinium trifluoroacetate in acetonitrile containing 2 equiv. water. The hydrolysis takes less than one minute to give the H-phosphonate diester, 2, which is immediately treated with tert-butylamine to remove the cyanoethyl group. This reaction is complete within ten minutes and the solution is then concentrated to remove tert-butylamine prior to detritylation with 3% dichloroacetic acid (DCA) and 10 equiv. of water in methylene chloride. The detritylation requires less than 10 minutes. The water prevents retritylation, which otherwise occurs to a significant extent upon quenching the DCA with pyridine. After changing the solvent to dry acetonitrile, the coupling with 1.3 equiv. of a second portion of 1 to give 3 is complete within two minutes.
Scheme 1.

Syntheses of c-di-GMP, 7, and the [Rp,Rp], 8a, and [Rp,Sp], 8b, thiophosphate analogs.
Oxidation using tert-butylhydroperoxide, or sulfurization using 3-((dimethylaminomethylidene)-amino)-3H-1,2,4-dithiazole-5-thione (DDTT),19 is carried out immediately after the coupling. The oxidation or sulfurization requires no more than 30 minutes. The solution is concentrated to change the solvent back to methylene chloride for the second detritylation, again using DCA/water, to give the linear dimer 4a or 4b. The reaction is quenched by addition of pyridine, followed by concentration to remove methylene chloride and water. Cyclization is then effected by addition of 3–3.5 equiv. of 5,5-dimethyl-2-oxo-2-chloro-1,3,2-dioxaphosphinane (DMOCP)20 to the pyridine solution of 4a or 4b, and is complete within 5–10 min. Pivaloyl chloride or adamantoyl chloride also work well, but we have found that DMOCP gives slightly cleaner results. Significantly, we have found that high dilution is not necessary for this H-phosphonate cyclization, which is done at a concentration of 20 mL/mmol. The excess DMOCP is quenched with water, followed by addition of either iodine or 3H-1,2-benzodithiol-3-one.21,22 The oxidation or sulfurization requires 5 minutes. Excess iodine is consumed by addition of the mixture to a solution of aqueous sodium bisulfite, to which sodium bicarbonate is added after 10 min. For the thioates the reaction mixture is added directly to a sodium bicarbonate solution.
The protected mixed diester/triester derivatives 5a–c are isolated by extraction of the aqueous mixture with 1:1 diethyl ether and ethyl acetate. The thiophosphate diastereomers 5b and 5c are separated and purified by silica chromatography in yields from 1 of 27 and 23%, respectively. Only two of the three thiophosphate diastereomers are obtained because the H-phosphonate cyclization and sulfurization gives only the Rp diastereomer.16,23 Treatment of 5a with tert-butylamine in acetonitrile gives 6a, which is readily purified by crystallization from methylene chloride, in 35–45% yield from 1. If desired, 5b and 5c can be similarly converted to 6b and 6c, which can be crystallized from water. The tert-butylamine step, however, is not necessary, as subsequent methylamine treatment efficiently removes the cyanoethyl group.
Full deprotection is carried out in two steps beginning with treatment with a solution of methylamine in ethanol at room temp for 1–2 h. After concentration to dryness, the residue is dissolved in minimal pyridine and treated with TEA·HF at 50° for 60 min. The final products, 7, 8a, or 8b, crystallize from the mixture as the triethylamonium salts upon addition of acetone.
The method reported here is significantly faster and more convenient than other approaches reported to date, allowing facile preparation of gram scale amounts of crystalline c-di-GMP. Of the eight reactions leading to 5a–c, only the 30 min phosphite triester oxidation/sulfurization takes more than ten min. Because of the fast reactions, the overall time for preparation of 5a–c from 1, is 8–10 h, with most of the time spent in concentrations on a rotary evaporator to change solvents. The conversion of 5a to 6a takes only an hour, while the time for crystallization of 6a varies from 1–8 h. The silica separation of 5b and 5c takes most of a day, as does the final deprotection, so that the overall time for 7, or 8a/8b, is about three days with overall yields from 1 of 30% for 7, 19% for 8a, and 17% for 8b.
This route to c-di-GMP is based on the most common commercially available guanosine phosphoramidite, eliminating the need for preparation of this starting material. Moreover, since the phosphoramidites of many other nucleosides, and analogs, are also available, this route should be generally applicable to preparation of a wide variety of other cyclic dinucleotides, including unsymmetrical molecules, as well as their thiophosphate analogs.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM79760. We thank Edward Miranda, an ACS Project SEED student, for assistance.
Footnotes
Supporting Information Available: Synthetic procedures, HPLC, 1H, 13C, 31P NMR, and UV spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Romling U, Amikam D. Curr Opin Microbiol. 2006;9:218–228. doi: 10.1016/j.mib.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Jenal U, Malone J. Annu Rev Genet. 2006;40:385–407. doi: 10.1146/annurev.genet.40.110405.090423. [DOI] [PubMed] [Google Scholar]
- 3.Cotter PA, Stibitz S. Curr Opin Microbiol. 2007;10:17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Tamayo R, Pratt JT, Camilli A. Annu Rev Microbiol. 2007;61:131–148. doi: 10.1146/annurev.micro.61.080706.093426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Science (Wash) 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulshina N, Baird NJ, Ferré-D’Amaré AR. Nat Struct Mol Biol. 2009;16:1212–1217. doi: 10.1038/nsmb.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. Nat Struct Mol Biol. 2009;16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross P, Mayer R, Weinhouse H, Amikam D, Huggirat Y, Benziman M, Vroom Ed, Fidder A, Paus Pd, Sliedregt LAJM, Marel GAvd, Boom JHv. J Biol Chem. 1990;265:18933–18943. [PubMed] [Google Scholar]
- 9.Hayakawa Y, Nagata R, Hirata A, Hyodo M, Kawai R. Tetrahedron. 2003;59:6465–6471. [Google Scholar]
- 10.Zhang Z, Gaffney BL, Jones RA. J Am Chem Soc. 2004;126:16700–16701. doi: 10.1021/ja0449832. [DOI] [PubMed] [Google Scholar]
- 11.Hyodo M, Hayakawa Y. Bull Chem Soc Jpn. 2004;77:2089–2093. [Google Scholar]
- 12.Amiot N, Heintz K, Giese B. Synthesis. 2006:4230–4236. [Google Scholar]
- 13.Hyodo M, Sato Y, Hayakawa Y. Tetrahedron. 2006;62:3089–3094. [Google Scholar]
- 14.Yan H, Aguilar AL. Nucleosides Nucleotides Nucl Acids. 2007;26:189–204. doi: 10.1080/15257770601112762. [DOI] [PubMed] [Google Scholar]
- 15.Kiburu I, Shurer A, Yan L, Sintim HO. Mol BioSyst. 2008;4:518–520. doi: 10.1039/b719423d. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Veliath E, Kim S, Gaffney BL, Jones RA. Nucleosides Nucleotides Nucl Acids. 2009;28:352–378. doi: 10.1080/15257770903044523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Kim S, Gaffney BL, Jones RA. J Am Chem Soc. 2006;128:7015–7024. doi: 10.1021/ja0613714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Q, Jones RA. Tetrahedron Lett. 1999;40:4653–4654. [Google Scholar]
- 19.Lemaître MM, Murphy AS, Somers RL. The Glen Report. 2006;18:1–6. [Google Scholar]
- 20.Strömberg R, Stawinski J. Nucleic Acids Res. Symposium Series No 18. 1987:185–188. [PubMed] [Google Scholar]
- 21.Stawinski J, Thelin M. J Org Chem. 1991;56:5169–5175. [Google Scholar]
- 22.Ghisaidoobe ABT, de Koning MC, Duynstee HI, Ten Kortenaar PBW, Overkleeft HS, Filippov DV, van der Marel GA. Tetrahedron Lett. 2008;49:3129–3132. [Google Scholar]
- 23.Battistini C, Fustinoni S, Brasca MG, Borghi D. Tetrahedron. 1993;49:1115–1132. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.