

Abstract
Objective
Zinc deficiency is common among populations at high risk for sepsis mortality, including elderly, alcoholic, and hospitalized patients. Zinc deficiency causes exaggerated inflammatory responses to endotoxin but has not been evaluated during bacterial sepsis. We hypothesized that subacute zinc deficiency would amplify immune responses and oxidant stress during bacterial sepsis [i.e., cecal ligation and puncture (CLP)] resulting in increased mortality and that acute nutritional repletion of zinc would be beneficial.
Design
Prospective, randomized, controlled animal study.
Setting
University medical center research laboratory.
Subjects
Adult male C57BL/6 mice.
Interventions
Ten-week-old, male, C57BL/6 mice were randomized into three dietary groups: 1) control diet, 2) zinc-deficient diet for 3 weeks, and 3) zinc-deficient diet for 3 weeks followed by oral zinc supplementation for 3 days (n = 35 per diet). Mice were then assigned to receive either CLP or sham operation (n = 15 each per diet). CLP and sham-operated treatment groups were further assigned to a 7-day survival study (n = 10 per treatment per diet) or were evaluated at 24 hours (n = 5 per treatment per diet) for signs of vital organ damage.
Measurements and Main Results
Sepsis mortality was significantly increased with zinc deficiency (90% vs. 30% on control diet). Zinc-deficient animals subject to CLP had higher plasma cytokines, more severe organ injury, including increased oxidative tissue damage and cell death, particularly in the lungs and spleen. None of the sham-operated animals died or developed signs of organ damage. Zinc supplementation normalized the inflammatory response, greatly diminished tissue damage, and significantly reduced mortality.
Conclusions
Subacute zinc deficiency significantly increases systemic inflammation, organ damage, and mortality in a murine polymicrobial sepsis model. Short-term zinc repletion provides significant, but incomplete protection despite normalization of inflammatory and organ damage indices.
Keywords: inflammation, cell injury, oxidants, multiple organ dysfunction score, cecal ligation and puncture
Sepsis is a major cause of morbidity, mortality, and healthcare costs in hospitalized patients and is the tenth leading cause of death overall in the United States (1). Currently, it is expected that between 20% and 50% of patients who develop sepsis will die despite receiving the current standard of appropriate therapy. The basic cellular and molecular mechanisms accounting for sepsis-related morbidity and mortality remain poorly understood. However, a number of host factors, including immunosuppression, advanced age, chronic alcoholism, and poor nutritional status, are known to increase sepsis mortality (2, 3).
Zinc is an essential dietary micronutrient with beneficial functions that facilitate cytoprotection, improved wound healing, and tissue repair (4). Humans, in response to sepsis or endotoxin administration, experience a transient decrease in plasma zinc levels without a concomitant loss of whole body zinc content (5). This phenomenon is believed to be a component of the host defense response designed to increase cellular zinc bioavailability for cytoprotective functions that involve protein synthesis, neutralization of reactive oxygen species, and prevention of microbial invasion (6). Therefore, we postulate that individuals who are zinc deficient may be disadvantaged with respect to their ability to respond to systemic acute stress.
Zinc deficiency is underestimated worldwide and is predicted to affect millions (4). Recent studies conducted in the United States and Europe indicate that a significant segment of the population, particularly those with chronic illness requiring hospitalization, are at an increased risk of zinc deficiency (7–9). Furthermore, a significant subpopulation of critically ill patients are chronic alcoholics (10), a condition associated with malnutrition, including depletion of zinc stores (4, 11). Zinc deficiency can also occur after the onset of trauma (12) or burn injury (13). Body stores of zinc and other antioxidant nutrients are further influenced by diet, advanced age, smoking, and female gender (9). Thus, it is likely that a clinically significant number of intensive care unit patients are zinc deficient.
Zinc deficiency is shown to increase mortality in the context of pneumococcal pneumonia, presumably related to the impaired immune response to specific bacterial antigens that are associated with bacterial invasion (14). In this context, supplementation of zinc in at-risk patients reduces the overall incidence of respiratory infections in an elderly population shown to have significant zinc depletion (15). Paradoxically, zinc deficiency causes heightened inflammatory responses and more severe organ injury in response to bacterial endotoxin (16, 17). Many experts agree that a vigorous innate immune response to infection favors rapid resolution of infection and improved outcomes during sepsis (18).
Considering the plieotropic functions of zinc relating to inflammatory and antioxidant responses, we hypothesized that zinc deficiency would be associated with increased systemic and local inflammation, increased oxidant stress, more severe vital organ damage, and higher mortality in the context of severe sepsis. To test this hypothesis, young adult mice were randomly assigned to receive a zinc-depleted diet or a matching control diet for 3 weeks before the onset of sepsis induced by cecal ligation and puncture (CLP). A third group received acute dietary supplementation of zinc 3 days before the onset of sepsis. The effects of dietary zinc depletion and supplementation on sepsis mortality, systemic inflammatory cytokine release, and indices of multiple organ damage were assessed.
METHODS
Establishing a Mouse Model of Zinc Deficiency
All experiments were approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Ten-week-old, adult, male, C57BL/6 mice (~25 g) (Harlan Sprague Dawley; Indianapolis, IN) with fully developed lungs, were randomly placed on a zinc-deficient diet (Harlan Teklad, TD85419; 1 ppm) (n = 35) or a matched control diet (TD85420; 50 ppm) (n = 35) for 3 weeks, a sufficient time to establish subacute zinc deficiency and eliminate any requirement for pair-feeding. A zinc-free environment was carefully maintained using deionized water in zinc-free containers, stainless steel cages, along with daily cage changes. An additional group (n = 35) received a zinc-fortified diet (TD07129; 100 mg/kg) for 3 days following the 3-week zinc-deficient regimen and before CLP to assess the impact of zinc supplementation.
Measurement of Zinc Status
A cohort of animals (n = 5) within each dietary group was designated for zinc status measurements and thus killed at the end of the 3-week dietary period. Zinc analysis was carried out by measuring serum zinc concentrations using the QuantiChrom Zinc Assay kit (BioAssay Systems, Hayward, CA) according to the manufacturer’s recommendations as well as quantitatively assessing liver and lung metallothionein content to characterize zinc nutritional status as previously described (19).
CLP
At the end of the dietary period in all groups, mice (n = 15 for each diet) were randomly subject to CLP via laparotomy under general anesthesia (1% isoflurane) as described previously (20) with slight modifications. Briefly, through an upper midline abdominal incision, the cecum was delivered, ligated with a silk suture 1 cm from the tip, and doubly punctured with a 21-gauge needle. After puncture, the cecum was gently squeezed to extrude a small amount of feces and returned to the abdominal cavity. The laparotomy was then closed. This model reproducibly results in ~30% mortality within 7 days in our hands. Additionally, matched animals (n = 15 for each diet) were randomly assigned to undergo sham operation, which involved laparotomy and cecal deliverance without ligation or puncture. CLP and sham operation groups were then assigned to either a 7-day survival study (n = 10 for each treatment on each diet) or were euthanized at 24 hours (n = 5 for each treatment on each diet) to obtain blood and tissue samples for further analyses.
Macroscopic Tissue Analysis
Immediately following euthanasia, the right main bronchus was cross-clamped, and the left lung was filled with 10% formalin at a constant pressure equal to 25 cm H2O allowing homogeneous expansion of the lung parenchyma. The right lung was then removed and snap-frozen for later mRNA and protein analyses, whereas the left lung was paraffin-embedded following exposure to 10% buffered formalin for 48 hours. Tissue samples obtained from the liver, spleen, and ileum were also snap-frozen and fixed in a similar manner. All paraffin-embedded specimens were cut at 5 μm for further evaluation including hematoxylin and eosin (H&E) staining. Inflammation in the lung was quantified using histogram analysis in Adobe Photoshop CS2. All white areas in the lung were maintained while all tissue areas were converted to black pixels. The values indicated represent the percent of white space (alveolar space) per high-powered field from at least seven digital images per lung per mouse from three representative animals per treatment condition.
Transferase-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Assay
Detection of cell death was carried out using the TACS.XL-Blue label kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s instructions. A minimum of five animals per treatment condition were randomly selected, and TUNEL-positive cells were quantified in the lung, spleen, liver, and ileum using histogram analysis in Adobe Photoshop CS2 by converting all TUNEL-positive-stained pixels to black and all other tissue to white. The values indicated represent the percent of TUNEL-positive pixels per high-powered field from at least 10 digital images per organ per mouse.
Real-Time Quantitative PCR
Total RNA from lungs of mice exposed to a normal or zinc-deficient diet was reverse-transcribed into cDNA and then subject to relative quantitation by reverse transcriptase-polymerase chain reaction as previously reported (21). Prevalidated primer mixtures for target and housekeeping genes were obtained from SuperArray (Frederick, MD). For quantitation of data, the comparative Δ( ΔCT) method was used. ΔCT = CT(target gene) − CT(house-keeping gene), and this value was calculated for each sample, where CT = cycle number threshold. The ΔCT calculation involved finding the difference between each sample’s ΔCT and its mean control ΔCT. These values were transformed to absolute values, where comparative expression level = 2Δ Δ (ΔCT), statistically analyzed, and then converted to relative copy number (RCN) for presentation purposes.
Plasma Cytokine Analysis
Multiplex quantification of cytokine and chemokine levels was conducted in mouse plasma samples obtained 24 hours after CLP or sham operation. The concentration of each cytokine was analyzed from a 50 μL sample volume using a custom-made Bio-Plex Mouse Cytokine 9-Plex panel that included tumor necrosis factor-α, interleukin (IL)-1β, IL-6, IL-10, chemokine (C-X-C motif) ligand, macrophage chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, and Regulated on Activation, Normal T Expressed and Secreted according to the manufacturer’s recommendations using the Bio-Plex 200 Analysis System (Bio-Rad Laboratories, Hercules, CA).
Measurement of Tissue Superoxide Dismutase Activity
Tissue superoxide dismutase (SOD) activity was determined spectrophotometrically using an SOD assay kit (Cayman Chemical, Ann Arbor, MI). The kit uses a tetrazolium salt for the detection of superoxide radicals generated by xanthine oxidase and hypoxanthine (22). Briefly, liver and lung tissue samples (~1 g) were excised following euthanasia and immediately immersed in an ice-cold homogenization buffer, minced, and repeatedly rinsed to remove as much blood as possible (20). The liver tissue was then Dounce homogenized (multiple up and down passes) while on ice, whereas the lung tissue was subjected to bead homogenization. Lung tissue samples were added to 1.0 mm Zirconia/Silica beads (v/v) and 500 μL of cold homogenization buffer and then lysed using a Mini-Bead Beater cell disrupter (Biospec Products, Bartlesville, OK) at 5000 rpm for 20 seconds, repeated four times at 5-minute intervals.
Tissue homogenates were subjected to high-speed centrifugation (10,000 × g for 5 minutes at 4°C), and the supernatant protein concentrations were then determined spectrophotometrically using the Bradford assay. The samples were applied to the SOD assay kit according to the manufacturer’s instructions. The addition of 1–3 mM potassium cyanide, which inhibits Cu/Zn SOD, allowed for the measurement of manganese superoxide dismutase (MnSOD) activity. Absorbance was read at 450 nm, and reaction rates were calculated based on the linearized SOD standard rate. The results were normalized to those of the sham-operated controls and statistically analyzed.
Measurement of Tissue Protein Carbonylation
Carbonyl modified proteins were quantified in tissue homogenates using the method initially described by Levine et al (23) with minor modifications, as described previously (20). The results were normalized to those of the sham-operated controls and statistically analyzed.
Statistical Analyses
All data are expressed as mean ± SEM, and statistical significance was based on a minimum value of p ≤ 0.05 for single comparisons and for the adjusted p values after multiple comparisons. SigmaStat 5.0 software was used to carry out the statistical analyses. Comparisons of plasma zinc and cytokine and chemokine levels were made using the two-tailed Student’s t test. Mortality results were compared using the Log Rank (Mantel-Cox) test. Comparison of mRNA transcripts was conducted using a multiple comparison adjustment with the Holm’s procedure based on the raw CT values (standardized). Relative tissue SOD activity and protein carbonylation (treatment vs. diet), as well as pixel analysis of lung micrographs, were compared using two-way ANOVA. The p values of all pairwise comparisons were obtained using the Tukey multiple comparison method.
RESULTS
Plasma zinc concentrations and tissue metallothionein levels were evaluated in the lung and liver to confirm that animals achieved a zinc-deficient state. Mice placed on the zinc-deficient diet consistently exhibited >50% reduction in plasma zinc concentrations when compared with the matching control diet group (Fig. 1A). Zinc-deficient mice receiving zinc supplementation had plasma zinc concentrations comparable with those of the control diet group. A concomitant decrease in tissue metallothionein occurred in both the lung and liver in zinc-deficient animals, whereas animals on the control diet or receiving zinc supplementation had similar, higher levels (Fig. 1B). The observed correlation between plasma zinc and tissue metallothionein content indicated that systemic zinc deficiency was achieved through dietary regulation. In addition, following the 3-week dietary period, there were no observable differences in overall animal physical appearance among the three groups or in body weight between the control and zinc-deficient animals (24.3 ± 1.2 vs. 24.6 ± 0.5 g, respectively; starting weight = 24.7 ± 0.5 g).
Figure 1.

Alteration of zinc status following dietary modification. A, plasma zinc levels in mice receiving a control diet (Ctrl), a zinc-deficient diet (Zn−), or a zinc-deficient diet followed by acute zinc supplementation (Zn+) (n = 5 per dietary group) (*p < 0.05). B, Liver and lung tissue homogenates from the same animals analyzed for total tissue metallothionein content.
Zinc deficiency had a profound impact upon survival in response to sepsis. CLP-treated mice fed a control diet experienced 70% 7-day survival (Fig. 2), whereas zinc-deficient CLP-treated mice had a significantly lower survival of 10%. The time to death was also accelerated in the latter group, with the majority expiring within the first 48 hours. The CLP treatment group that received acute zinc supplementation had a delayed onset of death and a marked improval in survival compared with the zinc-deficient CLP group.
Figure 2.

Zinc deficiency increases sepsis mortality. Time-dependent survival following sham operation or and cecal ligation and puncture (CLP) in animals assigned to control (Ctrl), zinc deficient (Zn−), or zinc deficiency followed by short-term zinc supplementation (Zn+) groups (n = 10 for each group). Survival of 100% was observed in all sham-operated animals (resulting in an overlap of the mortality curves for the three sham-operated groups). A significant increase in mortality following CLP was associated with zinc deficiency (p < 0.05).
Histologic evaluation of tissue samples obtained from the lung, spleen, liver, and ileum of mice at 24 hours post-CLP were compared with matching sham operation groups to assess possible systemic organ damage and enhanced cell death. The lungs from animals placed on the zinc-deficient diet without further intervention (not shown) or subsequently subject to sham surgery (Fig. 3A) seemed normal, exhibiting no substantial evidence of tissue injury or inflammation. By comparison, CLP-treated mice on the control zinc diet exhibited mild patchy inflammatory cellular infiltrates without gross changes in parenchymal tissue structures (Fig. 3B). The effects were more dramatic in the lungs of CLP-treated zinc-deficient mice demonstrating extensive inflammatory cell infiltrates, perivascular edema, and epithelial damage (Fig. 3C). In contrast, the CLP-treated zinc-deficient mice receiving zinc supplementation had lung histology comparable to the sham-operated control diet group (Fig. 3D). The magnitude of open airspace, assessing lung injury, was significantly reduced in CLP-treated zinc-deficient mice compared with CLP-treated animals in the control diet and zinc supplementation groups (Fig. 3E).
Figure 3.

Zinc deficiency increases lung pathology. Histologic assessments of H&E-stained lung tissue from representative (A) zinc-deficient sham operation (Zn−/sham), (B) control diet cecal ligation and puncture (Ctrl/CLP), (C) zinc-deficient CLP (Zn−/CLP), and (D) zinc deficient followed by acute zinc supplementation before CLP (Zn+/CLP) animals. Focal areas of inflammatory cell infiltrate along with features consistent with perivascular edema and epithelial attenuation were most pronounced in the Zn−/CLP group, whereas the Zn+/CLP group exhibited lung histology similar to that of sham operation. E, Quantitative analysis of lung air space demonstrated a significant reduction in the Zn−/CLP group compared with the Zn−/sham, Zn+/CLP, and Ctrl/CLP groups (*p < 0.05). F, Relative copy number (RCN) of mRNA transcripts constitutively expressed by lung epithelia and endothelia in the setting of acute lung injury (see text), including the ATP-binding-cassette transporter (ABCA), solute carrier transporter 34A2 (SLC34A2), vascular endothelial growth factor A (VEGFA), and surfactant protein A (SFTPA) (*p < 0.05). KDR, kinase insertion domain receptor; SOX, Syr-related HMG box transcription factor; SFTPB, surfactant associated protein B.
Because the extent of inflammation in the lung was greater than in the other tissues, making it more difficult to estimate actual lung parenchymal injury, a validated, quantitative reverse transcriptase-polymerase chain reaction-based assay was used. A significant decrease in transcripts for the adenosine triphosphate-binding-cassette transporter, solute carrier family 34 (sodium phosphate) member 2, vascular endothelial growth factor A, and surfactant protein A was observed in the lungs of CLP-treated animals compared with matching shams. Furthermore, gene expression was further reduced in the CLP-treated, zinc-deficient group (Fig. 3F), consistent with lung epithelial and endothelial damage (21).
Histologic characteristics of the spleens of sham-operated, zinc-deficient animals were similar to controls (not shown). In contrast, CLP-treated animals exhibited mild hypocellularity of the lymphocytic follicles, whereas zinc-deficient CLP-treated animals exhibited marked hypocellularity (white arrows) (Fig. 4). Relative to sham-operated animals on the control and zinc-deficient diets, CLP-treated animals also exhibited marked hepatocyte edema, the severity of which was greatest in the zinc-deficient mice (Fig. 4). The ilea of CLP-treated mice demonstrated no significant changes in overall structure; however, there was a small but significant increase in the number of apoptotic-appearing cells in the epithelial layer of the zinc-deficient CLP group (Fig. 4).
Figure 4.
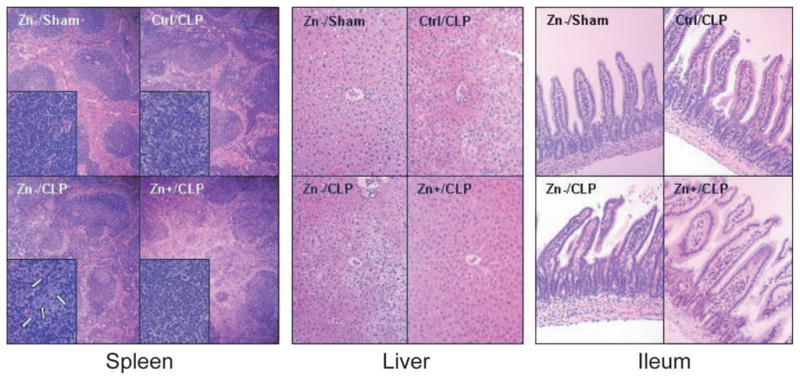
Morphology of the spleen, liver, and ileum. Morphologic evaluation of H&E-stained tissues in each of the treatment groups. Because all sham-operated animals had similar morphologic findings, for the sake of comparison, only the zinc-deficient sham operation (Zn−/sham) group is shown. Representative spleen samples were examined [magnification: ×4 and ×40 (insets)] in each treatment group. Minor hypocellularity of splenic lymphoid nodules (dark blue) was noted in the control diet cecal ligation and puncture (Ctrl/CLP) group. The hypocellularity was more exaggerated in the zinc-deficient CLP (Zn−/CLP) group and was associated with clusters of condensed apoptotic cells (inset, white arrows), whereas the zinc supplementation CLP (Zn+/CLP) group was similar to that of the Ctrl/CLP group. Relative to sham-operated (e.g., Zn−/sham) and Zn+/CLP animals, representative livers of Ctrl/CLP and Zn−/CLP (×10) animals demonstrated mild hepatocellular edema. There were no significant morphologic abnormalities of representative ilea (×10) in any of the treatment groups.
Further examination of these same vital organs was conducted using TUNEL analysis to detect apoptotic changes. A marked increase in TUNEL-positive cells occurred in all tissues from CLP animals and was most pronounced in the spleen and lung. Most striking were the findings in CLP-treated, zinc-deficient mice that exhibited a significant increase in the presence of TUNEL-positive cells across all tissues when compared with CLP-treated control diet animals within 24 hours of sepsis (Fig. 5). The observed pattern of TUNEL staining within the lymphoid follicles indicated that splenic lymphocytes were highly susceptible to apoptosis. Further, zinc supplementation decreased the occurrence of TUNEL-positive cells in all tissues, an effect that was most evident in the lung and spleen. Sham-operated animals on control and zinc-deficient diets did not have any evidence of increased cell death (data not shown).
Figure 5.

Modulation of cellular tissue injury by zinc status. Assessment of cell apoptosis in representative lung (A, B), spleen (C, D), ileal (E), and liver (F) tissues in each of the treatment groups. A consistent trend was observed in all tissues wherein zinc-deficient cecal ligation and puncture (Zn−/CLP) animals demonstrated an increased frequency of transferase-mediated dUTP-biotin nick end labeling-positive cells compared with the control diet CLP (Ctrl/CLP) group and that zinc supplementation (Zn+/CLP) reduced cell death. The extent of cell death was most apparent in lung and spleen tissues. Although a similar pattern was observed in the liver and ileum, the overall number of transferase-mediated dUTP-biotin nick end labeling-positive cells was notably less by comparison (*p < 0.05).
To determine the effects of dietary zinc upon the systemic inflammatory response in sepsis, plasma concentrations of cytokines (IL-1β, IL-6, IL-10, tumor necrosis factor-α), and chemokines (MIP-1α, MIP-1β, MCP-1, RANTES, KC) were measured 24 hours after CLP or sham operation. Overall, zinc deficiency increased the systemic inflammatory response to sepsis when compared with the control diet group. In particular, significant increases in plasma IL-6, IL-10 (Fig. 6), IL-1β, and MIP-1α (Table 1) were observed. Plasma MCP-1, KC, RANTES, and tumor necrosis factor-α levels were also noted to increase, whereas MIP-1β levels remained relatively unchanged (Table 1). Interestingly, zinc-depleted mice receiving acute zinc supplementation before CLP had plasma cytokine and chemokine concentrations comparable to CLP-treated animals on the control diet. With respect to the zinc-deficient diet, there were no differences in plasma cytokine or chemokine levels in the sham-operated animals. Thus, zinc deficiency amplified the systemic inflammatory response during polymicrobial sepsis within 24 hours, and acute zinc supplementation inhibited this response.
Figure 6.

Increased systemic inflammatory response during sepsis in response to zinc deficiency. Comparisons of plasma interleukin (IL)-6 (A) and IL-10 (B) concentrations in each of the treatment groups (n = 5 per dietary group) demonstrated a significant increase in both cytokines 24 hours after cecal ligation and puncture (CLP) in zinc-deficient animals (Zn−/CLP). By contrast, both cytokine levels were significantly reduced, similar to values observed in the control diet CLP (Ctrl/CLP) group, in animals receiving acute zinc supplementation (Zn+/CLP) (*p < 0.05).
Table 1.
Plasma cytokine/chemokine levels 24 hours after CLP in the three dietary group
| Cytokine/Chemokine Concentration (ng/mL) |
|||
|---|---|---|---|
| Cytokine/Chemokine | Zn−/CLP | Control/CLP | Zn+/CLP |
| IL-1β | 0.90 ± 0.24a | 0.47 ± 0.29 | 0.22 ± 0.08 |
| KC | 42.07 ± 10.18 | 20.65 ± 20.84 | 5.33 ± 3.89 |
| MCP-1 | 31.99 ± 7.33 | 17.09 ± 15.15 | 9.31 ± 2.71 |
| MIP-1α | 7.43 ± 1.86a | 3.40 ± 1.87 | 1.82 ± 0.47 |
| MIP-1β | 0.51 ± 0.12 | 0.58 ± 1.05 | 0.12 ± 0.02 |
| RANTES | 3.79 ± 0.87 | 2.12 ± 1.87 | 2.32 ± 0.50 |
| TNFα | 0.52 ± 0.09 | 0.46 ± 0.13 | 0.29 ± 0.09 |
CLP, cecal ligation and puncture; Zn−, a zinc-deficient diet; Zn+, zinc-deficient diet followed by acute zinc supplementation.
p < 0.05, compared with Control/CLP and Zn+/CLP groups.
The effects of zinc status upon total SOD and mitochondrial MnSOD activities were then assessed in snap-frozen lung and liver tissue homogenates (Table 2). Liver total SOD and MnSOD relative activities were significantly reduced in the zinc-deficient group and were partially restored by acute zinc replacement. Although total SOD activity was not different in the lungs in any of the groups, MnSOD activity was significantly lower in the animals fed the zinc-deficient diet. In keeping with the lower levels of SOD activity, tissue concentrations of carbonyl proteins, which reflect irreversible oxidative protein modification, were significantly higher in the lungs and livers of CLP-treated zinc-deficient animals. Acute zinc replacement completely normalized MnSOD activity in the lung, partially restored total and MnSOD activities in the liver, and attenuated protein carbonylation in the lungs and livers of CLP-treated animals.
Table 2.
Relative SOD activity and protein carbonylation in liver and lung tissue homogenates
| Tissue | Diet | Treatment | Total SOD (% of Control Sham) | MnSOD (% of Control Sham) | Protein Carbonylation (% of Control Sham) |
|---|---|---|---|---|---|
| Liver | Ctrl | CLP | 114.5 ± 8.8 | 100.6 ± 3.7 | 101.5 ± 11.8 |
| Sham | 100.0 ± 5.1 | 100.0 ± 6.1 | 100.0 ± 11.8 | ||
| Zn− | CLP | 67.7 ± 8.4a | 69.3 ± 9.1a | 128.3 ± 9.3a | |
| Sham | 67.4 ± 5.7a | 67.0 ± 8.4a | 108.9 ± 8.0 | ||
| Zn+ | CLP | 86.5 ± 3.0b | 97.6 ± 3.6c | 90.1 ± 9.2d | |
| Sham | 83.7 ± 5.8b | 79.9 ± 5.6a | 89.7 ± 5.9d | ||
| Lung | Ctrl | CLP | 112.1 ± 7.3 | 118.9 ± 9.8 | 106.4 ± 11.0 |
| Sham | 100.0 ± 6.7 | 100.0 ± 4.0 | 100.0 ± 9.4 | ||
| Zn− | CLP | 95.5 ± 6.0 | 92.2 ± 9.9a | 146.3 ± 4.7a | |
| Sham | 96.9 ± 7.9 | 86.1 ± 4.9a | 113.5 ± 10.0 | ||
| Zn+ | CLP | 99.7 ± 12.0 | 92.4 ± 12.7 | 93.7 ± 10.7d | |
| Sham | 91.5 ± 5.0 | 93.6 ± 6.5 | 98.7 ± 6.3 |
SOD, superoxide dismutase; Ctrl, Control; CLP, cecal ligation and puncture; Zn+, zinc-deficient diet followed by acute zinc supplementation; Zn−, a zinc-deficient diet.
p < 0.05, compared with matching Ctrl treatment;
p < 0.05, relative to matching Ctrl and Zn− treatments;
p < 0.05, compared with matching Zn− /CLP and corresponding Zn+/Sham;
p < 0.05, relative to matching Zn− treatment.
DISCUSSION
We report that zinc deficiency modifies oxidant status and increases inflammation, cell death, and overall mortality in a conventional murine model of polymicrobial sepsis. These findings represent an extension of previous observations wherein zinc deficiency was shown to increase the likelihood of developing respiratory infections in elderly humans (15) and promoted more invasive lung infection following Streptococcus pneumoniae inoculation, presumably relating to impaired antibacterial barrier (surfactant) defenses (14, 24). The CLP model represents an infection that has eluded typical barriers (e.g., lung or gut epithelium) and is confronted directly by the innate immune system. Based on acute endotoxemia models, one would have predicted a more robust innate immune response in zinc-deficient animals (16, 17), which could have effectively eradicated the infection resulting in a more favorable outcome. However, depletion of tissue antioxidant defenses attendant to zinc deficiency (25) would also promote oxidative organ damage under these conditions. Thus, the findings of these investigations were not self-evident, and this is the first study to show that zinc deficiency increases mortality in a relevant surgical, polymicrobial infection model.
Another novel feature of these investigations is the striking reversal of oxidant-related events, inflammation, and cell death in vital organs and spleens in the septic zinc-deficient mice following short-term oral zinc supplementation. The effect of diminished mortality was significant but remained elevated relative to the septic animals on the control diet. The cause for increased mortality in response to zinc deficiency remains unclear, but likely involves increased organ injury, as reflected by increased inflammation, cell death, and manifestations of oxidant stress in multiple tissues. This investigation extends previous work by evaluating individual target organs involved in MODS and identifying those that are adversely influenced by zinc deficiency. In particular, we observed that the ileum, liver, lung, and spleen had increased cell death. In addition, selective analysis of the liver and lungs revealed significant alterations of SOD activity associated with increased amounts of irreversibly oxidized (carbonylated) proteins. Together, these results suggest that zinc status in the critically ill population contributes to the development of multiple organ damage and consequent loss of function, the putative cause of sepsis mortality (26).
Zinc is a divalent metal cation and micronutrient that is essential for survival. The homeostasis of total body zinc stores in humans is tightly controlled with approximately 1% of total body zinc content replenished daily by dietary intake (6). When intake is low, homeostatic adjustment through tissue redistribution is insufficient to replace zinc losses, and a negative zinc balance occurs within weeks affecting multiple tissue compartments (27). In this study, the level of zinc deficiency achieved in blood samples would be considered mild to moderate. The concomitant alteration in metallothionein levels observed in both the lung and liver of zinc-deficient animals confirmed that zinc stores were likely depleted at the tissue level. Despite this, there were no distinguishable physical differences between zinc-deficient mice and their control diet counterparts before CLP.
The rapid onset of clinical symptoms observed in humans in response to extreme dietary zinc deficiency led to the discovery that in comparison to other metals, such as iron, zinc body stores are extremely labile. It is now realized that mammals primarily use their plasma pool, which represents approximately 10% of total body content, to acquire zinc in times of need (27). Furthermore, sepsis or endotoxin exposure in humans results in a transient decrease in plasma zinc (5) related to intracellular redistribution, which apparently bolsters the host’s barriers to bacterial defenses (28). This mechanism is in keeping with observed increases in the incidence of bacterial pneumonias in zinc-deficient children (29) and adults (15), and reduced rates of skin and pulmonary infections in burn-injured patients in whom deficiencies in zinc and other trace elements were corrected (30). However, these studies linking zinc deficiency to an increase in the incidence of sepsis provide little insight into the role of zinc during established infection wherein the potency of the innate immune response is balanced against the ability of the host to guard against “collateral damage” consequent to the immune response, which ultimately determines the clinical outcome (e.g., organ function, survival) (31).
The biological consequences of zinc deficiency may relate, in part, to its effects on nuclear factor (NF)-κB, a transcription factor central to many of the signaling networks involved in sepsis (32). Otsu et al (33) showed that depletion of zinc augments the DNA-binding activity of NF-κB such that immune responses are magnified, and others have shown that greater NF-κB activity correlates with higher sepsis mortality (34). Therefore, one plausible mechanism for the increased cytokine and chemokine production and higher mortality in the context of zinc deficiency is an increase in NF-κB activity. At higher concentrations, zinc interacts with proximal signaling intermediates thereby suppressing NF-κB activation (35). NF-κB activation is also directly linked to increased MnSOD expression and function (34); however, other variables such as changes in mitochondrial mass or inactivation by peroxynitrite also influence tissue MnSOD activity (36). The latter may account for the observed decrease in MnSOD activity in lung and liver tissues in the zinc-deficient septic mice. Regardless of the mechanism, it is likely that compromised antioxidant defenses and amplification of the immune response as a consequence of zinc deficiency contribute to the accumulation of oxidized proteins, and presumably other cell components (e.g., lipids and DNA) in vital organs.
It is interesting that acute zinc supplementation to reverse zinc deficiency did not completely restore sepsis mortality to that of the septic control diet group. Apparently, the magnitude of zinc supplementation was sufficient, based on normalized plasma zinc levels and tissue metallothionein content, and there were significant benefits in terms of attenuation of systemic cytokine/chemokine release and tissue injury. These findings are consistent with those of Waelput et al (37) who observed potent anti-inflammatory effects of zinc supplementation in a model of tumor necrosis factor-α-induced lethality in mice. However, if altered immune and antioxidant responses were primarily responsible for organ damage and mortality in zinc-deficient septic animals, one would expect that the mortality observed in the zinc-supplemented animals would approximate that of animals on the control zinc diet. The failure of acute zinc repletion to revert sepsis mortality to that observed in control animals implies that our measurements were not sensitive enough to detect relevant changes in immune or antioxidant status or that critical molecular events attendant to zinc deficiency are not readily reversible (38). In this regard, genome-level expression of genes relating to zinc homeostasis were over-represented, and zinc levels were found to be lower in nonsurvivors of pediatric sepsis (39). It is interesting to postulate that changes in zinc concentration influence gene expression in a way that promotes unfavorable sepsis outcomes. Further investigations are needed to fully elucidate the mechanisms by which zinc deficiency predisposes the host to sepsis mortality.
In summary, this study identifies dietary zinc as an important determinant of inflammation, organ damage, and mortality in a relevant model of polymicrobial sepsis (CLP). Zinc deficiency results in the augmentation of systemic and regional inflammatory responses, more severe tissue damage, as reflected by increased cell death and oxidant stress, and increased mortality. Although acute zinc repletion immediately before the onset of sepsis is protective in terms of normalizing the inflammatory response and reducing tissue damage, it only partially improves mortality compared with the animals maintained on the matching control (normal zinc) diet. These findings suggest that the destabilizing effects of zinc depletion are not readily reversible or that the approach to restoring zinc stores must be modified (e.g., longer duration or higher levels of supplementation) to completely normalize cell and organ functions. Whether zinc supplementation offers any benefit after the onset of sepsis in zinc-deficient subjects remains to be determined and is further complicated by challenges inherent in accurately measuring and correcting zinc nutrient status in humans during acute illness (40). In view of the prevalence of zinc deficiency in the adult population, particularly in those with comorbid disease (e.g., alcoholism) and to the extent that the rodent model is relevant to humans, these investigations have important implications for influencing sepsis outcomes in humans by identifying zinc deficiency and replenishing zinc stores in those who are at high risk for sepsis.
Acknowledgments
We thank Louise Fong and Elizabeth Josephs for assistance with the modification of zinc nutrition in the CLP model, Timothy Eubanks for assistance in data interpretation and presentation, and Gary Phillips for assistance with statistical analyses.
Supported by NIH RO3-AI62740 (to EDC) and HL R01 HL086981-01 (to DLK).
Footnotes
The authors have not disclosed any potential conflicts of interest.
References
- 1.Alberti C, Brun-Buisson C, Goodman SV, et al. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med. 2003;168:77– 84. doi: 10.1164/rccm.200208-785OC. [DOI] [PubMed] [Google Scholar]
- 2.Guidet B, Aegerter P, Gauzit R, et al. Incidence and impact of organ dysfunctions associated with sepsis. Chest. 2005;127:942–951. doi: 10.1378/chest.127.3.942. [DOI] [PubMed] [Google Scholar]
- 3.Moss M. Epidemiology of sepsis: Race, sex, and chronic alcohol abuse. Clin Infect Dis. 2005;41:S490 –S497. doi: 10.1086/432003. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT, Sandstead HH, Prasad AS, et al. Zinc: Health effects and research priorities for the 1990s. Environ Health Perspect. 1994;102:5– 46. doi: 10.1289/ehp.941025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaetke LM, McClain CJ, Talwalkar RT, et al. Effects of endotoxin on zinc metabolism in human volunteers. Am J Physiol. 1997;272:E952–E956. doi: 10.1152/ajpendo.1997.272.6.E952. [DOI] [PubMed] [Google Scholar]
- 6.Cousins RJ, Liuzzi JP, Lichten LA. Mammalian zinc transport, trafficking, and signals. J Biol Chem. 2006;281:24085–24089. doi: 10.1074/jbc.R600011200. [DOI] [PubMed] [Google Scholar]
- 7.Goode HF, Penn ND, Kelleher J, et al. Evidence of cellular zinc depletion in hospitalized but not in healthy elderly subjects. Age Ageing. 1991;20:345–348. doi: 10.1093/ageing/20.5.345. [DOI] [PubMed] [Google Scholar]
- 8.Schmuck A, Roussel AM, Arnaud J, et al. Analyzed dietary intakes, plasma concentrations of zinc, copper, and selenium, and related antioxidant enzyme activities in hospitalized elderly women. J Am Coll Nutr. 1996;15:462– 468. doi: 10.1080/07315724.1996.10718625. [DOI] [PubMed] [Google Scholar]
- 9.Galan P, Viteri FE, Bertrais S, et al. Serum concentrations of beta-carotene, vitamins C and E, zinc and selenium are influenced by sex, age, diet, smoking status, alcohol consumption and corpulence in a general French adult population. Eur J Clin Nutr. 2005;59:1181–1190. doi: 10.1038/sj.ejcn.1602230. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien JM, Jr, Lu B, Ali NA, et al. Alcohol dependence is independently associated with sepsis, septic shock, and hospital mortality among adult intensive care unit patients. Crit Care Med. 2007;35:345–350. doi: 10.1097/01.CCM.0000254340.91644.B2. [DOI] [PubMed] [Google Scholar]
- 11.Joshi PC, Guidot DM. The alcoholic lung: Epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol. 2007;292:L813–L823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- 12.Berger MM, Cavadini C, Chiolero R, et al. Copper, selenium, and zinc status and balances after major trauma. J Trauma. 1996;40:103–109. doi: 10.1097/00005373-199601000-00019. [DOI] [PubMed] [Google Scholar]
- 13.Voruganti VS, Klein GL, Lu HX, et al. Impaired zinc and copper status in children with burn injuries: Need to reassess nutritional requirements. Burns. 2005;31:711–716. doi: 10.1016/j.burns.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 14.Strand TA, Briles DE, Gjessing HK, et al. Pneumococcal pulmonary infection, septicaemia and survival in young zinc-depleted mice. Br J Nutr. 2001;86:301–306. doi: 10.1079/bjn2001399. [DOI] [PubMed] [Google Scholar]
- 15.Prasad AS, Beck FW, Bao B, et al. Zinc supplementation decreases incidence of infections in the elderly: Effect of zinc on generation of cytokines and oxidative stress. Am J Clin Nutr. 2007;85:837– 844. doi: 10.1093/ajcn/85.3.837. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Wang L, Song Z, et al. Abrogation of nuclear factor-kappaB activation is involved in zinc inhibition of lipopolysaccharide-induced tumor necrosis factor-alpha production and liver injury. Am J Pathol. 2004;164:1547–1556. doi: 10.1016/s0002-9440(10)63713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shea-Budgell M, Dojka M, Nimmo M, et al. Marginal zinc deficiency increased the susceptibility to acute lipopolysaccharide-induced liver injury in rats. Exp Biol Med (Maywood) 2006;231:553–558. doi: 10.1177/153537020623100509. [DOI] [PubMed] [Google Scholar]
- 18.Beutler B, Poltorak A. Sepsis and evolution of the innate immune response. Crit Care Med. 2001;29:S2–S6. doi: 10.1097/00003246-200107001-00002. [DOI] [PubMed] [Google Scholar]
- 19.Scheuhammer AM, Cherian MG. Quantification of metallothionein by silver saturation. Methods Enzymol. 1991;205:78 – 83. doi: 10.1016/0076-6879(91)05088-d. [DOI] [PubMed] [Google Scholar]
- 20.Crouser ED, Julian MW, Huff JE, et al. Carbamoyl phosphate synthase-1: A marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med. 2006;34:2439 –2446. doi: 10.1097/01.CCM.0000230240.02216.21. [DOI] [PubMed] [Google Scholar]
- 21.Hardie WD, Korfhagen TR, Sartor MA, et al. Genomic profile of matrix and vasculature remodeling in TGF-alpha induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2007;37:309 –321. doi: 10.1165/rcmb.2006-0455OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beauchamp C, Fridovich I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276 –287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 23.Levine RL, Garland D, Oliver CN, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464 – 478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 24.Strand TA, Hollingshead SK, Julshamn K, et al. Effects of zinc deficiency and pneumococcal surface protein A immunization on zinc status and the risk of severe infection in mice. Infect Immun. 2003;71:2009 –2013. doi: 10.1128/IAI.71.4.2009-2013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jing MY, Sun JY, Zi NT, et al. Effects of zinc on hepatic antioxidant systems and the mRNA expression levels assayed by cDNA microarrays in rats. Ann Nutr Metab. 2007;51:345–351. doi: 10.1159/000107677. [DOI] [PubMed] [Google Scholar]
- 26.Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546 –1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 27.King JC, Shames DM, Woodhouse LR. Zinc homeostasis in humans. J Nutr. 2000;130:1360S–1366S. doi: 10.1093/jn/130.5.1360S. [DOI] [PubMed] [Google Scholar]
- 28.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–266. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 29.Brooks WA, Santosham M, Naheed A, et al. Effect of weekly zinc supplements on incidence of pneumonia and diarrhoea in children younger than 2 years in an urban, low-income population in Bangladesh: Randomised controlled trial. Lancet. 2005;366:999 –1004. doi: 10.1016/S0140-6736(05)67109-7. [DOI] [PubMed] [Google Scholar]
- 30.Berger MM, Baines M, Raffoul W, et al. Trace element supplementation after major burns modulates antioxidant status and clinical course by way of increased tissue trace element concentrations. Am J Clin Nutr. 2007;85:1293–1300. doi: 10.1093/ajcn/85.5.1293. [DOI] [PubMed] [Google Scholar]
- 31.Crouser E, Exline M, Knoell D, et al. Sepsis: Links between pathogen sensing and organ damage. Curr Pharm Des. 2008;14:1840–1852. doi: 10.2174/138161208784980572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 33.Otsu K, Ikeda Y, Fujii J. Accumulation of manganese superoxide dismutase under metal-depleted conditions: Proposed role for zinc ions in cellular redox balance. Biochem J. 2004;377:241–248. doi: 10.1042/BJ20030935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abraham E. Alterations in cell signaling in sepsis. Clin Infect Dis. 2005;41:S459 –S464. doi: 10.1086/431997. [DOI] [PubMed] [Google Scholar]
- 35.von Bulow V, Dubben S, Engelhardt G, et al. Zinc-dependent suppression of TNF-alpha production is mediated by protein kinase A-induced inhibition of Raf-1, I kappa B kinase beta, and NF-kappa B. J Immunol. 2007;179:4180 – 4186. doi: 10.4049/jimmunol.179.6.4180. [DOI] [PubMed] [Google Scholar]
- 36.Muscoli C, Cuzzocrea S, Ndengele MM, et al. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530 –3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waelput W, Broekaert D, Vandekerckhove J, et al. A mediator role for metallothionein in tumor necrosis factor-induced lethal shock. J Exp Med. 2001;194:1617–1624. doi: 10.1084/jem.194.11.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haase H, Mazzatti DJ, White A, et al. Differential gene expression after zinc supplementation and deprivation in human leukocyte subsets. Mol Med. 2007;13:362–370. doi: 10.2119/2007-00049.Haase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong HR, Shanley TP, Sakthivel B, et al. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007;30:146 –155. doi: 10.1152/physiolgenomics.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowe NM. In search of a reliable marker of zinc status—Are we nearly there yet? Nutrition. 2005;21:883– 884. doi: 10.1016/j.nut.2004.12.011. [DOI] [PubMed] [Google Scholar]