

Abstract
Thirteen novel seco-DCK analogs (4–16) with several new skeletons were designed, synthesized and screened for in vitro anti-HIV-1 activity. Among them, three compounds (5, 13, and 16) showed moderate activity, and compound 9 exhibited the best activity with an EC50 value of 0.058 μM and a therapeutic index (TI) of 1000. The activity of 9 was better than that of 4-methyl DCK (2, EC50: 0.126 μM, TI: 301.2) in the same assay. Additionally, 9 also showed antiviral activity against a multi-RT inhibitor-resistant strain (RTMDR), which is insensitive to most DCK analogs. Compared with 2, compound 9 has a less complex structure, fewer hydrogen-bond acceptors, and a reduced log P value. Therefore, it is likely to exhibit better ADME, and appears to be a promising new lead for further development as an anti-HIV candidate.
Keywords: Seco-DCK analogs, Anti-HIV agents
1. Introduction
In our prior studies, 3′R,4′R-di-O-(−)-camphanoyl-(+)-cis-khellactone (DCK, 1) and 4-methyl DCK (2) showed remarkable potency against human immunodeficiency virus type 1 (HIV-1) replication in H9 lymphocytes.1, 2 Subsequent modifications of DCK focused on the coumarin substituents, ring isomers, and bioisosteric replacements.3 Hundreds of 1-derivatives were designed and synthesized, and the resulting data led to the following structure-activity relationship (SAR) conclusions. (a) A 4-methyl is favorable for enhancing anti-HIV activity and decreasing toxicity.2 (b) Certain bioisosteric DCK analogs, including 1-thia, 1-aza, 1′-thia and 1′-carbon DCKs, show comparable anti-HIV activity with 1.4–7 (c) Two bulky cis-(S)-(−)-camphanoyl groups at the 3′- and 4′-positions are preferred to other substituents; however, both of them are not essential. The 4′R-camphanoyl is more important.8 (d) gem-Dimethyl substitution at the 2′-position is greatly preferable to larger alkyl substituents or hydrogen atoms.9 (e) 3′R,4′R-Di-O-(−)-camphanoyl-2′,2′-dimethyl-dihydropyrano[2,3-f]chromone (DCP) analogs (such as 3), which are positional ring isomers, also exhibit significant anti-HIV activity.8, 10 However, development of 1-analogs as effective anti-AIDS drugs has been hindered by problems of low water solubility and bioavailability. DCP analogs, with the carbonyl group positioned differently in ring-A, retained high potency against both non-drug resistant and multi-RT-inhibitor-resistant viral strains. This result motivated us to change the skeleton of 1 by opening the A or C ring, which may represent a new break-through in developing DCK analogs as clinical trial candidates.
Therefore, in the current study, we explored a series of simplified DCK analogs with an opened A or C ring, termed seco-DCKs. With only two rings, seco-DCK compounds have less complex structures, fewer hydrogen-bond acceptors, and reduced log P values, which may result in better ADME than those of 2. Herein, we report the design, synthesis, and bio-evaluation of these new DCK analog series.
2. Results
2.1. Design
Compound 4, the first A ring-opened derivative (Figure 2) synthesized, lacks certain structural features found in the A ring of 1–3, such as a phenoxy oxygen at position-1. Therefore, we also designed compound 5 (Figure 2), which contains an ethoxy group that can freely rotate to adopt an optimal position and orientation for binding in the target site, as well as an acetyl moiety that can correspond to both the 4-oxo moiety of 3 and the 4-methyl group of 2.
Figure 2.

Structures of newly designed seco-DCK analogs 4–16.
In addition, to investigate whether the integrity of the C ring impacts antiviral activity, we further designed several additional seco-C ring DCKs, including 4′,8-seco (6), 2′,3′-seco (7) and bis (2′,3′- and 3′,4′-) seco DCK (8–12) analogs (Figure 2). Although both bulky camphanoyl moieties are retained in 6 and 7, their spatial positions and 3D directions could be changed significantly from those of 1 due to the absence of the C ring. Only the 4′-camphanoyl moiety is retained in 8–12, because prior SAR demonstrated the importance of this moiety.8 In addition, 9–12 completely lack a 3′-carbon, while 8 has a methyl group at the corresponding position of the seco-C ring.
In addition, previous bioisosteric replacement study demonstrated that 1-thia, 1′-thia, and 1-aza DCKs maintained potent antiviral activity or had better activity against HIV compared with 2.4,5,7 These results motivated us to synthesize compounds 13–16 with sulfur or nitrogen rather than oxygen linked to the camphanoyl group of the seco-DCKs
2.2. Chemistry
Scheme 1 describes the synthesis of compound 4. Commercially available 17 was reacted with 3-chloro-3-methyl-1-butyne in DMF in the presence of K2CO3 and KI to afford 18. The crude product was heated in refluxing N,N-diethylaniline to yield 19 with a pyrano C ring. Compound 19 was asymmetrically dihydroxylated using (DHQ)2-PHAL as a chiral catalyst to afford corresponding 3′R,4′R-dihydroxy compound 20. Compound 20 was then reacted with (−)-(S)-camphanic chloride at room temperature for 2 days to obtain the target compound 4.
Scheme 1.

Synthesis of seco-A ring DCK (4). Reagents and conditions: (i) 3-chloro-3-methyl-1-butyne, K2CO3, KI; (ii) N,N-diethylaniline; (iii) K2OsO2(OH)4, K3Fe(CN)6, (DHQ)2-PHAL, K2CO3; (iv) camphanoyl chloride, DMAP, CH2Cl2.
Compound 5 was synthesized from resorcinol via eight steps as shown in Scheme 2. Treatment of resorcinol 21 with ZnCl2-HOAc afforded a highly regioselective acetylation product, 1-(2,4-dihydroxyphenyl)ethanone (22). Selective protection of the 4-hydroxy group with benzyl bromide (23) and ethylation of the 2-hydroxy group with bromoethane gave 1-[2-ethoxy-4-(phenylmethoxy)phenyl]ethanone (24), which then was reacted with BBr3 in CH2Cl2 to yield 1-(2-ethoxy-4-hydroxyphenyl)ethanone (25). Compound 26 was prepared by reaction of 25 with 3-chloro-3-methyl-1-butyne. This crude product (26) directly underwent thermal ring closure upon refluxing in DMF and N,N-diethylaniline to afford the desired key intermediate 27 as the main product. After Sharpless asymmetric dihydroxylation (AD) of 27, followed by esterification of diol 28 with camphanic chloride, target compound 5 was obtained.
Scheme 2.

Synthesis of seco-A ring DCK (5). Reagents and conditions: (i) ZnCl2, HOAc, boiling, 30 min; (ii) K2CO3, KI, BnCl, acetone, reflux, 8h; (iii) CH3CH2Br, K2CO3, acetone, reflux; (iv) BBr3, CH2Cl2, −40 °C; (v) 3-chloro-3-methyl-1-butyne, K2CO3, KI, acetone, reflux; (vi) DMF, N,N-diethylaniline, reflux; (vii) K2OsO2(OH)4, K3Fe(CN)6, (DHQ)2-PHAL, n-BuOH, H2O, 0 °C; (viii) camphanoyl chloride, DMAP, CH2Cl2.
Scheme 3 illustrates the syntheses of C ring-opened compounds 6–9 and 13. 4′,8-Seco DCK (6) was prepared by alkylation of commercially available 4-methyl-7-hydroxycoumarin (29) with 3-chloro-3-methyl-1-butene, followed by Sharpless AD of 30 and esterification of the resulting diol with camphanic chloride (31). Key intermediates (32a and 32b) in the synthetic route to 2′,3′-seco and bis (2′,3′- and 3′,4′-) seco DCKs (7–9) were obtained from 29 by Duff formylation (32a) or Fries rearrangement (32b), respectively. After alkylation of the phenolic hydroxyl in 32 with isopropyl bromide, the carbonyl in 33 was reduced to a hydroxyl with NaBH4. Target compounds 8 and 9 were obtained by esterification of 34b and 34a with camphanic chloride, respectively. Dehydration of 34b with conc. H2SO4, followed by AD and esterification gave target compound 7. Thioester 13 was synthesized by treatment of 34a with Lawesson reagent, and esterification with camphanic chloride.
Scheme 3.

Synthetic routes to seco-C ring DCKs (6–9) and bioisosteric seco DCK 13. Reagents and conditions: (i) 3-chloro-3-methyl-1-butene, K2CO3, KI, acetone, reflux; (ii) K2OsO2(OH)4, K3Fe(CN)6, (DHQ)2-PHAL, n-BuOH, H2O, 0 °C; (iii) camphanoyl chloride, DMAP, CH2Cl2; (iv) for 32a: hexamethylene tetramine, HOAc, 90 °C; (v) for 32b: (1) Py/Ac2O, 5 h; (2) AlCl3, 160 °C; (vi) isopropyl bromide, NaH, DMF, reflux; (vii) NaBH4, MeOH; (viii) conc. H2SO4, acetone; (viiii) Lawesson reagent/N2/toluene, rt, 20h.
Scheme 4 shows the synthetic pathway to compounds 10–12. Compound 38 was obtained from 32a by a condensation reaction with ethane-1,2-diol in a toluene solution containing p-toluenesulfonic acid. Intermediates 39a–c were obtained from 38 by alkylation with various halides in the presence of K2CO3 and KI in acetone, followed by deprotection of the ketal moiety under acidic conditions to afford 40a–c. Compounds 40a–c were reduced to 41a–c, which were then esterified with camphanic chloride to yield target compounds 10–12.
Scheme 4.

Synthetic routes to seco-C ring DCKs (10–12). Reagents and conditions: (i) HOCH2CH2OH/TsOH/toluene; (ii) a: CH3I/b: CH3CH2Br/c: BrCH2COOCH3, K2CO3, KI, acetone, reflux; (iii) 2N HCl; (iv) NaBH4, MeOH; (v) camphanic chloride, DMAP, CH2Cl2.
The preparations of compounds 14–16 are depicted in Scheme 5. Compound 42 was synthesized in a one-pot reaction by amination of 33a with (NH4)2CO3 in EtOH, followed by reduction in the presence of NaBH4 in the same solution. Imines 43 and 45 were provided by reaction of 33a with methylamine and 4-methoxyaniline, respectively, and reduced to give 44 and 46. Three target compounds 14–16 were obtained from the amidation of 42, 44, and 46, respectively, with camphanic chloride.
Scheme 5.

Synthesis of bioisosteric seco-DCKs (14–16). Reagents and conditions: (ia) a. (NH4)2CO3/EtOH/reflux, b. NaBH4; (ib) CH3NH2, EtOH, rt; (ic) p-CH3C6H4NH2, EtOH, rt; (ii) NaBH4, MeOH; (iii) camphanic chloride, DMAP, CH2Cl2.
3. Discussion
Target seco-DCK analogs 4–9 were evaluated for in vitro suppression of HIV-1IIIB replication in the MT-2 cell line in parallel with AZT and 2. Compounds 6–9 were further tested against HIV-1NL4-3 and RTMDR, respectively, in MT-4 lymphocytes with 3 as reference. In addition, compounds 10–16 were screened for antiviral activity against HIV-1NL4-3 with 9 as control. Due to the protocol differences in the assay systems, 9 showed variable activities in different screenings. Nevertheless, 9 still showed the most promising potency in the above assays, especially good activity against RTMDR. All bioassay data are summarized in Tables 1, 2, and 3.
Table 1.
Anti-HIV-1IIIB data of seco-DCK analogs (4–9) in MT-2 cell linea
| Compd | IC50 (μM) | EC50 (μM) | TI |
|---|---|---|---|
| 4 | 39.6 | NS | |
| 5 | 39.6 | 0.19 | 208.3 |
| 6 | 39.2 | NS | |
| 7 | 39.2 | NS | |
| 8 | 56.5 | NS | |
| 9 | 58.4 | 0.058 | 1000 |
| 2 | 39.2 | 0.126 | 301.2 |
| AZT | 1872 | 0.044 | 42700 |
All data presented are averages of at least three separate experiments performed by Panacos Pharmaceutical Inc., Gaithersburg, MD.
CC50: concentration that inhibits uninfected H9 cell growth by 50%. EC50: concentration that inhibits viral replication by 50%. TI = therapeutic index IC50/EC50. NS: no suppression at the highest tested concentration (20 μM).
Table 2.
Anti-HIV data of seco-DCK analogs (7–10) in MT-4 cell linea
| Compd | IC50 (μM) | HIV-1NL4-3 | RTMDR | ||
|---|---|---|---|---|---|
| EC50 (μM) | TI | EC50 (μM) | TI | ||
| 6 | 20 | 20 | NS | 20 | NS |
| 7 | 20 | 20 | NS | 20 | NS |
| 8 | 20 | 20 | NS | 20 | NS |
| 9 | 17.27 | 0.5 | 34.8 | 1.89 | 9.13 |
| 3 | 9.27 | 0.08 | 116 | 0.133 | 69.9 |
All data presented are averages of at least three separate experiments performed by Dr. Chin-Ho Chen, Duke University, NC.
CC50: concentration that inhibits uninfected MT-4 cell growth by 50%. EC50: concentration that inhibits viral replication by 50%. TI = therapeutic index IC50/EC50. NS: no suppression at the highest tested concentration (20 μM).
Table 3.
Anti-HIV-1NL4-3 data of seco-DCK analogs (10–16) in MT-4 cell linea
| Compd | IC50 (μM) | EC50 (μM) | TI |
|---|---|---|---|
| 10 | 62.4 | 36.61 | 1.71 |
| 11 | 60.3 | 32.3 | 1.87 |
| 12 | 54.5 | 44.65 | 1.22 |
| 13 | 56.24 | 4.67 | 12.04 |
| 14 | 25 | 25 | 1 |
| 15 | 56.62 | 5.66 | 8 |
| 16 | 34.72 | 4.69 | 15 |
| 9 | 44.7 | 1.98 | 22.53 |
All data presented are averages of at least three separate experiments performed by Dr. Chin-Ho Chen, Duke University, NC.
CC50: concentration that inhibits uninfected MT-4 cell growth by 50%. EC50: concentration that inhibits viral replication by 50%. TI = therapeutic index IC50/EC50. NS: no suppression at the highest tested concentration (20 μM).
Initially, we synthesized compound 4. However, it did not show anti-HIV activity, which implied certain structural features present in the A ring of 2 or 3, such as the polar oxygen center at position-1, but absent in the seco-A ring of 4, are necessary for activity. Therefore, in compound 5, an ethoxy moiety was incorporated at this position on the phenyl ring. As we expected, the newly designed compound 5, which has both ether and carbonyl groups in its seco-A ring, exhibited moderate antiviral activity with an EC50 value of 0.19 μM and a TI of 208. This result indicates that, with proper substitutions in the seco-A ring, a modified bicyclic system with only the coumarin B and C rings can retain moderate antiviral activity.
Seco-C ring analogs 6 (4′,8-seco) and 7 (2′,3′-seco) contain two bulky camphanoyl moieties, but were not active in the anti-HIV screening. These data indicated that the orientation of the two camphanoyls is important to the antiviral potency. In 1 and 2, the 3′R,4′R-dicamphanoyl groups are constrained close to each other because of the rigid C ring, which benefits the anti-HIV activity. However, in C ring-opened 6 and 7, the two bulky groups can rotate more freely and orient differently from each other and the rest of the molecule, leading to abolished activity. This observation is consistent with our prior SAR research that only 3′R,4′R-configured DCK analogs were active; the remaining three 3′,4′-diastereoisomers were inactive.8 These results confirm that proper spatial orientations of the camphanoyl groups and the remainder of the molecule are necessary to achieve a proper “fit” into the putative viral binding site.
Seco-C ring DCK analog 9 (bis 2′,3′ and 3′,4′) exhibited the best anti-HIV activity among the newly designed compounds with an EC50 value of 0.058 μM and TI of 1000, and was threefold more potent than 2 (EC50: 0.126 μM, TI: 301.2) in the same antiviral replication screening against HIV-1IIIB. Moreover, 9 also showed marginal activity against the RTMDR strain with EC50 of 1.89 μM and TI of 9.13, using 3 (EC50: 0.133μM and TI: 69.9) as control. Notably, 9 is the first DCK analog to exhibit anti-HIV replication activity against a multi-drug resistant viral strain. The removal of one camphanoyl moiety, with retention of good anti HIV activity, is important, as the metabolic instability of 2 and its derivatives is a consequence of 3′- and 4-acetoxy camphanoyl ester cleavage on microsomes.11 Interestingly, compound 8 lost anti-HIV activity completely in the same assay, even though 8 and 9 differ only by the presence (8) or absence (9) of a 4′-methyl group. The 3D structures of 2, 8, and 9 were generated using Sybyl (version 7.3) in the standard energy minimizing condition to predict the orientation of the 4′-camphanoyl group (Figure 3). The models clearly showed that the orientation of the 4′-camphanoyl in 9 is more similar to that in 2, while the orientation of this group in both stereoisomers of 8 is very different, perhaps due to repulsion caused by the 4′-methyl group. These results further confirmed that proper interaction of the 4′-camphanoyl group with the viral target is essential to the anti-HIV potency of DCK analogs. Compound 9, with only one camphanoyl group, has a less complex structure, fewer hydrogen-bond acceptors, and potentially better ADME than 2. It also has a reduced log P value (3.81), which is one log unit lower than that of 2 (log P: 4.96) (calculated by ACD/LogP DB software).
Figure 3.
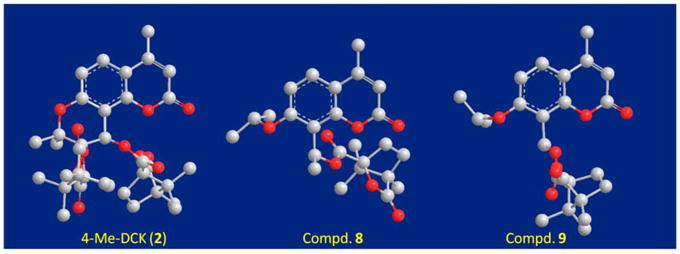
3D structures of 2, 8 and 9 by Sybyl (version 7.3) in the standard energy minimizing condition.
The promising seco-DCK hit compound 9 was further modified to build some initial SAR. The 7-isopropoxy group was changed to other alkoxy groups and the 4′-oxygen of 9 was changed to sulfur or nitrogen. The resulting analogs 10–16 were screened and compared with 9. From the data (Table 1 to Table 3), it can be seen that, although the activity of 9 varied due to the assay protocol change, it still showed the best anti-HIV potency among the newly synthesized compounds. Reducing the size of the 7-alkyl ether, as in compounds 10 (methyl) and 11 (ethyl), or enlarging it, as in 12 (methoxycarbonylmethyl), decreased the antiviral potency of the compounds (EC50 values 36.61, 32.3, and 44.65 μM, respectively). These results indicate that the substituent size at the 7-position is critical to the compound’s anti-HIV activity, with an isopropoxy moiety (as in 9) being optimal. The data also confirm our previous SAR conclusion, based on DCK analogs, that there is an important region of the binding site corresponding to the 2′- and 8-position of DCKs for viral target interaction.9
Compounds 13–16 with sulfur or nitrogen rather than oxygen attached to the camphanoyl group were also synthesized, because bioisosteric replacement is a common medicinal chemistry approach. Compounds 13 and 16 exhibited moderate anti-HIV activity with EC50 values of 4.67 and 4.69 μM, respectively, indicating that the antiviral activity of 9 can be retained upon heteroatom replacement. The inhibitory potency increase from 14 to 16, which corresponded to the enlarging size of the nitrogen substituent (H, methyl, p-methoxyphenyl, respectively), suggested that a bigger moiety is favored to interact with the binding pocket.
In summary, data from our newly designed seco-DCKs led to the following SAR conclusions. (a) Integrity of the A ring is not essential for anti-HIV activity; however, both ether (1-oxy) and carbonyl (4-oxo) features are needed in the seco-A ring. (b) One 4′-camphanoyl moiety is sufficient for anti-HIV activity of DCK analogs, and the 3′-position can be deleted completely. (c) A proper spatial orientation of the 4′-camphanoyl with respect to the rest of the molecule is critical for enhanced anti-HIV activity. (d) A suitably sized alkoxy group at position-7 is important for enhanced anti-HIV activity. (e) Converting the 4′-oxygen to sulfur or nitrogen can result in retained inhibitory activity, and a bulkier substituent on nitrogen is preferred.
Bis (2′,3′- and 3′,4′-) seco modification in the DCK series opens a significant new research area. Not only can such analogs exhibit enhanced activity against wild-type HIV strain while gaining some potency against RTMDR strain, they can also be synthesized and isolated more easily, due to their simplified structure and deletion of a chiral center. In addition, they have fewer hydrogen-bond acceptors and lower log P values, and may have better ADME.
4. Experimental procedures
4.1. Materials
All reagents and solvents used were analytical grade. Melting points were determined in open capillary tubes and are uncorrected. 1H and 13C NMR spectra were recorded in a Bruker-DPX 400 MHz. Mass spectra were measured with HP5973N analytical mass spectrometers. High resolution mass spectra (HRMS) were measured on a Shimadzu LCMS-IT-TOF with ESI interface. HPLC for purity determinations were conducted using a Shimadzu LCMS-2010 with a Grace Alltima 2.1 mm × 150mm HP C18 5μM column and Shimadzu SPD-M20A detector at 254 nm wavelength. HPLC purity analyses were determined by using two different solvent conditions. The first was 70% MeCN as solvent A and 30% H2O as solvent B with 0.2 mL/min flow rate; the second was 80% MeOH as solvent A and 20% H2O as solvent B with 0.1 mL/min flow rate. The HPLC model was an isocratic system. All target compounds had purity greater than 95%. Thin-layer chromatography (TLC) was performed on PLC silica gel 60 F254 plates. Commercially available silica gel H was used for column chromatography.
4.2. Synthesis of seco-A ring DCK (4)
4.2.1. 2,2,5-Trimethyl-6-acetyl-2H-chromene (19)
Commercially available 17, K2CO3 (2.5 equiv), KI (1 equiv), and excess 3-chloro-3-methyl-1-butyne (2~3 equiv) in dry DMF were heated to 70–80 °C with stirring until the reaction was complete as monitored by TLC. After filtering the solid, the filtrate was concentrated in vacuo. The residue 18, without purification, was directly heated to reflux in 10 mL of N,N-diethylaniline for 4–6 h. The reaction mixture was cooled to rt, diluted with EtOAc, and washed with 10% aqueous HCl, water, and brine. The organic layer was separated and concentrated. The residue was purified by column chromatography with an eluant of hexane:EtOAc = 5:1 to afford intermediate 19 as a brown oil; 60% yield (starting with 450 mg of 17); 1H NMR δ 7.47 (1H, d, J = 8.4 Hz, H-7), 6.40 (1H, d, J = 8.4 Hz, H-8), 6.60 (1H, d, J = 10.2 Hz, H-4), 5.69 (1H, d, J = 10.2 Hz, H-3), 2.50 (3H, s, -COCH3), 2.49 (3H, s, CH3-5), 1.40 (6H, s, 2×CH3-2).
4.2.1. 3R,4R-Dihydroxy-2,2,5-trimethyl-6-acetyl-chroman (20)
A mixture of K3Fe(CN)6 (3 equiv), K2CO3 (3 equiv), (DHQ)2-PYR or (DHQ)2PHAL (2% equiv), and K2OsO2(OH)4 (2% equiv) was dissolved in t-BuOH/H2O (v/v, 1:1) at rt. The solution was cooled to 0 °C and methanesulfonamide (1 equiv) was added with stirring. When the solution turned from light yellow to orange, 19 was added. The mixture was stirred for 1–2 days at 0°C and monitored by TLC. At completion, Na2S2O5 (excess), water, and CHCl3 were added, and stirring continued 0.5 h at rt. The mixture was extracted with CHCl3 three times, the combined organic layer was dried over K2SO4, and then solvent was removed. The residue was separated by column chromatography (hexane:EtOAc = 3:1) to afford pure dihydroxy compound with 58% yield (starting with 108 mg of 19). mp 106–108°C; 1H NMR δ 7.53 (1H, d, J = 8.5 Hz, H-7), 6.69 (1H, d, J = 8.5 Hz, H-8), 4.84 (1H, d, J = 4.8 Hz, H-4), 3.75 (1H, d, J = 4.8 Hz, H-3), 2.54 (3H, s, COCH3-6), 2.50 (3H, s, CH3-5), 1.38, 1.36 (3H each, s, CH3-2).
4.2.1. 3R,4R-Dicamphanoyloxy-2,2,5-trimethyl-6-acetyl-2H-chroman (4)
Compound 20, (S)-(−)-camphanic chloride (3 equiv), and DMAP (0.1~1 equiv) were reacted 1–2 days in CH2Cl2 at rt and monitored by TLC. At completion, the reaction mixture was concentrated and the residue was purified by PTLC with an eluant of hexane/EtOAc = 3:1 to afford the final compound 4 with yield 39% (starting from 55 mg of 20); mp 88–90 °C; MS-ESI+ (m/z, %): 628 (M++NH4+, 100); 1H NMR δ 7.66 (1H, d, J = 8.7 Hz, H-7), 7.77 (1H, d, J = 8.7 Hz, H-8), 6.43 (1H, d, J = 4.8 Hz, H-4), 5.30 (1H, d, J = 4.8 Hz, H-3), 2.52 (3H, s, COCH3-6), 2.29 (3H, s, CH3-5), 2.42, 2.17, 1.90, 1.68 (2H each, m, camphanoyl CH2), 1.43, 1.39 (3H each, s, CH3-2), 1.08, 1.07, 1.05, 1.04, 0.97, 0.93 (3H each, camphanoyl CH3).
4.3. Synthesis of seco-A ring DCK (5)
4.3.1. 1-(5-Ethoxy-2,2-dimethyl-2H-chromen-6-yl)ethanone (27)
Intermediate 26 is a known compound and was prepared from resorcinol as shown in Scheme 2. The crude product 26 (1.3 g, 5.3 mmol) was added to DMF/N,N-diethylaniline (15 mL/1 mL), and the mixture heated at reflux under nitrogen for 5 h to give 27 (1.2 g) as a colorless oil in 92% yield. 1H NMR δ 1.39 (t, 3H, 5-CH3), 1.41 (s, 6H, 2×-CH3), 2.58 (s, 3H, 6-CH3), 3.89 (q, 2H, 5-CH2), 5.66 (d, J = 10.17 Hz, 1H, 3-H), 6.58 (d, J = 10.17 Hz, 1H, 4-H), 6.58 (d, J = 8.22 Hz, 1H, 7-H), 7.54 (d, J = 8.61 Hz, 1H, 8-H). MS-EI+ (m/z, %): 246 (M+, 21.92) 231 (100) 203 (58.19) 149 (39.83).
4.3.2. 1-[(3R,4R)-Dihydroxy-5-ethoxy-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl]-ethanone (28)
A mixture of K3Fe(CN)6 (5 equiv), K2CO3 (5 equiv), (DHQ)2PHAL (5% equiv), and K2OsO2(OH)4 (5% equiv) in t-BuOH/H2O (v/v, 1:1) was cooled to −5–0 °C. Under stirring, compound 27 (268 mg, 1.01 mmol) was added. The mixture was stirred for 14 h at 0 °C, and then the reaction temperature was gradually raised to rt over 24 h. When TLC monitoring showed the reaction was completed, an excess of aqueous Na2S2O3 was added and stirring continued for 0.5 h. The mixture was extracted with EtOAc, the combined organic layer was dried over Na2SO4, and the solvent was removed. The residue was purified by chromatography on a silica column with petroleum ether: acetone = 10:1 as eluent to afford pure 28 as a clear oil (119 mg, 39%). 1H NMR δ 1.32 (s, 3H, 2-CH3), 1.48 (s, 3H, 2-CH3), 1.48 (t, 3H, 5-CH3), 2.58 (s, 3H, 6-CH3), 3.16 (d, J = 3.62 Hz, 1H, 3-CH), 3.81 (dd, J = 4.63 3.63 Hz, 1H, 4-CH2), 3.94 (m, 1H, 5-CH2), 4.13 (m, 1H, 5-CH2), 4.35 (s, 1H, 3-OH), 5.04 (d, J = 4.70 Hz, 1H, 4-OH), 6.67 (d, J = 8.96 Hz, 1H, 4-H), 7.59 (d, J = 8.75 Hz, 1H, 7-H). MS-EI+ (m/z, %): 280 (M+, 9.76) 263 (12.69) 209 (36.00) 181 (45.31) 163 (67.42) 137 (33.42) 43 (100).
4.3.3. 1-[(3R,4R-Di-O-(−)-camphanoyloxy)-5-ethoxy-2,2-dimethyl-3,4-dihydro-2H-chromen-6-y l)]-ethanone (5)
A mixture of compound 28 (1 equiv), (S)-(−)-camphanic chloride (2 equiv), and DMAP (3 equiv) was stirred for 2 h in CH2Cl2 at rt and monitored by TLC. After the reaction was completed, the mixture was purified by chromatography on silica column with petroleum ether: EtOAc = 2:1 as eluent to afford 5 (white powder) (146 mg, 75%). mp 230–232 °C. 1H NMR δ 7.64 (1H, d, J = 9.0 Hz, H-8), 6.07 (1H, d, J = 9.0 Hz, H-7), 6.48 (1H, d, J = 4.69 Hz, CHO-4), 5.51 (1H, d, J = 5.09Hz, CHO-3), 3.94 (1H, m, CH2O-5), 3.82 (1H, m, CH2O-5), 2.54 (3H, s, CH3CO-6), 2.42, 2.38, 1.90, 1.69 (each 2H, m, camphanoyl CH2), 1.45, 1.43 (each 3H, s, CH3-2) 1.11, 1.10, 1.09, 1.09, 0.97, 0.95 (each 3H, s, camphanoyl CH3). MS –ESI+ (m/z, %) 663 (M++Na, 100).
4.4. Synthetic routes to seco-C ring DCKs (6–9) and bioisosteric seco DCK 13
4.4.1. 7-(2-Methylbut-3-en-2-yloxy)-4-methyl-2H-chromen-2-one (30)
A mixture of 29 (2.50 g, 14.1 mmol), K2CO3 (20.00 g, 0.15 mol), KI (2.55 g, 9.59 mmol), and 3-chloro-3-methyl-1-butene in acetone (100 mL) was refluxed for 4 h. After filtering, the residue was recrystallized from EtOAc to yield 30 as colorless crystals (2.68 g, 77%), mp 94–97 °C. 1H NMR δ 7.48 (1H, d, J = 8.61 Hz, H-5), 6.86 (1H, dd, J = 8.61, 2.35 Hz, H-6), 6.81 (1H, d, J = 2.34 Hz, H-8), 6.12 (1H, s, H-3), 5.47 (1H, m, CHCH2-7), 4.57 (2H, d, J = 6.65 Hz, CH2CH-7), 2.39 (3H, s, CH3-4), 1.80, 1.76 (each 3H, s, (CH3)2C-7). MS-EI+ (m/z, %) 244 (M+, 4.65), 176 (M-68 100).
4.4.2. 7-(2R,3-Dihydroxy-1,1-dimethylpropoxy)-4-methyl-chromen-2-one (31)
Same synthetic procedure as for 28 but starting from 30. Compound 31 was crystallized from EtOAc as white crystals (266 mg, 58%). mp 172–173 °C. 1H NMR δ 7.67 (1H, d, J = 8.55 Hz, H-5), 6.96 (1H, dd, J = 8.55 2.56 Hz, H-6), 6.98 (1H, d, J = 2.56 Hz, H-8), 6.19 (1H, d, J = 0.85 Hz, H-3), 5.10 (1H, d, J = 5.56 Hz, HOCH-7), 4.48 (1H, s, HOCH2-7), 4.32 (1H, dd, J = 1.71 10.25 Hz, CHCH2OH-7), 3.90 (1H, dd, J = 8.12 9.73 Hz, CHCH2OH-7), 3.55 (1H, m, HOCHCH2-7), 2.39 (3H, s, CH3-4), 1.14, 1.08 (each 3H, s, (CH3)2C-7). MS-EI+ (m/z, %) 278 (M+, 20.04), 176 (M-102 100).
4.4.3. 7-(2R,3-Di-O-(−)-camphanoyloxy-1,1-dimethylpropoxy)-4-methyl-2H-chromen-2-one (6)
Same synthetic procedure as for 5 but starting from 31. Chromatography (CHCl3-MeOH, 50:1) to yield 6 as a white solid (410 mg, 93%). mp 255–260 °C. 1H NMR δ 7.56 (1H, d, J = 8.77 Hz, H-8), 6.86 (1H, dd, J = 2.63 8.77 Hz, H-7), 6.76 (1H, d, J = 2.63 Hz, H-8), 6.16 (1H, s, CH-3), 5.65 (1H, dd, J = 2.63 8.63 Hz, CHO-7), 4.41 (1H, dd, J = 2.63 10.52 Hz, CH2O-7), 4.23 (1H, dd, J = 8.33 10.53 Hz, CH2O-7), 2.45, 2.02, 1.92, 1.69 (each 2H, m, camphanoyl CH2), 2.39 (3H, s, CH3-4), 1.11, 1.10, 1.06, 1.06, 1.00, 0.94 (each 3H, s, camphanoyl CH3), 1.62 (each 3H, s, CH3C-7). 13C NMR δ: 9.67, 16.59, 16.66, 16.74, 16.93, 18.62, 22.36, 22.84, 28.75(2C), 30.43, 30.71, 54.17, 54.38, 54.87, 66.38, 75.84, 83.19, 91.00(2C), 102.05, 111.57, 112.41, 114.30, 126.04, 152.37, 155.08, 160,64, 160.98, 166.10, 166.83, 178.10(2C). MS-ESI+ (m/z, %) 639 (M+ + 1, 100). HRMS Calcd. for C35H42O11+Na 661.2619, found 661.2592(M+Na).
4.4.4. 7-Hydroxy-4-methyl-2-oxo-2H-chromene-8-carbaldehyde (32a)
A reaction mixture of 29 (20 g, 114 mmol) and hexamethylenetetramine (40 g, 285 mmol) in HOAc (150 mL) was stirred for 5.5 h at 95 °C. Thereafter, aq HCl solution (300 mL, conc. HCl:H2O=84:100, v/v) was added, and the reaction mixture was stirred for 0.5 h at 70 °C. After cooling, the reaction mixture was poured into ice-water (1.5 L) and extracted with EtOAc four times. The combined organic layer was dried over Na2SO4, and the solvent was removed. The residue was recrystallized from EtOAc to provide 32a as a light yellow solid (5.23 g, 22%). mp 120–122 °C.
4.4.5. 7-Isopropoxy-4-methyl-2-oxo-2H-chromen-8-carbaldehyde (33a)
Compound 32a (1 g, 5 mmol), ethane-1,2-diol (435 mg, 7.02 mmol), and p-toluenesulfonic acid (30 mg, 0.17 mmol) were dissolved in toluene (40 mL). The reaction mixture was refluxed for 2 h with removal of water. After adjusting the pH to 7–8 with triethylamine and washing with brine, the organic layer was dried over Na2SO4, filtered, and evaporated in vacuo. The residue was recrystallized from EtOAc to give a light yellow solid (630 mg, 51%, mp 214–218 °C). A mixture of this intermediate (500 mg, 2.02 mmol), K2CO3 (836 mg, 6.05 mmol), KI (50 mg, 0.30 mmol), and 2-bromopropane in acetone (50 mL) was refluxed for 6 h. After filtering and removing the solvent, the residue was recrystallized from EtOAc to yield a white solid (473 mg, 80%, mp: 150–152 °C). This solid was added to 2N HCl (30 mL), stirred for 5 h at rt, extracted with EtOAc four times, and dried over Na2SO4. Filtration and removal of the solvent in vacuo gave 33a as a white solid (390 mg, 97%). mp 166–168 °C. 1H NMR δ 10.64 (1H, s, CHO), 7.71 (1H, d, J = 9.00 Hz, H-6), 6.94 (1H, d, J = 9.00 Hz, H-5), 6.20 (1H, d, J = 1.17 Hz, H-3), 4.77 (1H, m, (CH3)2CHO-7), 2.41 (3H, d, J = 0.78 Hz, CH3-4), 1.44, 1.44 (each 3H, d, J = 6.65 Hz, CH3CH-7). MS-EI+ (m/z, %) 246 (M+, 6.48), 148 (M-98, 100).
4.4.6. 8-Hydroxymethyl-7-isopropoxy-4-methyl-2H-chromen-2-one (34a)
A mixture of 33a (200 mg, 0.81 mmol) and NaBH4 (116 mg, 3.07 mmol) in MeOH (10 mL) was stirred for 1.5 h at rt. After the pH was adjusted to 3–4 using 2N HCl, MeOH was removed in vacuo. The residue was extracted with EtOAc three times and the organic layer was dried over Na2SO4. After filtration and removal of the solvent, 34a was obtained as a colorless oil (240 mg, 100%). Crystals of 34a were obtained by recrystallization from EtOAc, mp: 118–121 °C. 1H NMR δ 7.51 (1H, d, J = 9.00 Hz, H-6), 6.89 (1H, d, J = 9.00 Hz, H-5), 6.14 (1H, d, J = 1.17 Hz, H-3), 4.96 (2H, s, CH2O), 4.73 (1H, m, (CH3)2CHO-7), 2.57 (1H, broad, OH), 2.40 (3H, d, J = 1.17 Hz, CH3-4), 1.42, 1.42 (each 3H, d, J = 5.87 Hz, CH3CH-7). MS-EI+ (m/z, %) 248 (M+, 10.76), 188 (M-60, 100).
4.4.7. 8-[O-(−)-Camphanoyloxymethyl]-7-isopropoxy-4-methyl-2H-chromen-2-one (9)
Same synthetic procedure as for 5 but starting from 34a. White solid (yield, 100%); mp 64–65 °C. 1H NMR δ 7.58 (1H, d, J = 9.00 Hz, H-6), 6.88 (1H, d, J = 8.61 Hz, H-5), 6.15 (1H, d, J = 1.17 Hz, H-3), 5.51 (2H, dd, J = 11.35 19.56 Hz, CH2O), 4.70 (1H, m, (CH3)2CHO-7), 2.43, 2.03, 1.89, 1.65 (each H, m, camphanoyl CH2), 2.39 (3H, s, CH3-4), 1.08, 1.02, 0.97 (each 3H, s, camphanoyl CH3), 1.36, 1.37 (each 3H, d, J = 4.70 Hz, CH3C-7). 13C NMR δ 9.67, 16.69, 18.70, 21.43, 21.98, 28.97, 30.61, 54.26, 54.71, 56.15, 66.02, 71.47, 91.29, 109.10, 111.44, 112.11, 113.46, 126.29, 152.39, 153.84, 159.67, 160.49, 167.37, 178.35. MS-ESI+ (m/z, %) 429 (M++1, 100). HRMS Calcd. for C24H28O7 – H 427.1762, found 427.1748(M-H).
4.4.8. 8-Acetyl-7-hydroxy-4-methyl-2H-chromen-2-one (32b)
Compound 29 (5 g, 22.9 mmol) and AlCl3 (4.59 g, 34.4 mmol) was stirred vigorously for 3 h, then poured into 2N HCl and extracted with EtOAc four times. The organic layer was dried over Na2SO4, filtered, and the solvent removed. The residue was purified by column chromatography on silica gel (hexane-EtOAc, 3:1) to provide 32b as a light yellow solid (1.2 g, 24%), mp: 207–209 °C.
4.4.9. 8-Acetyl-7-isopropoxy-4-methyl-chromen-2-one (33b)
Same synthetic procedure as for 33a but starting from 32b. White crystalline needles (yield, 47%); mp 139–140 °C. 1H NMR δ 7.53 (1H, d, J = 8.79 Hz, H-6), 6.87 (1H, d, J = 8.79 Hz, H-5), 6.13 (1H, d, J = 1.10 Hz, H-3), 4.67 (1H, m, CHO-7), 2.56 (3H, s, CH3-4), 1.36, 1.36 (each 3H, d, J = 10.55 Hz, CH3CH-7). MS-EI+ (m/z, %) 260 (M+, 8.46), 203 (M-57, 100).
4.4.10. 8-(1-Hydroxyethyl)-7-isopropoxy-4-methyl-2H-chromen-2-one (34b)
Same synthetic procedure as for 34a but starting from 33b. White crystals (yield, 100%); mp 126–127 °C. 1H NMR δ 7.46 (1H, d, J = 8.79 Hz, H-6), 6.88 (1H, d, J = 8.97 Hz, H-5), 6.14 (1H, d, J = 1.10 Hz, H-3), 5.54 (1H, m, CH3CHOH), 4.76 (1H, m, (CH3)2CHO-7), 3.71 (1H, d, J = 1.91 Hz, OH), 2.39 (3H, d, J = 1.10 Hz, CH3-4), 1.58 (3H, d, J = 6.59 Hz, CH3CH-8), 1.43, 1.43 (each 3H, d, J = 5.86 Hz, CH3CH-7). MS-EI+ (m/z, %) 262 (M+, 11.08), 202 (M-60, 100).
4.4.11. 8-[2-O-(−)-Camphanoyloxyethyl]-7-isopropoxy-4-methyl-2H-chromen-2-one (8)
Same synthetic procedure as for 5 but starting from 34b. White oil (yield, 100%). 1H NMR δ 7.50 (1H, d, J = 8.67 Hz, H-6), 6.87 (1H, dd, J = 8.94 2.89 Hz, H-5), 6.67 (1H, dd, J = 6.71 13.54 Hz, CH3CHO-8), 6.13 (1H, s, CH-3), 4.71 (1H, m, (CH3)2CHO-7), 2.49, 2.00, 1.91, 1.66 (each 1H, m, camphanoyl CH2), 2.39 (3H, s, CH3-4), 1.74 (3H, d, J = 6.57 Hz, R:S 1:1, CH3CH-8), 1.15, 1.09, 1.07, 1.01, 0.96, 0.87 (each 1.5H, s, R:S 1:1, camphanoyl CH3). MS-ESI+Na (m/z, %) 465 (M+ + Na, 100). HRMS Calcd. for C25H30O7 + Na 465.1884, found 465.1870 (M+Na).
4.4.12. 7-Isopropoxy-4-methyl-8-vinyl-2H-chromen-2-one (35)
A solution of 34b (75 mg, 0.29 mmol) and two drops of conc. H2SO4 in acetone (5 mL) was refluxed for 20 min. The reaction mixture was neutralized with 2N aqueous Na2CO3. After removal of the solvent and extraction with EtOAc, the organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography with an eluent of petroleum ether:EtOAc = 8:1 to provide pure 35 as a white crystalline solid in 17% yield; mp 102–104 °C. 1H NMR δ 7.43 (1H, d, J = 9.00 Hz, H-6), 7.08 (1H, dd, J = 12.13, 18.00 Hz, CHCH2-8), 6.88 (1H, d, J = 9.00 Hz, H-5), 6.33 (1H, dd, J = 2.35 18.00 Hz, CH2CH-8), 6.15 (1H, s, H-3), 5.61 (1H, dd, J = 2.35 12.13 Hz, CH2CH-8), 4.71 (1H, m, (CH3)2CHO-7), 2.40 (3H, d, J = 1.17 Hz, CH3-4), 1.41, 1.40 (each 3H, s, CH3CH-7). MS-EI+ (m/z, %) 244 (M+, 24.98), 174 (M-70, 100).
4.4.13. 8-(1R,2-Dihydroxyethyl)-7-isopropoxy-4-methyl-2H-chromen-2-one (36)
Same synthetic procedure as for 28 but starting from 35. Crystalline needles (yield, 70%); mp: 102–104 °C. 1H NMR δ 7.50 (1H, d, J = 9.00 Hz, H-6), 6.88 (1H, d, J = 9.00 Hz, H-5), 6.14 (1H, d, J = 1.17 Hz, H-3), 5.46 (1H, dd, J = 4.27 8.54 Hz, HOCH2CH-8), 4.75 (1H, m, (CH3)2CHO-7), 3.94 (1H, dd, J = 8.85 11.29 Hz, HOCHCH2-8), 3.83 (1H, m, HOCH2-8), 3.72 (1H, dd, J = 4.27 11.29 Hz, HOCH2CH-8), 2.39 (3H, d, J = 1.22 Hz, CH3-4), 1.79 (1H, s, HOCH-8), 1.42, 1.41 (each 3H, d, J = 5.60 Hz, CH3CH-7). MS-EI+ (m/z, %) 278 (M+, 100).
4.4.14. 8-(1R,2-Di-O-(−)-camphanoyloxyethyl)-7-isopropoxy-4-methyl-2H-chromen-2-one (7)
Same synthetic procedure as for 5 but starting from 36. White solid (yield, 100%); mp 64–66 °C. 1H NMR δ 7.57 (1H, d, J = 9.00 Hz, H-6), 6.88 (1H, d, J = 9.00 Hz, H-5), 6.89 (1H, dd, J = 8.61 3.91 Hz, CH2CHO-8), 6.15 (1H, d, J = 1.17 Hz, H-3), 5.02 (1H, dd, J = 11.74 8.61 Hz, CHCH2O-8), 4.71 (1H, m, (CH3)2CHO-7), 4.69 (1H, dd, J = 11.73 3.91 Hz, CHCH2O-8), 2.46, 2.02, 1.91, 1.66, (each 2H, m, camphanoyl CH2), 2.40 (3H, d, J = 1.18 Hz, CH3-4), 1.44, 1.43 (each 3H, d, J = 6.26 Hz, (CH3)2CHO-7), 1.09, 1.07, 1.02, 1.01, 0.91, 0.87 (each 3H, s, camphanoyl CH3). 13C NMR δ 9.68, 16.61, 16.69, 18.84, 21.90, 21.96, 28.95, 30.66(2C), 54.17, 54.79(2C), 64.63, 67.10, 71.96, 91.00(2C), 109.44, 111.42, 112.16, 113.54, 126.62, 152.41, 153.33, 159.17, 159.88, 166.60, 167.06, 177.94, 178.14. MS-ESI+ (m/z, %) 639 (M+ + 1, 100). HRMS Calcd. for C35H42O11 – H 637.2654, found 637.2634(M-H).
4.4.15. 7-Isopropoxy-8-(mercaptomethyl)-4-methyl-2H-chromen-2-one (37)
Under nitrogen, a mixture of compound 34a (150 mg, 0.61 mmol) and Lawesson reagent (367 mg, 0.91 mmol) in toluene (10 mL) was stirred for 20 h and monitored by TLC (hexane:EtOAc/1:1). After filtration, the solvent was removed in vacuo. The residue was purified by silica gel chromatography (eluent: hexane/EtOAc =1/1) to obtain 37 as a yellow solid (43 mg), yield:27%; mp 113–116 °C. 1H NMR δ 7.45 (1H, d, J = 9.00 Hz, H-6), 6.86 (1H, d, J = 8.99 Hz, H-5), 6.15 (1H, s, H-3), 4.73 (1H, m, 7-CH), 3.93 (2H, d, J = 8.59 Hz, 8-CH2), 2.40 (3H, s, 4-CH3), 2.13 (1H, t, J = 8.6 Hz, 8-OH), 1.42 (6H, d, J = 5.87 Hz, 2×7-CH3). MS-ESI + (m/z, %) 265 (M+ + 1, 100).
4.4.16. 8-[S-(−)-Camphanoyl)mercaptomethyl]-7-isopropoxy-4-methyl-2H-chromen-2-one (13)
Same synthetic procedure as for 5 but starting from 37. Light yellow crystals (yield, 91%); mp 166–168 °C. 1H NMR δ 7.47 (1H, d, J = 8.99 Hz, H-6), 6.83 (1H, d, J = 9.00 Hz, H-5), 6.13 (1H, s, H-3), 4.68 (1H, m, 7-CH), 4.47 (2H, s, 8-CH2), 2.50, 1.95, 1.68, 1.37 (each H, m, camphanoyl CH2), 2.39 (3H, s, CH3-4), 1.09, 1.09, 0.96 (each 3H, s, camphanoyl CH3), 1.37, 1.37 (each 3H, d, J = 2.47 3.13 Hz, 2×CH3C-7). 13C NMR δ 9.69, 16.53, 16.65, 18.69, 20.89, 22.01, 28.94, 31.08, 54.59, 55.44, 71.28, 96.30, 108.85, 112.09, 112.76, 113.53, 124.61, 152.39, 152.97, 158.86, 160.62, 177.95, 195.75. MS –ESI+ (m/z, %) 445 (M+ + 1).
4.5. Synthetic routes to seco-C ring DCKs (10–12)
4.5.1. 8-(1,3-Dioxolan-2-yl)-7-hydroxy-4-methyl-2H-chromen-2-one (38)
Using a water separator, a mixture of 32a (1 g, 5 mmol), ethane-1,2-diol (335 mg, 5.4 mmol) and p-toluenesulfonic acid (30 mg) in toluene (40 mL) was refluxed 2 h with removal of water. The reaction mixture was cooled to rt and the pH adjusted to 7~8 with triethylamine. After being washed with brine, the organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford a yellow solid, which was recrystallized from EtOH to yield a light yellow solid (630 mg, 51%): mp 214–218 °C. 1H NMR δ 2.39 (s, 3H, 4-CH3), 4.14, 4.26 (m, 4H, 2×-CH2), 6.13 (d, J = 1.18 Hz, 1H, 3-H), 6.40 (m, 1H, 8-CH), 6.85 (d, J = 8.60 Hz, 1H, 5-H), 7.52 (d, J = 8.60 Hz, 1H, 6-H), 9.15 (s, 1H, 7-OH). MS-EI+ (m/z, %) 248 (M+, 46.01).
4.5.2. Synthesis of intermediates 41a–c
Compound 38 (1 equiv), K2CO3 (5 equiv), and halide (3 equiv) were added into acetone (50 mL) and refluxed for 6 h. After filtration, the solvent was removed in vacuo to give crude product (39a–c), which was stirred for 5h in 2 N HCl solution (30 mL) at rt. The reaction mixture was extracted with EtOAc four times. The organic layer was dried over Na2SO4 and the solvent distilled to give a white solid (40a–c). The intermediate (1 equiv) and NaBH4 (1.5 equiv) in MeOH (10 mL) were allowed to react for 1 h at rt, then acidified to pH 3~4 with 2N HCl. After removal of MeOH, the residue was extracted with EtOAc three times. The organic layer was dried over Na2SO4 and concentrated to provide the desired product.
4.5.2.1. 8-Hydroxymethyl-7-methoxy-4-methyl-2H-chromen-2-one (41a)
Light yellow crystals from EtOAc, MS-ESI+, (m/z, %) 221.0 (M+ + 1, 100).
4.5.2.2. 8-Hydroxymethyl-7-ethoxy-4-methyl-2H-chromen-2-one (41b)
Light yellow crystals from MeOH, yield 68%, mp 169–172 °C. 1H NMR δ 7.52 (1H, d, J = 8.99 Hz, H-6), 6.88 (1H, d, J = 9.00 Hz, H-5), 6.15 (1H, d, J = 0.78 Hz, H-3), 4.98 (2H, d, J = 6.65 Hz, 8-CH2), 4.19 (2H, dd, J = 6.65 7.05 Hz, 7-CH3CH2O), 2.58 (1H, t, J = 3.52 3.13 Hz, 8-OH), 2.40 (3H, d, J = 1.17 Hz, CH3-3), 0.83 (3H, t, J = 6.05 7.04 Hz, 7-CH3).
4.5.2.3. Methyl 2-(8-hydroxymethyl-4-methyl-2-oxo-2H-chromen-7-yloxy)acetate (41c)
White crystals from MeOH, yield 79%, mp 142–144 °C. 1H NMR δ 7.52 (1H, d, J = 9.00 Hz, H-6), 6.78 (1H, d, J = 8.60 Hz, H-5), 6.18 (1H, d, J = 1.17 Hz, H-3), 5.02 (2H, s, 7-CH2), 4.81 (2H, d, 8-CH2), 3.81 (3H, s, 7-CH3), 2.55 (1H, broad s, 8-OH), 2.40 (3H, d, J = 1.17 Hz, CH3-3).
4.5.3. Synthesis of target compounds 10–12
Same synthetic procedure as for 5 but starting from 41a–c, respectively.
4.5.3.1. 8-[O-(−)-Camphanoyloxymethyl]-7-methoxy-4-methyl-2H-chromen-2-one (10)
White solid from hexane/acetone=1/1, yield 78%, mp 185–188 °C. 1H NMR δ 7.61 (1H, d, J = 8.61 Hz, H-6), 6.90 (1H, d, J = 9.00 Hz, H-5), 6.16 (1H, s, H-3), 5.51 (2H, dd, J = 13.7 14.08 Hz, 8-CH2), 3.93 (3H, s, 7-CH3), 2.45, 2.03, 1.88, 1.66 (each H, m, camphanoyl CH2), 2.42 (3H, s, 4-CH3), 1.62, 1.08, 1.03, 0.97 (each 3H, s, camphanoyl CH3). 13C NMR δ 9.66, 16.53, 16.61, 18.71, 28.96, 30.55, 54.30, 54.72, 55.95, 56.16, 91.29, 107.07, 110.60, 112.29, 113.90, 126.54, 152.38, 153.51, 160.35, 160.95, 167.37, 178.33. MS-ESI+ (m/z, %) 401 (M+ + 1, 100). HRMS Calcd. for C22H24O7 + Na 423.1414, found 423.1402(M+Na).
4.5.3.2. 8-[O-(−)-Camphanoyloxymethyl]-7-ethoxy-4-methyl-2H-chromen-2-one (11)
White solid from hexane/EtOAc=1/1, yield 48%, mp 161–163 °C. 1H NMR δ 7.59 (1H, d, J = 9.00 Hz, H-6), 6.88 (1H, d, J = 9.00 Hz, H-5), 6.15 (1H, d, J = 1.17 Hz, H-3), 5.53 (2H, dd, J = 1.34 17.60 Hz, 8-CH2), 4.16 (2H, dd, J = 7.04 13.69 Hz, 7-CH2), 2.45, 2.04, 1.89, 1.65 (each H, m, camphanoyl CH2), 2.41 (3H, s, CH3-4), 1.08, 1.02, 0.97 (each 3H, s, camphanoyl CH3), 1.45 (3H, t, J = 7.94 7.07 Hz, 7-CH3). 13C NMR δ 9.66, 14.65, 16.60, 16.65, 18.70, 28.96, 30.59, 54.28, 54.72, 56.03, 64.70, 91.29, 107.89, 110.63, 112.16, 113.70, 126.46, 152.42, 153.60, 160.44, 167.38, 178.35. MS-ESI+ (m/z, %) 415 (M+ + 1, 100). HRMS Calcd. for C23H26O7 + Na 437.1571, found 437.1470(M+Na).
4.5.3.3. 8-[O-(−)-Camphanoyloxymethyl]-7-(methoxycarbonylmethoxy)-4-methyl-2H-chromen-2-one (12)
White solid from EtOAc, yield 83%, mp 155–157 °C. 1H NMRδ 7.59 (1H, d, J = 8.60 Hz, H-6), 6.76 (1H, d, J = 8.60 Hz, H-5), 6.19 (1H, d, J = 1.18 Hz, H-3), 5.58 (2H, dd, J = 14.09 13.69 Hz, 8-CH2), 4.78 (2H, s, 7-CH2), 3.80 (3H, s, 7-CH3), 2.47, 2.04, 1.89, 1.65 (each H, m, camphanoyl CH2), 2.41 (3H, d, J =1.18 Hz, CH3-4), 1.08, 1.04, 0.96 (each 3H, s, camphanoyl CH3). 13C NMR δ 9.64, 16.52, 16.60, 18.68, 28.95, 30.55, 52.42, 54.28, 54.71, 55.91, 65.71, 91.28, 107.87, 111.55, 112.85, 114.75, 126.43, 152.15, 153.60, 159.14, 160.07, 167.28, 168.26, 178.32. MS-ESI+ (m/z, %) 459 (M+ + 1, 100). HRMS Calcd. for C24H26O9 + Na 481.1469, found 481.1468(M+Na).
4.6. Synthesis of bioisosteric seco-DCKs (14–16)
4.6.1. 8-Aminomethyl-7-isopropoxy-4-methyl-2H-chromen-2-one (42)
A mixture of 33a (246 mg, 1 mmol) and (NH4)2CO3 (1 g, 10.4 mmol) in EtOH (50 mL) was refluxed for 48 h, then NaBH4 (50 mg, 1.32 mmol) was added, and reflux continued for 15 min. After removal of the solvent, the residue was separated by silica gel chromatography to afford a yellow oil (164 mg). Light yellow crystals were obtained from hexane/acetone=1/1, yield 66%: mp 196–198.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.60 (1H, d, J = 9.00 Hz, H-6), 7.07 (1H, d, J = 8.70 Hz, H-5), 6.19 (1H, d, J = 1.8 Hz, H-3), 4.78 (1H, m, 7-CH), 3.78 (2H, s, 8-CH2), 2.37 (3H, s, CH3-4), 1.54 (2H, broad s, NH2), 1.29 (6H, d, J = 6.00 Hz, 2×CH3C-7). MS (ESI) (m/z, %) 495(2M+ +1, 100).
4.6.2. 8-[(N-(−)-Camphanoyl)aminomethyl]-7-isopropoxy-4-methyl-2H-chromen-2-one(14)
Same synthetic procedure as for 5 but starting from 42. Light yellow crystals (yield 87%); mp 236–237.5 °C. 1H NMR (400 MHz, CDCl3) δ 7.30 (1H, d, J = 9.16 Hz, Ar-H), 7.28 (1H, d, J = 9.17 Hz, Ar-H), 6.86 (1H, d, J = 9.17 Hz, Ar-H), 6.85 (1H, d, J = 9.16 Hz, Ar-H), 5.80 (1H, s, H-3), 5.76 (1H, s, H-3), 4.68 (5H, m), 4.52 (1H, d, J = 14.36 Hz), 2.58, 2.33, 1.94, 1.72 (each H, m, camphanoyl CH2), 2.22, 2.21 (3H, s, 2×CH3-4), 1.51, 1.48, 1.42, 1.37 (each 3H, d, 4×-CH3), 1.30, 1.16, 1.25 (each 3H, s, camphanoyl CH3). 13C NMR δ 9.73, 17.47, 17.94,18.49, 21.97, 22.14, 22.60, 29.50, 31.32, 37.51, 39.61, 53.81, 55.47, 70.80, 93.11, 108.75, 111.02, 112.05, 124.75, 152.34, 153.32, 160.15, 167.37, 179.06. MS-ESI+ (m/z, %) 428 (M+ + 1). HRMS Calcd. for C24H29NO6 + Na 450.1887, found 450.1874 (M+Na).
4.6.3. Synthesis of 43 and 45
Compound 33a (1 equiv) and amine (1.1 equiv) were dissolved in EtOH (10 mL). The mixture reaction was stirred for 2 h at rt and monitored by TLC (hexane/acetone=2/1). After removal of the solvent, yellow crystals were obtained from EtOAc.
4.6.3.1. 7-Isopropoxy-4-methyl-8-[(methylimino)methyl]-2H-chromen-2-one (43)
Yield 91%, mp 160–161 °C. 1H NMR δ 8.71 (1H, s, 8-CH), 7.03 (1H, d, J = 8.55 Hz, H-6), 6.15 (1H, d, J = 8.55 Hz, H-5), 5.97 (1H, s, H-3), 4.59 (1H, m, 7-CH), 3.44 (3H, s, 8-CH3), 2.49 (3H, d, J = 4.89 Hz, CH3-4), 1.36, 1.35 (6H, s, 2×CH3C-7). MS-EI+ (m/z, %) 260 (M+ +1, 65.87), 232 (76.43), 190 (100).
4.6.3.2. 7-Isopropoxy-8-[(4-methoxyphenylimino)methyl]-4-methyl-2H-chromen-2-one (45)
Yield 100%, mp 157–159 °C. 1H NMR δ 10.62, 8.84 (1H, s, 8-CH), 7.70, 7.59 (1H, d, J = 9.00 Hz, H-6), 7.30 (1H, d, J = 8.99 Hz, H-5), 7.28 (1H, d, J = 7.44 Hz, 8-Ar-H), 6.95, 6.74, 6.65 (3H, m, 8-Ar-H), 6.18 (1H, s, H-3), 4.73 (1H, m, 7-CH), 3.84 (3H, s, 8-OCH3), 2.42 (3H, s, CH3-4), 1.41 (6H, s, 2×CH3C-7). MS (EI) (m/z, %) 351 (M+ + 1, 14.23), 229 (100).
4.7.4. Synthesis of 44 and 46
A mixture of imine (1 equiv) and NaBH4 (2.3 equiv) in MeOH (10 mL) was stirred for 30 min at rt. After removal of the solvent, the residue was extracted with EtOAc three times. The organic layer was dried over anhydrous Na2SO4, and concentrated to get a light yellow solid, which was recrystallized from EtOAc to provide white crystals.
4.7.4.1. 7-Isopropoxy-4-methyl-8-[(N-methyl)aminomethyl]-2H-chromen-2-one (44)
Yield 98%, mp 128–130 °C. 1H NMR δ 7.46 (1H, d, J = 9.00 Hz, H-6), 6.86 (1H, d, J = 9.00 Hz, H-5), 6.13 (1H, s, H-3), 4.68 (1H, m, 7-CH), 4.02 (2H, s, 8-CH2), 2.41 (3H, s, CH3-3), 2.39 (3H, d, J = 0.78 Hz, 8-CH3), 1.73 (1H, broad, 8-NH), 1.40, 1.39 (each 3H, d, CH3CH-7). MS-EI+ (m/z, %) 261 (M+, 8.05), 246 (100).
4.7.4.2. 7-Isopropoxy-8-[(N-4-methoxyphenyl)aminomethyl]-4-methyl-2H-chromen-2-one (46)
Yield 96 %: mp 137.5–140 °C. 1H NMR δ 7.43 (1H, d, J = 9.00 Hz, H-6), 6.84 (1H, d, J = 9.00 Hz, H-5), 6.74 (4H, dd, J = 9.00 Hz, 8-Ar-H), 6.13 (1H, s, H-3), 4.73 (1H, m, 7-CH), 4.56 (2H, s, 8-CH2), 3.71 (3H, s, 8-OCH3), 2.37 (3H, s, CH3-3), 2.39 (3H, d, J = 0.78 Hz, 8-CH3), 1.59 (1H, br, 8-NH), 1.44, 142 (each 3H, d, CH3CH-7). MS (EI) (m/z, %) 353 (M+, 44.59), 310 (5.53), 231 (19.07), 189 (100).
4.7.6. Synthesis of 15 and 16
Same synthetic procedure as for 5 but starting from 44 and 46, respectively.
4.7.6.1. 8-[(N-Methyl-N-(−)-camphanoyl)aminomethyl]-7-isopropoxy-4-methyl-2H-chromen-2-one(15)
Yield 86%. 1H NMR δ 7.50 (1H, d, J = 9.00 Hz, H-6), 6.84 (1H, d, J = 8.61 Hz, H-5), 6.09 (1H, d, J = 1.17 Hz, H-3), 5.09, 4.72 (2H, dd, J = 11.35 19.56 Hz, CH2N-8), 4.65 (1H, m, (CH3)2CHO-7), 2.91 (3H, s, CH3N), 2.39, 2.03, 1.89, 1.62 (each H, m, camphanoyl CH2), 2.37 (3H, s, CH3-4), 1.07, 1.05, 1.02 (each 3H, s, camphanoyl CH3), 1.31, 1.23 (each 3H, d, J = 6.30 Hz, CH3C-7). MS-ESI+ (m/z, %) 442 (M+ + 1, 100).
4.7.6.2. 8-[(N-4-Methoxyphenyl-N-(−)-camphanoyl)aminomethyl]-7-isopropoxy-4-meth yl-2H-chromen-2-one (16)
Yield 100%, colorless oil. 1H NMR δ 7.39 (1H, d, J = 8.61 Hz, H-6), 6.72 (1H, d, J = 8.61 Hz, H-5), 7.00–6.49 (5H, m, Ar-H), 5.97 (1H, s, H-3), 5.46 (1H, d, J = 3.3 Hz, CH2N-8), 4.91 (1H, d, J = 3.59 Hz, CH2N-8), 4.60 (1H, m, (CH3)2CHO-7), 3.68 (3H, s, CH3O), 1.75,1.55, 1.40, 1.10 (each H, m, camphanoyl CH2), 2.33 (3H, s, CH3-4), 1.20, 1.03, 0.97 (each 3H, s, camphanoyl CH3), 1.33, 1.25 (each 3H, d, J = 5.87 Hz, CH3C-7). MS-ESI+ (m/z, %) 534 (M+ + 1, 100).
4.8. Anti-HIV replication assay against wild-type HIV-1IIIB in MT-2 cell lines
This assay was performed by Panacos Pharmaceuticals, Inc as follows. The human T-cell line, MT-2, was maintained in continuous culture with L-glutamine at 5% CO2 and 37 °C. Test samples were first dissolved in dimethyl sulfoxide. The following were the final drug concentrations routinely used for screening 100, 20, 4 and 0.8 μg/mL. For agents found to be active, additional dilutions were prepared for subsequent testing so that an accurate EC50 value could be determined. Test samples were prepared, and to each sample well was added 90 μL of media containing H9 cells at 3 × 105 cells/mL and 45 μL of virus inoculum (HIV-1 IIIB isolate) containing 125 TCID50. Control wells containing virus and cells only (no drug) and cells only (no virus or drug) were also prepared. A second set of samples was prepared identical to the first and were added to cells under identical conditions without virus (mock infection) for toxicity determinations (IC50 defined below). In addition, AZT and 2 were also assayed during each experiment as a positive drug control. On days 1 and 4 post-infection (PI), spent media was removed from each well and replaced with fresh media. On day 6 PI, the assay was terminated and cultured supernatants were harvested for analysis for virus replication by p24 antigen capture. The compound toxicity was determined by XTT using the mock-infected sample wells. If a test sample inhibited virus replication and was not toxic, its effects were reported in the following terms: IC50, the concentration of test sample that was toxic to 50% of the mock-infected cells; EC50, the concentration of test sample that was able to suppress HIV replication by 50%; and the therapeutic index (TI), the ratio of the IC50 to EC50.12
4.9. Anti-HIV replication assay against HIV-1NL4-3 and RTMDR strains assays in MT-4 cell lines
A previously described HIV-1 infectivity assay was used in the experiments.13 A 96-well cell culture plate was used to set up the virus replication screening assay. HIV-1 NL4-3 or RTMDR at a multiplicity of infection (MOI) of 0.001 was used to infect MT-4 cells. Culture supernatants were collected on day 4 post-infection for a p24 assay using an ELISA kit from Perkin Elmer.
Supplementary Material
Figure 1.

Structures of previously synthesized DCK analogs (1, 2) and 2-Et-DCP analog (3).
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China awarded to P. Xia (No. 20272010) and Y. Chen (No. 30200348 and 30873164), respectively. It was also supported by a research grant (2002046069) for PhD program from the National Education Administration of China awarded to P. Xia, and Grant AI-33066 from the National Institute of Allergies and Infectious Diseases awarded to K. H. Lee.
Abbreviations
- DCK
(3′R,4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone
- HIV-1
human immunodeficiency virus type 1
- DCP
3′R,4′R′-di-O-(−)-camphanoyl-2′,2′-dimethyldihydropyranol[2,3-f]chromone
- RTMDR
multi-RT inhibitor-resistant strain
- ADME
Absorption, Distribution, Metabolism, and Elimination
- AZT
zidovudine
- SAR
structure-activity relationships
- DMF
dimethyl formamide
- AD
asymmetric dihydroxylation
- DMAP
4-(dimethylamino)pyridine
- (DHQ)2PHAL
hydroquinine 1,4-phthalazinediyl diether
- TLC
Thin-layer chromatography
Footnotes
Additional information on compound purity, high-resolution mass spectral data, and HPLC analysis results of the seco DCK analogs 4–9.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, Fujioka T, Mihashi K, Lee KH. J Med Chem. 1994;37:3947. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 2.Xie L, Takeuchi Y, Cosentino LM, Lee KH. J Med Chem. 1999;42:2662. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 3.Yu D, Suzuki M, Xie L, Morris-Natschke SL, Lee KH. Med Res Rev. 2003;23:322. doi: 10.1002/med.10034. [DOI] [PubMed] [Google Scholar]
- 4.Yang ZY, Xia Y, Xia P, Cosentino LM, Lee KH. Bioorg Med Chem Lett. 2000;10:1003. doi: 10.1016/s0960-894x(00)00126-8. [DOI] [PubMed] [Google Scholar]
- 5.Xia P, Yin ZJ, Chen Y, Zhang Q, Zhang BN, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2004;14:3341. doi: 10.1016/j.bmcl.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Huang SX, Xia P, Xia Y, Yang ZY, Yu DL, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2007;17:4316. doi: 10.1016/j.bmcl.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Zhang Q, Zhang BN, Xia P, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg Med Chem. 2004;12:6383. doi: 10.1016/j.bmc.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 8.Yu D, Chen C, Brossi A, Lee KH. J Med Chem. 2004;47:4072. doi: 10.1021/jm0400505. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q, Chen Y, Xia P, Xia Y, Yang ZY, Yu DL, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2004;14:5855. doi: 10.1016/j.bmcl.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 10.Yu D, Brossi A, Kilgore N, Wild C, Allaway G, Lee KH. Bioorg Med Chem Lett. 2003;13:1575. doi: 10.1016/s0960-894x(03)00201-4. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki M, Li Y, Smith PC, Swenberg JA, Martin DE, Morris-Natschke SL, Lee KH. Drug Metab Dispos. 2005;33:1588. doi: 10.1124/dmd.105.004218. [DOI] [PubMed] [Google Scholar]
- 12.Xie L, Yu D, Wild C, Allaway G, Turpin J, Smith PC, Lee KH. J Med Chem. 2004;47:756. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 13.Evers M, Poujade C, Soler F, Ribelli RY, James C, Lelièvre Y, Gueguen JC, Reisdorf D, Morize I, Pauwels R, De Clercq E, Hénin Y, Bousseau A, Mayaux JF, Le Pecq JB, Dereu N. J Med Chem. 1996;39:1056. doi: 10.1021/jm950670t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.