

Abstract
Axon growth is regulated by many proteins, including adhesion molecules, which need to be trafficked correctly to axons. The adhesion molecule L1/neuron–glia cell adhesion molecule (NgCAM) travels to axons via an endocytosis-dependent pathway (transcytosis), traversing somatodendritic endosomes. The Eps15 homology domain (EHD) family proteins (EHD1–EHD4) play important roles in endosomal recycling and possibly in endocytosis. We investigated whether EHD1 regulates L1/NgCAM trafficking in neurons. Both short hairpin-mediated downregulation and overexpression of EHD1 led to dendritic mistargeting of NgCAM. Downregulation of EHD1 showed increased endosomal accumulation of NgCAM, whereas, surprisingly, overexpression of EHD1 led to impairment of L1/NgCAM internalization in neurons but not in fibroblasts. Transferrin internalization, however, was unaffected. At longer overexpression times of EHD1, NgCAM endocytosis returned to normal, suggesting rapid upregulation of compensatory endocytic pathways. EHD1 is capable of hetero-oligomerization, and an endogenous complex of EHD1 and EHD4 was identified previously. We therefore tested whether short-term overexpression of other EHD family members showed a similar endocytosis defect. Expression of EHD4, but not of EHD3, also caused a defect in L1/NgCAM endocytosis. Oligomerization of EHD1 was required to cause NgCAM endocytosis defects, and simultaneous expression of EHD1 and EHD4 rescued NgCAM endocytosis. Therefore, balanced levels of EHD1–EHD4 are important for NgCAM endocytosis in neurons. Our data suggest that EHD1 plays roles in both endosomal recycling and a specialized endocytosis pathway in neurons used by NgCAM. We propose that EHD1 and EHD4 act as hetero-oligomeric complexes in this pathway.
Introduction
Endosome function is important for neurite growth (Hirling et al., 2000; Alberts and Galli, 2003), but the exact role of endosomes in neurite growth is unclear (Sann et al., 2009). One role of the endosomal system might be to regulate levels of adhesion molecules at the cell surface by endocytic uptake and subsequent recycling (Allen and Chilton, 2009; Lasiecka et al., 2009).
L1 is a cell adhesion molecule highly enriched on the axonal surface. Neuron–glia cell adhesion molecule (NgCAM) is the name for the chick homolog of L1. L1 is involved in axonal pathfinding and guidance, axonal branching, and myelination (Maness and Schachner, 2007). The L1 cytoplasmic tail binds a number of intracellular partners (Kamiguchi et al., 1998; Dickson et al., 2002; Nagaraj and Hortsch, 2006; Maness and Schachner, 2007), including the clathrin adaptor AP2. Endocytosis correlates with growth cone advance on L1 substrate (Kamiguchi and Yoshihara, 2001). Our laboratory uncovered that endosomal trafficking is also required to target L1 properly to the growing axon. L1/NgCAM reaches the axon indirectly by transcytosis, involving initial somatodendritic targeting followed by endocytosis and trafficking to the axon from somatodendritic endosomes (Wisco et al., 2003; Yap et al., 2008a,b). Other cargos, such as transferrin (Tfn) receptors, also endocytose and traverse somatodendritic endosomes but are not ultimately sorted to axons (Park et al., 2004; Yap et al., 2008b).
The Eps15 homology domain (EHD) family, comprising EHD1–EHD4, plays crucial roles in endosomal trafficking. The EHD proteins have a highly conserved domain structure. The N-terminal G domain is required for ATP binding and membrane recruitment, the central region for dimerization/oligomerization, and the C-terminal EH domain for binding to NPF motifs in a number of proteins (Grant and Caplan, 2008). The EH domain also contains a lipid binding site (Naslavsky et al., 2007; Grant and Caplan, 2008; Jovic et al., 2009). Structural studies of EHD2 suggest that oligomers of EHD2 bind to lipids and deform them into tubules, initiating budding (Daumke et al., 2007). EHD proteins also associate with preexisting tubular membranes (Jovic et al., 2009). It is likely that different EHD oligomers regulate distinct transport steps, but which oligomer acts where remains essentially unknown. In nonpolarized cells, EHD1 (Rme1), EHD3, and EHD4 (pincher) function in endosomes and regulate trafficking through early and recycling endosomes (Grant and Caplan, 2008). EHD2, conversely, appears to regulate endocytosis (Guilherme et al., 2004). EHD1 has also been suggested to regulate endocytosis of IGF1 receptor (Rotem-Yehudar et al., 2001).
In neurons, EHD4 (Shao et al., 2002) has been implicated in endocytosis rather than (or in addition to) recycling (Sharma et al., 2008). EHD4 was identified as an NGF-induced protein in PC12 cells, and it mediates Trk receptor endocytosis via a novel, neurotrophin-stimulated macroendocytosis pathway (Valdez et al., 2005, 2007). In this study, we propose that L1/NgCAM, but not transferrin, is a cargo for EHD1/EHD4-sensitive uptake pathways and that hetero-oligomers of EHD1 and EHD4 are involved in regulating the endocytosis of NgCAM in neurons.
Materials and Methods
Cell culture
Hippocampal neurons.
Primary cultures of hippocampal neurons are obtained from embryonic day 18 rat hippocampi as described previously (Yap et al., 2008b). Cells were plated on poly-l-lysine coverslips and incubated with DMEM with 10% horse serum. After 4 h, the cells were transferred into serum-free medium supplemented with B27 (Invitrogen) and cultured for 9–12 d in vitro (DIV).
COS cells and NRK cells.
COS and NRK (normal rat kidney) cells were maintained in DMEM plus 10% fetal bovine serum using standard cell culture methods.
PC12 cells.
PC12 cells were maintained in DMEM plus 10% horse serum plus 5% fetal bovine serum, and all transfections were conducted using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer.
Reagents
Antibodies and plasmids.
Anti-EHD1 antibodies were raised in rabbits against the peptide ADLPPHLIPPSKRRHE. The 8D9 anti-NgCAM hybridoma developed by Dr. Vance Lemmon was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology (Iowa City, IA). Rabbit anti-FLAG, anti-hemagglutinin (HA), and anti-myc antibodies were purchased from Sigma and Santa Cruz Biotechnology, respectively, whereas mouse anti-rab5 antibody was from Abcam. EHD family plasmids [myc–EHD1, myc–EHD1V203P, green fluorescent protein (GFP)–EHD1V203P, myc–EHD1G65R, myc–EHD3, HA–EHD4, and HA–EHD4G68R] and affinity-purified anti-EHD1 and anti-EHD4 antibodies were described previously (Caplan et al., 2002; Naslavsky et al., 2006, 2009; Sharma et al., 2008). FLAG–Rme1 and FLAG–Rme1G429R were kindly provided by Dr. Fred Maxfield (Weill Cornell Medical College, New York, NY) and Dr. Barth Grant (Rutgers University, Piscataway, NJ). Low-density lipoprotein receptor (LDLR) plasmid was a gift from Dr. Heike Folsch (Northwestern University, Evanston, IL). Anti-rat L1 rabbit polyclonal antibody raised against rat L1 was kindly provided by Dr. Vance Lemmon (University of Miami, Coral Gables, FL). Hybridoma producing anti-LDLR antibodies was purchased from American Type Culture Collection.
Short-hairpin RNA plasmids.
To specifically knockdown the EHD1 protein expression, we used short-hairpin RNA (shRNA) targeting the rat EHD1 sequences obtained from Super Array Bioscience. The shRNAs were expressed from the U1 promoter cassette of pGeneclip hMGFP vector containing GFP under the regulation of the cytomegalovirus promoter. The sequences of the four short-hairpin RNA targeting the rat EHD1 were as follows: shEHD1#1, GCC CAC CTA TGA TGA GAT CTT; shEHD1#2, AAG GAG ATG CCC AAT GTC TTT; shEHD1#3, CTG CGT AAG ATG CAG GAA CTT; and shEHD1#4, CTG CTA GAT GAC GAG GAG TTT. The scrambled sequence used as a control (shRandom) was GGA ATC TCA TTC GAT GCA TAC.
Transfection of neuronal cultures
Neuronal cultures at 9–12 DIV were transfected using Lipofectamine2000 with 1 μg of DNA, 3 μl of Lipofectamine2000 for 60–90 min, washed, and incubated for 16–20 h. To reduce the number of overexpressing cells, NgCAM plasmid was mixed with empty plasmid containing no insert. When indicated by the experimental design, empty plasmid was included to maintain the same experimental plasmid concentrations across multiple experimental conditions. Nucleofection of freshly dissociated neurons was performed as described previously (Yap et al., 2008) using a Nucleofector device (Amaxa Biosystems). At 1 and 3 DIV after nucleofection, endocytosis assay with acid strip was performed to visualize endocytosed L1 in transfected neurons.
Immunocytochemistry
Cells were fixed in 2% paraformaldehyde/3% sucrose/PBS in 50% conditioned medium at room temperature for 30 min, quenched in 10 mm glycine/PBS for 10 min. The fixation conditions used do not introduce holes into the overwhelming majority of cells. Coverslips were then blocked in 5% horse serum/1% BSA/PBS with or without 0.05% Triton X-100 for 30 min. Antibodies were diluted in 1% BSA/PBS and incubated for 1–2 h. Coverslips were mounted in Vectashield (Vector Laboratories) and viewed on a Carl Zeiss Axiophot with a 40× objective. Images were captured with the Orca cooled CCD camera (Hamamatsu) using Openlab software (ImproVision) and processed identically in Adobe Photoshop (Adobe Systems). For all surface staining, live cells were incubated for 20 min at 37°C in primary antibody before fixation, and secondary antibody was applied before permeabilization.
Endocytosis assay
Neurons expressing NgCAM for 18 h were incubated with 8D9 anti-NgCAM antibodies for 20 min at 37°C and washed several times, and all antibody remaining on the surface was stripped by treatment with pH 2.0, MEM for 2 min (Fourgeaud et al., 2003), washed extensively, and returned to the incubator for various amounts of times before fixation in 2% paraformaldehyde/3%sucrose/PBS, pH 7.4. Internalized NgCAM antibody was detected by applying Alexa-568 goat anti-mouse antibody after permeabilization. Permeabilization was achieved either with 0.2% saponin or 0.2% Triton X-100 for 10 min at room temperature as in a previous study (Wisco et al., 2003).
Polarity index
Axon and dendrite fluorescence intensities were measured along a 1-pixel-wide line in NIH ImageJ after background subtraction and normalized to length. The axonal polarity of L1/NgCAM is determined by dividing average axonal intensity of L1/NgCAM surface staining by the average intensity of surface L1/NgCAM on dendrites (Wisco et al., 2003). The axon/dendrite polarity index (A/D PI) is a very sensitive readout of NgCAM/L1 polarization, and even small effects because of partial downregulation can be detected.
Quantification and statistical analysis
For quantifying axonal polarity or somatic endosomal NgCAM, 15–20 cells were quantified per conditions in each of minimally three experiments. For determining the percentage of cells endocytosing NgCAM, 150–200 cells per condition and experiment were counted in minimally three separate experiments. Expression levels of transfected proteins were compared for differences in expression levels. If necessary, plasmid concentrations were adjusted to equalize expression levels. We routinely transfect in excess of 150 cells per coverslips, easily enough for scoring sufficient numbers of cells. Cells were excluded from scoring if their morphology is abnormal, indicating poor health. Statistical tests were performed using SPSS version 13 Statistics software.
Results
Overexpression of EHD1 or dominant-negative EHD1 diminishes axonal polarization of NgCAM
EHD1 plays a role in endosomal recycling in non-neuronal cells. Likewise, interference with EHD1 function using a dominant-negative EHD1 impedes the recycling of AMPA receptors in neurons (Park et al., 2004). Because axonal localization of NgCAM requires endosomal trafficking, we asked whether the same dominant-negative EHD1 (EHD1G429R) interfered with proper axonal localization of NgCAM. Cultured hippocampal neurons at 10–12 DIV were transfected with NgCAM plasmid and simultaneously with GFP as control, wild-type FLAG–EHD1, or FLAG–EHD1G429R. At 18 h after transfection, live neurons were incubated with anti-NgCAM antibody before fixation to visualize the surface distribution of NgCAM. NgCAM in control neurons (coexpressing GFP) accumulated preferentially on the axon at steady state and was only faintly visible on the soma and dendrites (Fig. 1A). To quantify the extent of polarized expression, we determined the average pixel intensity along the distal part of axons and along the dendrites and calculated a polarity index by dividing the average axon intensity by the average dendrite intensity (A/D PI, as in the study by Wisco et al., 2003). In GFP-expressing control cells, NgCAM was fivefold to sixfold enriched on the axon compared with dendrites (Fig. 1D). In contrast, neurons coexpressing EHD1G429R showed an increased presence of NgCAM on the somatodendritic surface (Fig. 1C), resulting in a significant decrease of A/D PI (Fig. 1D). Surprisingly, coexpressing wild-type EHD1 also decreased A/D PI of surface NgCAM (Fig. 1B,D). As reported previously (Park et al., 2004), overexpression of EHD1 induced many short dendritic protrusions resembling filopodia and spines. These results indicated that changing the levels and/or function of EHD1 disrupted correct targeting of NgCAM to the axon.
Figure 1.

Overexpression and downregulation of EHD1 diminishes NgCAM polarization to the axon. A–C, The surface pool of NgCAM (red) was stained in live cells coexpressing (in green) GFP as control (A), wild-type (WT) FLAG–EHD1 (B), or FLAG–EHD1G429R (C). The merged green and red channels are shown in the right column. The axon is indicated by an arrow. D, The A/D PI was determined and plotted. A/D PI is diminished for NgCAM in the presence of EHD1G429R or wild-type EHD1. **p < 0.001, statistically significant from GFP control. Error bars indicate SEM. n = 20 cells for NgCAM plus GFP, 38 cells for NgCAM plus EHD1G429R, and 33 cells for NgCAM plus wild-type EHD1. A/D PI of NgCAM Y33A is unaffected by overexpression of wild-type EHD1 or EHD1G429R. n = 20 cells for NgCAM Y33A plus GFP, 25 cells for NgCAM Y33A plus EHD1G429R, and 26 cells for NgCAM Y33A plus wild-type EHD1. E, The average fluorescence intensity of surface NgCAM was determined along dendrites for cells expressing shRandom-GFP, shEHD1#1–GFP, or shEHD1#2–GFP for 4 d. Average fluorescence intensities were normalized to the values of shRandom–GFP. n = 35 cells for shRandom–GFP, 44 cells for shEHD1#1–GFP, and 35 cells for shEHD1#2–GFP. Error bars indicate SEM. *p < 0.05, **p < 0.00001. The measurements used for D (GFP, EHD, and EHDG429R) were also displayed as normalized dendrite intensity for comparison.
To test whether NgCAM missorting occurred on the endocytic or the biosynthetic post-Golgi pathway, we coexpressed a point mutant of NgCAM, NgCAMY33A, together with GFP as control, EHD1, or EHD1G429R. We showed previously that NgCAMY33A did not use the transcytosis pathway but instead traveled to the axon directly from the trans-Golgi network on an endocytosis-independent pathway (Wisco et al., 2003). Impairment of the endocytic pathway would therefore not disturb axonal polarization of NgCAMY33A, whereas impairment of post-Golgi sorting would. NgCAMY33A showed normal axonal polarization (A/D PI) regardless of the coexpression of EHD1 (Fig. 1D), placing the EHD1-dependent defect in the endocytic (uptake per se) or endosomal (post-uptake) pathway.
Downregulation of EHD1 diminishes axonal polarization of NgCAM
Our finding that wild-type EHD1 and EHD1G429R overexpression caused a decrease in NgCAM axonal polarity raised the possibility that NgCAM polarization to the axon was sensitive to levels of EHD1. We therefore performed experiments using small-hairpin-interfering plasmids targeting EHD1 to knockdown EHD1 expression levels. The efficiency of downregulating endogenous EHD1 was tested for four different shEHD1 plasmids in NRK cells compared with a control plasmid containing a random sequence (shRandom–GFP). All shRNA plasmids also encoded GFP for identification of transfected cells. Three of the four shRNA plasmids gave consistent downregulation of endogenous EHD staining in NRK cells. Quantification of endogenous EHD1 in NRK cells revealed that expression of shEHD1#1 or shEHD1#2 plasmids resulted in an average of 40–60% decrease of endogenous EHD1 levels (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The levels of another EHD family member, EHD4, conversely, were not decreased by shEHD1 plasmids in NRK cells (data not shown). We then transfected neuronal cultures at 7 DIV with NgCAM plasmid and either one of two shEHD–GFP plasmids, shEHD1#1–GFP or shEHD1#2–GFP or with control shRandom–GFP. To determine whether the polarized distribution of L1/NgCAM was affected when shEHD1 plasmids were expressed, we measured the average fluorescence intensities of surface NgCAM along dendrites at 11 DIV. We usually determine an A/D PI to quantify the relative localization of NgCAM to axons and dendrites. We were unable to determine the A/D PI in cells expressing shEHD1 plasmids because the crisscrossing of NgCAM-expressing axons became so extensive after 4 d of NgCAM and shEHD1 expression that the axon tips could not be unambiguously traced back to their cell bodies. We therefore measured the intensities of surface NgCAM on dendrites only. In control cells expressing NgCAM and shRandom–GFP control plasmid, NgCAM dendrite intensity was 756 ± 35 arbitrary fluorescent units (AFU) (mean ± SEM). Cells expressing NgCAM and shEHD1#1 or shEHD#2 plasmids showed increased NgCAM intensity on dendrites (shEHD1#1–GFP dendrite intensity, 1233 ± 44 AFU, mean ± SEM, p < 0.0001; shEHD1#2–GFP dendrite intensity, 921 ± 35 AFU, mean ± SEM, p < 0.05), indicating that NgCAM showed mislocalization to the dendritic surface when EHD1 was downregulated by two different shRNA plasmids. Normalized data are shown in Figure 1E. Average dendrite levels of NgCAM are also shown for EHD1 and EHD1G429R overexpression for comparison. The effect of shEHD1#1–GFP was larger than that of shEHD1#2–GFP but qualitatively the same. We thus find that both increased and decreased levels of EHD1 causes increased accumulation of NgCAM on the dendritic surface.
Upregulation and downregulation of EHD1 levels lead to different effects on endosomal accumulation of NgCAM
So far, we found that expression of EHD1 or EHD1G429R led to missorting of NgCAM to the somatodendritic surface. Similarly, there are increased levels of NgCAM on dendrites in cells expressing shEHD1 plasmids. Because EHD family members play roles in both endocytosis and endosomal recycling and localize to both endosomes and the plasma membrane, the increased accumulation of NgCAM on the dendritic surface could be attributable to decreased endocytosis from the somatodendritic domain (possibility 1) or missorting to the somatodendritic surface from endosomes during recycling (possibility 2). To distinguish between these possibilities, we performed NgCAM endocytosis assays (as in the study by Yap et al., 2008b). We predicted that there would be either lower levels of NgCAM in endosomes because of decreased endocytosis (possibility 1) or increased levels of NgCAM in endosomes because more NgCAM was available on the somatodendritic surface for antibody uptake (possibility 2). Control cells (expressing GFP or shRandom–GFP) showed robust uptake of NgCAM into somatodendritic endosomes (Fig. 2A, red; D; F, top). We quantified the fluorescence of endocytosed NgCAM associated with somatic endosomes using NIH ImageJ. Knockdown of EHD1 using two different shEHD1 plasmids increased somatodendritic endosomal accumulation of NgCAM (Fig. 2F, bottom; G). This observation is consistent with possibility 2, potentially reflecting missorting during recycling. The increased accumulation of endosomes could additionally be attributable to slowed recycling from endosomes and/or defects in transport of endosomes to the axon.
Figure 2.

Overexpression of EHD1 diminishes endosomal accumulation of NgCAM, whereas downregulation of EHD1 increases endosomal accumulation. A–C, Endocytosis of NgCAM (after 20 min of anti-NgCAM antibody uptake; red) in the somatodendritic domain in neurons expressing (in green) GFP as control (A), FLAG–EHD1G429R (B), or wild-type (WT) FLAG–EHD1 (C). The merged green and red channels are shown in the right column. The dendrites are indicated by arrowheads. D shows a magnified view of endocytosed NgCAM from the cells shown in A–C. E, The percentage of neurons showing endocytic uptake of NgCAM was determined for 150–200 cells each in four independent experiments. Error bars indicate SEM. *p < 0.01, statistically significant from GFP. F, Endocytosis of NgCAM (after 20 min of anti-NgCAM antibody uptake) in the somatodendritic domain in neurons expressing shRandom–GFP as control (top) or shEHD1#1–GFP (bottom). The GFP channel is shown in the inset. G, Endosomal levels of NgCAM (after 20 min of anti-NgCAM antibody uptake) were quantified in NIH ImageJ for cells expressing shRandom–GFP or two different shEHD1 plasmids targeting different regions of EHD1 (shEHD1#1–GFP and shEHD1#2–GFP). Values are normalized to shRandom–GFP. Two independent experiments were quantified and their averages combined in the graph. n = 43 cells for shRandom–GFP, 47 cells for shEHD1#1–GFP, and 32 cells for shEHD1#2–GFP. Error bars are SEM. *p < 0.05, **p < 0.001.
In contrast, when EHD1 or EHD1G429R were expressed, NgCAM labeling of somatic endosomes was greatly decreased (Fig. 2B–D) compared with GFP–control cells. Quantifying the percentage of transfected cells that showed NgCAM endocytosis revealed that both EHD1 and EHD1G429R showed a significant decrease in the percentage of cells showing endocytosis (Fig. 2E). We noted that cells still showing NgCAM endocytosis usually expressed lower levels of EHD1, whereas endocytosis was not or barely detectable in cells with high overexpression levels. In addition to determining the percentage of endocytosing cells, we also quantified the fluorescence of endocytosed NgCAM associated with somatic endosomes in GFP-expressing control cells, EHD1G429R-expressing cells, and EHD1-expressing cells (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Cells coexpressing EHD1 showed on average less than half of NgCAM soma fluorescence of GFP cells, whereas EHD1G429R was reduced to 65% of control.
Because many cells showed no detectable uptake of NgCAM into somatic endosomes when EHD1 was overexpressed, we conclude that NgCAM either (1) does not endocytose efficiently in the first place or (2) undergoes endocytosis followed by rapid recycling from a pre-early endosome (pre-EE) compartment that is membrane-near and very small in size, so that it would not appear as distinct puncta by immunofluorescence. These two possibilities are not mutually exclusive. Because it is clear that NgCAM does not accumulate in the large and easily visualized EEA1-positive endosomes in most EHD1-expressing cells, we refer to the combined possibilities of inefficient endocytosis and rapid pre-EE recycling as “endocytosis defect” in the remainder of this work.
Loss-of-function phenotype of EHD1 in neurons?
Surprisingly, we find that knockdown of EHD1 showed a different phenotype from EHD1 and EHD1G429R overexpression and reduced endocytosis was not apparent after 4 d of shEHD1 expression. The different phenotypes of the knockdown and dominant-negative experiments could arise if the level of downregulation achieved was not sufficient to disturb NgCAM endocytosis but was sufficient to disturb endosomal recycling.
In addition, because the time courses of the two experiments were strikingly different (18 h for EHD1 overexpression and 4 d for shEHD1 expression), the time course could affect the phenotype. We therefore repeated the EHD1 overexpression experiment with a longer time course. Strikingly, despite the continued presence of overexpressed EHD1, NgCAM endocytosis after 4 d of overexpression was not statistically different from that of GFP control cells, whereas it was statistically significant after 18 h of expression (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The disappearance of the NgCAM endocytosis defect after 4 d of EHD1 expression suggested that compensatory endocytic pathways were upregulated over time that were not operative in 18 h. Such compensatory mechanisms are frequently observed when other endocytic proteins are inhibited or downregulated (Damke et al., 1995; Kim and Ryan, 2009). In agreement with our observations, compensation of L1 uptake was also observed in previous work using L1 with mutations in the endocytosis motif (Long et al., 2001). We therefore document here another example that longer-term inhibition of endocytic pathways by knockdown or knock-out approaches can cause fairly rapid compensatory mechanisms to emerge in cells. Because the acute effect of changing EHD1 levels was the impairment of NgCAM endocytosis, we focused our analysis in this work on the more acutely occurring phenotype. The recycling defects evident after 4 d of EHD1 knockdown will be analyzed in more detail elsewhere (manuscript in preparation).
Expression of EHD1G429R, but not wild-type EHD1, leads to defective recycling of transferrin
The apparent decrease in somatodendritic endocytosis of NgCAM caused by EHD1 constructs was unexpected because no defect in endocytosis had been observed for Tfn in non-neuronal cells (Lin et al., 2001; Caplan et al., 2002; Braun et al., 2005), or neurons, or for AMPA receptors in neurons (Park et al., 2004). Rather, recycling of Tfn and AMPA receptors was impaired by expression of EHD1G429R, leading to increased endosomal accumulation. Wild-type EHD1 overexpression, conversely, had no effect on Tfn or AMPA receptor recycling. Consistent with the published literature, we found that steady-state loading of Tfn in cells coexpressing either EHD1 or EHD1G429R was the same as in untransfected controls (supplemental Fig. 4A,B, “t0,” available at www.jneurosci.org as supplemental material), but Tfn showed slowed recycling (t = 45 min chase) when EHD1G429R was coexpressed (supplemental Fig. 4B, “t45,” available at www.jneurosci.org as supplemental material). Tfn recycling, but not steady-state endocytosis, was therefore impaired by EHD1G429R but not by wild-type EHD1. Surprisingly then, our results showed that the effects of EHD1 overexpression appeared to be specific to the cargo, affecting Tfn and NgCAM differently.
Overexpression of EHD1 in non-neuronal cells does not inhibit NgCAM endocytosis
To investigate whether the EHD1 effect on NgCAM endocytosis was specific to neuronal cells, we repeated NgCAM uptake experiments with or without coexpressed EHD1 or EHD1G429R in two non-neuronal cell lines, COS cells (Fig. 3) and NRK cells (data not shown). In both of these non-neuronal cells, NgCAM uptake was easily observed in control cells expressing GFP as well as in cells coexpressing EHD1 or EHD1G429R (Fig. 3A–C), and levels of endocytosed NgCAM were not statistically different between control cells and EHD1-expressing cells (Fig. 3D). L1/NgCAM uptake is therefore affected in a cell-type-specific manner by overexpression of EHD1.
Figure 3.

Overexpression of EHD1 in non-neuronal cells does not lead to NgCAM endocytosis impairment. A–C, COS cells were cotransfected with NgCAM and GFP as control (A), wild-type FLAG–EHD1 (B), or FLAG–EHD1G429R (C). Anti-NgCAM antibody uptake was performed for 20 min before fixation and immunostaining. Endocytosed NgCAM is shown in the left column (red), and the FLAG tag is shown in the middle column (green). Merged red and green channel are shown in the right column. Robust accumulation of NgCAM in endosomes is observed for all conditions. D shows quantification of endocytosed NgCAM intensity normalized to GFP controls. n = 29 cells for GFP, 32 cells for EHD1, and 42 cells for EHD1G429R. The differences were not statistically significant.
EHD4 affects NgCAM endocytosis and polarity
EHD1 is part of a gene family with four closely related members, EHD1–EHD4 (Grant and Caplan, 2008). EHD2 is minimally expressed in mammalian brain (Blume et al., 2007; George et al., 2007). We therefore determined the extent of NgCAM endocytosis in cells coexpressing myc–EHD3 or HA–EHD4. Overexpression of EHD3 did not cause impairment of NgCAM endocytosis (Fig. 4B,E). A second quantification method is shown in supplemental Figure 2B (available at www.jneurosci.org as supplemental material). Intriguingly, overexpression of EHD4 significantly impaired NgCAM endocytosis (Fig. 4C,E) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material) compared with GFP-expressing cells (Fig. 4A,E). An inactive mutant of EHD4 (EHD4G68R), conversely, which has no ATPase activity, does not bind to membranes, and does not dimerize/oligomerize (Sharma et al., 2008), showed normal levels of NgCAM endocytosis (Fig. 4D).
Figure 4.

EHD4, but not EHD3, affects NgCAM endocytosis and axonal polarity. A–D, Endocytosis of NgCAM (after 20 min of anti-NgCAM antibody uptake; red) in the somatodendritic domain in neurons expressing (in green) GFP as control (A), myc–EHD3 (B), HA–EHD4 (C), or the inactive mutant HA–EHD4G68R (D). The “overlay” column shows the merged red and green channels. The “zoom” column shows a magnified view of endocytosed NgCAM from the cells shown in A–D. E, myc–EHD3 or HA–EHD4 were expressed in neurons for 18 h, and NgCAM endocytosis was determined. The percentage of neurons showing endocytic uptake of NgCAM was determined counting 150–200 cells each in three to four independent experiments. Error bars indicate SEM. *p < 0.01, statistically significant from GFP. F, A/D PI of surface NgCAM was determined for neurons coexpressing GFP as control, myc–EHD3, or HA–EHD4. Error bars indicate SEM. **p < 0.001, statistically significant from GFP. n = 20 for GFP, 29 for EHD3, and 36 cells for EHD4.
Next, we determined whether NgCAM surface polarity was also disturbed in cells overexpressing EHD3 or EHD4. The A/D PI for NgCAM surface localization was not changed by coexpression of EHD3 compared with GFP-expressing control cells, but coexpression of EHD4 led to a significant decrease in NgCAM axon/dendrite polarity (Fig. 4F), similar to that found for EHD1 overexpression.
The lack of phenotype for EHD3 was surprising given the close sequence homology with EHD1. We therefore checked whether myc–EHD3 expression levels were in a similar range to EHD1 overexpression. For this purpose, we repeated the NgCAM polarity index determination for myc–EHD1 and myc–EHD3. The fluorescence intensity ranges for the two myc-tagged constructs was not different, and NgCAM A/D PI remained high even at high expression levels of myc–EHD3 (data not shown).
Overexpression of EHD4 impairs endocytosis of endogenous L1
We next determined whether the endocytosis of endogenous L1 was also impaired by EHD4 overexpression. The neuroendocrine cell line PC12 highly expresses L1 endogenously. We therefore tested the effects of EHD4 overexpression in PC12 cells, using endocytosis of endogenous L1 as an assay. Similarly to NgCAM in mature primary neurons, L1 uptake was inhibited in PC12 cells highly overexpressing EHD4 (marked with asterisks) but not in cells expressing GFP or low levels of EHD4 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Next, we asked whether endocytosis of endogenous L1 in primary hippocampal neurons was sensitive to EHD4 overexpression. For this experiment, we electroporated dissociated hippocampal neurons before plating with EHD4–HA plasmid and then measured endocytosis of endogenous L1 either the next day or after 3 d. For technical reasons, we were not able to perform this experiment in more mature cultures (as detailed in the study by Yap et al., 2008b). Endogenous L1 endocytosis was significantly inhibited by EHD4 overexpression after 1 d (Fig. 5A,C) (43% of control levels) but not after 3 d (Fig. 5B,C) (94% of control levels) of overexpression. Similarly to NgCAM endocytosis therefore, L1 endocytosis was inhibited at 18 h of EHD4 expression but mostly restored after 3 d of expression.
Figure 5.

Endocytosis of endogenous L1 is inhibited by EHD4 overexpression in cultured hippocampal neurons. A, B, HA–EHD4 was electroporated into dissociated hippocampal neurons before plating (left column), and endocytosis of L1was assayed (right column) after 1 d (A) or 3 d (B) of expression using anti-L1 antibody uptake for 20 min. Arrows point at transfected neurons. Untransfected neurons show robust anti-L1 uptake (right column). C, The fluorescence intensity of endocytosed L1 in the soma of untransfected control cells (black bars) and HA–EHD4-expressing cells (gray bar) was quantified and normalized to control cells. Error bars indicate SEM. ***p < 0.0001. n = 55 transfected cells for day 1; n = 29 transfected cells for day 3. One representative experiment is shown.
Localization of EHD1 and EHD4 in neurons
The distribution of EHD proteins for both endogenous and exogenous protein has been characterized in several non-neuronal cell types (Blume et al., 2007; George et al., 2007) but has not been investigated in neurons. In non-neuronal cells, EHD1 is found in endosomes as well as long tubules that are part of the recycling endosomes. A subpopulation of EHD1 also localizes to dispersed early endosomes and to the plasma membrane (Lin et al., 2001). EHD3 and EHD4 are also found in endosomes throughout the cell but are more heavily concentrated in early endosomes (Galperin et al., 2002; Sharma et al., 2008). In PC12 cells and in primary neuronal cultures, EHD4 is also found at the plasma membrane in addition to endosomes (Valdez et al., 2005). We raised an antibody in rabbits against the C terminus of EHD1. The antibody recognized multiple members of the EHD family on Western blots of tagged EHD proteins (data not shown).
To determine whether EHD family proteins localize to endomembranes in neurons, neuronal cultures were stained with the anti-Ct EHD antibody after permeabilization. Numerous puncta reminiscent of endosomes were stained in both neurons and glial cells (Fig. 6A–E). Preabsorption of the antibody with the immunizing peptide abolished the staining (data not shown). In glial cells, short EHD-positive tubules were frequently detected (Fig. 6E), similar to staining reported for other non-neuronal cells. In neurons, tubules were not prominent and only occasionally could short tubules be found. The most prominent staining in neurons was clusters of organelles in the soma. Additionally, dispersed presumptive endosomes were stained in both dendrites and axons, albeit more faintly in axons (Fig. 6A,C). Often, EHD-positive compartments could also be detected in axonal growth cones (Fig. 6D). These compartments were mostly round. In addition to the punctate staining in the cytoplasm, the nucleus was also stained in neuronal cultures in many instances (Fig. 6A). Nuclear staining was competed by incubation with the immunizing peptide (data not shown). No nuclear staining was observed with the same antibody and staining protocol in fibroblast cell lines in which only the presumptive endosomal staining was visible (data not shown). The significance of the nuclear staining in neurons is not known, but a nuclear localization signal has been identified in EHD1 from the sequence (Haider et al., 1999).
Figure 6.

Localization of endogenous EHD family proteins. A, EHD proteins localize to bright puncta in dendrites (counterstained with MAP2 in B) and more faintly in axons. Additionally, diffuse cytoplasmic or membrane staining is detectable. In addition, the nucleus is stained brightly. C and D show zoomed images of a dendrite (C) and an axonal growth cone (D). E, Glial cells also show numerous round puncta as well as elongated tubules. F, Neuronal cultures were stained with an anti-EHD1 affinity-purified antibody. A zoomed image of the boxed area is shown in F′. The nucleus stains brightly with only faint staining elsewhere. G, H, Neuronal cultures were stained with an anti-EHD4 affinity-purified antibody (H) and the somatodendritic marker MAP2 (G). A zoomed image of the boxed area is shown in H′.
We additionally used affinity-purified anti-EHD1 and anti-EHD4 antibodies that are specific to their respective EHD family members (Sharma et al., 2008). These specific reagents are difficult to obtain and show only faint staining in neurons. We again see strong nuclear staining with the anti-EHD1 antibody (Fig. 6F) as well as punctate staining of presumptive endosomes throughout the soma, dendrites, and axons (Fig. 6F′). This staining pattern is consistent with EHD1 distribution in non-neuronal cells with the notable exception of the nuclear staining, which we only observe in the neuronal cultures. Affinity-purified anti-EHD4 antibody did not stain the nucleus of neurons, but staining was found prominently on punctate structures in the soma and dendrites (Fig. 6H,H′).
EHD1 oligomerization is necessary for inhibition of NgCAM endocytosis
EHD family members dimerize with themselves and with other members of the EHD family and form higher oligomeric structures that are crucial to their function (Grant and Caplan, 2008). Notably, the only dimer currently known to exist endogenously is the EHD1/EHD4 dimer (Sharma et al., 2008). Proteins participating in multimeric functional complexes are often sensitive to expression levels because the balance of subunits is crucial for correctly assembling the oligomeric structure (Papp et al., 2003). Because overexpression of wild-type EHD1 or EHD4 had the same phenotype for NgCAM endocytosis, we hypothesized that the mechanism of the overexpression phenotype of EHD1 or EHD4 might be that the proper balance of components required for NgCAM endocytosis in neurons was changed. Because EHD4 overexpression phenocopied EHD1 overexpression, we tested whether the capability for dimerization/oligomerization was necessary for the effect of EHD1 overexpression on NgCAM endocytosis. To test this idea, we used a previously characterized EHD1 mutant (myc–EHD1V203P) (Fig. 7A), which shows a significantly decreased capacity for dimerization/oligomerization with EHD family members by coimmunoprecipitation and two-hybrid analysis (Naslavsky et al., 2006; Sharma et al., 2008).
Figure 7.
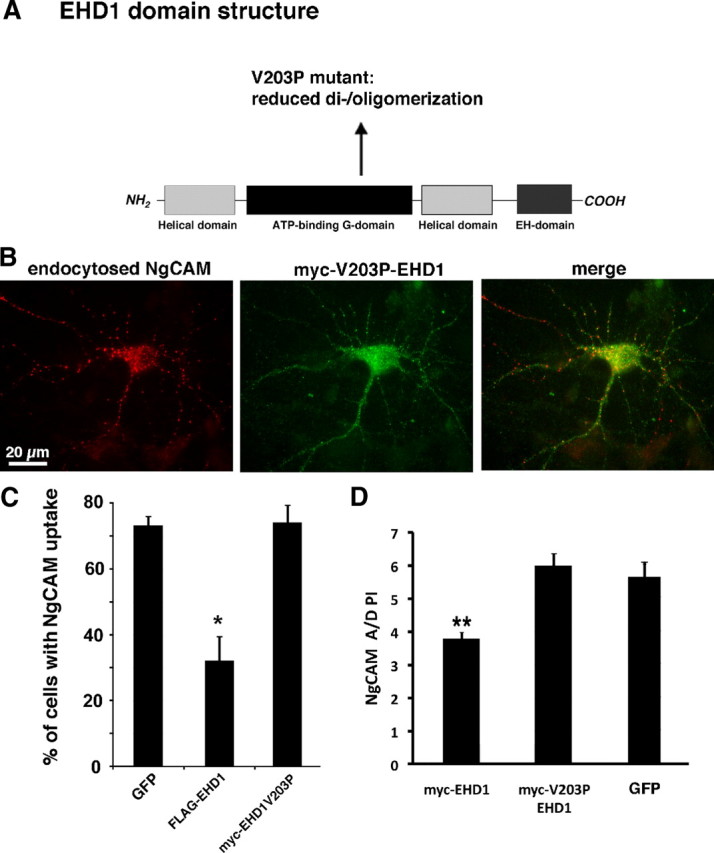
EHD1 oligomerization is necessary for inhibition of NgCAM endocytosis. A, Schematic diagram of the domain organization of EHD1. The position of the V203 mutant is indicated. B, Endocytosis of NgCAM (after 20 min of anti-NgCAM antibody uptake; red) in the somatodendritic domain in a neuron coexpressing myc–EHD1V203P (green). Endocytosis of NgCAM is high in cells expressing myc–EHD1V203P. C, The percentage of neurons showing endocytic uptake of NgCAM was determined for 150–200 cells each in four independent experiments. The oligomerization-impaired EHD1V203P mutant does not impair NgCAM endocytosis. Error bars indicate SEM *p < 0.01, statistically significant from GFP. D, A/D PI of surface NgCAM was determined for neurons coexpressing myc–EHD1, or myc–V203P EHD1 or GFP as control. Error bars indicate SEM. **p < 0.0001, statistically significant from GFP. n = 32 for myc–EHD1 and 25 for myc–V203P EHD1. The expression range of the two myc-tagged constructs is comparable (data not shown).
In contrast to wild-type EHD1, expression of EHD1V203P did not interfere with NgCAM uptake (Fig. 7B,C). Likewise, NgCAM axonal polarity (A/D PI) was not diminished by EHD1V203P overexpression (Fig. 7D). As before, we checked the expression levels of myc–EHD1V203P compared with myc–EHD1 as well as GFP–EHD1V203P compared with GFP–EHD1 and found them to be in the same range (data not shown). We therefore conclude that interactions disrupted by the V203P point mutation (most likely dimerization/oligomerization with another EHD family member) were required for the decrease in NgCAM endocytosis observed with EHD1 expression.
Coexpression of EHD1 rescues the endocytosis and polarity defects of EHD4
Because both EHD1 and EHD4 overexpression caused impairment of NgCAM endocytosis and the dimerization/oligomerization capability of EHD1 was necessary for the observed effect, a balanced expression of EHD1 and EHD4 might be required for normal NgCAM endocytosis. We argued that overexpression of EHD1 or EHD4 would change the relative levels of EHD1 and EHD4 and thereby disturb the required balance of EHD1 versus EHD4 subunits for formation of functional EHD1/EHD4 oligomers. If this were correct, simultaneous overexpression of both EHD1 and EHD4 should restore the proper balance of EHD1 and EHD4 and lead to rescue of the NgCAM endocytosis defects observed during overexpression of only one of the proteins. We therefore expressed NgCAM together with EHD1 and EHD4 and performed NgCAM endocytosis assays. Figure 8A shows an example of a triple-transfected cell expressing FLAG–EHD1, HA–EHD4, and endocytosed NgCAM. High levels of endocytosed NgCAM (right panel) could be observed. Endocytosed NgCAM partially overlapped with cytoplasmic puncta of EHD1 and EHD4 staining (Fig. 8B), in agreement with the endosomal localization of EHD1 and EHD4 observed in fibroblasts (Grant and Caplan, 2008). Endosomal localization of EHD1 and EHD4 was also confirmed by costaining with the endosomal protein rab5 (Fig. 8C, arrows).
Figure 8.

EHD1/EHD4 coexpression rescues NgCAM endocytosis and axonal polarity. A Coexpression of HA–EHD4 (left) and FLAG–EHD1 (middle) rescues endocytosis of NgCAM (right). A single confocal section is shown. B, The area boxed in A is displayed as two-color (1–3) or triple-color (4) overlays. The colors are as indicated below: red is endocytosed NgCAM, blue is HA–EHD4, and green is myc–EHD1. C, Neurons were transfected with either FLAG–EHD1 (left column) or HA–EHD4 (right column) and costained with antibodies against rab5. Merged images are shown in the top row (red, anti-HA or anti-FLAG; aqua, rab5). The corresponding single-channel images are shown in the middle row (rab5) and the bottom row (FLAG or HA). D, The percentage of neurons showing endocytic uptake of NgCAM was determined for cells coexpressing FLAG–EHD1 and HA–EHD4, myc–EHD1V203P and HA–EHD4, or myc–EHD3 and HA–EHD4. Only wild-type EHD1 rescued NgCAM endocytosis. One hundred fifty to 200 cells were counted each in three to four independent experiments. Error bars indicate SEM. *p < 0.05, statistically significant from EHD1 plus EHD4.
Quantification of the percentage of cells with NgCAM endocytosis in cultures expressing both EHD1 and EHD4 showed that NgCAM endocytosis was returned to normal levels (Fig. 8D). Simultaneous expression of EHD1 thus rescued the NgCAM endocytosis defect caused by expression of EHD4 alone. To test whether dimerization/oligomerization was necessary for the observed rescue, we coexpressed NgCAM with EHD4 and the oligomerization-impaired EHD1V203P mutant. Simultaneous expression of EHD4 and EHD1V203P did not rescue NgCAM endocytosis (Fig. 8D), arguing that dimerization/oligomerization of EHD1 and EHD4 was indeed necessary in this process. EHD3 coexpression also could not substitute for EHD1 and did not rescue the endocytosis defect caused by EHD4 overexpression (Fig. 8D).
Last, we tested whether coexpression of EHD1 and EHD4 also rescued the loss of axonal polarity observed for overexpression of EHD1 or EHD4 alone. Surface distribution of NgCAM was determined using the A/D PI, as before. Axonal polarity of NgCAM was restored: A/D PI of NgCAM in GFP-expressing controls was 5.5 ± 0.25 (mean ± SEM; n = 40 cells) and in EHD1 plus EHD4 coexpressing cells 5.0 ± 0.20 (mean ± SEM; n = 40 cells).
We noted that coexpression of EHD1 and EHD4 led to increased membrane-near staining for both markers, whereas coexpression of EHD1 and EHD3 was overwhelmingly found on internal endosomes with no obvious membrane-near staining (supplemental Fig. 6A,B, available at www.jneurosci.org as supplemental material). In addition, the membrane protein LDLR was coexpressed and stained live with an antibody against an extracellular epitope. Line scans across the soma showed coinciding intensity peaks at the position of the plasma membrane for EHD1 and EHD4, but coinciding intensity peaks were not apparent for EHD3 (supplemental Fig. 6C–E, available at www.jneurosci.org as supplemental material).
Discussion
Our experiments suggest that the axonal cell adhesion molecule L1/NgCAM uses a specialized endocytosis pathway in neurons, dependent on EHD1 and EHD4, either directly or indirectly. The other receptors currently known to use EHD4-mediated endocytosis are the neurotrophin receptors (Valdez et al., 2005) and most recently Nogo A (Joset et al., 2010). In very young cultured neurons, bulk membrane retrieval is also EHD4 dependent (Bonanomi et al., 2008), but this pathway disappears with maturation. Furthermore, we propose that the relative levels of EHD1 to EHD4 need to be regulated for the continued functioning of NgCAM endocytosis, suggesting the model that EHD1 and EHD4 might act as hetero-oligomers in this pathway.
A cell-type-specific phenotype for EHD1 in NgCAM endocytosis
In non-neuronal cells, no effects of EHD1 on endocytosis of multiple cargos have been found. Rather, EHD1 has been strongly implicated in endosomal trafficking from recycling endosomes (Rapaport et al., 2006; Grant and Caplan, 2008). EHD1 was reportedly found in a complex with the clathrin adaptor AP2 (Rotem-Yehudar et al., 2001), but no direct experiments of endocytosis were reported. Our work now shows that NgCAM endocytosis was not perturbed by EHD1 overexpression in non-neuronal cells but strongly inhibited in neurons, uncovering an unexpected cell-type specificity for EHD1 in endocytosis. This specificity might be attributable to the expression of genes in fibroblasts that can functionally compensate for EHD1/EHD4 in endocytosis of NgCAM.
A novel cargo for the EHD4 pathway
In non-neuronal cells, EHD4 plays a role in endosomal trafficking from early endosomes (Sharma et al., 2008) but not in endocytosis per se. In neurons, conversely, EHD4 is involved in endocytosis of neurotrophin receptors (Shao et al., 2002; Valdez et al., 2005, 2007): EHD4 mediates uptake of Trk receptors after BDNF stimulation via a novel pathway, coined “macroendocytosis.” Immunoelectron microscopy showed strong labeling of plasma membranes and ruffled internalized membranes by EHD4–HA (Shao et al., 2002), reminiscent of macropinosomes. Adjoining multivesicular bodies are also labeled and might in fact be generated directly from the macropinosomes (Valdez et al., 2005). Our work now adds the axonal cell adhesion molecule L1/NgCAM as a new putative EHD4 cargo. TrkA actually contains an NPF motif that could in principle bind directly to the EH domain. NgCAM, conversely, does not have an NPF motif.
A model: a role in NgCAM endocytosis for EHD1/EHD4 oligomers
Several lines of evidence lead us to propose that, in neurons, EHD1 and EHD4 operate as oligomers with a certain optimal stoichiometry to regulate NgCAM endocytosis: (1) EHD1 and EHD4 overexpression have identical phenotypes, inhibition of NgCAM endocytosis, whereas EHD3 overexpression does not; (2) the oligomerization-defective EHD1V203P mutant does not cause NgCAM endocytosis defects; (3) simultaneous overexpression of EHD1 and EHD4 at comparable levels rescues the NgCAM endocytosis defect observed by overexpression of either one alone; and (4) the oligomerization-defective EHD1V203P mutant does not rescue EHD4-mediated NgCAM endocytosis defects.
Our work raises the intriguing possibility that, in neurons, EHD1 and EHD4 play a role directly in NgCAM endocytosis at the plasma membrane. It is striking that low-level coexpression of EHD1 and EHD4 recruits them both to the plasma membrane, whereas EHD1 and EHD3 are recruited onto endosomes when coexpressed. In addition, EHD4 has been localized in neurons to the plasma membrane as well as to endosomes by immunoelectron microscopy (Valdez et al., 2005). Interestingly, the balance of EHD1 and EHD4 levels appears crucial because overexpression of either one inhibits endocytosis. This observation suggests that oligomers can become dysfunctional by inclusion of too much EHD1 or EHD4. EHD3 overexpression, conversely, does not negatively impact the EHD oligomers functioning in NgCAM endocytosis. Therefore, we propose the model that EHD1–EHD4 oligomers over a certain range of stoichiometries are functional in NgCAM endocytosis in neurons. This role could involve direct regulation of endocytosis at the plasma membrane, as suggested by the membrane-near localization, but an indirect effect of EHD1/EHD4 overexpression on NgCAM endocytosis cannot be completely ruled out at this point. For instance, balanced EHD1/EHD4 levels might be needed for the proper recycling of a crucial putative endocytosis protein required specifically for NgCAM endocytosis in neurons.
Our results show strong parallels between the effects of overexpressing EHD1 and EHD4 on NgCAM endocytosis in neurons. Strikingly, EHD3 overexpression does not result in the same phenotypes. This is surprising given the close homology of the proteins (87% between EHD1 and EHD3; 75% between EHD3 and EHD4). Furthermore, EHD3 and EHD4 have been implicated in the same trafficking step, early to recycling endosome transport (Grant and Caplan, 2008). Our results, in contrast, suggest that, in neurons, EHD1 and EHD4 might regulate the same step, namely NgCAM endocytosis. Consistent with this notion, it has been shown that overexpressed EHD1 strongly interacts with EHD4. Furthermore, a complex of endogenous EHD1 and EHD4 can be immunoisolated from cells (Sharma et al., 2008). This complex might perform distinct functions in neuronal cells. Other dimer/oligomer complexes of EHD family proteins likely exist in vivo as well but have not been demonstrated because of the lack of sufficiently specific antibodies.
Cargo specificity of EHD1/EHD4 pathway
In addition, not all cargos are equally affected by EHD1, even in neurons. Our data and that of others demonstrate that transferrin endocytosis is not disturbed by EHD1 overexpression, nor is endocytosis of AMPA receptors (Lin et al., 2001; Caplan et al., 2002; Park et al., 2004; Braun et al., 2005). Our work thus uncovered an unexpected cargo selectivity. In addition, we found that transferrin recycling is disrupted by the dominant-negative EHD1G429R mutant but not by wild-type EHD1. We argue that this observation suggests the hypothesis that transferrin recycling might be mediated by EHD1 homodimers/oligomers in neurons. If EHD1/EHD3 or EHD1/EHD4 heterodimers/oligomers were functioning in transferrin recycling, we would expect to see disruption of the EHD oligomer stoichiometry by overexpression of wild-type EHD1.
Dosage sensitivity of EHD family members
Our results show that NgCAM endocytosis is highly dosage sensitive to the expression of EHD1 and EHD4 but not of EHD3. Overexpression-sensitive genes have been identified in Saccharomyces cerevisiae and make up ∼18% of the yeast genome (Sopko et al., 2006). In the majority of these genes, the overexpression phenotype does not phenocopy the loss-of-function phenotype. Similarly, overexpression and downregulation of EHD1 give distinct phenotypes with regard to NgCAM endocytosis. What do these distinct phenotypes tell us about the putative functions of the endogenous protein? It is generally assumed that a loss-of-function phenotype tells us about what the protein does do, whereas a gain-of-function phenotype tells us about what the protein can do but not necessarily what the protein does do. In many instances, this interpretation is warranted, but more complex scenarios are not uncommon. As an example, downregulation of the endocytosis clathrin adaptor AP2 unexpectedly led to only a partial decrease in endocytosis of synaptic vesicle proteins (Kim and Ryan, 2009). The residual endocytosis showed complex kinetics and was attributable to compensatory upregulation of other endosomal pathways. Some of these compensatory pathways could be mostly eliminated by additional knockdown of the TGN-resident AP1 adaptor. The loss-of-function phenotype therefore also told us what another protein (i.e., AP1) can do rather than what the protein in question (i.e., AP2) does do. In the case of the EHD1 and EHD4 overexpression phenotype, we also found that compensatory endosomal pathways were upregulated after 3–4 d and L1/NgCAM endocytosis was no longer significantly impaired. Interestingly, compensatory changes in expression levels of EHD1 were found in EHD4 knock-out mice (Sengupta et al., 2009).
One of the proposed mechanisms for dosage sensitivity is that proteins that participate in protein complexes are sensitive to the balance of components (“balance hypothesis”) and overexpression of one of the components can lead to misassembly of the whole complex (Semple et al., 2008). Alternatively, overexpressed proteins could potentially participate in off-target interactions causing defects in other pathways. Our data support the balance hypothesis for the EHD proteins because (1) overexpressing the dimerization partner of EHD1 rescues the phenotype, ostensibly restoring the balance, (2) the rescue can be abrogated by a single point mutation, V203P, which causes a decrease in dimerization, (3) EHD3 is not dosage sensitive with respect to NgCAM endocytosis despite very high sequence homology, and (4) we observe cargo and cell-type specificity. These observations taken together argue against nonspecific toxic effects of the overexpression.
We conclude that NgCAM does not share the same endocytosis machinery used by other cargos, such as transferrin and AMPA receptors. Rather, L1/NgCAM uses EHD1/EHD4-dependent pathways for endocytosis, specifically in neuronal cells. It is currently not known whether the EHD4 pathway uses clathrin and clathrin adaptors or is molecularly distinct. The fact that Trk receptors also use EHD4-dependent endocytosis pathways raises intriguing future questions about the potential regulatory crosstalk between neurotrophins and trafficking of cell adhesion molecules to axons. This question was just made more relevant by the recent discovery that TrkB receptors in commissural neurons travel from the soma to axons via transcytosis (Ascaño et al., 2009), possibly on the same pathway that we discovered for NgCAM.
Footnotes
This work was supported by National Institutes of Health Grant GM086913 (B.W.) and by Grants GM074876 and P20 RR018759 from the National Center for Research Resources (both to S.C.). We thank Drs. Kevin Pfister (University of Virginia, Charlottesville, VA), Frank Solomon (Massachusetts Institute of Technology, Cambridge, MA), and Heike Folsch (Northwestern University, Evanston, IL) for critical reading of this manuscript. Special thanks go to Dr. Frank Solomon (Massachusetts Institute of Technology) for always engaging in rigorous discussions and debate. We are grateful to Drs. Fred Maxfield, Barth Grant, and Vance Lemmon for generously supplying reagents. Last, we thank the members of the Winckler Laboratory for helpful advice.
References
- Alberts P, Galli T. The cell outgrowth secretory endosome (COSE): a specialized compartment involved in neuronal morphogenesis. Biol Cell. 2003;95:419–424. doi: 10.1016/s0248-4900(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Allen J, Chilton JK. The specific targeting of guidance receptors within neurons: who directs the directors? Dev Biol. 2009;327:4–11. doi: 10.1016/j.ydbio.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Ascaño M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume JJ, Halbach A, Behrendt D, Paulsson M, Plomann M. EHD proteins are associated with tubular and vesicular compartments and interact with specific phospholipids. Exp Cell Res. 2007;313:219–231. doi: 10.1016/j.yexcr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Fornasiero EF, Valdez G, Halegoua S, Benfenati F, Menegon A, Valtorta F. Identification of a developmentally regulated pathway of membrane retrieval in neuronal growth cones. J Cell Sci. 2008;121:3757–3769. doi: 10.1242/jcs.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Pinyol R, Dahlhaus R, Koch D, Fonarev P, Grant BD, Kessels MM, Qualmann B. EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol Biol Cell. 2005;16:3642–3658. doi: 10.1091/mbc.E05-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, Bonifacino JS. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 2002;21:2557–2567. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Baba T, van der Bliek AM, Schmid SL. Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J Cell Biol. 1995;131:69–80. doi: 10.1083/jcb.131.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- Dickson TC, Mintz CD, Benson DL, Salton SR. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J Cell Biol. 2002;157:1105–1112. doi: 10.1083/jcb.200111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Bessis AS, Rossignol F, Pin JP, Olivo-Marin JC, Hémar A. The metabotropic glutamate receptor mGluR5 is endocytosed by a clathrin-independent pathway. J Biol Chem. 2003;278:12222–12230. doi: 10.1074/jbc.M205663200. [DOI] [PubMed] [Google Scholar]
- Galperin E, Benjamin S, Rapaport D, Rotem-Yehudar R, Tolchinsky S, Horowitz M. EHD3: a protein that resides in recycling tubular and vesicular membrane structures and interacts with EHD1. Traffic. 2002;3:575–589. doi: 10.1034/j.1600-0854.2002.30807.x. [DOI] [PubMed] [Google Scholar]
- George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, Band V, Band H. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans. BMC Cell Biol. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Caplan S. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic. 2008;9:2043–2052. doi: 10.1111/j.1600-0854.2008.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Soriano NA, Bose S, Holik J, Bose A, Pomerleau DP, Furcinitti P, Leszyk J, Corvera S, Czech MP. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem. 2004;279:10593–10605. doi: 10.1074/jbc.M307702200. [DOI] [PubMed] [Google Scholar]
- Haider NB, Searby C, Galperin E, Mintz L, Horowitz M, Stone EM, Sheffield VC. Evaluation and molecular characterization of EHD1, a candidate gene for Bardet-Biedl syndrome 1 (BBS1) Gene. 1999;240:227–232. doi: 10.1016/s0378-1119(99)00395-9. [DOI] [PubMed] [Google Scholar]
- Hirling H, Steiner P, Chaperon C, Marsault R, Regazzi R, Catsicas S. Syntaxin 13 is a developmentally regulated SNARE involved in neurite outgrowth and endosomal trafficking. Eur J Neurosci. 2000;12:1913–1923. doi: 10.1046/j.1460-9568.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- Joset A, Dodd DA, Halegoua S, Schwab ME. Pincher-generated Nogo-A endosomes mediate growth cone collapse and retrograde signaling. J Cell Biol. 2010;188:271–285. doi: 10.1083/jcb.200906089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jović M, Kieken F, Naslavsky N, Sorgen PL, Caplan S. Eps15 homology domain 1-associated tubules contain phosphatidylinositol-4-phosphate and phosphatidylinositol-(4,5)-bisphosphate and are required for efficient recycling. Mol Biol Cell. 2009;20:2731–2743. doi: 10.1091/mbc.E08-11-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Yoshihara F. The role of endocytic l1 trafficking in polarized adhesion and migration of nerve growth cones. J Neurosci. 2001;21:9194–9203. doi: 10.1523/JNEUROSCI.21-23-09194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V. Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci. 1998;21:97–125. doi: 10.1146/annurev.neuro.21.1.97. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Synaptic vesicle recycling at CNS snapses without AP-2. J Neurosci. 2009;29:3865–3874. doi: 10.1523/JNEUROSCI.5639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiecka ZM, Yap CC, Vakulenko M, Winckler B. Compartmentalizing the neuronal plasma membrane from axon initial segments to synapses. Int Rev Cell Mol Biol. 2009;272:303–389. doi: 10.1016/S1937-6448(08)01607-9. [DOI] [PubMed] [Google Scholar]
- Lin SX, Grant B, Hirsh D, Maxfield FR. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat Cell Biol. 2001;3:567–572. doi: 10.1038/35078543. [DOI] [PubMed] [Google Scholar]
- Long KE, Asou H, Snider MD, Lemmon V. The role of endocytosis in regulating L1-mediated adhesion. J Biol Chem. 2001;276:1285–1290. doi: 10.1074/jbc.M006658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Nagaraj K, Hortsch M. Phosphorylation of L1-type cell-adhesion molecules: ankyrins away! Trends Biochem Sci. 2006;31:544–546. doi: 10.1016/j.tibs.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Sharma M, Jovic M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–177. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Chenavas S, Sorgen PL, Caplan S. EHD1 and Eps15 interact with phosphatidylinositols via their Eps15 homology domains. J Biol Chem. 2007;282:16612–16622. doi: 10.1074/jbc.M609493200. [DOI] [PubMed] [Google Scholar]
- Naslavsky N, McKenzie J, Altan-Bonnet N, Sheff D, Caplan S. EHD3 regulates early-endosome-to-Golgi transport and preserves Golgi morphology. J Cell Sci. 2009;122:389–400. doi: 10.1242/jcs.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pál C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Rapaport D, Auerbach W, Naslavsky N, Pasmanik-Chor M, Galperin E, Fein A, Caplan S, Joyner AL, Horowitz M. Recycling to the plasma membrane is delayed in EHD1 knockout mice. Traffic. 2006;7:52–60. doi: 10.1111/j.1600-0854.2005.00359.x. [DOI] [PubMed] [Google Scholar]
- Rotem-Yehudar R, Galperin E, Horowitz M. Association of insulin-like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem. 2001;276:33054–33060. doi: 10.1074/jbc.M009913200. [DOI] [PubMed] [Google Scholar]
- Sann S, Wang Z, Brown H, Jin Y. Roles of endosomal trafficking in neurite outgrowth and guidance. Trends Cell Biol. 2009;19:317–324. doi: 10.1016/j.tcb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Semple JI, Vavouri T, Lehner B. A simple principle concerning the robustness of protein complex activity to changes in gene expression. BMC Syst Biol. 2008;2:1. doi: 10.1186/1752-0509-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, George M, Miller KK, Naik K, Chou J, Cheatham MA, Dallos P, Naramura M, Band H, Zheng J. EHD4 and CDH23 are interacting partners in cochlear hair cells. J Biol Chem. 2009;284:20121–20129. doi: 10.1074/jbc.M109.025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Akmentin W, Toledo-Aral JJ, Rosenbaum J, Valdez G, Cabot JB, Hilbush BS, Halegoua S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J Cell Biol. 2002;157:679–691. doi: 10.1083/jcb.200201063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Naslavsky N, Caplan S. A role for EHD4 in the regulation of early endosomal transport. Traffic. 2008;9:995–1018. doi: 10.1111/j.1600-0854.2008.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, Boone C, Andrews B. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Valdez G, Akmentin W, Philippidou P, Kuruvilla R, Ginty DD, Halegoua S. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J Neurosci. 2005;25:5236–5247. doi: 10.1523/JNEUROSCI.5104-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Philippidou P, Rosenbaum J, Akmentin W, Shao Y, Halegoua S. Trk-signaling endosomes are generated by Rac-dependent macroendocytosis. Proc Natl Acad Sci U S A. 2007;104:12270–12275. doi: 10.1073/pnas.0702819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisco D, Anderson ED, Chang MC, Norden C, Boiko T, Fölsch H, Winckler B. Uncovering multiple axonal targeting pathways in hippocampal neurons. J Cell Biol. 2003;162:1317–1328. doi: 10.1083/jcb.200307069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CC, Nokes RL, Wisco D, Anderson E, Fölsch H, Winckler B. Pathway selection to the axon depends on multiple targeting signals in NgCAM. J Cell Sci. 2008a;121:1514–1525. doi: 10.1242/jcs.022442. [DOI] [PubMed] [Google Scholar]
- Yap CC, Wisco D, Kujala P, Lasiecka ZM, Cannon JT, Chang MC, Hirling H, Klumperman J, Winckler B. The somatodendritic endosomal regulator NEEP21 facilitates axonal targeting of L1/NgCAM. J Cell Biol. 2008b;180:827–842. doi: 10.1083/jcb.200707143. [DOI] [PMC free article] [PubMed] [Google Scholar]