

Abstract
The subcellular localization of membrane Ca2+ channels is crucial for their functioning, but is difficult to study because channels may be distributed more closely than the resolution of conventional microscopy is able to detect. We describe a technique, stochastic channel Ca2+ nanoscale resolution (SCCaNR), employing Ca2+-sensitive fluorescent dyes to localize stochastic openings and closings of single Ca2+-permeable channels within <50 nm, and apply it to examine the clustered arrangement of inositol trisphosphate receptor (IP3R) channels underlying local Ca2+ puffs. Fluorescence signals (blips) arising from single functional IP3Rs are almost immotile (diffusion coefficient <0.003 μm2 s−1), as are puff sites over prolonged periods, suggesting that the architecture of this signaling system is stable and not subject to rapid, dynamic rearrangement. However, rapid stepwise changes in centroid position of fluorescence are evident within the durations of individual puffs. These apparent movements likely result from asynchronous gating of IP3Rs distributed within clusters that have an overall diameter of ∼400 nm, indicating that the nanoscale architecture of IP3R clusters is important in shaping local Ca2+ signals. We anticipate that SCCaNR will complement superresolution techniques such as PALM and STORM for studies of Ca2+ channels as it obviates the need for photoswitchable labels and provides functional as well as spatial information.
Introduction
The ubiquitous functioning of calcium as an intracellular second messenger requires that its actions are spatially and temporally restricted to regulate specific signaling cascades (1). Thus, the subcellular localization of Ca2+ channels is crucial for their appropriate functioning, and the range of action of Ca2+ in the cytosol is restricted to micrometer or even submicrometer scales by binding to endogenous immobile Ca2+ buffers (2,3). An important example, on which we focus, concerns Ca2+ liberation from the endoplasmic reticulum (ER) through inositol trisphosphate receptor (IP3R) channels. These are Ca2+-permeable channels that are gated both by the second messenger IP3 and (biphasically) by Ca2+ itself. Activation of IP3Rs by Ca2+ ions diffusing from neighboring channels thus results in a regenerative amplification via Ca2+-induced Ca2+ release (CICR) (4–6). The extent of this functional coupling depends strongly upon the spacing between IP3Rs, so that spatial localization of these channels is a major determinant of cellular Ca2+ signals. IP3Rs are distributed in clusters, leading to a hierarchical organization of Ca2+ signals (7) that may involve openings of only single channels (i.e., blips) (8,9); concerted opening of several channels in a cluster via CICR that remains restricted to a single cluster (i.e., puffs) (9,10); and Ca2+ waves that propagate throughout a cell by successive cycles of Ca2+ release, diffusion, and CICR between clusters (11). The density and spatial arrangement of IP3Rs within a cluster determine the probability of Ca2+-mediated channel-channel interactions, and thus the ultimate translation of stochastic single-channel activity into global whole-cell responses. However, crucial questions concerning the architecture and stability of IP3R clusters and the diffusional mobility of functional IP3R remain unanswered, because the dimensions of the clusters are close to or below the resolution limit of classical microscopy.
Diffraction limits the resolution of conventional light microscopy to ∼250 nm—a value not improved upon for a century after the original observation of Abbe (12). However, novel far-field fluorescence imaging approaches now allow the diffraction limit to be circumvented (13,14), and recent techniques such as PALM (15), FPALM (16), and STORM (17) theoretically have unlimited spatial resolution. These methods rely on the principle that a single emitter (e.g., a fluorescent molecule) can be localized with a precision that depends only on the number of photons collected; in practice, this would be a few tens of nanometers or better (18). The task is then to ensure that the density of fluorescent molecules is sufficiently low that two fluorophores are unlikely to lie indistinguishably close to one another. This has been accomplished by utilizing photoswitchable fluorophores, and accumulating superresolution images across repeated imaging cycles during which only sparsely distributed fluorophores are stochastically switched on. Here, we introduce a variant of this approach that enables simultaneous monitoring of the function and superresolution localization of active membrane ion channels in intact cells, by imaging the Ca2+ flux resulting from stochastic openings of individual Ca2+-permeable channels.
By utilizing total internal reflection fluorescence (TIRF) microscopy to image highly localized fluorescence signals from freely-diffusible organic Ca2+ indicator dyes, we had previously developed an optical patch-clamp technique (19) that reports the gating activity of single Ca2+ channels in the plasma (19,20) and ER membranes (21). When a channel opens, a microdomain of elevated cytosolic [Ca2+] is rapidly established around the channel pore, resulting in localized, stepwise fluorescence signals—i.e., single-channel Ca2+ fluorescence transients (SCCaFTs). SCCaFTs thus resemble patch-clamp records of single-channel currents, but with the major advantage of being able to simultaneously and independently monitor activity from hundreds of channels. We now extend this technique to localize channel positions by determining the centroid position of SCCaFTs with much higher precision than the spatial width of the fluorescence signal itself. Moreover, channel gating is stochastic, so that overlap of signals between closely adjacent channels can be minimized by employing situations where the mean open probability is low. We therefore name this technique “stochastic channel Ca2+ nanoscale resolution” (i.e., SCCaNR). We first establish the precision of the SCCaNR technique using expressed Ca2+-permeable plasmalemmal nicotinic receptor/channels and endogenous IP3Rs, and then address the submicrometer organization and dynamics of the IP3/Ca2+ signaling pathway.
Materials and Methods
Cell culture and loading
Human neuroblastoma SH-SY5Y cells were loaded with caged iIP3, fluo-4, and EGTA as previously described (22). Xenopus oocytes were injected cDNA clones coding for the muscle nicotinic receptor and fluo-4-dextran before imaging SCCaFTs, which was performed while voltage-clamping at −120 mV with 6 mM extracellular [Ca2+] to increase the electrochemical driving force for Ca2+ influx (23).
Total internal reflection microscopy
Imaging of local [Ca2+]i signals was accomplished using a home-built TIRF microscope system based around an IX 70 microscope equipped with an X60 TIRFM objective (Olympus, Melville, NY) as described (22). Fluorescence of cytosolic fluo-4 was excited within the ∼100-nm evanescent field formed by total internal reflection of a 488-nm laser beam at the coverglass/aqueous interface. Images of emitted fluorescence (λ >510 nm) were captured at a resolution of 128 × 128 pixels (1 pixel = 0.33 μm) at 420 frames s−1 by a Cascade 128 electron-multiplied charge-coupled device camera (Roper Scientific, Trenton, NJ). Native IP3Rs in SH-SY5Y cells were activated by photorelease of IP3 from a caged precursor by flashes of ultraviolet (350–400 nm) light focused uniformly across the imaging field.
Reagents
The membrane-permeant caged IP3 analog ci-IP3/PM was purchased from SiChem (Bremen, Germany). EGTA-AM, fluo-4 AM, fluo-4 dextran, and pluronic F-127 were from Invitrogen (Carlsbad, CA). All other reagents were purchased from Sigma (St. Louis, MO).
Image processing and analysis
Fluorescence pseudo-ratio image stacks (ΔF/F0) were created as described (21). Local Ca2+-fluorescence signals were fit to a circularly-symmetrical Gaussian function using a custom routine in Slidebook (Intelligent Imaging Innovations, Santa Monica, CA) to localize events in x and y dimensions by least-squares fitting of a symmetrical two-dimensional Gaussian function to a resolution of 0.2 pixels. The amplitude was allowed to vary to achieve the best fit whereas the standard deviation was preset to three pixels (∼1 μm). Finally, the x-y coordinates were dithered by adding uniformly distributed random numbers within the range ±0.1 pixel.
The displacement of fluorescence signals over time was established by calculating the distance between the mean centroid position during a given event and its average position during subsequent events. The displacements of a given channel during all openings were then squared and plotted versus the time between the respective channel openings. The diffusion coefficient (D) was calculated from a regression fit to the data as D = d2/4t, where d = mean distance from origin at time t.
Results
Superresolution localization of individual ion channels by Ca2+ imaging
Optical patch-clamping by TIRF microscopy (Fig. 1 a) reveals Ca2+ flux through individual ion channels through binding of Ca2+ ions to a diffusible, high-affinity fluorescent probe (fluo-4) loaded into the cytosol (19), and is applicable to plasmalemmal channels (19,20) (Movie S1 in the Supporting Material) and to ER channels close to the cell membrane (21) (Movie S2). Owing to the cytosolic diffusion of free Ca2+ ions and of Ca2+-bound dye, the fluorescence (SCCaFT) generated by the opening of an individual Ca2+-permeable channel approximates a circularly symmetrical Gaussian distribution (24) (Fig. 1 b). Fitting a symmetrical two-dimensional Gaussian function to the image data then allows us to localize the position of the single channel as the centroid of the Gaussian fit (see Materials and Methods). The precision with which optical point sources can be localized improves with decreasing spatial width of the observed signal. On that basis it is expected to be lower for SCCaFTs, which have typical widths of ∼700 nm (19), full width at half-maximum (FWHM) amplitude, as compared to single fluorescent molecules, where the width is determined by the point-spread function of the microscope objective (typical FWHM ∼300 nm). Our first objective was, therefore, to determine the precision of the SCCaNR technique, and to explore the factors limiting its precision.
Figure 1.
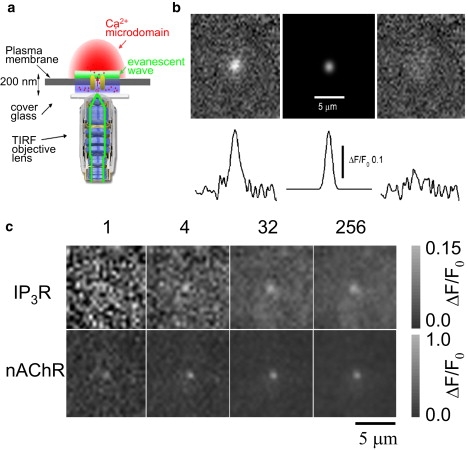
Imaging and localizing fluorescence calcium signals from single channels. (a) Schematic diagram (not to scale) of the TIRFM setup utilized to visualize Ca2+ flux through plasmalemmal nicotinic channels and IP3-mediated Ca2+ release events near the plasma membrane. (b) Two-dimensional Gaussian fitting of single-channel calcium fluorescence signals. (Top panels, left to right) Fluorescence image of a SCCaFT from a nAChR; the synthetic two-Gaussian fit to the data (SD ≈ 2.8 pixels = 1.12 μm); and the residual remaining after subtracting the Gaussian function from the experimental data. (Bottom traces) Corresponding fluorescence ratio measurements through the center of these images. (c) Ca2+ fluorescence images obtained during individual openings of a single IP3R (top) and a single nAChR (bottom). (Leftmost panels) Representative single image frames (2 ms exposure time), and (remaining panels) averages of increasing numbers of successive frames, as indicated.
Precision of localization
Localization of true point-sources, such as single fluorescent molecules, improves inversely as the width of the microscope point-spread function, and as the square-root of the number of photons collected, and can be directly estimated from these parameters (15,25). In light of factors including background fluorescence, diffusion, and variations in Ca2+ flux due to channel flickering, this method of determining localization precision is less applicable for SCCaFTs. Instead, we took advantage of the bright fluorescence arising from multiple Ca2+-bound fluo-4 molecules (24) to image at a fast rate (420 frames/s) to obtain repeated measurements during an individual channel opening. The precision of single-channel localization could then be calculated as the standard error of the mean.
This procedure is illustrated in Fig. 1 c, showing representative examples of fluorescence ratio images (ΔF/F0) captured during single, relatively long openings of a native IP3R channel in the ER of a cultured SH-SY5Y neuroblastoma cell and of a Ca2+-permeable nAChR channel expressed in the plasma membrane of a Xenopus oocyte. Panels show single image frames on the left, and images formed by averaging over increasing numbers of sequential frames (4, 32, and 256) captured during the opening. As more frames were incorporated into the average image, the fluorescence profile became more symmetrical and the overall noise was reduced. Fig. 2 a shows frame-by-frame measurements of local fluorescence and corresponding x-y centroid positions of SCCaFTs arising at a blip site (left) and through a nAChR channel (right). The fluorescence signals resulting from Ca2+ flux through the nAChR channel were approximately four-times greater than for the IP3R channel, and the scatter in centroid localizations was correspondingly smaller. In both cases, there was little or no apparent overall shift in SCCaFT location throughout the record.
Figure 2.
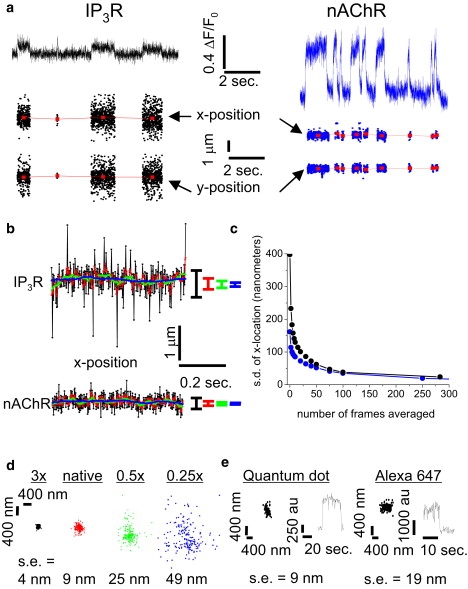
Estimating the precision of single-channel localization. (a) Localizing Ca2+ signals from individual channels. (Top) Representative traces showing the average fluorescence (ΔF/F0) measured over time from a 3 × 3 pixel (1 × 1 μm) region surrounding a single IP3R (left; black) and nAChR (right; blue). (Bottom) The fluorescence resulting from Ca2+ flux through each channel was fit to a two-dimensional Gaussian function for every frame (420 s−1) during which the channels were open. The x and y center positions of the Gaussian fits are plotted versus time. (b) Precision of channel localization as a function of frame averaging. The x positions localized during a single opening of an IP3R (top) and nAChR (bottom) are plotted versus time. The noisy black traces show locations derived from individual frames. Colored traces show mean locations calculated from running boxcar averages of increasing numbers of adjacent fames (red, 2, green, 10, blue, 50). Colored bars at the right indicate the corresponding standard deviations. (c) Standard deviation of the estimated x location of the channels in panel b (IP3R, black; nAChR, blue) as a function of the number of frames averaged. (d) Precision of channel localization as a function of SCCaFT intensity. The fluorescence signal formed by averaging 256 frames during a single nAChR opening was scaled and added to each of 200 successive frames of background noise recorded during a typical imaging session to create a synthetic test image sequence. Each frame was then fit to a two-dimensional Gaussian function, and the resulting center positions are plotted in x-y space. Panels show locations derived from image sequences formed after scaling the averaged SCCaFT image by different factors, as indicated. (e) Localization of a single quantum dot (left) and a single Alexa 647 molecule, for comparison with the SCCaNR technique. (Traces) Fluorescence during a single blink of a 565-nm quantum dot and stepwise bleaching of a single Alexa 647 molecule from which localizations were obtained. (Dot-plots) x-y positions derived from successive imaging frames (500-ms exposure time for quantum dot, 200 ms for Alexa 647).
To quantify how the precision of single channel localization improves as a function of the number of frames captured, we monitored the x position of individual channels (Fig. 2 b: IP3R, top; nAChR, bottom) throughout selected, prolonged (∼1 s) single opening events. Given the apparent lack of movement over longer records (see Fig. 2 a), we assume the channels were stationary during this interval. Black traces indicate frame-by-frame estimates of x position, and colored traces show the result of smoothing by a running box-car average over increasing numbers of frames, as indicated. Fig. 2 c plots the standard deviation of the x position versus the number of frames averaged. The brighter signal from the nAChR channel produced an appreciably greater precision when determined over only a few tens of frames, but both the nAChR and IP3R channels could be located to within <60 nm (±1 SD) from 50 frame (100 ms) averages, improving to ∼20 nm when averaged over the full duration (∼1 s) of the channel openings.
The precision of localization is also affected by the relative brightness of the signal with respect to uncorrelated background noise (18). To explore how this ratio affects our localization precision, we created an average fluorescence image from 256 frames during a single-channel opening of an nAChR. We then took this virtually noise-free image and added it to 200 sequential frames of background recorded from the same region while the channel was closed. Fig. 2 d shows scatter plots of frame-by-frame localizations of this idealized signal in the presence of varying relative levels of background noise. The plot marked as native preserved the original amplitude of the averaged SCCaFT relative to background fluorescence, and yielded a localization precision of 9 nm (mean +1 SE) over the 200 frames. This precision improved to ∼4 nm when the SCCaFT amplitude was magnified threefold, and degraded to ∼50 nm when the amplitude was reduced to one-quarter.
Finally, we were interested to compare our method of localizing single channels via their Ca2+ flux with other superresolution techniques, such as PALM and STORM, which image single fluorescent molecules. Fig. 2 e shows scatter plots obtained by localizing positions of a quantum dot (left) and a fluorescent Alexa 647 dye molecule (right) immobilized on a coverslip and imaged at 500 or 200 ms per frame, respectively. The position of the Alexa molecule was localized with a precision of ∼20 nm (mean ± SE) after averaging over ∼50 frames (10 s), and the brighter quantum dot was localized to within ∼10 nm over an equivalent period. Thus, SCCaNR can achieve a single molecule localization precision very similar to PALM and STORM (15,17).
Estimating the diffusion coefficient of blip sites in intact cells
Having established the validity and precision of SCCaNR localization, we then applied this technique to study the functional architecture of the IP3 signaling system in intact cells at submicrometer scales. We first investigated the motility of functional IP3Rs within intact SH-SY5Y cells by tracking blip sites. IP3Rs have been reported to redistribute in cells after agonist stimulation (26,27), and it has been proposed that diffusional motility of IP3Rs over scales of ∼1 μm within a few seconds may underlie their dynamic aggregation into clusters (28). However, existing measurements of the diffusion coefficient of IP3Rs in the ER membrane derive largely from FRAP (fluorescence recovery after photobleaching) studies of overexpressed GFP-tagged IP3R (27,29–31), which may not faithfully replicate the behavior of the functional native receptors, and which may report only population measurements and have given, widely varying values ranging between 0.01 and 0.45 μm2 s−1.
As shown in Fig. 2 c, the centroid location of the fluorescence signal arising from a single opening of an IP3R can be localized within a few tens of nm. To examine whether functional IP3R channels move over longer periods of several seconds, we determined the mean x-y locations of blips during successive openings (Fig. 3 a; inset). The graph in Fig. 3 a plots the square of the displacement from origin for each of the successive events as a function of time. The data fall on a roughly linear relationship on these axes, as expected for a random-walk diffusion process, and the slope of the regression line corresponds to a diffusion coefficient of ∼0.006 μm2 s−1. Fig. 3 b (inset) shows a scatter plot of diffusion coefficients estimated in this way for 15 blip sites, with diffusion coefficients of zero assigned in cases where a negative slope was obtained for the displacement-squared versus time-plot. Values ranged between zero and 0.012 μm2 s−1, with a mean of 0.003 μm2 s−1. To place these results in context with the proposal that functional IP3R channels may diffuse over 1 μm in a few seconds (28), we illustrate in Fig. 3 b the mean distances that blip sites would move as a function of time based on the diffusion coefficients given in Fig. 3 b (inset).
Figure 3.
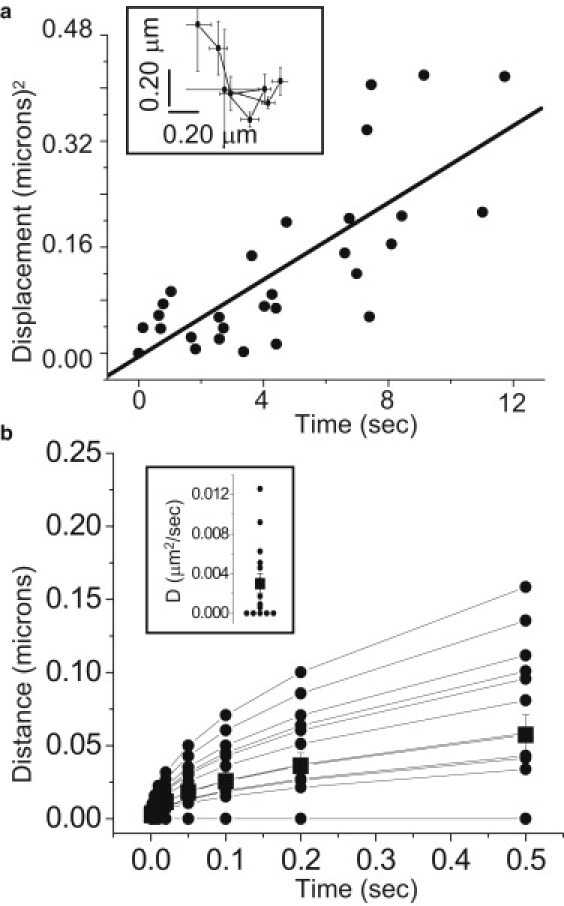
The diffusion coefficient of single IP3R in intact living cells estimated by tracking SCCaFT positions during successive channel openings. (a, Inset) Mean locations ±1 SE determined from eight sequential events arising at a single isolated site throughout a 10 s recording. The graph plots the mean-square displacement from origin of these locations as a function of time. (Line) Linear regression fit to the data. The slope of the line corresponds to a two-dimensional diffusion coefficient of 0.006 μm2 s−1. (b, Inset) Diffusion coefficients estimated from 15 separate records (solid circles) as in panel a. Data points plotted as zero correspond to negative regression slopes of displacement squared versus time. The square symbol shows the mean and standard error of the diffusion coefficient estimated for all 15 channels. Plot illustrating the mean displacement that would be reached after different times for each of the individual channels in panel b (inset), together with that of a channel diffusing with the mean diffusion coefficient of 0.003 μm2 s−1 (square symbol).
Puff sites show little movement over long timescales
Puffs are transient, local Ca2+ signals (Movie S3) that arise from discrete clusters of IP3R (10). However, the spatial arrangement of channels underlying a calcium puff is not well understood. We first investigated whether puff sites show appreciable movement over many seconds by tracking the mean centroid locations of fluorescence signals during several successive puffs at individual sites. Fig. 4 a illustrates averaged images of six puffs over ∼10 s after photorelease of iIP3 in an SH-SY5Y cell, and Fig. 4 b shows corresponding measurements of fluorescence intensity from a 1 × 1 μm region centered on the puff site (upper), together with frame-by-frame localizations of x and y positions of the fluorescence centroid (lower). Mean x and y locations averaged over each event are marked in red, and for the puff site illustrated the locations varied by <150 nm throughout the record. To obtain an upper bound for the diffusion coefficient of puff sites, we again plotted the square of displacements from origin of mean puff centroids as a function of time for successive puffs at given sites (Fig. 4 c). The correlation is weak (R = 0.12), and the slope of the line corresponds to a mean diffusion coefficient of only 0.002 μm2 s−1. Fig. 4 d shows a scatter plot of diffusion coefficients estimated individually from each of 14 sites; negative regression slopes are assigned a value of zero. The greatest value observed was 0.0028 μm2 s−1.
Figure 4.
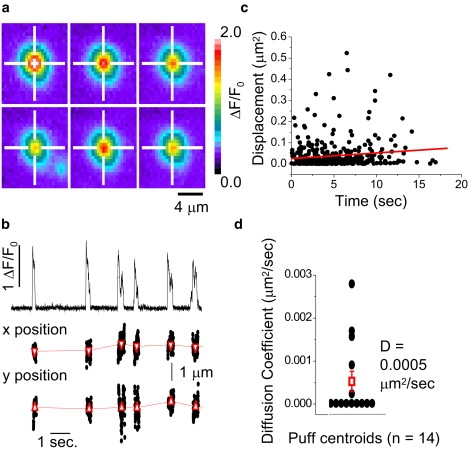
Puff sites show little or no movement over periods of several seconds. (a) Panels show average fluorescence images obtained during six successive Ca2+ puffs occurring at the same site. Each image is an average of 61–151 frames (420 frames s−1) captured during individual events. Cross-hairs mark the centroid location of the first puff. (b) The top trace plots average fluorescence ratio (ΔF/F0) measured within a 3 × 3 pixel region (1 × 1 μm) centered on the same puff site as in panel a, showing six discrete events within ∼10 s. The fluorescence images during these puffs were fit to a two-dimensional Gaussian function, and the center positions of the Gaussian fits are plotted as x and y locations at corresponding times (lower traces). Solid circles show locations derived from individual frames, and triangles mark the mean locations (mean ± SE) during each of the six puffs. (c) Plot of the mean-square displacement from origin versus time for all puff centroids analyzed (n = 14 puff sites). The line is a linear regression fit, with a slope corresponding to a two-dimensional diffusion coefficient of 0.002 μm2 s−1. (d) Diffusion coefficients calculated for 14 separate puff sites (solid circles), and overall mean ±1 SE (open square).
Resolving closely adjacent puff sites
Ca2+ puffs were originally proposed to be unitary events, limited to a single, discrete cluster of IP3Rs (10). However, this definition has been muddied by findings that closely adjacent sites may show correlated behavior, such that a puff at one site is likely to trigger a nearly simultaneous puff at the other site (10,32). The distinction of what is a unitary event then becomes a matter of experimental resolution; that is to say, whether two sites are separated sufficiently to be distinguished in comparison to the diffusional spread of the Ca2+ fluorescence signal. We now show that it is possible to resolve puffs arising from discrete clusters within < 1 μm, even though their Ca2+ fluorescence signals overlap in space and time.
Fig. 5 shows an example of such a binary situation, which on cursory examination appears to represent only a single puff site. The traces in Fig. 5 a show fluorescence profiles during four successive puffs, recorded simultaneously from two 3 × 3 pixel (1 × 1 μm) regions of interest (color-coded as red and black), displaced by two pixels from one another in the y direction. Puffs had a spatial width (FWHM) of ∼2 μm, and as expected from this wide distribution, signals were broadly similar from both regions of interest. However, it was noteworthy that at differing times during individual puffs, the fluorescence was greater in one or other of the regions, suggesting that the events involved asynchronous Ca2+ release from different sites. This notion is reinforced by localization of centroids of fluorescence signals, which distributed at two distinct sites separated by ∼700 nm in the y dimension (Fig. 5 b). Corresponding scatter plots of frame-by-frame x-y locations for each event are shown in Fig. 5 c, again showing two sites, displaced in y but little in x, that lie roughly in the centers of the two regions of interest used to measure the fluorescence profiles (as indicated in the third panel of Fig. 5 c). In the first panel, a puff is initiated first at the red site (open circles), and then triggers concurrent Ca2+ release from the black site (solid circles) so that the centroid location then likely reflects a weighted mean between the two sites: in the second panel, only the red site is active, and in the remaining panels, it appears that initial, transient activation of the black site resulted in sequential activation of the red site. Thus, it is possible to unambiguously identify signals arising from two adjacent IP3R clusters, even when spaced so that their fluorescence signals largely overlap (Fig. 5 d).
Figure 5.
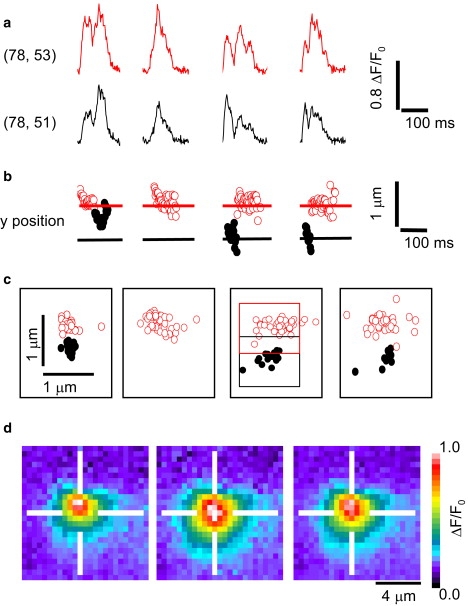
Functional coupling between discrete but closely adjacent puff sites. (a) Traces show simultaneous measurements of fluorescence during four puff events obtained from two overlapping 3 × 3 pixel regions centered two-pixels (0.66 μm) apart in the y dimension. Differences between the two regions suggest that signals arise from independent Ca2+ release at two discrete sites, but are contaminated by bleedthrough of fluorescence between the two regions. (b) Plots show the frame-by-frame y locations of fluorescence signals derived from two-dimensional Gaussian fits to the events shown in panel a. Points are coded according to whether the fluorescence was greater in the upper (open circles) region of interest, or the lower (solid circles) region. (c) Scatter plots show corresponding locations in x-y space. The positions of the regions of interest used to derive the fluorescence traces in panel a are outlined in the third panel. (d) Fluorescence images obtained by averaging selected frames at times corresponding to open circles in panel b (left), frames corresponding to solid circles (middle), and all frames during puffs (right).
Estimating IP3R cluster dimensions via movements of puff centroids during single events
Even though the mean position of puff sites changed very little over tens of seconds, we observed systematic changes in centroid locations of the Ca2+ signal within the durations of individual puffs that we consider to arise at a single site (Fig. 6 a). These deviations cannot be attributed to noise in the localization, because relatively consistent readings (SD of ∼100 nm) were generally obtained over several successive frames, followed by abrupt steps of as much as 300 nm to some new mean position. Given the long-tem stability of puff locations, together with the very low motility of blip sites, it is improbable that movements of the Ca2+ centroid arose through physical movement of the underlying active IP3R channels. Instead, given that puffs in SH-SY5Y cells typically involve simultaneous opening of <10 IP3R channels (21), we interpret the movement of the Ca2+ centroid to arise as different channels at different locations within a cluster stochastically open and close. The centroid locations then represent weighted mean positions among those channels that happen to be open at any given time. Puffs imaged by TIRF microscopy in the presence of cytosolic EGTA show distinct, stepwise quantal transitions, reflecting openings and closings of individual IP3Rs (21). Different fluorescence-amplitude step levels were often associated with differing centroid locations, and in Fig. 6 a we divided the event into three time segments, indicated by differing colors. A scatter plot (upper right, Fig. 6 a) of frame-by-frame x-y centroid positions showed distinctly different locations during these segments; this was demonstrated also by the lower right panel, showing means and standard errors of locations during each segment.
Figure 6.
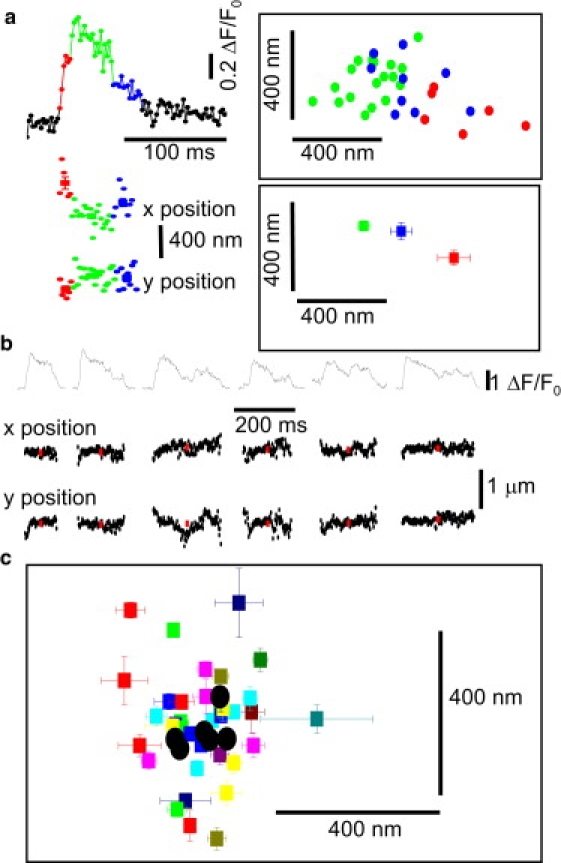
Movement of centroid locations of the Ca2+ signal within individual puffs. (a) Representative example of shifts in centroid location of the fluorescence signal during a single puff. (Traces, left) A puff divided into several time segments (indicated by different colors) based on the fluorescence amplitude level. (Scatter plots, right) Centroid locations of the fluorescence signals at different times during this puff (color-coded as for the traces at the left). (Upper plot) Locations determined from individual image frames; (lower plot) locations (mean ±1 SE) averaged over each color-coded time segment. (b) Fluorescence records (upper traces) and frame-by-frame x and y locations of six Ca2+ puffs replotted from Fig. 4b on enlarged scales. (Red) Puff locations (mean ±1 SE) averaged within each event. (c) Scatter of puff centroid locations throughout the six successive events shown in panel b. (Squares) Locations (mean ±1 SE) determined within discrete amplitude steps as illustrated in panel a. (Black circles) Mean locations averaged from the entire duration of each puff.
Excepting instances when only a single channel is open, the centroids do not map the physical locations of functional IP3Rs within a cluster. Nevertheless, the Ca2+ centroid positions throughout repeated puffs yield valuable information about the architecture of that site, allowing us to define a minimal bound for the dimensions of the cluster. Our procedure is illustrated in Fig. 6 b, showing the fluorescence profiles during repeated puffs (upper trace), and corresponding measurements of x and y centroid locations (lower traces). Fig. 6 c shows pooled data after dividing all six successive puffs at this site into segments as in Fig. 6 a. Colored symbols represent x-y locations (mean ± 1 SE) derived from individual time segments within puffs, and black circles represent mean locations throughout each puff. The individual segment locations scatter across a range of 300–400 nm; appreciably greater than the standard error associated with each measurement, and greater than the change in mean position of the puff site throughout successive puffs during the six-second duration of the record. We thus conclude that IP3Rs at this puff site are distributed throughout a cluster with a diameter of at least 300–400 nm.
Discussion
SCCaNR
We describe and characterize a superresolution imaging technique termed “stochastic channel Ca2+ nanoscale resolution” (SCCaNR) based upon Ca2+ flux through individual Ca2+ permeable ion channels. The ability of SCCaNR to localize the positions of single ion channels with high precision is analogous to other techniques such as PALM and STORM that involve fitting a two-dimensional Gaussian to the effective point spread function (PSF) of individual, sparsely distributed fluorescent sources. The precision of such techniques depends principally upon the number of photons that can be collected from each emitter. In this respect, SCCaNR has the advantage that the single-channel fluorescence signal arises from numerous Ca2+-bound indicator molecules (24). Moreover, the signal is not limited by dye bleaching, because there is a continuous diffusional turnover of indicator molecules from beyond the evanescent field. On the other hand, the localization precision is degraded by the wider diffusional spread of Ca2+-bound indicator as compared to the microscope PSF, and depends upon both the single-channel Ca2+ flux (current) and the channel open duration. In practice, we achieved a precision of ∼60 nm for channel openings persisting for 100 ms.
In previous reports, we highlighted the applicability of the optical patch-clamp technique (the basis of the SCCaNR technique) to the study of various Ca2+-permeable ion channels ranging from voltage-gated channels in the plasma membrane to IP3Rs within the ER (19–24). Even channels that show only a small fractional conductance to Ca2+, such as nAChR (∼2 pS) (20), can be localized with high precision. Thus, we believe that SCCaNR will be widely applicable to a range of ion channels, including TRP channels, NMDA and AMPA receptors, and many others (23). Because SCCaFTs from the several voltage- and ligand-gated Ca2+-permeable channels we have thus far examined appear to be essentially immobile (23), it should be possible to accumulate high-resolution images over time, employing stimuli (voltage or ligand concentration) giving a low open probability so as to achieve an appropriately sparse activation of channels during each imaging frame.
Spatial distribution and motility of functional IP3Rs
The interactions between individual IP3Rs within a cluster to generate puffs, and interactions between puff sites to generate waves, are mediated by Ca2+ diffusion and CICR. Therefore, the spatial organization of functional IP3Rs must be tightly regulated to ensure appropriate physiological signaling. The ability of the SCCaNR technique to directly localize Ca2+ flux through IP3Rs on submicrometer scales casts new light on the stability and nanoscale architecture of this signaling pathway.
One topical question concerns whether IP3R clusters are preformed or are dynamically regulated after agonist stimulation. Taufiq-Ur-Rahman et al. (28) and Rahman and Taylor (33) recently reported that patch-clamp recordings from IP3Rs on the nuclear envelope showed these channels to be spaced ∼1 μm apart at rest, but that upon agonist stimulation become rapidly recruited into clusters wherein their properties are modified by direct intermolecular contact. On this basis, dynamic regulation of clustering by IP3 was proposed to facilitate hierarchical recruitment of the elementary events that underlie all IP3-evoked cellular Ca2+ signals (28). Against this, our results indicate that the motility of active IP3R is too low to underlie such dynamic clustering in the ER membrane. Approximately one-third of the blip sites we examined showed no detectable movement over 20 s, and we obtained an overall mean diffusion coefficient of <0.003 μm2 s−1. Stochastic simulations indicate that aggregation of channels at puff sites by a diffusional trapping mechanism would then require many seconds if constrained by this low diffusion coefficient, whereas puffs involving activation of multiple IP3R are already apparent within <200 ms of photorelease of IP3 (34).
In addition to our observation that blip sites are almost immotile, we confirm that puff sites also remain at stable locations. Previous observations with lower resolution in Xenopus oocytes and mammalian cells (21,35–37) have demonstrated that puff sites move <1 or 2 μm over several minutes, and we now show that centroid positions of Ca2+ fluorescence averaged over individual puffs show displacements of <150 nm during repeated events over ∼20 s. However, we did observe marked changes (as great as 300 nm in 100 ms) in centroid position throughout the evolution of individual puffs. In light of our finding that blip sites are almost immotile, it is improbable that such movements of the Ca2+ centroid could result from physical movement of functional IP3R channels within a cluster, as this would imply an apparent diffusion coefficient as high as 1 μm2 s−1. Instead, we interpret them to arise from stochastic openings and closings of channels situated at different locations within a cluster—a hypothesis supported by observations that changes in puff centroid position often corresponded with stepwise changes in fluorescence amplitude that reflect openings and closings of individual channels (21). The fluorescence centroid will thus represent a weighted mean position between those channels that are open at any given time, and the scatter of centroid positions from many observations at a single puff site thereby provides a minimum estimate of the spatial extent over which IP3Rs are distributed throughout a cluster. In the future, we anticipate achieving a more precise mapping of individual functional IP3Rs within a cluster by localizing only those instances where a single channel is open at a time—a superresolution imaging approach analogous to PALM or STORM, but capitalizing on the stochastic gating of channels rather than stochastic photoswitching of fluorophores. Nevertheless, our present results already indicate that, in concordance with earlier modeling studies (38), puff sites (clusters) have a diameter ∼400 nm. Given that puffs in SH-SY5Y cells typically involve the simultaneous opening of fewer than 10 channels (21), the mean spacing between functional IP3Rs will be >100 nm. This distance is appreciable in relation to the predicted fall-off of [Ca2+] around a single open channel (24) (half-decline in ∼40 nm), implying a relatively loose coupling between IP3Rs via Ca2+ diffusion and CICR. The nano-architecture of functional IP3R within a cluster is thus likely to be important in determining the spatio-temporal evolution of puffs—a fact that is missing in some model simulations that approximate puff sites as a point source with uniform [Ca2+] (39).
Localizing channel activity versus channel proteins
SCCaNR localizes Ca2+ signals arising from single open channels, whereas PALM and STORM utilize the PSF generated from a fluorescently tagged protein or conjugated antibody to localize the position of the channel protein itself. Thus, we are able to image native channels in a live cell rather than an engineered construct overexpressed in a living cell (PALM), or native proteins in a fixed cell (STORM). Furthermore, we can be sure that we are localizing only functional channels under minimally perturbed conditions (presence of a Ca2+ indicator dye), and better localization of specific channel subtypes could be achieved by use of appropriate stimuli (e.g., specific agonists for ligand-gated channels) in conjunction with standard pharmacological approaches as used for electrophysiological isolation of particular channel currents.
It is important, nonetheless, to make a distinction between sites of single channel activity and localization of channel proteins. In particular, various labeling techniques in diverse mammalian cell types show a high density of IP3R distributed extensively throughout the cytoplasm, whereas imaging studies reveal relatively few IP3R channels at just a few discrete puff sites (34), suggesting that only a fraction of IP3R may be active under our experimental conditions. Moreover, we find that blip and puff sites appear almost immotile, in apparent contradiction to FRAP experiments where photobleaching of GFP-tagged IP3Rs indicate appreciable greater diffusion coefficients for IP3Rs, with estimates varying from ∼0.01 μm2 s−1 to 0.45 μm2 s−1 (27,29–31). We thus hypothesize that cells may contain two different populations of IP3Rs:
-
1.
A subset of receptors that are anchored by association with static cytoskeletal structures and which, possibly as a consequence of this tethering, display high sensitivity to IP3; and
-
2.
A population of mobile IP3Rs that is functionally unresponsive under our experimental conditions (34).
In the second case, our localizations of blip and puff signals would reflect the localization of the anchoring sites, rather than of IP3R per se. Future attempts to distinguish between these two possibilities will be aided by the complimentary application of superresolution techniques to image both structure (PALM/STORM) and function (SCCaNR) in parallel. Regardless of underlying mechanism, however, our results already point to a relatively stable architecture of functional IP3-mediated Ca2+ signaling at the single-channel level.
Supporting Material
Three movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00535-7.
Supporting Material
Acknowledgments
We thank Karl Kilborn (Intelligent Imaging Innovations) for writing the custom localization routine, Angelo Demuro for data gathered from Xenopus studies, and Neil Beri for assistance with image analysis.
This work was supported by grants (No. GM48071 and No. GM065830) from the National Institutes of Health (Bethesda, MD), and by a training grant from the University of California Systemwide Biotechnology Research & Education Program GREAT to S.W.
References
- 1.Berridge M.J., Lipp P., Bootman M.D. The versatility and universality of calcium signaling. Nat. Rev. Mol. Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Allbritton N.L., Meyer T., Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 3.Marchant J.S., Parker I. Functional interactions in Ca2+ signaling over different time and distance scales. J. Gen. Physiol. 2000;116:691–696. doi: 10.1085/jgp.116.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezprozvanny I., Watras J., Ehrlich B.E. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 6.Parker I., Ivorra I. Inhibition by Ca2+ of inositol trisphosphate-mediated Ca2+ liberation: a possible mechanism for oscillatory release of Ca2+ Proc. Natl. Acad. Sci. USA. 1990;87:260–264. doi: 10.1073/pnas.87.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callamaras N., Marchant J.S., Parker I. Activation and co-ordination of InsP3-mediated elementary Ca2+ events during global Ca2+ signals in Xenopus oocytes. J. Physiol. 1998;509:81–91. doi: 10.1111/j.1469-7793.1998.081bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker I., Choi J., Yao Y. Elementary events of InsP3-induced Ca2+ liberation in Xenopus oocytes: hot spots, puffs and blips. Cell Calcium. 1996;20:105–121. doi: 10.1016/s0143-4160(96)90100-1. [DOI] [PubMed] [Google Scholar]
- 9.Parker I., Yao Y. Ca2+ transients associated with openings of inositol trisphosphate-gated channels in Xenopus oocytes. J. Physiol. 1996;491:663–668. doi: 10.1113/jphysiol.1996.sp021247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Y., Choi J., Parker I. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J. Physiol. 1995;482:533–553. doi: 10.1113/jphysiol.1995.sp020538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson S.P., Keizer J., Pearson J.E. Fire-diffuse-fire model of dynamics of intracellular calcium waves. Proc. Natl. Acad. Sci. USA. 1999;96:6060–6063. doi: 10.1073/pnas.96.11.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbe E. Contributions to the Theory of the Microscope and of Microscopic Resolution. [Beitrage zur theorie des mikroskopsund der mikroskopischen wahrnehmung]. Arch. f. Mikr. Anat. 1873;9:413–420. [Google Scholar]
- 13.Huang B., Bates M., Zhuang X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hell S.W. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 15.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 16.Hess S.T., Girirajan T.P., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández-Suárez M., Ting A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 19.Demuro A., Parker I. Optical single-channel recording: imaging Ca2+ flux through individual N-type voltage-gated channels expressed in Xenopus oocytes. Cell Calcium. 2003;34:499–509. doi: 10.1016/s0143-4160(03)00154-4. [DOI] [PubMed] [Google Scholar]
- 20.Demuro A., Parker I. “Optical patch-clamping”: single-channel recording by imaging Ca2+ flux through individual muscle acetylcholine receptor channels. J. Gen. Physiol. 2005;126:179–192. doi: 10.1085/jgp.200509331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith I.F., Parker I. Imaging the quantal substructure of single IP3R channel activity during Ca2+ puffs in intact mammalian cells. Proc. Natl. Acad. Sci. USA. 2009;106:6404–6409. doi: 10.1073/pnas.0810799106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith I.F., Wiltgen S.M., Parker I. Localization of puff sites adjacent to the plasma membrane: functional and spatial characterization of Ca2+ signaling in SH-SY5Y cells utilizing membrane-permeant caged IP3. Cell Calcium. 2009;45:65–76. doi: 10.1016/j.ceca.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demuro A., Parker I. Imaging single-channel calcium microdomains. Cell Calcium. 2006;40:413–422. doi: 10.1016/j.ceca.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuai J., Parker I. Optical single-channel recording by imaging Ca2+ flux through individual ion channels: theoretical considerations and limits to resolution. Cell Calcium. 2005;37:283–299. doi: 10.1016/j.ceca.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tateishi Y., Hattori M., Mikoshiba K. Cluster formation of inositol 1,4,5-trisphosphate receptor requires its transition to open state. J. Biol. Chem. 2005;280:6816–6822. doi: 10.1074/jbc.M405469200. [DOI] [PubMed] [Google Scholar]
- 27.Chalmers M., Schell M.J., Thorn P. Agonist-evoked inositol trisphosphate receptor (IP3R) clustering is not dependent on changes in the structure of the endoplasmic reticulum. Biochem. J. 2006;394:57–66. doi: 10.1042/BJ20051130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taufiq-Ur-Rahman, Skupin A., Taylor C.W. Clustering of InsP3 receptors by InsP3 retunes their regulation by InsP3 and Ca2+ Nature. 2009;458:655–659. doi: 10.1038/nature07763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukatsu K., Bannai H., Mikoshiba K. Lateral diffusion of inositol 1,4,5-trisphosphate receptor type 1 is regulated by actin filaments and 4.1N in neuronal dendrites. J. Biol. Chem. 2004;279:48976–48982. doi: 10.1074/jbc.M408364200. [DOI] [PubMed] [Google Scholar]
- 30.Cruttwell C., Bernard J., Mauger J.P. Dynamics of the Ins1,4,5P3 receptor during polarization of MDCK cells. Biol. Cell. 2005;97:699–707. doi: 10.1042/BC20040503. [DOI] [PubMed] [Google Scholar]
- 31.Ferreri-Jacobia M., Mak D.O., Foskett J.K. Translational mobility of the type 3 inositol 1,4,5-trisphosphate receptor Ca2+ release channel in endoplasmic reticulum membrane. J. Biol. Chem. 2005;280:3824–3831. doi: 10.1074/jbc.M409462200. [DOI] [PubMed] [Google Scholar]
- 32.Demuro A., Parker I. Multi-dimensional resolution of elementary Ca2+ signals by simultaneous multi-focal imaging. Cell Calcium. 2008;43:367–374. doi: 10.1016/j.ceca.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahman T., Taylor C.W. Dynamic regulation of IP3 receptor clustering and activity by IP3. Channels (Austin) 2009;3:226–232. doi: 10.4161/chan.3.4.9247. [DOI] [PubMed] [Google Scholar]
- 34.Smith I.F., Wiltgen S.M., Parker I. Ca2+ puffs originate from preestablished stable clusters of inositol trisphosphate receptors. Sci. Signal. 2009;2:ra77. doi: 10.1126/scisignal.2000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchant J.S., Parker I. Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 2001;20:65–76. doi: 10.1093/emboj/20.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dargan S.L., Parker I. Buffer kinetics shape the spatiotemporal patterns of IP3-evoked Ca2+ signals. J. Physiol. 2003;553:775–788. doi: 10.1113/jphysiol.2003.054247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas D., Lipp P., Bootman M.D. Hormone-evoked elementary Ca2+ signals are not stereotypic, but reflect activation of different size channel clusters and variable recruitment of channels within a cluster. J. Biol. Chem. 1998;273:27130–27136. doi: 10.1074/jbc.273.42.27130. [DOI] [PubMed] [Google Scholar]
- 38.Shuai J., Rose H.J., Parker I. The number and spatial distribution of IP3 receptors underlying calcium puffs in Xenopus oocytes. Biophys. J. 2006;91:4033–4044. doi: 10.1529/biophysj.106.088880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swaminathan D., Ullah G., Jung P. A simple sequential-binding model for calcium puffs. Chaos. 2009;19:037109. doi: 10.1063/1.3152227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.