

Abstract
Soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE)-mediated lipid mixing can be efficiently recapitulated in vitro by the incorporation of purified vesicle membrane (-v) SNARE and target membrane (t-) SNARE proteins into separate liposome populations. Despite the strong correlation between the observed activities in this system and the known SNARE physiology, some recent works have suggested that SNARE-mediated lipid mixing may be limited to circumstances where membrane defects arise from artifactual reconstitution conditions (such as nonphysiological high-protein concentrations or unrealistically small liposome populations). Here, we show that the previously published strategies used to reconstitute SNAREs into liposomes do not significantly affect either the physical parameters of the proteoliposomes or the ability of SNAREs to drive lipid mixing in vitro. The surface density of SNARE proteins turns out to be the most critical parameter, which controls both the rate and the extent of SNARE-mediated liposome fusion. In addition, the specific activity of the t-SNARE complex is significantly influenced by expression and reconstitution protocols, such that we only observe optimal lipid mixing when the t-SNARE proteins are coexpressed before purification.
Introduction
The maintenance of intracellular compartments and the precise transportation of proteins and lipids between them are essential elements in eukaryotic cellular life. Both processes are dependent upon regulated intracellular membrane fusion events orchestrated by a highly conserved family of proteins called soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) (1–4). The energy released during the formation of a compact four-helix bundle between the conserved SNARE motifs of the vesicle membrane SNARE (v-SNARE) and the target membrane SNARE (t-SNARE) brings the two cognate membranes into close apposition and drives their fusion (5–8).
The minimal machinery of intracellular membrane fusion, the SNAREpin, was first functionally identified using a lipid mixing assay where fusion was observed between two separate populations of artificial liposomes bearing reconstituted cognate v- and t-SNARE proteins (7), and has since been used to follow artificial liposomes containing t-SNARE proteins fusing with purified synaptic vesicles (9,10). This robust and versatile lipid mixing assay based on fluorescence resonance energy transfer (FRET) measurements has been widely used to study membrane fusion events, including protein-free or protein-assisted fusion reactions (11–14). We list here some of the many insights into SNARE function gained from this assay.
-
1.
In general, liposome fusion can only be driven by the topologically restricted assembly of a three-helix t-SNARE in one liposome population and a single-helix v-SNARE in the second population (15). Although this topological restriction is resolutely maintained across most systems (16), it is less essential in homotypic fusion systems, where both SNAREs are expected to reside on each membrane (17,18).
-
2.
SNARE-driven membrane fusion is sensitive to the immediate lipid environment, and can be stimulated by an asymmetric distribution of cholesterol or acidic lipids, such as phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidic acid (PA), across the fusing membranes (19,20).
-
3.
Intermediate states in SNAREpin formation can be captured in this assay when SNARE assembly is manipulated via temperature (7), through the action of SNARE-derived and SNARE-targeted peptides (21), or indirectly, through the introduction of lipidic fusion antagonists that halt membrane fusion at a late assembly stage (22).
-
4.
Variations on the basic FRET-based assay have been used to separately monitor both total and inner-leaflet lipid mixing (23–25), allowing an increasingly comprehensive understanding of the intrinsic capacity of SNAREs to drive hemifusion.
Physiologically, SNARE-mediated membrane fusion is often under strict spatial and temporal regulation (e.g., during Ca2+-triggered insulin or neurotransmitter release). The in vitro liposome fusion assay using recombinant SNAREs and regulatory proteins has proven very useful in establishing how various proteins exert a role in fusion. For example, the SM proteins, Sec1 and Munc18-1, strongly accelerate SNARE-mediated liposome fusion by interacting with both t- and v-SNARE proteins (26,27). The stimulation of membrane fusion by Munc18-1 was only observed with cognate SNARE pairs for synaptic transmission, indicating a role for SM proteins in enhancing fusion specificity (27). Tucker et al. observed that the cytoplasmic domain of Synaptotagmin 1 (SYT1) could stimulate liposome fusion in the presence, but not in the absence, of Ca2+, which is consistent with a key role of SYT1 in Ca2+-triggered exocytosis (28). Whether SYT1 is soluble or membrane-anchored (and to which membrane, target or vesicle) ultimately dictates the sensitivity of the assay to free Ca2+ (29,30). Finally, addition of the soluble regulator Complexin (CPX) to this assay activated SNAREpin zippering (31) and revealed an intermediate assembly state that appears to favor hemifusion (32). It came as no surprise that the precise activity of the regulatory proteins is influenced by physical parameters such as the electrostatic charge and the lipid composition of fusing membranes (33–37).
FRET-based lipid mixing studies have thus provided significant insights into the molecular mechanisms of fusion events mediated by SNARE proteins, and have led to a minimal model for SNARE-mediated membrane fusion that is now widely accepted (3,4,38). However, two recent articles have raised concerns about the overall relevance of the assay. One group argued that the reconstitution method has a profound impact upon how and whether SNAREs drive lipid mixing, and suggested that only very small proteoliposomes fuse efficiently (39). The second group failed to observe any SNARE-mediated liposome fusion and concluded that only extraordinarily high SNARE densities can give rise to lipid mixing (40). In this article, we intend to comprehensively characterize the determinants of SNARE-mediated lipid mixing in the context of the FRET-based liposome fusion assay using active proteins. We carry out systematic titrations with proteoliposomes prepared at various protein densities and using different reconstitution strategies. We determine the liposome size and the orientation of the proteins after incorporation, and we examine protein and lipid recoveries in each proteoliposome preparation, so that instead of using theoretical values, actual lipid/protein ratios are known for each sample.
Materials and Methods
Protein expression and purification
Unless otherwise noted, all proteins were expressed in the BL21-CodonPlus (DE3)-RIL Escherichia coli bacterial strain from Stratagene (La Jolla, CA). Full-length mouse VAMP2-His6 was expressed and purified from pTW2 (7), and the full length t-SNARE complex including mouse His6-SNAP25 and rat Syn1A was expressed and purified from the polycistronic plasmid pTW34 (41). In a subset of experiments aimed at comparing the activity of t-SNARE proteins made from coexpressed or separately expressed t-SNARE subunits, the full length t-SNARE complex was made from pTW12 (7) and pJM37, which encode for rat Syn1A-His6 and mouse GST-SNAP25, respectively. Coexpressed t-SNAREs pTW12 and pJM37 were cotransformed into Rosetta DE3 cells (Novagen, Madison, WI), and the Syn1A-His6/GST-SNAP25 complex was isolated using glutathione sepharose 4B (GE Healthcare, Piscataway, NJ) and cleaved off using PreScission protease (GE Healthcare) overnight at 4°C. Separately expressed t-SNAREs, either pTW12 or pJM37, were transformed into Rosetta cells. Syn1A-His6 was eluted off Ni-NTA beads (Invitrogen, Carlsbad, CA), and GST-SNAP25 was again cleaved off glutathione beads using PreScission protease. Syn1A-His6 was then incubated overnight on ice with SNAP25 in a 1:3 molar ratio, and the assembled complex was run over a MonoQ ion exchange column for final purification.
Proteoliposome reconstitution
The lipids used in this study were purchased from Avanti Polar Lipids as chloroform solutions. In all titration experiments, VAMP2 was reconstituted with the donor lipid mix comprised of 82 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 15 mol % 1,2-dioleoyl-sn-glycero-3-(phospho-L-serine) (sodium salt) (DOPS), 1.5 mol % 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (DPPE-RHO), and 1.5 mol % 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (DPPE-NBD); the t-SNARE complex was reconstituted with the acceptor lipid mix made of 85 mol % POPC and 15 mol % DOPS. SNARE proteins were reconstituted into liposomes by two different means: the standard method or the direct method.
Dry lipid films were produced by evaporating the chloroform solution with a nitrogen stream for 30 min and under vacuum for 2 h.
In the standard method, the thin lipid films were hydrated with SNARE proteins diluted at the appropriate concentration in buffer A (25 mM HEPES pH 7.4, 100 mM KCl, 1 mM dithiothreitol, 10% (v/v) glycerol, 1% (w/v) n-octyl-β-D-glucopyranoside (β-OG)). The solution was vortexed vigorously for 1 h at room temperature. The direct method was performed as previously described by Chen et al. (39). The thin lipid films were hydrated in buffer B (25 mM HEPES, pH 7.4, 100 mM KCl, and 10% (v/v) glycerol) by vigorously vortexing for 1 h at room temperature. The multilamellar liposome suspension was homogenized by seven freeze-thaw cycles and forced through two polycarbonate membranes with the desired pore size (50 or 100 nm) at least 19 times. To prepare liposomes with smaller size (sonication method), the lipid suspension was disrupted with a microtip sonicator in 30 s pulses for ∼10 min until the solution appeared clear. Metal particles and large liposomes in these sonicated samples were removed by centrifugation for 30 min at 100,000 × g. SNARE proteins diluted in buffer A were added to these preformed purely lipidic liposomes for a final β-OG concentration of 0.66% (w/v), and the solution was incubated for 1 h at room temperature under gentle vortexing.
The detergent concentration in all proteoliposome preparations (standard or direct method) was next reduced to 0.33% (w/v) by rapid dilution in buffer C (25 mM HEPES, pH 7.4, 100 mM KCl, 1 mM dithiothreitol, and 10% (v/v) glycerol), and then removed by overnight flow dialysis against 4 L of buffer C. Proteoliposomes were isolated on a Nycodenz flotation gradient as previously described (7) and preserved on ice for up to 2 weeks.
SNARE-mediated lipid-mixing assay
For each assay, 45 μl of t-SNARE liposomes (a final concentration of ∼3 mM of lipids) were added to 96-well FluoroNunc plates (Nalge Nunc, Rochester, NY) and prewarmed at 37°C for 7 min. The fusion reaction was initiated by adding 5 μl of v-SNARE liposomes (∼0.2 mM of lipids final) at room temperature. Fusion between t- and v-SNARE liposomes was measured by following the dequenching of the DPPE-NBD fluorescence resulting from its dilution into the fused liposomes (7). The NBD fluorescence was monitored at 2-min intervals for 160 min (excitation at 460 nm; emission at 538 nm) by a Fluoroskan II (Thermo Labsystems, Göteborg, Sweden) or a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) plate reader equilibrated to 37°C; fusion kinetics were indistinguishable on the two machines. After 120 min, 10 μl of 2.5% (w/v) n-dodecylmaltoside (Boehringer, Ingelheim, Germany) was added to completely dissolve the liposomes and thus measure the NBD fluorescence at infinite dilution; the data were then normalized as previously described (7).
Protein and lipid recovery
The concentrations of SNARE proteins before and after reconstitution into liposomes were quantified by amido-black staining (42); comparison between these two numbers led to protein recovery. In a similar way, lipid recovery was measured by comparing the RHO fluorescence intensity of the isolated proteoliposomes to that of the starting material. Recovery of lipid in t-SNARE samples (samples without fluorescent lipid) was approximated based on recovery of lipid in a subset of t-SNARE liposomes containing fluorescent lipids. This approximation is likely to be valid, as protein recoveries in each sample (with and without fluorescence) were essentially equivalent. Protein and lipid recoveries were occasionally less efficient, notably for samples prepared by the standard or the direct method with sonicated liposomes. However the final lipid/protein ratios remained similar to that of other liposome preparations, indicating ordinary protein incorporation into these samples.
Protein topology assay
A chymotrypsin cleavage assay was used to determine the orientation of SNAREs after reconstitution into liposomes. Proteoliposomes were treated with 2 μM chymotrypsin at room temperature for 30 min before the reaction was quenched by the addition of 10 mM HCl. All SNAREs facing toward the outside of the liposomes were thus proteolyzed, whereas those facing the lumen were protected. The reaction mixtures were next loaded onto an SDS-PAGE gel, together with the same volume of corresponding untreated samples, and the band intensities were measured with the software ImageJ (43). The percentage of protected proteins was quantified by comparing the amount of intact SNAREs in the chymotrypsin-treated sample to that in the nontreated sample.
Size determination by cryo-electron microscopy
The liposomes were diluted fivefold with buffer C, and 5 μl was applied to perforated carbon-coated grids (R2/4, Quantifoil, Jena, Germany), which had been glow-discharged in the presence of amylamine to reduce the tendency of the liposomes to stick to the carbon support film and to provide generally thicker ice regions. Samples were quickly frozen into vitreous ice by plunging into liquid ethane. Images were recorded on a 20 FEG Cryo-Electron Microscope (Tecnai, Hillsboro, OR) operating in low-dose mode. Both freezing and imaging were conducted at the New York Structural Biology Center. Typical imaging conditions included 50,000× magnification and 3- to 5-μm defocus. Images were analyzed with the software ImageJ.
Results and Discussion
The two properties suggested to play an additional role in the propensity of SNAREs to drive lipid mixing are protein density and liposome size, either of which may be manipulated by the choice of approaches used to reconstitute proteins into liposomes. In their study, Chen et al. used two different reconstitution methods (39): 1), the standard reconstitution method, where the proteoliposomes were formed by cosolubilizing lipids and proteins with detergent before detergent removal by dialysis; and 2), the direct reconstitution method, where the proteins were incorporated into preformed purely lipidic liposomes in the presence of lower amounts of detergent. At physiological protein densities, they observed lipid mixing with liposomes prepared by the standard method but not with those prepared by the direct method. Based on the observation that the standard method yielded more heterogeneous liposome populations than the direct method, this result was accounted for by the significant accumulation of very small proteoliposomes (<30 nm in diameter)—which are prone to fusion due to their high curvature stress—when using the standard method. Using the direct method and a PEG-mediated fusion assay, Dennison et al. observed only efficient docking, instead of lipid mixing, when the proteoliposomes were reconstituted at low protein densities (40). Both studies thus concluded that SNARE-induced lipid mixing observed in previous reports was likely dependent upon the reconstitution method and the physical state of the proteoliposomes.
Effect of SNARE density on liposome fusion
We tested side by side the fusion activity of proteoliposomes bearing SNAREs at various surface densities, and prepared by several reconstitution methods (direct incorporation of proteins into purely lipidic liposomes made by extrusion or sonication, or standard comicellization of lipids and proteins).
A typical fusion titration matrix obtained with proteoliposomes prepared by incorporating SNAREs into purely lipidic liposomes of 50 nm is shown in Fig. 1. Both the initial fusion rate and the final fusion extent are obviously SNARE-density-dependent. In addition, liposome fusion remains very efficient over a wide range of lipid/protein ratios. The increase in fluorescence is reduced to a very low background level when t-SNARE liposomes are incubated with the cytoplasmic domain of VAMP2 before adding v-SNARE liposomes. This serves as a control for the fluorescence drift, which we observe in every lipid mixing assay independent of SNARE density, and confirms that the observed lipid mixing depends on SNARE complex formation.
Figure 1.
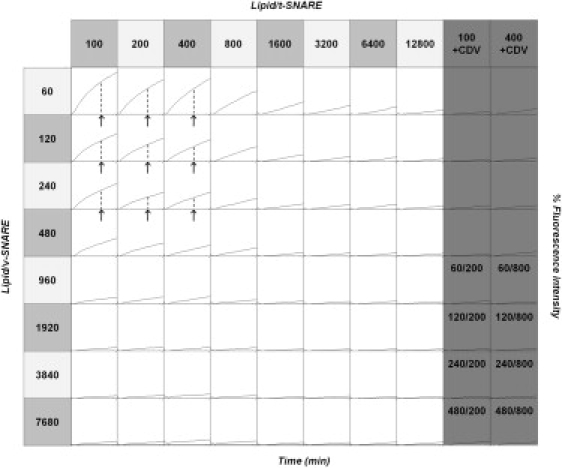
Effect of SNARE density on the kinetics and extent of lipid mixing. Proteoliposomes were prepared by the direct incorporation of SNARE proteins into preformed 50-nm protein-free liposomes. The lipid/protein ratios are theoretical values, derived from starting amounts of protein and lipid. Lipid mixing is measured by monitoring the dequenching of DPPE-NBD lipid probes upon fusion of the fluorescently labeled v-SNARE liposomes with the unlabeled t-SNARE liposomes (x axis range, 0–120 min; y axis range, 0–20% of maximum fluorescence signal; arrows and dashed lines indicate the extent of lipid mixing at t = 80 min, as used in Fig. 2). The last two columns are control experiments in which fusion is inhibited by addition of the cytoplasmic domain of VAMP2 (CDV), which prevents SNAREpin formation by binding to t-SNAREs. The numbers in the last four rows of the control experiments give the v-SNARE and t-SNARE densities used in these reactions. Lipid mixing remains efficient over a wide range of lipid/protein ratios (lipid/t-SNARE ≤ 1600 and lipid/v-SNARE ≤ 480) and, importantly, for SNARE densities consistent with physiology (see main text).
To examine whether liposome fusion was similarly dependent upon v- and t-SNARE surface densities, we compared the extent of lipid mixing 80 min after initiation of the assay across all SNARE densities tested. For a given t-SNARE density, the lipid/v-SNARE ratio required to achieve half of the maximum fusion efficiency (i.e., the fusion obtained with the highest copy number of v-SNAREs in the liposomes) is always between 240 and 480 (Fig. 2). In a similar way, for a given v-SNARE density, one requires 1 t-SNARE/800–1600 lipids to reach half of the maximum fusion efficiency (Fig. S1 in the Supporting Material). Liposome fusion is therefore more sensitive to the variation of the v-SNARE surface density.This may be due in part to the higher percentage of v-SNAREs that face the lumen of the liposomes after reconstitution and are thus not available for fusion (see below).
Figure 2.
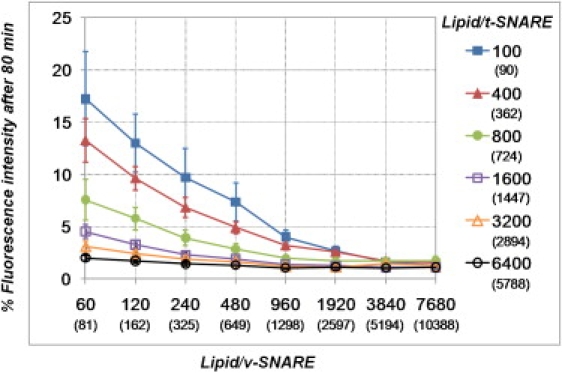
Fusogenicity of SNARE liposomes prepared by the direct incorporation of proteins into preformed 50-nm purely lipidic liposomes (n = 12; error bars indicate standard errors). The extent of lipid mixing is measured as the normalized fluorescence intensity 80 min after initiation of the reaction (Fig. 1, arrows and dashed lines) and plotted against the lipid/v-SNARE ratio (see Fig. S1 for fusion data plotted against the lipid/t-SNARE ratio, and Fig. S3 and Fig. S4 for fusogenicity of SNARE liposomes prepared according to other methods). The lipid/SNARE ratios are depicted either as theoretical (i.e., those expected based on protein and lipid inputs, as in Fig. 1) or as actual (i.e., taking into account average measures of lipid and protein recoveries, as well as protein orientation) values (see section entitled Proteoliposome characterization).
It is interesting to note that very similar results were obtained with proteoliposomes prepared using other, previously published, reconstitution strategies (Fig. S2, Fig. S3, and Fig. S4), suggesting that SNARE density, not reconstitution approach, is the most important variable.
A recent quantitative description of synaptic vesicle composition by Takamori et al. (44) established that there are ∼70 copies of VAMP2/42-nm synaptic vesicle, which would correspond to proteoliposomes with a lipid/VAMP2 ratio of ∼120 (when considering only the outer lipid leaflet since, in a synaptic vesicle, most of the VAMP2s are facing outside). We found that, irrespective of the preparation method, there is always significant lipid mixing when the v-SNARE is at its physiological density, and the lipid/t-SNARE ratio is <800–1600 (Fig. S2). The physiological density of t-SNAREs is not yet precisely known, but many independent studies have shown that these proteins concentrate in small (∼100-nm) domains. A recent work by Sieber et al. using PC12 cells predicted a local (clustered) lipid/Syn1A ratio of ∼50. Further, they found an average plasma membrane Syn1A density of 1800 molecules per μm2, which would correspond to proteoliposomes with a lipid/Syn1A ratio of 800 (45). In addition, the Syn1A clusters were shown to partially colocalize with SNAP25 clusters at sites where secretory vesicles dock and fuse (46,47). The physiological lipid/t-SNARE ratio will thus not be greater than 800 and will very likely be much lower. Therefore, we observed efficient SNARE-mediated lipid mixing at SNARE densities consistent with the physiology of synaptic vesicle fusion.
Why could other groups not observe more robust and systematic SNARE-mediated lipid mixing in their assay? There are two ways to prepare t-SNAREs for in vitro reconstitution experiments: 1), by coexpression of Syn1A and SNAP25 (the approach used here); and 2), by incubating separately purified Syn1A and SNAP25 (the approach used by Chen et al. and Dennison et al.). Several labs, including ours, have reported efficient membrane fusion when using coexpressed t-SNARE complexes (7,28,48). To further confirm this, we directly compared the fusion activity of the coexpressed t-SNAREs used here to that of t-SNAREs made by separately expressing and subsequently assembling Syn1A and SNAP25. The t-SNARE complexes made from individually expressed t-SNARE subunits clearly led to a dramatic reduction in lipid mixing efficiency (Fig. 3), despite the fact that t-liposome properties were independent of the t-SNARE reconstitution strategy (see below). To make sure that this drop in fusion was not due to the use of new plasmids (those coding for individually expressed t-SNARE subunits), we also performed liposome fusion experiments involving t-SNAREs made from coexpression of these new plasmids. Again, coexpressed t-SNAREs led to much higher lipid mixing efficiency than separately expressed t-SNAREs (Fig. S5). This suggests that the means by which the t-SNARE complex is manipulated is crucial to maintaining its activity. For example, it is known that 2:1 Syn1A/SNAP25 complexes, where the second Syn1A molecule occupies the position of VAMP2, are possible products of incubation of Syn1A and SNAP25, which reduces the rate of SNARE complex assembly and thus fusion efficiency (49,50).
Figure 3.
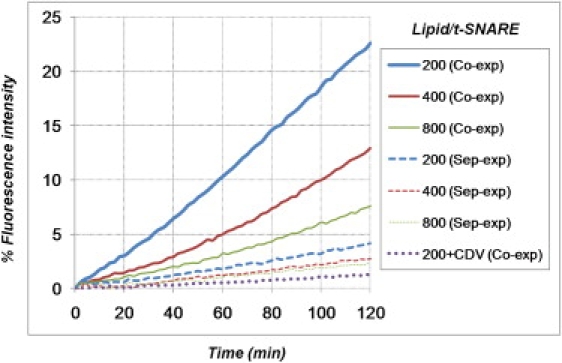
Coexpressed t-SNAREs are more fusogenic than separately expressed t-SNAREs. v-SNARE liposomes 50 nm in size (prepared by the direct method, with lipid/protein = 120) were fused with 50-nm liposomes (direct method) containing t-SNAREs at various surface densities (lipid/protein = 200, 400, or 800) and prepared by two different means: coexpression of Syn1A and SNAP25 as in Figs. 1 and 2 (solid lines) or incubation of separately purified Syn1A and SNAP25 (dashed lines). The extent of lipid mixing is reduced at least fourfold when using separately expressed t-SNARE subunits.
Proteoliposome characterization
Surface density of SNAREs available for fusion
Instead of using theoretical lipid/protein ratios, as in previous works, which assume both proper (i.e., topologically correct) and comprehensive incorporation of purified proteins, we wanted to determine the actual lipid/protein ratios (i.e., in our final gradient-floated liposomes). This requires determination of the recovery of both lipids and proteins after liposome reconstitution. The final protein concentration was examined using the amido-black method (42), and lipid recovery was quantified by comparing the Rhodamine fluorescence signal of the proteoliposomes with that of the starting material. By these measures, we found that the final lipid/protein ratios never differ by >25% from the target values (Table 1 and Table S1).
Table 1.
Main physical parameters of 50-nm proteoliposomes prepared by the direct method
| Liposome size (nm) | Protein recovery (%) | Lipid recovery (%) | Proteins facing out (%) | |
|---|---|---|---|---|
| t-SNARE liposomes | 54 ± 11 | 56 ± 9 | 78 ± 9 | 77 ± 7 |
| v-SNARE liposomes | 57 ± 12 | 70 ± 21 | 89 ± 9 | 47 ± 13 |
Total number of proteoliposome preparations was 12. Error bars indicate standard deviations.
Next, we established how many SNAREs were incorporated with their binding domain oriented toward the outside of the liposomes (SNAREs that are actually available for mediating fusion). To do this, we used a chymotrypsin cleavage assay, in which the protease selectively cleaves SNAREs facing outside, whereas SNAREs facing the lumen of the liposomes remain protected (Fig. 4 and Fig. S6). t-SNARE and v-SNARE proteins displayed different orientation properties. In t-SNARE liposomes, ∼65–85% of the proteins were exposed to the extravesicular medium, whereas in v-SNARE liposomes, ∼45–60% of the proteins were facing outside (Table 1, Table S1, and Table S2). This difference may result from the size difference between the bulky t-SNARE complex and the single-chain v-SNARE protein and is consistent with our very first reconstitutions more than a decade ago (7). We were surprised to learn that the standard comicellization scheme and the direct incorporation method, as previously used in other SNARE reconstitution studies (39,40), gave rise to similar protein insertion topology. This can be explained by the high detergent/lipid ratio used in the direct method employed here (39). In a systematic study of detergent-assisted transmembrane protein incorporation into preformed liposomes, the authors recommended incorporating the proteins into detergent-saturated liposomes at the onset of solubilization (51). In the case of the detergent used here (β-OG), this corresponds to a detergent/lipid (molar) ratio of 1.3, which is lower than the ratio of 4.5 used here. At such a high detergent/lipid ratio, proteoliposomes are formed, at least in part, by micellar coalescence and proteins can thus insert into the bilayers from both sides.
Figure 4.
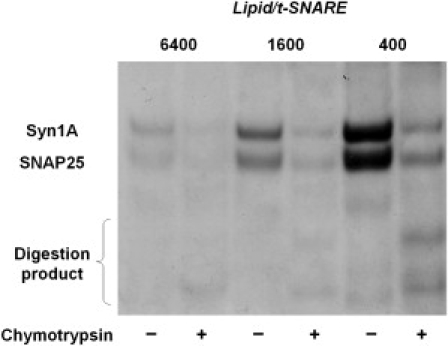
Orientation of SNAREs in the liposome membrane. In this example, proteoliposomes were prepared by direct incorporation of t-SNAREs into 50-nm liposomes. Upon addition of chymotrypsin, t-SNAREs facing outside were proteolyzed, whereas those facing the lumen of the liposomes were protected. Four t-SNARE liposomes with different lipid/protein ratios were exposed to chymotrypsin for 30 min at room temperature and then loaded onto an SDS-PAGE gel, juxtaposed with the same amount of corresponding untreated samples. The percentage of unprotected t-SNAREs, i.e., those exposing their cytosolic domain to the outside, was calculated by comparing the band intensity of the chymotrypsin-treated sample to that of the untreated sample. In this case, ∼75% of the t-SNAREs have their cytoplasmic domain oriented toward the outside of the liposomes. Statistics and results for other SNARE liposomes are displayed in Table 1, Table S1, and Table S2.
Taken together, these results show that the surface densities of active SNAREs in our proteoliposomes (SNAREs in the outer monolayer of liposomes that are thus available for binding and fusion) are very close to theoretical values (Fig. 2 and Table S3).
Highly fusogenic subpopulations
Given the complex mixture of protein, lipid, and detergent from which liposomes are ultimately reconstituted, an absolutely homogeneous distribution of materials is an impossible standard. At the single-liposome level there may be significant variability in protein density or liposome size, and it is therefore important to establish whether a small proportion of liposomes with unusual composition may in fact be responsible for the bulk of the signal.
One such argument holds that only the smallest liposomes, with extreme and nonphysiological curvatures, are prone to SNARE-mediated fusion (since the diameter of synaptic vesicles ranges from 30 nm to 60 nm (44), we will consider that an artificial liposome is abnormally small when its size is <30 nm). To address this concern, we asked two questions: 1), Are the liposomes we prepared composed of disparate or highly variable sizes, such that some could be considered highly curved? and 2), Under what circumstances could the signal we observe originate from a small population? That is, is the amount of signal we observe consistent with only a fraction of the liposomes participating in the fusion reaction?
To consider size, we measured the diameters of liposomes deriving from each of the reconstitution approaches using cryoelectron microscopy (Fig. 5 and Fig. S7). We did not observe any significant difference in the physical parameters of the proteoliposomes prepared by each of the reconstitution approaches (Table 1 and Table S1). In all cases, the average diameter of t-SNARE liposomes was 50–65 nm and that of v-SNARE liposomes was 35–55 nm. In addition, the size distributions were comparable, with <5% of these liposomes having a diameter <30 nm. The only exceptions were v-liposomes prepared by the standard method, which were smaller on average compared to those prepared by other methods (average diameter of 35 nm compared to 45–55 nm, with ∼20% having a diameter <30 nm). It is worthy of note that this size shift toward a somewhat smaller population of v-liposomes did not have a dramatic impact on either the kinetics or extent of lipid mixing. Thus, the preparation method does not appear to drastically impact the size of our liposomes, and the average size of the population does not appear to be an indicator of overall fusogenicity.
Figure 5.
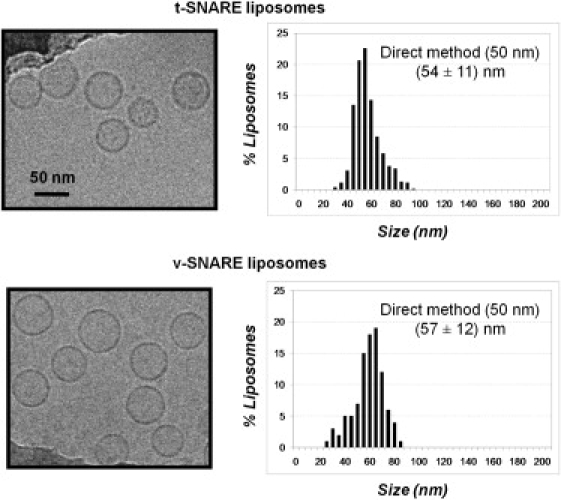
Size distribution of SNARE liposomes measured by cryoelectron microscopy. Proteoliposomes were made by direct incorporation of SNAREs into 50-nm protein-free liposomes (as in Figs. 1 and 2). Histograms of t-SNARE and v-SNARE liposomes (lipid/protein = 400 and 60, respectively) were obtained from n = 446 and n = 627 liposomes, respectively; error bars indicate standard deviations. The size distributions of SNARE liposomes prepared by other methods are displayed in Fig. S7.
But is there a possibility of a particularly active minor fraction of liposomes contributing most of the signal? The magnitude of our fluorescence change renders this interpretation unlikely, even in the case of v-liposomes prepared by the standard method (which display the largest subpopulation of highly curved liposomes). Fusion data in this article are plotted in percent maximum fluorescence observed after detergent addition, that is, after complete solubilization of the liposomes in which the FRET pairs are maximally separated and quenching has been reduced to zero. This is a common and convenient notation used not only in the SNARE field, but across many fusion paradigms. However, since the extent of quenching is related to the surface density of fluorophores in a nonlinear way (52), the relationship between percent maximum fluorescence and extent of liposome fusion is not always intuitive. For this reason, we also introduced the metric rounds of fusion (41), in which we determine the absolute extent to which the total fluorescent lipid would have to be diluted (by fusion with nonfluorescent liposomes) to achieve the fluorescence intensity gains observed. Thus, in our most active (i.e., highest surface density) preparations using the standard method, the total fluorescence increase we observe (17% in Fig. S3 D) is consistent with a 70% dilution of the original fluorescence or, to put it another way, it is as if 70% of the v-liposomes each fused once with nonfluorescent t-liposomes (41). Further, because the relationship between the increase in fluorescence and dilution is nonlinear, it would not be sufficient for 20% of the v-liposomes (the subpopulation of highly curved liposomes) to instead fuse three to four times each. If all of the fluorescence increase in our sample were to arise from just 20% of the v-liposomes, these v-liposomes would have to fuse enough to generate >100% of their detergent maximum signal, which is of course impossible (see Supporting Material for more details). Even if one included the contribution of 30-nm liposomes to the fluorescence increase, v-liposomes would still have to fuse on average 10 times with comparably sized fluorophore-free t-liposomes. Finally, if one were to suppose that such a pool of hyperactive liposomes existed, it could not arise simply from very small highly curved liposomes, since after even just one fusion event, the resulting v-t liposome would now have a diameter >30 nm.
In short, although it is likely that individual liposomes display varying propensities to fuse, and some subpopulations might be particularly fusogenic, our bulk measures are dominated by a behavior that represents a very large fraction of the total liposome pool.
Conclusions
The methods previously described to reconstitute transmembrane SNAREs into artificial liposomes do not significantly affect either the physicochemistry of the proteoliposomes (size, actual lipid and protein compositions, and fraction of the proteins available for binding and fusion) or the ability of SNAREs to drive lipid mixing in vitro. Forming the t-SNARE complex from coexpressed Syn1A and SNAP25 subunits is, however, crucial to maintaining its fusogenic activity. An important finding is that significant lipid mixing is always observed at SNARE densities consistent with the physiology of synaptic vesicle fusion.
Supporting Material
Text, three tables, and seven figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00558-8.
Supporting Material
Acknowledgments
We thank Frédéric Pincet for critical reading of the manuscript and many fruitful discussions.
This work was supported by the Human Frontier Science Program, a travel grant from the National Science Foundation, and National Institutes of Health grants to James E. Rothman. David Tareste is funded by an ANR Jeunes Chercheurs grant.
Contributor Information
James E. Rothman, Email: james.rothman@yale.edu.
David Tareste, Email: david.tareste@inserm.fr.
References
- 1.Söllner T., Whiteheart S.W., Rothman J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 2.Brunger A.T. Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- 3.Jackson M.B., Chapman E.R. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 2006;35:135–160. doi: 10.1146/annurev.biophys.35.040405.101958. [DOI] [PubMed] [Google Scholar]
- 4.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 5.Hanson P.I., Roth R., Heuser J.E. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 6.Sutton R.B., Fasshauer D., Brunger A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 7.Weber T., Zemelman B.V., Rothman J.E. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 8.Li F., Pincet F., Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 9.Hu K., Carroll J., Davletov B. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 2002;415:646–650. doi: 10.1038/415646a. [DOI] [PubMed] [Google Scholar]
- 10.Holt M., Riedel D., Jahn R. Synaptic vesicles are constitutively active fusion machines that function independently of Ca2+ Curr. Biol. 2008;18:715–722. doi: 10.1016/j.cub.2008.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Struck D.K., Hoekstra D., Pagano R.E. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 12.Düzgüneş N., Goldstein J.A., Felgner P.L. Fusion of liposomes containing a novel cationic lipid, N-[2,3-(dioleyloxy)propyl]-N,N,N-trimethylammonium: induction by multivalent anions and asymmetric fusion with acidic phospholipid vesicles. Biochemistry. 1989;28:9179–9184. doi: 10.1021/bi00449a033. [DOI] [PubMed] [Google Scholar]
- 13.Hoekstra D., Klappe K. Fluorescence assays to monitor fusion of enveloped viruses. Methods Enzymol. 1993;220:261–276. doi: 10.1016/0076-6879(93)20088-k. [DOI] [PubMed] [Google Scholar]
- 14.Chan Y.H., van Lengerich B., Boxer S.G. Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides. Proc. Natl. Acad. Sci. USA. 2009;106:979–984. doi: 10.1073/pnas.0812356106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parlati F., McNew J.A., Rothman J.E. Topological restriction of SNARE-dependent membrane fusion. Nature. 2000;407:194–198. doi: 10.1038/35025076. [DOI] [PubMed] [Google Scholar]
- 16.Paumet F., Brügger B., Rothman J.E. A t-SNARE of the endocytic pathway must be activated for fusion. J. Cell Biol. 2001;155:961–968. doi: 10.1083/jcb.200104092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwilling D., Cypionka A., Jahn R. Early endosomal SNAREs form a structurally conserved SNARE complex and fuse liposomes with multiple topologies. EMBO J. 2007;26:9–18. doi: 10.1038/sj.emboj.7601467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mima J., Hickey C.M., Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J. 2008;27:2031–2042. doi: 10.1038/emboj.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vicogne J., Vollenweider D., Pessin J.E. Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc. Natl. Acad. Sci. USA. 2006;103:14761–14766. doi: 10.1073/pnas.0606881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong J., Borbat P.P., Shin Y.K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. USA. 2009;106:5141–5146. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melia T.J., Weber T., Rothman J.E. Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J. Cell Biol. 2002;158:929–940. doi: 10.1083/jcb.200112081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melia T.J., You D., Rothman J.E. Lipidic antagonists to SNARE-mediated fusion. J. Biol. Chem. 2006;281:29597–29605. doi: 10.1074/jbc.M601778200. [DOI] [PubMed] [Google Scholar]
- 23.Lu X., Zhang F., Shin Y.K. Membrane fusion induced by neuronal SNAREs transits through hemifusion. J. Biol. Chem. 2005;280:30538–30541. doi: 10.1074/jbc.M506862200. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y., Zhang F., Shin Y.K. Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2005;12:417–422. doi: 10.1038/nsmb921. [DOI] [PubMed] [Google Scholar]
- 25.Bhalla A., Chicka M.C., Chapman E.R. Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- 26.Scott B.L., Van Komen J.S., McNew J.A. Sec1p directly stimulates SNARE-mediated membrane fusion in vitro. J. Cell Biol. 2004;167:75–85. doi: 10.1083/jcb.200405018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen J., Tareste D.C., Melia T.J. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Tucker W.C., Weber T., Chapman E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 29.Mahal L.K., Sequeira S.M., Söllner T.H. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J. Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stein A., Radhakrishnan A., Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat. Struct. Mol. Biol. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- 31.Malsam J., Seiler F., Söllner T.H. The carboxy-terminal domain of complexin I stimulates liposome fusion. Proc. Natl. Acad. Sci. USA. 2009;106:2001–2006. doi: 10.1073/pnas.0812813106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaub J.R., Lu X., McNew J.A. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat. Struct. Mol. Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 33.Dai H., Shen N., Rizo J. A quaternary SNARE-synaptotagmin-Ca2+-phospholipid complex in neurotransmitter release. J. Mol. Biol. 2007;367:848–863. doi: 10.1016/j.jmb.2007.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martens S., McMahon H.T. Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- 35.Paddock B.E., Striegel A.R., Reist N.E. Ca2+-dependent, phospholipid-binding residues of synaptotagmin are critical for excitation-secretion coupling in vivo. J. Neurosci. 2008;28:7458–7466. doi: 10.1523/JNEUROSCI.0197-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon T.Y., Lu X., Shin Y.K. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2008;15:707–713. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seiler F., Malsam J., Söllner T.H. A role of complexin-lipid interactions in membrane fusion. FEBS Lett. 2009;583:2343–2348. doi: 10.1016/j.febslet.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malsam J., Kreye S., Söllner T.H. Membrane fusion: SNAREs and regulation. Cell. Mol. Life Sci. 2008;65:2814–2832. doi: 10.1007/s00018-008-8352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X., Araç D., Rizo J. SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys. J. 2006;90:2062–2074. doi: 10.1529/biophysj.105.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dennison S.M., Bowen M.E., Lentz B.R. Neuronal SNAREs do not trigger fusion between synthetic membranes but do promote PEG-mediated membrane fusion. Biophys. J. 2006;90:1661–1675. doi: 10.1529/biophysj.105.069617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parlati F., Weber T., Rothman J.E. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl. Acad. Sci. USA. 1999;96:12565–12570. doi: 10.1073/pnas.96.22.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaffner W., Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 1973;56:502–514. doi: 10.1016/0003-2697(73)90217-0. [DOI] [PubMed] [Google Scholar]
- 43.Abramoff M.D., Magelhaes P.J., Ram S.J. Image processing with ImageJ. Biophotonics Intl. 2004;11:36–42. [Google Scholar]
- 44.Takamori S., Holt M., Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 45.Sieber J.J., Willig K.I., Lang T. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- 46.Lang T., Bruns D., Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieber J.J., Willig K.I., Lang T. The SNARE motif is essential for the formation of syntaxin clusters in the plasma membrane. Biophys. J. 2006;90:2843–2851. doi: 10.1529/biophysj.105.079574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu T., Tucker W.C., Weisshaar J.C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys. J. 2005;89:2458–2472. doi: 10.1529/biophysj.105.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao W., Poirier M.A., Shin Y.K. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat. Struct. Biol. 2001;8:308–311. doi: 10.1038/86174. [DOI] [PubMed] [Google Scholar]
- 50.Pobbati A.V., Stein A., Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 51.Rigaud J.L., Lévy D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003;372:65–86. doi: 10.1016/S0076-6879(03)72004-7. [DOI] [PubMed] [Google Scholar]
- 52.Fung B.K., Stryer L. Surface density determination in membranes by fluorescence energy transfer. Biochemistry. 1978;17:5241–5248. doi: 10.1021/bi00617a025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.