

Abstract
Most dsRNA viruses have a genome-enclosing capsid that comprises 120 copies of a single coat protein (CP). These 120 CP subunits are arranged as asymmetrical dimers that surround the icosahedral fivefold axes, forming pentamers of dimers that are thought to be assembly intermediates. This scheme is violated, however, in recent structures of two dsRNA viruses, a fungal virus from family Partitiviridae and a rabbit virus from family Picobirnaviridae, both of which have 120 CP subunits organized as dimers of quasisymmetrical dimers. In this study, we report the CP backbone trace of a second fungal partitivirus, determined in this case by electron cryomicroscopy and homology modeling. This virus also exhibits quasisymmetrical CP dimers that are connected by prominent surface arches and stabilized by domain swapping between the two CP subunits. The CP fold is dominated by α-helices, although β-strands mediate several important contacts. A dimer-of-dimers assembly intermediate is again implicated. The disordered N-terminal tail of each CP subunit protrudes into the particle interior and likely interacts with the genome during packaging and/or transcription. These results broaden our understanding of conserved and variable aspects of partitivirus structure and reflect the growing use of electron cryomicroscopy for atomic modeling of protein folds.
Introduction
Encapsidated double-strand (ds)RNA viruses are currently classified into seven taxonomic families (Birna-, Chryso-, Cysto-, Partiti-, Picobirna-, Reo-, and Totiviridae) with natural hosts from bacteria to humans (1). Among these viruses are many significant pathogens including rotavirus, bluetongue virus, and rice dwarf virus. Their genomes comprise a varying number of dsRNA segments, from 1 in totiviruses to 12 in some reoviruses. Virions from each family have been subject to structure determinations by x-ray crystallography and/or by transmission electron cryomicroscopy (cryoTEM) and 3D image reconstruction. In five of the families (all but birna- and chrysoviruses), the virions possess a uniquely organized, genome-enclosing capsid that comprises 120 copies of a single coat protein (CP) (2–8). In each case, this 120-subunit T=1 (so-called T=2) capsid, together with the viral genome, the viral RNA-dependent RNA polymerase (RdRp), and any other viral enzymes required for RNA synthesis and processing, is designed to remain intact throughout infection and functions as an elegant nanomachine for transcription and replication (9,10).
Penicillium stoloniferum viruses S and F (PsV-S and -F) are small dsRNA viruses from the saprophytic, filamentous fungus P. stoloniferum (11) and contributed some of the first evidence that dsRNA is a potent inducer of interferon (12). Both PsV-S and -F are classified in the family Partitiviridae, genus Partitivirus (13). Both possess two essential genome segments, dsRNA1 and -2, which are individually packaged into separate virus particles (14). The larger (dsRNA1: 1754 bp for PsV-S, 1677 bp for PsV-F) encodes the RdRp (539 aa for PsV-S, 538 aa for PsV-F), whereas the smaller (dsRNA2: 1582 bp for PsV-S, 1500 bp for PsV-F) encodes the CP (434 aa for PsV-S, 420 aa for PsV-F) (15,16). PsV-S and -F are distantly related: the primary sequences of RdRp and CP are respectively 27% and 19% identical between the two viruses in pairwise alignments with T-Coffee (17) generated for this study. PsV-F, but not PsV-S, contains a satellite segment, the 677-bp dsRNA3, which is unrelated in sequence to the other two segments (16). Both viruses can coinfect P. stoloniferum at the same time (11), but the CP of each associates only with itself in forming capsids (18) and packages only its own RNAs (11). Purified virions of both viruses exhibit semiconservative transcription activity (8,19). Like most other fungal viruses, PsV-S and -F do not naturally undergo efficient extracellular transmission and are instead transmitted intracellularly during cell division and cell-cell fusion (13).
We have determined previously the structure of PsV-S virions to 7.3-Å resolution by cryoTEM and 3D image reconstruction (20). One obvious feature of the PsV-S cryoreconstruction is that 60 unusual surface arches, each formed by a quasisymmetrical CP dimer, protrude from the capsid shell. We have also determined recently the structure of PsV-F virions to 3.3 Å resolution by x-ray crystallography (8). The main surface features of PsV-F are quite similar to those of PsV-S, though distinguishable in specific details. Unexpectedly, we discovered a domain-swapping mechanism that produces extensive interactions between the two subunits in each preferred CP dimer. These two subunits are related by almost-perfect local twofold (2f) symmetry, consistent with the observation of quasisymmetrical dimers in the cryoreconstruction of PsV-S (20). Also unexpectedly, the capsid assembly of PsV-F appears to involve the formation of a dimer-of-dimers intermediate, with an arrangement similar to the unrelated flavivirus E glycoprotein (21) and different from the pentamer-of-dimers intermediate suggested for several other dsRNA viruses (2,3,9,22). The crystal structure of a rabbit picobirnavirus determined recently also shows quasisymmetrical CP dimers and domain swapping (7), although many details of this structure differ from those of PsV-F.
Recent advances in cryoTEM reconstruction have led to several near-atomic structure models from single-particle analyses of thousands or tens of thousands of randomly oriented, symmetrical particles (23–26). Here we report the backbone structure of PsV-S virions, determined by homology modeling from a cryoreconstruction to ∼4.5 Å resolution. Our PsV-S model shows capsid organization and domain-swapping mechanisms very similar to those in PsV-F. The PsV-S CP comprises two domains: one forming the capsid shell and the other a surface arch. Superposition of the PsV-S model onto that of PsV-F indicates that the shell domains of the two CPs assume almost identical shape and topology, whereas the arches differ substantially in both orientation and structural details. Although the structure is strongly α-helical, four 4-strand β-sheets, two of which are made of β-strands from both subunits, play an important role in stabilizing the CP dimers related by local 2f symmetry. A compact, diamond-shaped dimer of these dimers is centered on each icosahedral 2f axis and represents a likely assembly intermediate. Examination of densities in the central cores of both viruses reveals less compact RNA layers in PsV-S compared to PsV-F. Rod-like densities appear at the inner capsid surface in cryoreconstructions computed at lower resolutions and are probably formed by the positively charged, disordered N-terminal peptide in PsV-S, as also in PsV-F. These observations suggest that divergent members of family Partitiviridae use strongly similar principles of capsid architecture, particle assembly, and RNA organization.
Materials and Methods
Due to space limitations, these are described in the Supporting Material. References 27–41 pertain to the methods.
Results and Discussion
Subnanometer cryoTEM maps of PsV-F and comparisons with crystal structure
Using established methods (28,42), we refined the cryoreconstruction of PsV-F virions from 8.0 Å resolution (8) to 6.0 Å resolution (Fig. 1, A and B). For each map, resolution was estimated using the conservative, FSC score of 0.5 (31). An atomic model of the PsV-F capsid derived from the 3.3 Å PsV-F crystal structure (8) was then fitted into the 6.0 Å cryoTEM map and found to match remarkably well except in a few surface loops where the crystal structure exhibits high B factors.
Figure 1.
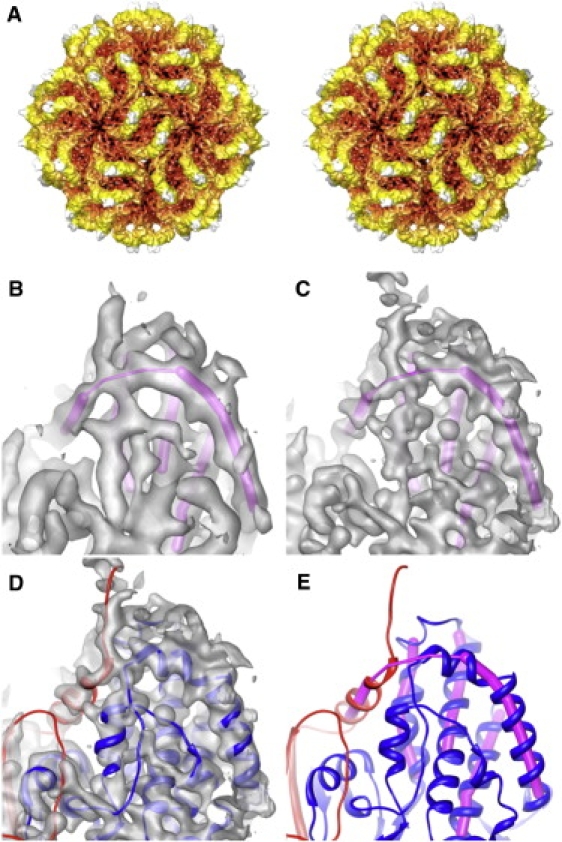
PsV-F capsid structure. (A) Stereo view along a 2f direction of a cryoTEM reconstruction at 6.0 Å resolution. The particle is radially depth-cued, from red (low radii) to white (high radii). (B and C) The same magnified view of cryoTEM reconstructions respectively calculated at 6.0 and 4.7 Å resolution. Superimposed in magenta is an interpretation of the main-chain directionality solely from these maps. (D) Crystal-derived atomic model of PsV-F CP dimer (8) fitted into the 4.7 Å map. (E) Atomic model of PsV-F CP dimer with the two chains shown in red and blue, indicating the close contact between these two chains that leads to the wrong interpretation of main-chain directionality in B and C.
The fitted atomic model was used as a visual guide to assess the quality of results from further steps of refinement (see Materials and Methods in the Supporting Material), which led to an improved, higher-resolution cryoreconstruction of PsV-F. This further-refined cryoTEM map correlated well with a crystallographic map calculated at comparable resolution by noncrystallographic symmetry averaging and phase extension, and all α-helices and β-strands fit nicely within the cryoTEM densities. Based on the appearances of features in the cryoTEM map, especially the ridges and troughs of α-helical elements (Fig. 1, C and D), we estimated the effective resolution to be slightly better than 5.0 Å. Visual comparisons of this map to crystallographic or cryoTEM maps of other similarly resolved structures (23,24) supported this estimate.
The improved quality of the PsV-F cryoTEM map made it much easier to identify secondary-structure elements, as illustrated in Fig. 1, B–E. In Fig. 1, B, C, and E, magenta rods are used to represent elements interpreted as α-helices in the 6.0 Å map (Fig. 1 C), including the extended boomerang-like feature near the top of each view. With reference to the cryoTEM map alone, whether at 6.0 Å or even at higher resolution (∼5.0 Å, Fig. 1 D), the densities contributing to this feature were most readily interpreted to be continuous and hence formed by contiguous regions of the same peptide chain. However, on fitting the crystal-derived atomic model, it was obvious that the boomerang is not continuous and instead is formed by two different polypeptide chains in close proximity (Fig. 1, D and E). This represents part of the domain swapping that occurs in the PsV-F CPA-CPB dimer as first described by Pan et al. (8). Thus, correct de novo modeling of the backbone path of PsV-F CP from cryoTEM maps alone, even at slightly better than 5.0 Å resolution, was confounded by particular, complex aspects of the capsid structure.
Resolution estimate for PsV-F cryoreconstruction
The resolution of our final PsV-F map was estimated to be 5.0 Å or slightly better based on visual assessment of characteristic features (e.g., appearances of α-helices and β-sheets). To obtain a more objective estimate of resolution, we computed the correlation between structure factors computed from the full-atom, crystal-derived model and the cryoTEM map as a function of spatial frequency (see Materials and Methods in the Supporting Material). Careful analysis of these and several other correlations suggested that the effective resolution of the PsV-F cryoTEM map was ∼4.7 Å. At this resolution the correlation coefficient between model and cryoreconstruction is similar to that observed when the model is compared to the final, crystallographically averaged map at 3.3 Å resolution (Fig. 2 A). Hence, our systematic determination that the resolution limit of the PsV-F cryoreconstruction reached 4.7 Å is consistent with our previous estimate of slightly >5.0 Å based on less objective, visual comparisons. These results also support the notion that the commonly used FSC = 0.5 criterion may underestimate the effective resolution of some cryoTEM maps or portions thereof (43).
Figure 2.
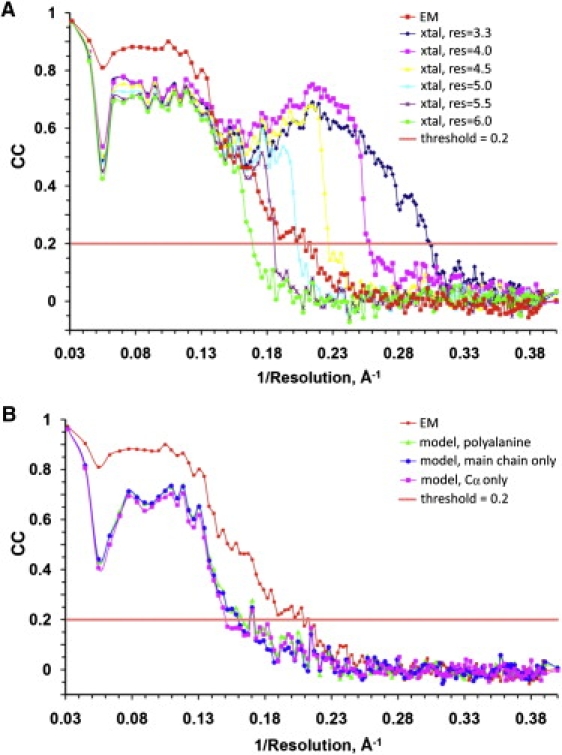
Resolution estimation of the PsV-F cryoTEM map based on crystallographic maps. (A) CC plots comparing the cryoTEM map (EM) with the 3.3 Å x-ray crystallographic map (xtal) calculated at different resolution (res) limits, in Å. (B) CC plots comparing the final cryoTEM map (EM) with different versions of the PsV-F atomic model.
In the above analysis, we used the full atomic model of PsV-F to compute correlations with the cryoreconstruction. Subsequently, to assess how much detail the model must include to make meaningful resolution calculations, we repeated the systematic correlation analysis with three, progressively less-detailed models. These included a polyalanine model and models containing only backbone or Cα atoms. CC plots computed for each of these models versus the cryoreconstruction showed that the inclusion of side-chain information produces higher correlations compared with less-detailed models and that these correlations extend over a wide range of spatial frequencies (Fig. 2 B). Even at moderate resolutions such as 6.0 Å where side chains are not resolved, the full atomic model showed better correlations than any of the less-detailed models, all of which yielded correlations at the noise level. We interpret these findings to indicate that, even though side chains may not be well resolved in our maps, they made important contributions to the observed electron scattering.
CryoTEM maps of PsV-S at higher resolution
Our success with refining the cryoreconstruction of PsV-F virions from 8.0 Å to >5.0 Å encouraged us to attempt the same for PsV-S, which was resolved previously to 7.3 Å (20) and for which there is yet no crystal structure. Using the same basic refinement procedures that worked well with the PsV-F image data, we improved the cryoreconstruction of PsV-S virions, first to ∼6.0 Å resolution and ultimately to an effective resolution at least as good as for PsV-F (Fig. 3). In this case, high-resolution features indicative of secondary structures (e.g., ridges and troughs on all α-helices) and also backbone groups and even some side chain densities were clearly visible (Fig. 3 C). Though most side chains were not resolved, density knobs regularly spaced ∼3.8 Å along the backbone were visible. This interval closely matches the distance between adjacent Cα atoms in protein structures. Based on such observations, we estimated the effective resolution of the final map to approximate 4.5 Å (see Materials and Methods in the Supporting Material). As was true for PsV-F, visual comparisons of features present in the PsV-S map to other, similarly resolved structures (23,24) supported this estimate.
Figure 3.
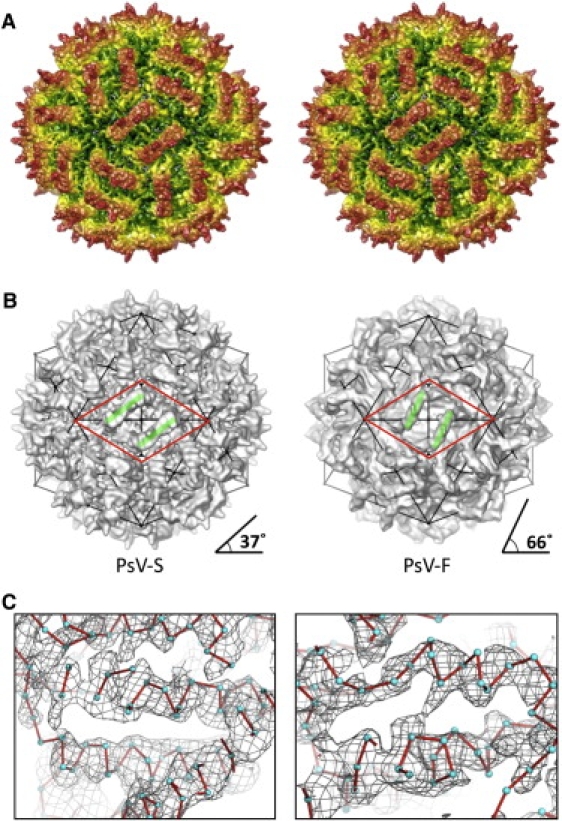
PsV-S capsid structure. (A) Stereo view along a 2f direction of a cryoTEM reconstruction at ∼4.5 Å resolution (estimated). The particle is radially depth-cued, from green (low radii) to red (high radii). (B) The CPA-CPB dimer assumes different directionality (green lines; angle measurements estimated at bottom) in PsV-S (left) and -F (right). Both particles are viewed along a 2f direction. Symmetry elements are evident from the icosahedral line drawing superimposed on each particle. The approximate boundaries of the putative assembly intermediate, a dimer of CPA-CPB dimers, are outlined by a red diamond in each virus. (C) Magnified views of two regions of the PsV-S cryoTEM map, represented as a wire mesh and with positions of Cα atoms modeled as cyan spheres and connected by red lines to indicate chain path. For C, the map is displayed at a high density-contour level (2.0 σ) to exclude low-density features.
The PsV-S and -F cryoTEM maps display many similar features but also some distinct differences. Both virus capsids are constructed from 60 CPA-CPB dimers, each of which has a prominent surface arch with local 2f symmetry. Also, four large, pleat-like densities occur at the dimer interface in both virions, which indicates that PsV-S uses a domain-swapping mechanism for CPA-CPB dimer formation as first reported for PsV-F (8). Despite these striking similarities, the conformations of the surface arches are quite different in the two viruses. In PsV-F, the two tips of each arch are in close contact, whereas in PsV-S they splay apart. The opening beneath the PsV-S arch (Fig. 3 A) is also smaller than that in PsV-F. In addition, the surface arches of the two viruses are oriented quite differently: the long axis of the arch of the CPA-CPB dimer assumes an ∼37° angle in PsV-S, versus an ∼66° angle in PsV-F, relative to the line between neighboring 5f vertices of the icosahedral capsids (Fig. 3 B). Such a marked difference in orientation, however, is not observed in the shell domains, which assume much more similar placements in the two viruses. Moreover, despite the different morphologies and orientations of the PsV-S and -F arches, the local 2f axes that relate the two subunits in a CPA-CPB dimer in the two viruses are located at similar, though not identical, positions in the icosahedral capsids.
Modeling the PsV-S capsid dimer
The CPs of PsV-F and PsV-S exhibited only 19% sequence identity in an initial T-Coffee alignment (17) for this study, so it was not surprising that the crystal-derived atomic model of PsV-F CP fitted poorly into the PsV-S cryoTEM map. However, several features in PsV-S, particularly in the shell domain and ascribed to α-helices, were shifted by only a few Å from apparently homologous elements in PsV-F. Given this, and the fact that backbone features and a number of partial side chains were evident in the PsV-S map (Fig. 3 C), we used the PsV-F atomic model to guide the construction of an accurate backbone model of the PsV-S CPA-CPB dimer.
We initiated modeling based on the assumption that the CPs of both viruses adopt a similar fold. This made it relatively easy to connect all, clearly visible, secondary-structure elements in PsV-S despite some loops not being well resolved in the cryoTEM map. This led to an initial, Cα backbone model for the CPA subunit of PsV-S. Assignment of the PsV-S sequence along this backbone was guided by secondary-structure predictions generated by JPred (35) (Fig. 4) and comparisons of the distribution pattern of predicted secondary structures to that of the initial backbone model. These predictions suggested that, as with PsV-F, the PsV-S CP is predominantly α-helical, which is completely consistent with the large number of twisted, tube-like density features present in the cryoTEM map. Sequence assignment along the modeled PsV-S Cα backbone was further facilitated by taking into account the predicted alignment of the PsV-S and -F CP sequences (Fig. 4). Although these two viruses exhibit low primary-sequence identity in the respective CPs, the alignment of the sequences revealed close correspondence in the predicted helices and strands (Fig. 4).
Figure 4.
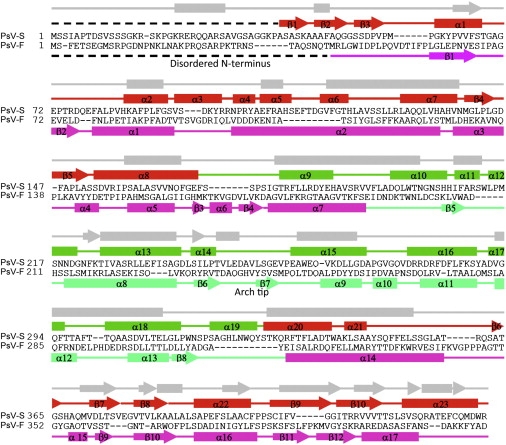
Secondary-structure assignments. The secondary structures of PsV-S are shown above the sequence (shell domain in red, arch domain in green), and those of PsV-F below the sequence (shell domain in magenta, arch domain in bluish-green). The predicted secondary structure of PsV-S is shown in gray at top. α-helices are represented by rods, β-strands by arrows, random coils by solid lines, and disordered regions by broken lines.
The ∼4.5 Å PsV-S cryoreconstruction shows that nearly 30% of the aa residues can be associated with knob-like features, which appear to represent partial side chains in the regions of well-defined α-helices and β-strands (see Fig. 3 C). Taking into account the size, shape, and location of each knob, we were able to fine-tune the assignment of sequence along the PsV-S Cα backbone. We also placed Cα atoms next to each knob within density ascribed to the main chain and, during model building and rebuilding, fine-tuned the model to assure that proper stereochemical constraints were obeyed (avoiding steric clashes and regularizing interatomic distances between consecutive turns in α-helices and between β-strands in sheets). The model was then refined in real space to reinforce ideal bond lengths, bond angles, and main-chain dihedral angles (44). Though side chains were not included in our final PsV-S model, they were incorporated during refinement of the model by using rotamers that best accounted for the knobs. The side chains, which may not be accurately modeled at this resolution, nevertheless helped to maintain proper distances between main chains during refinement of the model.
The above procedure was used to model subunit A of the PsV-S CP dimer. We derived an initial model of subunit B by simply applying a local 2f-symmetry operation to the CPA model. Several subsequent rounds of rigid-body refinement were carried out to adjust the relative positions of the two subunits in the cryoTEM density map. Thereafter, the conformation and position of individual aa residues in subunit B were manually adjusted to account for local structural differences between subunits A and B. Any steric clashes between the two subunits in the resulting model were also removed manually, and the stereochemistry of the entire dimer was improved via real-space refinement using RSRef (37,38).
The PsV-S model we present here consists of the main-chain atoms for aa 39–434 in both subunits of the CP dimer (Fig. 5). Only the N-terminal 38 aa are missing in the model for each subunit, presumably because this highly charged region is flexible or disordered and therefore leads to weak or missing density in the symmetrized cryoreconstructions of virions. This region contains nine basic aa (Arg and Lys) and extends toward the interior of the particle where it likely interacts with the viral genome during assembly and/or transcription. Similarly, the first 41 aa of the PsV-F subunit are positively charged and disordered. Other than the disordered N-terminal region, the interior wall of the PsV-S capsid seems to be populated with a large number of acidic aa residues (Asp76, Glu111, Asp114, Asp146, Glu352, and Glu377, from each CP subunit). Because PsV-F (8) and other dsRNA viruses (9) have capsids with a negatively charged inner surface, and it is generally believed that electrostatic repulsion facilitates movement of the negatively charged genome inside transcriptionally active particles, the presence of a negatively charged interior wall in PsV-S is consistent with the modeled CP backbone having largely the correct sequence register.
Figure 5.
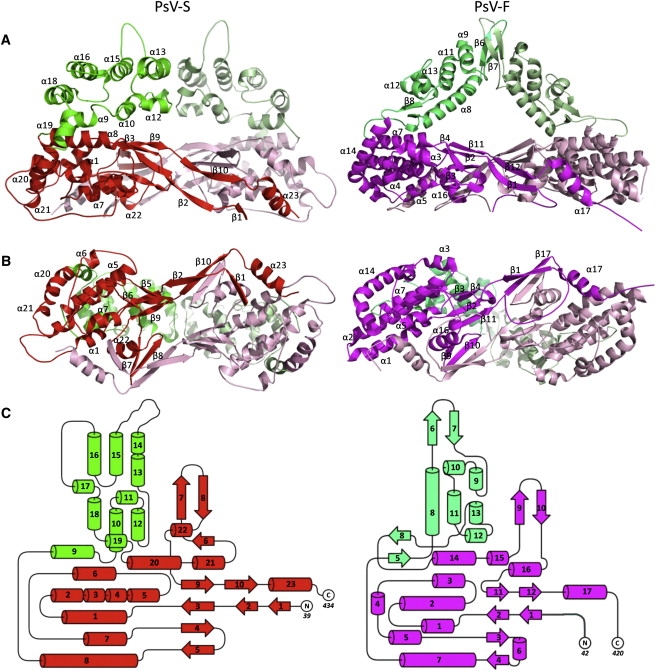
3D structures of PsV-S and -F CP dimers. (A) Side view and (B) inside-out view of a CPA-CPB dimer of (left) PsV-S and (right) -F. The color scheme is identical to that in Fig. 4. The two subunits from a CP dimer are colored in different shades for better differentiation. Secondary structures are labeled as defined in Fig. 4. (C) Topology diagram of (left) PsV-S and (right) -F. Secondary structures are labeled by numbers only.
Comparisons of PsV-F and -S capsid structures
Our cryoreconstruction of PsV-S virions to 7.3 Å resolution reported previously showed distinctive surface arches formed by quasisymmetrical CP dimers (20). At that resolution, however, it was not clear how the individual CPA and CPB subunits interacted to form the arches or the capsid shell. The 3.3 Å crystal structure of PsV-F virions shows a similar capsid architecture to PsV-S, and at that higher resolution a domain-swapping mechanism responsible for CPA-CPB dimer formation in the capsid shell domain was newly shown (8). Here, a new cryoreconstruction of PsV-S virions at much-improved resolution, combined with homology modeling, reveals greater details of the PsV-S capsid organization and suggests a common architecture and assembly pathway for PsV-S and -F, and perhaps for partiti- and picobirnaviruses in general.
The PsV-S CP, like that of PsV-F, is a two-domain structure (Fig. 5). The shell and arch domains respectively form the continuous capsid and the dimeric surface protrusions. The shell domain comprises aa 39–171 and 327–434. The arch domain (aa 172–326) is effectively a large polypeptide insertion in the shell domain. In each virus the two icosahedrally independent subunits, CPA and CPB, adopt almost identical structures. The root mean-square deviation distance between all 396 Cα atoms in CPA and CPB of PsV-S is only 0.8 Å (after a 178.5° rotation and a 0.3 Å translation to superimpose the two subunits maximally) according to our model, compared to 1.2 Å (after a 180° rotation and a 0.2 Å translation) as noted previously for PsV-F (8). The three most structurally divergent regions between CPA and CPB in PsV-S closely match the most divergent regions in PsV-F and include both the N- and the C-termini of each polypeptide. In the absence of experimentally determined absolute scaling of the PsV-S image data, the absolute dimensions of the PsV-S and -F capsids cannot be robustly compared. However, it appears that the outermost diameter of the PsV-S capsid is almost the same as that of PsV-F (∼380 Å), whereas the inner diameter of the capsid may be somewhat larger in PsV-S (255–260 Å) than in PsV-F (245–250 Å).
The shell domain of PsV-S CP has a strikingly similar structural fold to that in PsV-F (Fig. 5). Four β-sheets, two from each monomer, occur at the dimer interface. Two of these sheets contain β-strands from two different subunits (i.e., β7 and β8 from one, β2 and β10 from the other), suggesting that the domain-swapping mechanism, as originally observed in PsV-F (8), plays a major role in stabilizing the quasisymmetrical dimer interaction in PsV-S as well. The organization of PsV-S CPA-CPB dimers within the capsid shell moreover appears almost identical to that in PsV-F (8). As in PsV-F, the interface between parallel dimers across the icosahedral 2f axes in PsV-S is much larger than the pairwise interfaces around the icosahedral 3f and 5f axes, suggesting that a dimer of CPA-CPB dimers is a likely intermediate during particle assembly (see Fig. 3 B). Sequence identity in the shell-domain region of the T-Coffee alignment is 22% (vs. 19% overall).
The structure of the PsV-S arch domain differs substantially from that in PsV-F (Fig. 5). First, the PsV-S arch is bulkier. According to our model, the PsV-S arch consists of roughly two 3-helix bundles arranged in two separate layers and contains 167 aa. In contrast, the PsV-F arch consists of both α-helices and β-sheets and contains only 124 aa. Second, the main part of each arm of the arch runs almost tangentially to the capsid shell in PsV-S, but runs upward at almost 50° with respect to the shell in PsV-F. This difference leads to the smaller opening beneath the arch in PsV-S. Third, the most radially extended portions (tips) of the two arms in the PsV-S arch are separated by ∼32 Å and point away from one another; contacts between the two arms are made instead by the two 3-helix bundles. In contrast, in PsV-F the radially extended tips fold into two β-hairpins that twist around each other and provide most of the contacts between the arms. Lastly, the long axis of each dimeric arch assumes a less acute angle in PsV-S than in PsV-F, ∼37° vs. ∼66°, relative to the line between neighboring 5f axes in the capsid (see Fig. 3 B). From the CP structure models (Fig. 5), it is evident that these differing orientations reflect differences in the arch “supports,” where each CP subunit in PsV-F contains a unique, small β-sheet (strands β5 and β8) that lies tangential to the shell and swivels the positions of the supports, counterclockwise as viewed from above, relative to those in PsV-S. Sequence identity in the arch-domain region of the T-Coffee alignment is only 16%. The function of these surface arches in partitivirus replication remains unknown, but their structures in both virions suggest a role in capsid assembly or stabilization.
Internal densities and genome organization
The PsV-S cryoTEM map includes two concentric layers of RNA densities in the particle interior (Fig. 6 A). These two layers appear to be less closely spaced in PsV-S (∼35 Å) than do the three layers reported for PsV-F (∼25 Å) (8). To highlight the RNA organization and other internal features, the cryoTEM maps were computed to a resolution cutoff of 8–10 Å. This procedure reduced the high-frequency noise seen as discontinuous densities in the high-resolution maps, but had no effect on the interlayer RNA spacing in either virus. Partitiviruses are known to package their two essential genome segments, dsRNA1 and -2, into separate virus particles. Because the two segments of PsV-S are very similar in size to those of PsV-F (see Introduction), the apparently larger RNA spacing in PsV-S might be explained by a somewhat larger internal capsid diameter. Another possibility is that the satellite segment, dsRNA3, unique to PsV-F (16) might be copackaged with the genome segments, thereby increasing the per-particle RNA content of PsV-F relative to PsV-S.
Figure 6.
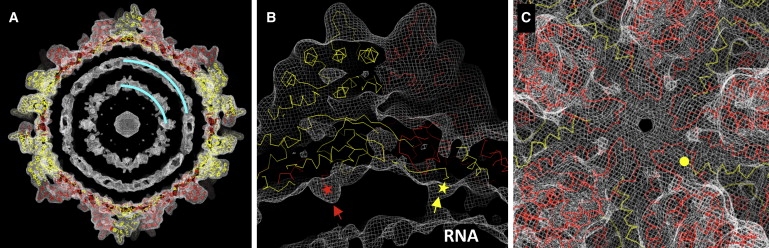
Genome organization and transcription. The PsV-S cryoreconstruction was computed to a resolution cutoff of (A) 10 Å or (B and C) 8 Å to highlight less-ordered components in the particle interior. Density-contour levels for the three panels were 1.05, 0.85, and 1.5 σ. The cryoTEM map is represented by a wire mesh, within which the Cα backbone of the PsV-S CP subunits (CPA, red; CPB, yellow) are fitted. (A) Internal genomic RNA densities. Cyan curves highlight the two RNA density layers. (B) Rod-like densities on the inner capsid surface. The disordered N-termini of CPA and CPB are highlighted by red and yellow stars, respectively. (C) PsV-S capsid structure around the 5f symmetry axis. The N-terminus of one CPB subunit is highlighted by a yellow circle.
The outermost RNA layer in the PsV-S cryoreconstruction, particularly when the contour level is reduced to display lower-density features, appears to form connections to the capsid shell via 120 rod-like structures (Fig. 6 B). These rods extend from the extreme end of the ordered N-termini of both CPA and CPB and thus seem likely to be formed by some portion of the first 38 aa, which are disordered and hence not visible at high resolution, and were therefore not included in the backbone models. It is also evident that the rod density is stronger in CPB (Fig. 6 B). The overall PsV-S CP sequence contains only 9% basic aa, but the 28-aa stretch between residues 15 and 43 is 32% Arg and Lys. This basic nature suggests that the N-terminal end of PsV-S CP may interact with the viral genome to facilitate packaging and/or transcription. The PsV-F CP N-terminus is also rich in basic aa, with eight Arg and Lys (24%) between residues 11 and 43. Interestingly, despite the apparently larger spacing between the RNA layers in PsV-S, the distance between the outermost RNA layer and the inner capsid surface appears to be about the same as that in PsV-F (∼25 Å). This greater consistency between the two viruses might be determined by RNA interactions with the N-terminal CP peptides in each.
In PsV-F, the two rod-like densities projecting toward the virion interior from each CP dimer lie ∼15 Å apart, which is close enough for the N-terminal peptides potentially to form dimers within the particle interior. In contrast, the rod densities from each PsV-S CP dimer lie ∼40 Å apart and form a different pattern of connections between the shell and the underlying genome. The N-terminal peptide of PsV-S CP makes a sharp turn toward the particle interior at Ala39. Therefore, the N-terminal peptides of PsV-S and -F may both be more likely to remain monomeric, instead of forming dimers as we originally proposed (8). Assuming a monomeric form, the structure of the N-terminal peptide is likely to be rather flexible. In particular, residues 15–28 contain seven of the nine Arg and Lys from the PsV-S N-terminus, and secondary-structure predictions suggest that this region is likely to form a loop-helix motif that might interact with dsRNA.
At the icosahedral 5f axes where newly synthesized mRNAs might emerge, we observe a helix–turn–helix feature in each CP monomer of PsV-S, which gives rise to a thin layer of density surrounding the symmetry axis (Fig. 6 C). This is distinct from what occurs in PsV-F, where the extreme C-terminus of the CPB molecule snakes from underneath the helix-turn-helix feature (formed by CPA) toward the 5f axis. By comparison, the C-terminal tail of PsV-S CPB folds into an α-helix and occupies a location away from the 5f axis (Fig. 6 B). Although no pore is apparent on the 5f axis of the PsV-S capsid, we expect the helix-turn-helix motif may undergo large-scale conformational changes to permit the release of nascent transcripts.
The RdRp molecules in most dsRNA viruses are generally believed to be tethered via noncovalent interactions to the interior of the capsid near the 5f symmetry axes (9,45). Notably, the PsV-S cryoTEM map displayed at reduced contour level shows a density-deficient feature, surrounded by RNA-attributable densities, underneath each 5f vertex (Fig. S1). This feature might reflect exclusion of the RNA densities by one or more copies of the PsV-S RdRp, as similarly suggested from high-resolution structures of other dsRNA viruses (9). Another density-deficient feature is seen at each 3f symmetry axis, but appears smaller and less continuous than that at the 5f axis (Fig. S1).
Conclusions
The findings in this report broaden our understanding of conserved and variable aspects of partitivirus structure. They also reflect the growing use of electron cryomicroscopy for atomic modeling of protein folds. Concluding remarks relate to the latter outcome.
Ab initio backbone modeling, in our opinion, remains extremely challenging at 4.5 Å resolution, even for highly averaged cryoTEM maps such as we obtained for PsV-S. There are two main problems with such maps. The first is discontinuous densities caused in many cases by mobile surface loops between less-mobile secondary structures. The second is misleadingly continuous densities caused by poorly resolved side- and main-chain groups that are closely positioned in space. Without prior information about the overall fold and topology of the protein, it is therefore likely that ab initio modeling would result in erroneous assignment of connectivity at numerous positions in the backbone trace. On the other hand, any homologous protein structure that is available for fitting, as long as it folds similarly, would assist with backbone modeling. Moreover, a homologous structure with substantial sequence identity would help in registering the sequences of the fitted and modeled polypeptides.
Continued procedural improvements to extend cryoTEM maps beyond 4.5 Å resolution—resolving additional side- and main-chain elements and thereby eliminating both discontinuous and misleadingly continuous densities—would of course aid in ab initio modeling. In an attempt to fine-tune the fitting of our PsV-S model with best-guessed side chains, the crystallographic refinement program CNS and the structure factors calculated from the cryoreconstruction were used to refine the structural model. Although this pseudocrystallographic refinement was able to produce an atomic model with side chains that better fit the cryoTEM densities, the stereochemistry of the refined model was poor, as indicated by a Ramachandran plot, with large deviations in bond lengths and angles as well as many close atom-atom contacts (data not shown). In addition, due to the high degree of noncrystallographic symmetry in virions (60-fold icosahedral symmetry in this case), Rfree could not be effectively used to monitor for overrefinement. Based on these experiences, we conclude that efforts to develop more robust low-resolution refinement methods are warranted. Such refinement methods could conceivably use both amplitude and phase information for the structure factors, different from typical crystallographic refinement in which only amplitude information is available. In the absence of high-resolution data and due to the inherently low observation/parameter ratio, it is essential to include as many real-space stereochemistry restraints as possible (e.g., individual secondary-structure elements as rigid bodies, hydrogen-bonding constraints, and main-chain rotamers) when attempting ab initio modeling and refinement with current cryoTEM maps.
Supporting Material
Materials and methods and a figure are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00556-4.
Supporting Material
Acknowledgments
We thank R. Sinkovits and N. Olson for expert advice and ideas on ways to enhance the computer processing and imaging aspects of this study. The San Diego Supercomputer Center provided access to TeraGrid computing, and support from the University of California-San Diego and the Agouron Foundation to T.S.B. was used to establish and equip cryoTEM facilities at the University of California-San Diego.
This work was supported by the National Institutes of Health (R37 GM033050 and 1S10 RR020016 to T.S.B., R01 AI067638 to Y.J.T.), the Department of Agriculture National Research Initiative Competitive Research Program (2001-35319-10010 to S.A.G.), and by the Robert A. Welch Foundation (C-1565 to Y.J.T.).
Footnotes
Junhua Pan's present address is Laboratory of Molecular Medicine, Children's Hospital, 320 Longwood Ave., Boston, MA 02115.
Wendy F. Ochoa's present address is Burnham Institute for Medical Research, La Jolla, CA 92037.
References
- 1.Mertens P. The dsRNA viruses. Virus Res. 2004;104:3–13. doi: 10.1016/j.virusres.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Grimes J.M., Burroughs J.N., Stuart D.I. The atomic structure of the bluetongue virus core. Nature. 1998;395:470–478. doi: 10.1038/26694. [DOI] [PubMed] [Google Scholar]
- 3.Naitow H., Tang J., Johnson J.E. L-A virus at 3.4 Å resolution reveals particle architecture and mRNA decapping mechanism. Nat. Struct. Biol. 2002;9:725–728. doi: 10.1038/nsb844. [DOI] [PubMed] [Google Scholar]
- 4.Castón J.R., Ghabrial S.A., Carrascosa J.L. Three-dimensional structure of Penicillium chrysogenum virus: a double-stranded RNA virus with a genuine T=1 capsid. J. Mol. Biol. 2003;331:417–431. doi: 10.1016/s0022-2836(03)00695-8. [DOI] [PubMed] [Google Scholar]
- 5.Coulibaly F., Chevalier C., Rey F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell. 2005;120:761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Huiskonen J.T., de Haas F., Butcher S.J. Structure of the bacteriophage ϕ6 nucleocapsid suggests a mechanism for sequential RNA packaging. Structure. 2006;14:1039–1048. doi: 10.1016/j.str.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 7.Duquerroy S., Da Costa B., Rey F.A. The picobirnavirus crystal structure provides functional insights into virion assembly and cell entry. EMBO J. 2009;28:1655–1665. doi: 10.1038/emboj.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan J., Dong L., Tao Y.J. Atomic structure reveals the unique capsid organization of a dsRNA virus. Proc. Natl. Acad. Sci. USA. 2009;106:4225–4230. doi: 10.1073/pnas.0812071106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinisch K.M., Nibert M.L., Harrison S.C. Structure of the reovirus core at 3.6 Å resolution. Nature. 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 10.Tao Y., Farsetta D.L., Harrison S.C. RNA synthesis in a cage—structural studies of reovirus polymerase λ3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 11.Bozarth R.F., Wood H.A., Mandelbrot A. The Penicillium stoloniferum virus complex: two similar double-stranded RNA virus-like particles in a single cell. Virology. 1971;45:516–523. doi: 10.1016/0042-6822(71)90352-7. [DOI] [PubMed] [Google Scholar]
- 12.Banks G.T., Buck K.W., Stone O.M. Viruses in fungi and interferon stimulation. Nature. 1968;218:542–545. doi: 10.1038/218542a0. [DOI] [PubMed] [Google Scholar]
- 13.Ghabrial S.A., Ochoa W.F., Nibert M.L. Partitiviruses: general features. In: Mahy B.W.J., van Regenmortel M.H.V., editors. Encyclopedia of Virology. 3rd ed. Elsevier; Oxford: 2008. pp. 68–75. [Google Scholar]
- 14.Buck K.W., Kempson-Jones G.F. Biophysical properties of Penicillium stoloniferum virus S. J. Gen. Virol. 1973;18:223–235. doi: 10.1099/0022-1317-18-3-223. [DOI] [PubMed] [Google Scholar]
- 15.Kim J.W., Kim S.Y., Kim K.M. Genome organization and expression of the Penicillium stoloniferum virus S. Virus Genes. 2003;27:249–256. doi: 10.1023/a:1026343831909. [DOI] [PubMed] [Google Scholar]
- 16.Kim J.W., Choi E.Y., Lee J.I. Genome organization and expression of the Penicillium stoloniferum virus F. Virus Genes. 2005;31:175–183. doi: 10.1007/s11262-005-1793-y. [DOI] [PubMed] [Google Scholar]
- 17.Poirot O., O'Toole E., Notredame C. Tcoffee@igs: a web server for computing, evaluating and combining multiple sequence alignments. Nucleic Acids Res. 2003;31:3503–3506. doi: 10.1093/nar/gkg522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buck K.W., Kempson-Jones G.F. Capsid polypeptides of two viruses isolated from Penicillium stoloniferum. J. Gen. Virol. 1974;22:441–445. [Google Scholar]
- 19.Buck K.W. Semi-conservative replication of double-stranded RNA by a virion-associated RNA polymerase. Biochem. Biophys. Res. Commun. 1978;84:639–645. doi: 10.1016/0006-291x(78)90753-2. [DOI] [PubMed] [Google Scholar]
- 20.Ochoa W.F., Havens W.M., Baker T.S. Partitivirus structure reveals a 120-subunit, helix-rich capsid with distinctive surface arches formed by quasisymmetric coat-protein dimers. Structure. 2008;16:776–786. doi: 10.1016/j.str.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn R.J., Zhang W., Strauss J.H. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakagawa A., Miyazaki N., Tsukihara T. The atomic structure of rice dwarf virus reveals the self-assembly mechanism of component proteins. Structure. 2003;11:1227–1238. doi: 10.1016/j.str.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Jiang W., Baker M.L., Chiu W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–1134. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 24.Ludtke S.J., Baker M.L., Chiu W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Yu X., Jin L., Zhou Z.H. 3.88 Å structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X., Settembre E., Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. USA. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suloway C., Pulokas J., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Yan X., Sinkovits R.S., Baker T.S. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J. Struct. Biol. 2007;157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowman V.D., Chase E.S., Smith T.J. An antibody to the putative aphid recognition site on cucumber mosaic virus recognizes pentons but not hexons. J. Virol. 2002;76:12250–12258. doi: 10.1128/JVI.76.23.12250-12258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X., Ji Y., Baker T.S. Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure. 2005;13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Heel M., Schatz M. Fourier shell correlation threshold criteria. J. Struct. Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Havelka W.A., Henderson R., Oesterhelt D. Three-dimensional structure of halorhodopsin at 7 Å resolution. J. Mol. Biol. 1995;247:726–738. doi: 10.1006/jmbi.1995.0176. [DOI] [PubMed] [Google Scholar]
- 33.Jones T.A. A set of averaging programs. In: Dodson E.J., Gover S., Wolf W., editors. Molecular Replacement. SERC Daresbury Laboratory; Warrington, UK: 1992. pp. 91–105. [Google Scholar]
- 34.Collaborative Computational Project No. 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Cole C., Barber J.D., Barton G.J. The JPred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brünger A.T., Adams P.D., Warren G.L. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Chapman M.S. Restrained real-space macromolecular refinement using a new resolution-dependent electron density function. Acta Crystallogr. A. 1995;51:69–80. [Google Scholar]
- 38.Korostelev A., Bertram R., Chapman M.S. Simulated-annealing real-space refinement as a tool in model building. Acta Crystallogr. D Biol. Crystallogr. 2002;58:761–767. doi: 10.1107/s0907444902003402. [DOI] [PubMed] [Google Scholar]
- 39.Kleywegt G.J., Read R.J. Not your average density. Structure. 1997;5:1557–1569. doi: 10.1016/s0969-2126(97)00305-5. [DOI] [PubMed] [Google Scholar]
- 40.Kraulis P. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 1991;24:946–950. [Google Scholar]
- 41.Goddard T.D., Huang C.C., Ferrin T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007;157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Ji Y., Marinescu D.C., Baker T.S. A model-based parallel origin and orientation refinement algorithm for cryoTEM and its application to the study of virus structures. J. Struct. Biol. 2006;154:1–19. doi: 10.1016/j.jsb.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenthal P.B., Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 44.Jones T.A., Zou J.Y., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X., Walker S.B., Baker T.S. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nat. Struct. Biol. 2003;10:1011–1018. doi: 10.1038/nsb1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.