

Abstract
Three basic proline-rich salivary proteins have been produced through the recombinant route. IB5 is a small basic proline-rich protein that is involved in the binding of plant tannins in the oral cavity. II-1 is a larger protein with a closely related backbone; it is glycosylated, and it is also able to bind plant tannins. II-1ng has the same polypeptidic backbone as II-1, but it is not glycosylated. Small angle x-ray scattering experiments on dilute solutions of these proteins confirm that they are intrinsically disordered. IB5 and II-1ng can be described through a chain model including a persistence length and cross section. The measured radii of gyration (Rg = 27.9 and 41.0 ± 1 Å respectively) and largest distances (rmax = 110 and 155 ± 10 Å respectively) show that their average conformations are rather extended. The length of the statistical segment (twice the persistence length) is b = 30 Å, which is larger than the usual value (18 Å − 20 Å) for unstructured polypeptide chains. These characteristics are presumably related to the presence of polyproline helices within the polypeptidic backbones. For both proteins, the radius of gyration of the chain cross-section is Rc = 2.7 ± 0.2Å. The glycosylated protein II-1 has similar conformations but the presence of large polyoside sidegroups yields the structure of a branched macromolecule with the same hydrophobic backbone and hydrophilic branches. It is proposed that the unusually extended conformations of these proteins in solution facilitate the capture of plant tannins in the oral cavity.
Introduction
Intrinsically disordered proteins (IDPs), often referred to as naturally unfolded proteins are proteins that lack well-structured 3D folds, and therefore do not have a stable tertiary structure (1–8). These proteins have long remained relatively obscure in our view of the protein universe because they do not crystallize, and therefore do not produce any diffraction spot in x-ray crystallography. More recently, it has been shown that ∼30% of all proteins in eukaryotic organisms are intrinsically unstructured (5,9). This discovery challenged the traditional protein structure paradigm, which stated that a specific well-defined structure was required for the correct function of a protein. Biochemical evidence has since shown that intrinsically unfolded proteins are functional and that their lack of folded structures is related to their functions (10).
Intrinsically unstructured proteins have particular sequences. The sequence signature of unfolded proteins (or unfolded regions of proteins) is as follows: 1), a bias toward polar and charged amino acids (Gln, Ser, Pro, Glu, Lys); 2), a bias away from bulky hydrophobic residues Val, Leu, Met, Phe, Trp, Tyr); and 3), in some cases a low sequence complexity with repeated short amino acid sequences.
Salivary proline rich proteins (sPRPs), which constitute about two-thirds of proteins secreted by the human parotid glands, have this type of sequence signature (11,12). They contain repeated sequences with high proportions of Pro, Gly and Gln, or Glu residues (1,3,4). Circular dichroism and NMR studies of some of these proteins give indications of largely disordered structures with short polyproline II helical sections (13–15). sPRPs are divided into glycosylated, acidic, and basic types that, despite their structural similarities, have different functions (16).
The function of basic salivary PRPs (bPRPs) is to bind polyphenolic plant compounds (e.g., tannins) present in food (17,18) and thus protect against their anti-nutritional effects (19). bPRPs are present in the saliva of primates and herbivorous animals but almost absent in that of carnivorous animals. They make it possible for herbivorous animals to consume foods that contain up to 5% tannins by weight. The capacity of bPRPs to bind and precipitate tannins has been ascribed to their proline rich sequences (20,21) that, together with their high glycine content (22), confers them an open structure providing a large binding surface and multiple contact points (3,4,20,23,24). Moreover, the proline residues may provide hydrophobic interactions and hydrogen bond acceptor sites for the binding of tannins.
Glycosylated proline rich proteins (PRPs) ensure oral lubrication, bind oral bacteria, and are also able to bind tannins (20,25,26). Earlier biophysical data suggest that oligosaccharide moieties attached to a protein backbone may be capable of assuming a variety of conformational geometries depending on the oral surface with which it interacts (25,27). Thus, the shape or conformation of these salivary molecules could play an important role in defining their functional role in the oral cavity. These proteins account for ∼17% of human parotid salivary proteins (12,28,29).
This study is part of a larger project focused on the interactions of plant tannins with salivary proteins. For this purpose, we have developed an heterologous expression system for the production of some human basic PRPs through integration of the gene coding for a human salivary proline-rich pro-protein, PRB4S, into a yeast genome (30). We obtained a nonglycosylated protein (IB5), a glycosylated protein (II-1), and the nonglycosylated form of the latter (II-1ng). Preliminary investigations indicate that these proteins do bind tannins (21,31). Moreover, we have shown the presence in solution of IB5-tannin soluble supramolecular structures with different stoichiometries that show the ability of bPRPs to bind and scavenge tannins (32). Increasing tannin concentration leads to the precipitation of IB5-tannin complexes, whereas only limited aggregation is observed with the glycosylated protein II-1 at the same protein/tannin ratio (33). These different behaviors may be related to the functions of these proteins, involving either complexation of tannin molecules in ingested foods and drinks, which may reduce their toxicity, or formation of precipitates, which may contribute to the perception of astringency in the oral cavity.
We report a small angle x-ray scattering (SAXS) study of these proteins (for general references see Receveur-Bréchot et al. (7) and Bernadó and Blackledge (8)). The aim is to gain a precise view of their conformations and to find out whether these conformations confer functional advantages like the ability to bind several tannin ligands. However, IDPs in solution explore an enormous number of conformations, and SAXS experiments only determine average correlations between the relative positions of atoms in the macromolecules. We addressed this problem in the following way. First, we acquired high quality spectra with a high resolution (10 Å), to maximize the information content of the data. Then we determined a set of geometrical parameters that characterize the average protein structure, such as the radius of gyration, persistence length and cross-section of the polypeptide chain, and we compared these structural parameters to those of other intrinsically disordered proteins. Finally we used mathematical algorithms to produce representative conformations that reproduce the experimental scattering curves.
Materials and Methods
Large-scale production and purification of PRPs
The Pichia pastoris system for expression of heterologous recombinant proteins has been used. It allows large yields of properly matured proteins and generally yields protein-bound oligosaccharides that are of much shorter chain length than found in Saccharomyces cerevisiae (34). As described previously, bPRPs were overexpressed on a large scale in P. pastoris using the methanol-inducible alcohol oxidase promoter (35). They were produced during the growth phase and secreted into the culture medium. Five majors bands around the expected molecular weight (apparent molar masses 45–15 KDa) were detected in the supernatant with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE) analysis (Fig. 1, lane 1). The SDS PAGE technique was carried out under reducing conditions using 12.5% acrylamide running gels, the protein bands were stained by R250 Coomassie brilliant blue (35,36) and the organic solvent was omitted from the acetic acid destaining solution. In these conditions the PRP bands appear pink-violet; IB5 and II-1ng give neat pink bands whereas II-1 gives a very diffuse one.
Figure 1.
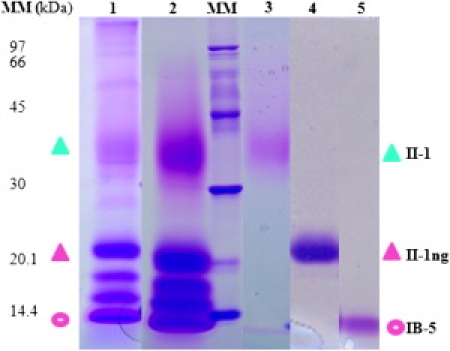
SDS-PAGE patterns of proteins obtained at successive stages of purification. (Lane 1) Crude supernatant. (Lane 2) PRP extract after streamline. (Lane 3) Purified fraction of the glycosylated protein II-1 (blue triangle). (Lane 4) Purified fraction of the nonglycosylated form of II-1 (II1ng) (pink triangle). (Lane 5) Purified fraction of IB-5 (pink circle). MM, molecular mass markers.
Ion-exchange chromatography
After centrifugation of the crude medium, the recombinant PRPs were recovered from the supernatant using a single step ion-exchange (cationic) chromatography. Their high isoelectric point (above pH = 11) allows binding to the silica surfaces of the ion exchange column at neutral pH and moderate ionic strength, whereas the other macromolecules (yeast proteins and polysaccharides) of the supernatant do not. We used a FPLC Biocad/Sprint system (Perseptive Biosystems, MA) with a Pharmacia Streamline 50 column (id 2 cm/L 40 cm). The matrix (Streamline SP XL) was equilibrated in buffer (50 mM Tris HCl, pH 8.0). A preparative chromatography technique called expanded bed adsorption was used. The gel was expanded using upside flow at 13 mL/min until the top of the bed was stable. The supernatant obtained after centrifugation of the crude culture (280 mL/run) was applied after dilution (1:2) in the same buffer to the column (upside flow). The flow was subsequently inverted (downside flow) and the plunger was lowered to 1 cm from the top of the bed. The proteins were eluted with 150 mL of the same buffer containing 1 M NaCl at a 5 mL × min−1 flow rate. The protein elution was monitored by measuring absorbance at 230 nm. The purified extract contained five PRPs, and was free of polysaccharides and yeast proteins (Fig. 1, lane 2).
Gel filtration chromatography and sample preparation
A partial separation of the five PRPs was carried out through size exclusion chromatography using the FPLC Biocad/Sprint system. Aliquots of the extract purified previously were applied on a Superdex 75 HR 3.2/30 PC or a Hiload 16/60 Superdex 75 column (Pharmacia), using ammonium acetate 50 mM, pH 5.5 buffer at a 0.8 mL × min−1 flow rate. The elution was monitored by absorbance at 230 nm. Collected fractions were checked by SDS-PAGE. Those containing either IB5, II-1, or II-1ng were freeze-dried until use. The purified proteins were checked by MS experiments. The masses obtained from spectra deconvolution were in agreement with the theoretical ones determined from their primary sequences (32, F. Canon, unpublished). For the SAXS experiments, the protein solutions were prepared by mixing carefully weighted volumes of protein powder and of buffer (ammonium acetate 50 mM pH 5.5 buffer) in such a way that the concentration was known precisely. The samples were then injected in a capillary located on the beam path, at a temperature regulated at 20°C. We checked that solutions made without and with freeze-drying give identical spectra.
SAXS instruments and methods
SAXS experiments were carried out using the Nanostar instrument (Bruker, Karlsruhe, Germany) at IBBMC in Orsay. The x-rays were produced by a rotating anode (Cu Kα, wavelength λ = 1.54 Å), and the scattered x-rays were collected using a 2D position sensitive detector (Vantec) positioned at 662 mm from the sample. The scattering vector range was 0.011 < q < 0.40 Å−1 where q = 4πsinθ/λ and 2θ is the scattering angle. Further experiments were carried out on the beamline SWING, at the Synchrotron SOLEIL. The incident beam energy was 12 keV, and the sample to detector (Aviex CCD) distance was set to 1927 mm. The scattering vector range was 0.008 < q < 0.49 Å−1. Several successive frames (typically 25) of 4 s each were recorded for both sample and pure solvent. We checked that x-rays did not cause irradiation damage by comparing the successive frames, before calculating the average intensity and experimental error. For protein IB5, identical spectra were obtained from the Nanostar instrument and from Swing; the data presented in this study are from Swing. For II-1ng and II-1, spectra were obtained with the Nanostar only. Scattering from the pure solvent was measured and subtracted from the corresponding protein spectra. Intensities were scaled using the scattering of water. For each protein, the original solutions (concentrations 5.8–8.7 g × L−1) were compared to solutions with concentrations two and four times lower. The concentration dependence was very small, indicating that interactions between dissolved proteins were weakly repulsive at such concentrations. Indeed, at this concentration the average distance between the centers of mass of proteins was >14 nm, which is much larger than the Debye screening length in 50 mM salt (1.4 nm). Nevertheless the data at low and high concentrations are spliced to obtain SAXS curves unaffected by interparticle interactions at small angles and to improve the statistics in the outer region. The scattered intensities were measured on an absolute scale; calculations of the molar mass of each protein from the intensity extrapolated to q → 0 gave values that were within 15% of those determined through ESI-MS, which is a usual accuracy given the uncertainties in the density of the proteins. This agreement confirmed that the dilute solutions contained independent macromolecules only.
Results
Primary structures
The amino acid primary structures were deduced from N-terminal sequencing and mass spectrometry analysis of the proteins referred to the sequence of the cloned PRB4S cDNA. They showed the presence of four main isoforms of IB5 (Fig. 2 a), which differ by a few N-terminal amino acids, with molar masses 7481.36 (IB5a, abundance 14.5%), 7238.09 (IB5b, 44.3%), 7079.93 (IB5c, 27.8%), and 6923.74 (IB5d, 13.4%). Note that IB5 cannot be glycosylated because it lacks the N-glycosylation signal on its amino acid sequence.
Figure 2.

Sequences of the recombinant PRP (a) IB5 and (b) II-1. For IB5, four isoforms, IB5a–d, have been copurified, and five for II-1 (II-1a–e). For the two proteins, the isoforms differ by just a few amino acids. Repeated sequences are underlined and stars indicate potential sites of glycosylation.
For II-1 and II-1ng, ESI-MS has shown that two main forms and three minor ones were copurified. The average molar masses of the peptide backbones were 14,479.10 (II-1nga, 38%), 14,093.63 (II-1ngb, 28%), 14,180.72 (II-1ngc, 13%), 14,566.19 (II-1ngd, 8%), and 14,722.38 (II-1nge, 13%), all showing five sites for potential glycosylation (Fig. 2 b).
However, mass spectra of EndoH deglycosylated II-1 indicate that the main form of glycosylated II-1 presents one glycosylation (F. Canon, unpublished). The localization of this glycosylation among the five N-X-S sites has not been determined; however, it is known that those closest to the N-terminal are available for glycosylation for a longer time, and therefore are more likely to be glycosylated (38). The total mass of the polyosides bound to the II-1 backbone is estimated to be 26% of the mass of the protein, corresponding to ∼20–30 mannose per protein. This is an average from monosaccharide assay after trimethylsilylation of polyosides (39) and aldolization methods (40). The broad band observed for protein II-1 in the SDS-PAGE experiments and crowded spectra obtained by ESI-MS indicate glycosylation heterogeneity classically observed during production of recombinant glycoproteins (41). Concerning the structure of these polyosides we can infer that they may be similar to those found in other recombinant glycosylated proteins arising from P. Pastoris. Its N-glycosylation synthesis pathway mirrors that of typical mammalian cells up to the point where Man8GlcNAc2 N-glycosylated proteins exit the endoplasmic reticulum. These proteins are a model of human salivary N-glycosylated PRPs (42–44).
Bioinformatic analysis
For proteins that may be partly or fully unstructured, there are some well established programs (http://www.disprot.org) that predict the extent of folding and structural order of their conformations in solution, on the basis of the amino acid sequence (45). All programs predicted no folding at all and a disorder index near maximum disorder (Fig. SI-1 in the Supporting Material). Such an extreme result is rather uncommon among unstructured proteins. Note, however, that these programs are not able to recognize the presence of short polyproline I (PPI) or polyproline II (PPII) structural elements that occur in proline rich proteins.
Conformations according to SAXS
For unstructured proteins that do not have a permanent secondary or tertiary structure, SAXS experiments determine the average conformation of the protein in solution. This average conformation is described by the pair distance distribution function P(r), i.e., the number of different electron pairs with a mutual distance between r and r + dr within the protein (46,47). For an isolated macromolecule, P(r) is a function that initially grows with the number of chain elements that can be found at a distance r from a given chain element, goes through a maximum value Pmax at the most populated distance and then decays to reach zero at the maximum distance rmax within the macromolecule. For the three proteins, IB5, II-1ng, and II-1, we calculated P(r) from SAXS spectra using the GNOM procedure with the q range 0.015–0.35 Å−1 (48) (Fig. 3). For comparison the calculated P(r) of a freely jointed chain are also traced. The P(r) of a dense sphere has a symmetrical shape that reflects the fact that the largest distance within a dense object is rather short; on the other hand, P(r) of a freely jointed chain (i.e., a chain in which the orientations of successive segments are uncorrelated) has a long tail at large distances, because the conformations of such a chain stretch to large distances. The pair distance distributions of the three salivary proteins are similar to that of a freely jointed chain. For IB5 and to a lesser extent for II-1ng, P(r) has a small shoulder around r = 10 Å, which may be tentatively ascribed to short and rigid structural elements within the chain (see below).
Figure 3.
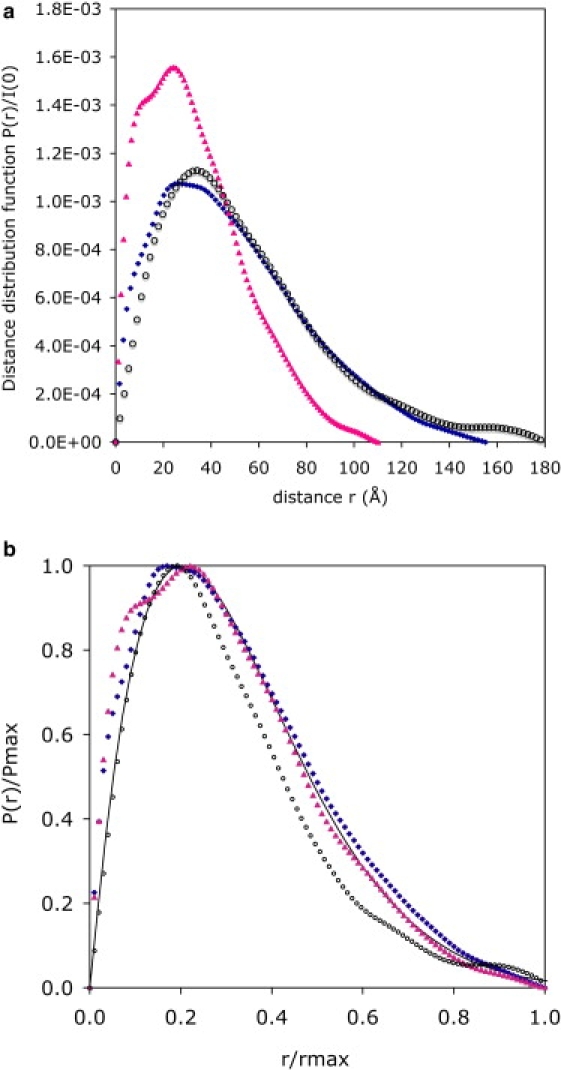
Pair distances distribution functions P(r) for the proteins IB5 (pink triangles), II-1ng (blue dots), and II-1 (open circles). (a) Horizontal axis, distance r; vertical axis, distance distribution P(r)/I(0). (b) Reduced coordinates r/ rmax where rmax is the largest distance within each protein and P(r)/Pmax where Pmax is the maximum value of P(r). (Solid line) Theoretical distributions of freely jointed chains with the same radii of gyration.
Overall dimensions
Table 1 presents the radii of gyration Rg and maximum distances rmax of the three PRPs. The values of Rg were calculated from the pair distance distribution P(r) according to the classical expression:
| (1) |
Table 1.
Characteristic dimensions of the salivary proteins
| Protein | M (Da) | L (Å)† | rmax (Å)∗ | Rg(Å)∗, Rg (Å)† | b (Å)† | Rc (Å)† |
|---|---|---|---|---|---|---|
| IB5 | 7481, | 188 ± 10 | 110 ± 10 | 27.9 ± 1∗ | 29.7 ± 1 | 2.7 ± 0.2 |
| 7238, | 27.5 ± 1† | |||||
| 7080, | ||||||
| 6923 | ||||||
| II-1ng | 14,480, | 364 ± 20 | 155 ± 10 | 41.0 ± 1∗ | 29.9 ± 1 | 2.7 ± 0.2 |
| 14,095 | 40.3 ± 1† | |||||
| II-1 | 20,000 (average) | 178 ± 10 | 45.9 ± 2 | 2.7 ± 0.2 |
The radius of gyration of the cross section, Rc, was determined through Eq. 5 and the radius of gyration of the whole protein, Rg, was calculated according to with given by Eq. 4.
Structural parameters for the nonglycosylated proteins IB5 and II-1ng according to the P(r).
Structural parameters for the nonglycosylated proteins IB5 and II-1ng according to fits by Eqs. 2, 4, and 5.
In the case of an extended protein it is more appropriate to use this relation rather than the well-known Guinier approximation that is valid only within a very restricted q range. These radii of gyration are much larger than those of globular proteins with comparable molar mass. Indeed, for globular proteins, the radius of gyration follows the law Rg ≈ 3(n)(1/3) where n is the number of residues in the polypeptide backbone. Given the sequences of IB5 (n ≈ 70) and II-1 (n ≈ 140), this would yield Rg = 12.4 and 15.6 Å respectively, instead of the much larger values found here. On the other hand, an analysis of literature values for IDPs yields Rg ≈ 2.54(n)0.522 (8). For IB5, this would yield Rg = 23.3 Å, and for II-1ng, Rg = 33.5 Å. The experimental values for IB5 and II-1ng are still larger, indicating that these proteins have strongly extended conformations.
It is also instructive to compare the maximum distances. These are quite large (about half the end-to-end distance of a polyproline PPII helix), indicating again that the conformations are quite extended. There is also an interesting effect of the glycosylation on the overall dimensions. Indeed, comparing II-1 with II-1ng, we find that the maximum distance is 11% larger and the radius of gyration 15% larger. This reflects the contribution of the polyoside sidegroup, and is consistent with a location near the end rather than near the center of the macromolecule. Indeed, if the sidegroup was located near the center of the macromolecule, the maximum distance rmax of the glycosylated protein would be the same as that of the nonglycosylated one, and its radius of gyration Rg would be shorter rather than larger.
Shorter distances
The very large average dimensions of these proteins in solution must result from properties of the amino acid sequence. This is a question regarding finer details of the chain conformations, and the relevant information is contained in the high q part of the experimental spectra. These features are best seen in the Kratky-Porod representation, which enhances the high q part of the spectra. In this representation, a freely jointed chain yields a plateau at high q values because its scattering curve decays according to a q−2 power law. Fig. 4 compares the spectra of the three proteins, plotted in reduced coordinates, i.e., I(q)/I(0) as a function of the reduced scattering vector x1/2 = qRg. For comparison, the theoretical scattering curves of a dense sphere and a freely jointed chain are also traced. The scattering curve of the freely jointed chain is given by the Debye function gD(x):
| (2) |
Figure 4.
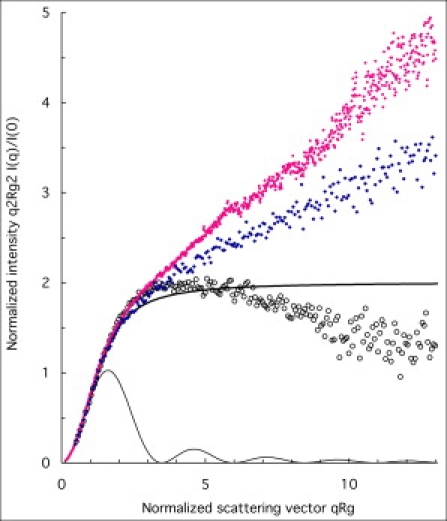
SAXS spectra for the proteins IB5 (pink dots), II-1ng (blue dots), and II-1 (circles), dissolved in ammonium acetate 50 mM pH 5.5 buffer. Horizontal axis: normalized scattering vector qRg. Vertical axis: normalized intensity (qRg)2I(q)/I(q → 0). (Thin full line) Scattering curve for a dense homogenous sphere. (Thick full line) Scattering curve for a freely jointed chain (Eq. 2).
At low q values, all spectra are superimposed, because of the use of reduced coordinates. At high q values, the spectra of the proteins IB5 and II-1ng rise above the theoretical curve for a freely jointed chain (Eq. 2). Accordingly, the scattered intensity I(q) has a decay that is in between that of a rod (q−1 power law) and that of freely jointed chain (q−2 power law). The classical way to describe such configurations is to introduce a persistence length that measures the orientational correlations between successive monomers. At large scales, a chain with a persistence length is equivalent to a random chain with a statistical element b that is twice the persistence length (49).
The spectrum of the glycosylated protein II-1 decays faster than the q−2 power law of a freely jointed chain. Because it has the same backbone as II-1ng, this faster decay must be an effect of the sugar groups. As indicated above (primary structures), II-1 has one large branched polyoside located at one of the potential glycosylation sites (most likely N35). Hence the structure of the glycosylated protein is a branched chain, because the polyoside branches out from the chain at one location and because the polyoside is itself a branched chain. It is well known that branched chains yield faster decays at high q (50).
Chain models
To characterize the conformations by average geometrical parameters, we attempted to fit the experimental spectra with specific models. For the nonglycosylated proteins, the Kratky-Porod plot (Fig. 4) suggests that the appropriate model is a chain with a persistence length (51). We used the model proposed by Sharp and Bloomfield (52), which yields the following scattering function:
| (3) |
where b is the length of the statistical element, L the contour length of the chain, x is equal to q2Lb/6, and gD(x) is the Debye function given in Eq. 2. In this model, the radius of gyration of the Debye function Rg = (Lb/6)(1/2) is corrected with a function of the ratio y = L/b:
| (4) |
L and b are used as fitting parameters. They are related by L = Nb, where N is the number of statistical elements. Note that the contour length L of a disordered chain is the length at maximum physically possible extension and is always larger than the largest dimension of the protein, rmax, unless the chain is a rigid rod (49). An upper limit for L is given by L = n × a × f, where n is the number of amino acids in the sequence, a = 3.78 Å is the length per amino acid, and f accounts for geometrical constraints of the polypeptide chain (f = 0.95) (53). If the polypeptide chain contains secondary structure elements, the contour length must be smaller than this value.
To fit the whole spectrum, it is also necessary to take into account the thickness of the chain through Rc, the radius of gyration of its cross section. The fitting function for a thick filament is then that of a thick worm-like chain (WLC):
| (5) |
However, L and Rc are not independent parameters. They are related to the dry volume of the chain, V, which is known from the protein molar mass, M, the density d = 1.4 ± 0.1 g/cm3 and Avogadro's number NAv:
| (6) |
Finally, the whole scattering curve was fitted according to Eqs. 3, 5, and 6 with Eq. 3 parameters only, i.e., I(q → 0), L and b. The range of validity of Eq. 3 is expected to be 0.01 < q < 0.1 Å−1. However, the fits are actually quite good over the whole range of q, i.e., up to q = 0.4 Å−1. The parameters extracted from the fits are listed in Table 1 and the fits are presented in Fig. 5.
Figure 5.
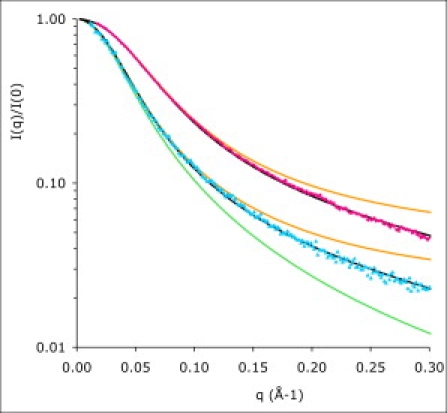
Scattering from the nonglycosylated proteins IB5 (pink diamonds) and II-1ng (blue triangles), compared with the scattering curves calculated according to the Debye model (Eq. 2, green line fit of II-1ng), the Sharp-Bloomfield model (Eq. 3, orange lines), and the WLC model with thickness (Eq. 5, black lines). The structural parameters extracted from the WLC fits are given in Table 1.
Rather satisfactorily, the radii of gyration listed in Table 1 and obtained from the fits of the experimental spectra by the WLC model are identical to those obtained from the pair distance distribution function P(r), which is model independent.
The fitting L values of 188 and 364 Å for IB5 and II-1ng respectively are significantly lower than the high bound values L = naf of 251 and 503 Å, which suggests strongly the presence of secondary structure elements that could be very short PPII- or PPI-type helical fragments. Indeed, the L/n values derived from curve fitting (2.6 Å for both IB5 and II-1ng respectively) are intermediary between the helical rise per residue values for a PPI-type helix (1.7 Å) and a PPII-type helix (3.1 Å) (54).
The existence of secondary structure elements is corroborated by the values of the statistical length of the order of 30 Å for both proteins, significantly higher than that expected for a completely unfolded polypeptide chain found in the literature, which is ∼18–20 Å.
Finally, the cross-section radius of gyration values Rc resulting from the fits was compared to the value directly calculated from the sequence using the expression
| (7) |
where the sum runs over all residues, and gives the position of the projection of atom i with mass mi on the plane normal to Cαi−Cαi+1 axis.
For both proteins, the calculated value for Rc is found close to 1.9 Å, whereas the fitting value is slightly higher (2.7 for both proteins). This is quite satisfactory in view of the purely atomic character of the calculation that does not take into account the excess of water molecules in the vicinity of the protein nor thermal motion of the protein atoms that entails a thermal volume around the protein. A thickness of at least 0.5 Å is generally found in the literature for this thermal volume (55). Furthermore, the presence of PPI- or PPII-type helices should increase the average Rc value with respect to the value of 1.9 Å calculated for a chain with the same sequence but no such secondary structure elements.
Glycosylated protein
For the glycosylated protein, II-1, the scattering curve can be fitted using the WLC model with the same contour length (325 Å) than for the nonglycosylated protein, indicating that both proteins have similar polypeptide backbones. On the contrary, the value of Rc is significantly higher (≈6.5 Å). This is of course an effect of the large polyoside sidegroups, which are not described properly by the WLC model. Another approach in this case is to describe this protein as a branched macromolecule. Accordingly, the spectrum has been compared with that of disordered structures that repeat at every scale with a fractal exponent df up to a scale characterized by a radius of gyration Rg. The simplest way of doing this is to use the Fisher-Burford approximation (56,57):
| (8) |
A good fit is obtained with the calculated scattering curve of fractal objects that have a radius of gyration of 45.9 ± 2 Å, a fractal dimension df = 2.43, which is indeed appropriate for branched macromolecules, and the same cross section as for the other proteins (Fig. 6). This is the most information that can be obtained without entering detailed information about the primary structure of the protein.
Figure 6.
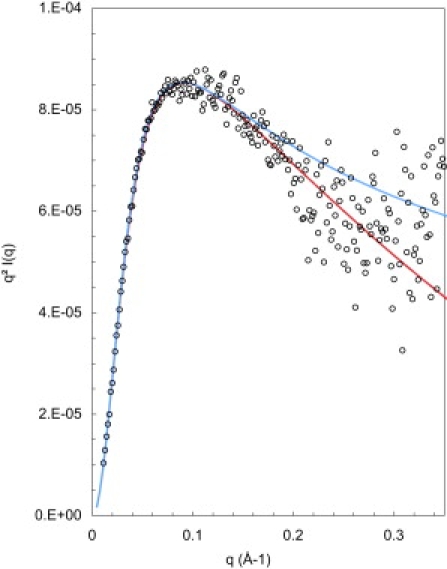
Scattering from the glycosylated protein II-1 in solution (open circles), compared with the calculated scattering curve for branched chains with a radius of gyration Rg = 45.9 Å, a fractal dimension df = 2.43 (blue line) or the same with a cross section Rc = 2.7 Å (red line).
Reconstruction of data-compatible conformations
The numbers given above for rmax, Rg, b, and Rc contain all the information that is available for the proteins in solution. Still, it is instructive to reconstruct some typical conformations that reproduce the experimental spectra, and therefore match these average parameters. Here there is a choice between constructing a single conformation that reproduces the experimental spectrum (58) (BUNCH approach), or choosing a subset of all possible conformations of the protein (59) (EOM approach). The first choice is somewhat restrictive, because a single conformation cannot give a fair view of the astronomical number of all conformations that are explored by the protein during its thermal motions. The second choice has the potential of better representing the variety of actual conformations. However, there is the possibility that the subset of chosen conformations results from a biased choice, and that it does not represent fairly the ensemble of actual conformations. We have tried both approaches, and found that both reproduce the data for protein IB5. In the second approach, however, the subset of conformations that was chosen by the program appeared to depend on the level of noise of the data and on the number of chosen conformations. Fig. SI-3 a shows various types of distributions of Rg, that are bimodal or monomodal depending on initial conditions. At present, we are not able to derive a physical interpretation from these distributions. In this article, we present the first type of reconstruction, and in the Supporting Material we present the second approach.
We used the program BUNCH that was developed by Petoukhov and Svergun to describe a protein as a combination of rigid bodies joined by flexible linkers (58). For IB5, three polyproline repeats in the sequence (PPPP, PPP, and PPPPP) were taken as rigid bodies and described as pieces of a polyproline II helix. The other residues were replaced with dummy residues centered at Cα positions, separated by 3.78 Å, and treated as linkers. The program adjusts the positions of the rigid bodies and of the dummy residues to obtain the best agreement with the experimental spectrum. Then we used another program (SABBAC) (60) to take into account steric constraints due to sidegroups of the amino acids, and add these sidegroups to the backbone. In this way, we obtained a proper polypeptidic chain with all the sidegroups. Finally, a last adjustment was made using the program CRYSOL (61) to verify that the conformation produced by SABBAC does reproduce the experimental spectrum.
We carried out 20 runs of BUNCH. The agreement between experimental data and the scattering curve calculated from the coordinates of the dummy atoms in the model is excellent in each run, with χ = 1.1 (Fig. SI-2). The results of each run provide an image of an equivalent conformation that reproduces the average distribution of distances within the protein (Fig. 7). These conformations are all different but share the common property of being extremely extended, in agreement with the overall dimensions listed in Table 1, more precisely compatible with a chain constituted by six or seven (=L/b) rigid elements of mean length 30 Å (=b).
Figure 7.
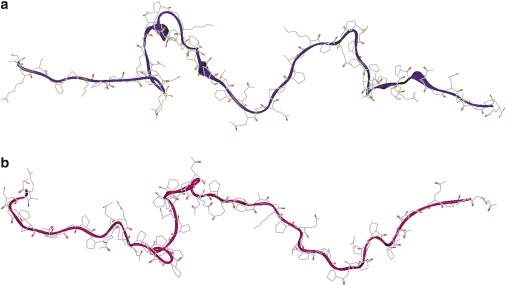
Two examples of data-compatible conformations obtained using the BUNCH program for the protein IB5.
Conclusions
The PRPs IB5 and II-1ng have conformations that are unusually extended, compared with other IDPs. Their values of Rg are significantly higher than those given by the expression Rg ≈ 2.54(n)0.522 valid for several IDP (8). The ratio of the radius of gyration to the contour length of the chain (Rg/L ≈ 0.11 for II-1ng) is also significantly higher than that found recently using the same formalism for a thermally denatured protein (Rg/L ≈ 0.08) (53).
This very strong extension is due to the length of the statistical segment (twice the persistence length), which is 30 Å, also unusually large for intrinsically disordered proteins (the usual value is on the order of 18 Å). The radius of gyration of the chain cross section is 2.7 ± 0.2 Å. These characteristics are presumably related to the numerous short polyproline repeats within the polypeptidic backbones. The glycosylated PRP II-1 has similar conformations but the presence of a large polyoside sidegroup yields an overall structure in solution that is closer to that of a self-similar branched macromolecule.
The conformations of these proteins in solution may reflect an evolutionary adaptation to the capture of plant tannins in the oral cavity. IB5 is known to precipitate when the tannin concentration in solution exceeds a threshold (31). This may be related to the feeling of astringency, which is a tactile perception associated with a loss of lubrication in the oral cavity. In this respect, the extended conformations in solution must optimize the accessibility of hydrophobic amino acids (mainly proline) to which the tannin molecules may bind. These extended conformations may also make it possible to bind stacks of tannin molecules, or polymerized tannins. II-1 is known to form limited aggregates on binding tannins, which do not precipitate. In previous work, we have shown that these aggregates are dense globules with an average radius of 100 Å, in between the radius of gyration (45 Å) and the largest distance (178 Å) of the free protein in solution (33). This type of aggregation may result from the amphiphilic nature of this protein because the hydrophobic residues of the protein backbone may form the core of the globule whereas the large polyoside sidegroups remain at the surface, as in the case of surfactant micelles. This behavior may have three possible functions: 1), a regulation of the tannin concentration in the oral cavity; 2), a reduction of the viscosity of saliva, due to the loss of macromolecules with extended conformations; and 3), a change in the interfacial properties of saliva, because this protein has an overall configuration that is characteristic of an amphiphile. These functions may contribute to the perception of astringency when the tannin concentration in the oral cavity exceeds a level that is safe for the host.
Uncited reference
37.
Supporting Material
Three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00548-5.
Supporting Material
Acknowledgments
We thank Patrice Vachette for illuminating discussions.
This work was supported by the French Agence Nationale de la Recherche (07-BLAN-02 to A.N.R.), and the European Commission Sixth Framework Programme (RIDS 011934 to J.P.).
References
- 1.Uversky V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 3.Tompa P. The functional benefits of protein disorder. J. Mol. Struct. THEOCHEM. 2003;666–667:361–371. [Google Scholar]
- 4.Tompa P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays. 2003;25:847–855. doi: 10.1002/bies.10324. [DOI] [PubMed] [Google Scholar]
- 5.Ward J.J., Sodhi J.S., Jones D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 7.Receveur-Bréchot V., Bourhis J.M., Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 8.Bernadó P., Blackledge M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys. J. 2009;97:2839–2845. doi: 10.1016/j.bpj.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J., Faeder J.R., Camacho C.J. Toward a quantitative theory of intrinsically disordered proteins and their function. Proc. Natl. Acad. Sci. USA. 2009;106:19819–19823. doi: 10.1073/pnas.0907710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 11.Edgar W.M. Saliva: its secretion, composition and functions. Br. Dent. J. 1992;172:305–312. doi: 10.1038/sj.bdj.4807861. [DOI] [PubMed] [Google Scholar]
- 12.Azen E.A., Amberger E., Niece R.L. PRB1, PRB2, and PRB4 coded polymorphisms among human salivary concanavalin-A binding, II-1, and Po proline-rich proteins. Am. J. Hum. Genet. 1996;58:143–153. [PMC free article] [PubMed] [Google Scholar]
- 13.Cid H., Vargas V., Bustos S. Secondary structure prediction of human salivary proline-rich proteins. FEBS Lett. 1986;198:140–144. doi: 10.1016/0014-5793(86)81200-5. [DOI] [PubMed] [Google Scholar]
- 14.Simon C., Pianet I., Dufourc E.J. Synthesis and circular dichroism study of the human salivary proline-rich protein IB7. J. Pept. Sci. 2003;9:125–131. doi: 10.1002/psc.438. [DOI] [PubMed] [Google Scholar]
- 15.Pascal C., Paté F., Delsuc M.A. Study of the interactions between a proline-rich protein and a flavan-3-ol by NMR: residual structures in the natively unfolded protein provides anchorage points for the ligands. Biopolymers. 2009;91:745–756. doi: 10.1002/bip.21221. [DOI] [PubMed] [Google Scholar]
- 16.Chan M., Bennick A. Proteolytic processing of a human salivary proline-rich protein precursor by proprotein convertases. Eur. J. Biochem. 2001;268:3423–3431. doi: 10.1046/j.1432-1327.2001.02241.x. [DOI] [PubMed] [Google Scholar]
- 17.Baxter N.J., Lilley T.H., Williamson M.P. Multiple interactions between polyphenols and a salivary proline-rich protein repeat result in complexation and precipitation. Biochemistry. 1997;36:5566–5577. doi: 10.1021/bi9700328. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y., Bennick A. Interaction of tannin with human salivary proline-rich proteins. Arch. Oral Biol. 1998;43:717–728. doi: 10.1016/s0003-9969(98)00040-5. [DOI] [PubMed] [Google Scholar]
- 19.Mehansho H., Butler L.G., Carlson D.M. Dietary tannins and salivary proline-rich proteins: interactions, induction, and defense mechanisms. Annu. Rev. Nutr. 1987;7:423–440. doi: 10.1146/annurev.nu.07.070187.002231. [DOI] [PubMed] [Google Scholar]
- 20.Hagerman A.E. Chemistry of tannin-protein complexation. In: Hemingway R.W., Karchesy J.J., editors. Chemistry and Significance of Condensed Tannins. Plenum Press; New York, NY: 1989. pp. 323–331. [Google Scholar]
- 21.Sarni-Manchado P., Canals-Bosch J.-M., Cheynier V. Influence of the glycosylation of human salivary proline-rich proteins on their interactions with condensed tannins. J. Agric. Food Chem. 2008;56:9563–9569. doi: 10.1021/jf801249e. [DOI] [PubMed] [Google Scholar]
- 22.Yokotsuka K., Singleton V.L. Interactive precipitation between phenolic fractions and peptides in wine-like model solutions: turbidity, particle size, and residual content as influenced by pH, temperature and peptide concentration. Am. J. Enol. Vitic. 1995;46:329–338. [Google Scholar]
- 23.Haslam E. Cambridge University Press; Cambridge, UK: 1998. Practical Polyphenolics: From Structure to Molecular Recognition and Physiological Action. [Google Scholar]
- 24.Jöbstl E., O'Connell J., Williamson M.P. Molecular model for astringency produced by polyphenol/protein interactions. Biomacromolecules. 2004;5:942–949. doi: 10.1021/bm0345110. [DOI] [PubMed] [Google Scholar]
- 25.Hatton M.N., Loomis R.E., Tabak L.A. Masticatory lubrication. The role of carbohydrate in the lubricating property of a salivary glycoprotein-albumin complex. Biochem. J. 1985;230:817–820. doi: 10.1042/bj2300817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asquith T.N., Uhlig J., Butler L. Binding of condensed tannins to salivary proline-rich glycoproteins: the role of carbohydrates. J. Agric. Food Chem. 1987;35:331–334. [Google Scholar]
- 27.Loomis R.E., Bergey E.J., Tabak L.A. Circular dichroism and fluorescence spectroscopic analyses of a proline-rich glycoprotein from human parotid saliva. Int. J. Pept. Protein Res. 1985;26:621–629. doi: 10.1111/j.1399-3011.1985.tb03220.x. [DOI] [PubMed] [Google Scholar]
- 28.Kauffman D.L., Keller P.J. The basic proline-rich proteins in human parotid saliva from a single subject. Arch. Oral Biol. 1979;24:249–256. doi: 10.1016/0003-9969(79)90085-2. [DOI] [PubMed] [Google Scholar]
- 29.Oho T., Rahemtulla F., Hjerpe A. Purification and characterization of a glycosylated proline-rich protein from human parotid saliva. Int. J. Biochem. 1992;24:1159–1168. doi: 10.1016/0020-711x(92)90387-g. [DOI] [PubMed] [Google Scholar]
- 30.Pascal C., Bigey F., Sarni-Manchado P. Overexpression and characterization of two human salivary proline rich proteins. Protein Expr. Purif. 2006;47:524–532. doi: 10.1016/j.pep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Pascal C., Poncet-Legrand C., Vernhet A. Interactions between a nonglycosylated human proline-rich protein and flavan-3-ols are affected by protein concentration and polyphenol/protein ratio. J. Agric. Food Chem. 2007;55:4895–4901. doi: 10.1021/jf0704108. [DOI] [PubMed] [Google Scholar]
- 32.Canon F., Paté F., Sarni-Manchado P. Characterization, stoichiometry, and stability of salivary protein-tannin complexes by ESI-MS and ESI-MS/MS. Anal. Bioanal. Chem. 2009;395:2535–2545. doi: 10.1007/s00216-009-3180-3. [DOI] [PubMed] [Google Scholar]
- 33.Pascal C., Poncet-Legrand C., Vernhet A. Aggregation of a proline-rich protein induced by epigallocatechin gallate and condensed tannins: effect of protein glycosylation. J. Agric. Food Chem. 2008;56:6724–6732. doi: 10.1021/jf800790d. [DOI] [PubMed] [Google Scholar]
- 34.Bretthauer R.K., Castellino F.J. Glycosylation of Pichia pastoris-derived proteins. Biotechnol. Appl. Biochem. 1999;30:193–200. [PubMed] [Google Scholar]
- 35.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Beeley J.A., Sweeney D., Khoo K.S. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis of human parotid salivary proteins. Electrophoresis. 1991;12:1032–1041. doi: 10.1002/elps.1150121207. [DOI] [PubMed] [Google Scholar]
- 37.Reference deleted at proof.
- 38.Tschopp J.F., Sverlow G., Grinna L. High-level secretion of glycosylated invertase in the methylotrophic yeast, Pichia Pastoris. Nat. Biotechnol. 1987;5:1305–1308. [Google Scholar]
- 39.Doco T., O'Neill M.A., Pellerin P. Determination of the neutral and acidic glycosyl-residue compositions of plant polysaccharides by GC–EI-MS analysis of the trimethylsilyl methyl glycoside derivatives. Carbohydr. Polym. 2001;46:249–259. [Google Scholar]
- 40.Harris P.J., Henry R.J., Stone B.A. An improved procedure for the methylation analysis of oligosaccharides and polysaccharides. Carbohydr. Res. 1984;127:59–73. doi: 10.1016/0008-6215(84)85106-x. [DOI] [PubMed] [Google Scholar]
- 41.Dennis J.W., Granovsky M., Warren C.E. Protein glycosylation in development and disease. Bioessays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Helmerhorst E.J., Oppenheim F.G. Saliva: a dynamic proteome. J. Dent. Res. 2007;86:680–693. doi: 10.1177/154405910708600802. [DOI] [PubMed] [Google Scholar]
- 43.Reddy M.S., Levine M.J., Tabak L.A. Structure of the carbohydrate chains of the proline-rich glycoprotein from human parotid saliva. Biochem. Biophys. Res. Commun. 1982;104:882–888. doi: 10.1016/0006-291x(82)91331-6. [DOI] [PubMed] [Google Scholar]
- 44.Gillece-Castro B.L., Prakobphol A., Fisher S.J. Structure and bacterial receptor activity of a human salivary proline-rich glycoprotein. J. Biol. Chem. 1991;266:17358–17368. [PubMed] [Google Scholar]
- 45.He B., Wang K., Dunker A.K. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19:929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 46.Guinier A., Fournet G. Wiley; New York, NY: 1955. Small Angle Scattering of X Rays. [Google Scholar]
- 47.Glatter O., Kratky O. Academic Press; New York, NY: 1982. Small Angle X-Ray Scattering. [Google Scholar]
- 48.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 49.Grosberg A.Y., Khokhlov A.R. AIP Press; New York: 1994. Statistical Physics of Macromolecules. [Google Scholar]
- 50.Burchard W. Statistics of star-shaped molecules. I. Stars with polydisperse side chains. Macromolecules. 1974;7:835–841. [Google Scholar]
- 51.Rawiso M., Duplessix R., Picot C. Scattering function of polystyrene. Macromolecules. 1987;20:630–648. [Google Scholar]
- 52.Sharp P., Bloomfield V.A. Light scattering from wormlike chains with excluded volume effects. Biopolymers. 1968;6:1201–1211. doi: 10.1002/bip.1968.360060814. [DOI] [PubMed] [Google Scholar]
- 53.Pérez J., Vachette P., Durand D. Heat-induced unfolding of neocarzinostatin, a small all-β protein investigated by small-angle x-ray scattering. J. Mol. Biol. 2001;308:721–743. doi: 10.1006/jmbi.2001.4611. [DOI] [PubMed] [Google Scholar]
- 54.Gu W., Helms V. Dynamical binding of proline-rich peptides to their recognition domains. Biochim. Biophys. Acta. 2005;1754:232–238. doi: 10.1016/j.bbapap.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 55.Bánó M., Marek J. How thick is the layer of thermal volume surrounding the protein? Biophys. Chem. 2006;120:44–54. doi: 10.1016/j.bpc.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 56.Fisher M.E., Burford R.J. Theory of critical-point scattering and correlations. I. The Ising model. Phys. Rev. 1967;156:583–622. [Google Scholar]
- 57.Kallala M., Sanchez C., Cabane B. Structures of inorganic polymers in sol-gel processes based on titanium oxide. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 1993;48:3692–3704. doi: 10.1103/physreve.48.3692. [DOI] [PubMed] [Google Scholar]
- 58.Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernadó P., Mylonas E., Svergun D.I. Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 60.Maupetit, J., R. Gautier, and P. Tuffery. 2006. SABBAC: online Structural Alphabet-based protein BackBone reconstruction from Alpha-Carbon trace. Nucleic Acids Res. 34(Web Server issue):W147-W51 (http://bioserv.rpbs.jussieu.fr/SABBAC.html). [DOI] [PMC free article] [PubMed]
- 61.Svergun D., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.