

Abstract
There is considerable interest in the signaling mechanisms of immunoreceptors, especially when triggered with membrane-bound ligands. We have quantified the spatiotemporal dynamics of the redistribution of immunoglobulin E-loaded receptors (IgE-FcɛRI) on rat basophilic leukemia-2H3 mast cells in contact with fluid and gel-phase membranes displaying ligands for immunoglobulin E, using total internal reflection fluorescence microscopy. To clearly separate the kinetics of receptor redistribution from cell spreading, and to precisely define the initial contact time (±50 ms), micropipette cell manipulation was used to bring individual cells into contact with surfaces. On ligand-free surfaces, there are micron-scale heterogeneities in fluorescence that likely reflect regions of the cell that are more closely apposed to the substrate. When ligands are present, receptor clusters form with this same size scale. The initial rate of accumulation of receptors into the clusters is consistent with diffusion-limited trapping with D ∼10−1μm2/s. These results support the hypothesis that clusters form by diffusion to cell-surface contact regions. Over longer timescales (>10 s), individual clusters moved with both diffusive and directed motion components. The dynamics of the cluster motion is similar to the dynamics of membrane fluctuations of cells on ligand-free fluid membranes. Thus, the same cellular machinery may be responsible for both processes.
Introduction
Understanding cellular transmembrane signaling—i.e., how chemical stimuli that are membrane impermeant can trigger cellular responses—is critical to almost all aspects of cell biology. Transmembrane signaling plays important roles in cancer and in immune responses, including allergic responses. Our principal interest is immune signaling by mast cells, for which the rat basophilic leukemia 2H3 (RBL) cell line is typically used as a model (1–3). When multivalent antigen binds to immunoglobulin E (IgE)-loaded receptors (IgE-R) on the cell surface, it causes aggregation of the receptors, leading to receptor phosphorylation (4,5) and a subsequent signaling cascade that results in degranulation, i.e., exocytosis of histamine, serotonin, and other mediators of inflammation (5).
Monovalent ligands in supported fluid bilayers (SLBs) are able to cause large-scale receptor clustering in T cells and B cells (6,7) in cytoskeleton-dependent processes; when the bilayers also contain adhesion ligands, a concentric pattern of clustered immunoreceptors surrounded by a ring of adhesion complexes is formed and is called the immunological synapse (IS). This structure is thought to play a critical role in immune cell signaling between contacting cells (8). The ability of monovalent fluid membrane-bound ligands to stimulate mast cells was recognized a decade before the immunological synapse was first identified. Using a haptenated (dinitrophenyl, DNP) lipid, Weis et al. (9) showed that modest concentrations of this lipid in supported lipid monolayers led to IgE receptor aggregation in microclusters and subsequent (although weak) mast cell degranulation. This observation has been confirmed in detail in a recent study (10), which also showed, for the first time, that the small receptor clusters coalesce within a few minutes to form a large central patch that is reminiscent of the T cell receptor (TCR) patch in the IS. Weis et al. (9) suggested that laterally mobile haptens aggregated IgE receptors by trapping receptors at points of close contact between the rough cell surface and the lipid monolayer. Despite a lack of quantitative confirmation, this hypothesis has become the accepted paradigm (4,11–13).
Although diffusion-mediated trapping is perhaps the simplest explanation of IgE receptor aggregation on fluid membranes, other hypotheses are tenable. For example, it has been proposed that some immunoreceptor signaling occurs through force transduction (14,15). If such signaling occurs with the IgE receptor, then aggregation could be a consequence. Given the recent interest in such novel signaling mechanisms, it is appropriate to revisit the behavior of RBL cells on fluid membranes, with the goal of determining quantitatively whether that behavior can be explained by simple trapping, or whether the spatiotemporal dynamics of aggregate formation require as yet unidentified additional signals or processes.
To make quantitatively useful measurements of receptor aggregation, the time of contact must be determined precisely. Previous observations of RBL-surface ligand interactions were done by allowing RBL cells to settle under gravity onto haptenated surfaces (16,10). As a consequence, the contact area between the cell and the surface is gradually increasing (as the cell spreads out), whereas IgE receptors are simultaneously redistributing into clusters. This makes a quantitative analysis of the clustering behavior exceedingly complex, as both new membrane area and new receptors are continually being added to the interface. To precisely fix the time of first cell contact with a substrate, we implemented a micropipette manipulation technique. Micropipette manipulation has been used extensively to measure the physical properties of biomembranes (17,18), and their interactions with cells (19–21). Other techniques have been used to control cell-surface contact: a film-thinning technique (22) allows control of the initial contact time and area, but offers no control of the position of the cell in the field of view, which is problematic for our total internal reflection fluorescence (TIRF) microscopy. Optical tweezers (23) apply small forces and thus give an uncertainty in the time at which full cell contact is achieved of ∼2 s, which is 40-fold less precise than we obtain using pipette manipulation.
To investigate receptor dynamics, RBL cells were primed with fluorescent anti-DNP IgE and then pipette-manipulated onto different ligand-containing and control surfaces in a TIRF microscope: fluid and gel-phase bilayers, chemically cross-linked multivalent ligands on glass, and bare glass. The characteristic sizes of fluorescence heterogeneities were analyzed using image correlation spectroscopy (ICS) (24,25) in which the rotationally averaged correlation peak was explicitly corrected for shot noise and camera read noise. Heterogeneities were seen on all cells, even on cells contacted with ligand-free surfaces. Moreover, the sizes of such heterogeneities were quite similar on ligand-bearing and ligand-free surfaces, suggesting the heterogeneities (initially) are simply points of close contact between the cell and the substrate, where the TIRF illumination is brightest. On ligand-bearing surfaces, the heterogeneities brighten over time, which we interpret as accumulation of receptors by binding to ligands at the points of close contact. (We use the term aggregation when the ligands are multivalent, and the more generic term clustering when ligands are monovalent.) A finite element diffusion model fit the dynamics of cluster brightening well, and gave a diffusion coefficient consistent with reported IgE receptor mobility. We believe these results quantitatively support a diffusion trapping model for the initial clustering of receptors.
Further analyses of the data also showed that clusters are initially randomly distributed over the cell; that the clusters do move over the cell surface, but with a combination of diffusive and directed transport, with a very slight centripetal bias. The dynamics of this motion is comparable to the fluctuations in RBL cell membranes that we observe on ligand-free fluid lipid bilayers. Finally, at later times (∼1 min), clusters begin to coalesce near the center of the contact area. This coalescence gives rise to the mast cell synapse (10). The possible biological role of a mast cell synapse is as yet unclear, but the ability of the mast cell to respond to mobile, monovalent haptens may be important in interactions with parasites or with other immune cells. It is noteworthy that mast cells may also act as antigen presenting cells (26) and form a classical immunological synapse with T helper cells (27).
Mathematical Background
ICS with explicit noise removal
ICS was used to quantify receptor distributions on cell surfaces (24,25). The two dimensional spatial autocorrelation function, g(ɛ,η) of an image i(x,y) is:
| (1) |
where the angular brackets denote spatial averaging over the image, and ɛ and η are spatial lag variables. In systems without orientational order, all the information in the correlation function is contained in its rotational average, g(r), which we use here. The peak of the autocorrelation at g(0) (the intensity variance) includes a significant contribution from spatially uncorrelated camera read noise and shot noise; the contribution from these noise sources can be directly measured and then subtracted from the autocorrelation (28,29). Because signal is and noise in are uncorrelated (25),
| (2) |
where . Then
| (3) |
and
| (4) |
using the fact that mean of the noise is zero so that . This also leads immediately to
| (5) |
so that
| (6) |
Noise variance for the EMCCD camera was measured by repeated imaging of a stationary test sample containing the full range of intensities. The single-pixel noise variance was then computed over the time series of images and plotted as a function of single-pixel mean intensity. This calibration line was then used to estimate for each pixel in the cell images; pixel averaging gave . Further information on camera noise calibration can be found in Lidke et al. (30).
Receptor cluster nearest-neighbor distribution
The nearest-neighbor distances of receptor clusters were studied to provide evidence as to the randomness of cluster positions. For randomly placed disks of diameter h, the nearest-neighbor distance distribution is given by Torquato et al. (31):
| (7) |
where P(r) is the probability of a disk having a nearest neighbor at a distance between r and r + dr, and ρ is the number density of disks. As not all clusters had the same diameter, Eq. 7 was weighted by the fraction of disks with each diameter h:
| (8) |
where H is the Heaviside step function.
Temporal image correlation spectroscopy
Temporal image correlation spectroscopy was adapted from Kolin et al. (32) to extract IgE receptor cluster dynamics. The normalized intensity fluctuation temporal autocorrelation function of an image series as a function of time lag τ is:
| (9) |
where the angular brackets denote spatial and temporal averaging. To characterize the mode of transport of IgE receptors, the normalized temporal correlation function was fit to a flow + diffusion model for a single population of particles (32) (fit parameters in bold):
| (10) |
The fit diffusion time τd was used to compute the diffusion constant D = h2/4τd and the flow time τf was used to calculate the flow speed v = h/τf (32) where h is the typical cluster diameter (measured from the spatial correlation function).
Materials and Methods
Cells
RBL-2H3 cells were maintained in minimal essential medium (MEM) (Invitrogen, Carlsbad, CA) with 10% fetal calf serum. Anti-DNP IgE was obtained from collaborators (33) and purified as described previously (34). Fluorescent IgE conjugates were created using N-hydroxysuccinimide esters of Alexa Fluor 488 (Invitrogen) or Dy-520XL (Dyomics GmbH, Jena, Germany). Before microscopy, cells were IgE primed by incubation with 0.5 μg/mL of fluorescent IgE overnight. At the day of the experiment, the cell media was exchanged with MEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% L-glutamine (MEM-FBS), and cells were removed to suspension. Then 0.5 mL aliquots (∼50 000 cells/aliquot) of primed cells were stored in 1 mL tubes at 37°C in a humidified chamber with 5% CO2 until later use.
Surface preparation
All glass slides were cleaned with piranha before use.
Cross-linked surfaces
DNP-conjugated bovine serum albumin (DNP24-BSA at 1 μg/mL; Invitrogen) was cross-linked to poly-L-lysine coated coverslips with two homobifunctional cross-linkers; ethylene glycol bis(succinimidylsuccinate) (Fisher, Rockford, IL) or glutaraldehyde (Sigma, St. Louis, MO). Reactions were quenched with 100 mM glycine in phosphate buffer saline and prepared coverslips were stored in buffer until use for up to 1 day.
SLBs
SLBs were made by spontaneous liposome fusion (35). Lipids (Avanti, Alabaster, AL) were dissolved in chloroform, dried under N2 then 1 h vacuum. The lipid film was then suspended in phosphate buffer saline + 2 mM Mg2+ to 1.3 mM and sonicated for 5 min using a probe sonicator. Laterally mobile bilayers were formed from 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and N-dinitrophenyl-aminocaproyl phosphatidylethanolamine (DNP-Cap PE), and immobile bilayers were formed using 1,2-dipalmitoyl-sn-lycero-3-phosphocholine (DPPC). Bilayers were formed on piranha-cleaned cover glass in 15 min on a slide warmer at 37°C (POPC) or in 8 min in an oven at 55°C (DPPC). Each bilayer coated coverslip was kept immersed during transfer to the imaging chamber; before adding cells to the bilayer, the chamber was flushed with 500 μL of MEM-FBS. Lipid mobility was checked using photobleaching and single-particle tracking as described elsewhere (10). To test the effect of pipette micromanipulation on POPC bilayers, a fluorescent lipid N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl)-1,2-dihexadecanoyl-sn-glycero-3-phospho-ethanolamine, triethylammonium salt (Invitrogen) at 1 mol % was incorporated in addition to 25 mol % DNP-lipid. The bilayer integrity was observed before and after one cell was pipette-pressed. No apparent bilayer defects were caused by micromanipulation as shown in Fig. S1 in the Supporting Material.
Cell micromanipulation
Glass micropipettes were pulled from soda lime glass No. 564 Micro-Hematocrit tubes (Carolina Biological Supply, Berlington, NC) on a DMZ-Universal Puller (Zeitz-Instruments, Martinsried, Germany), using a four-step program to achieve ∼2 μm tip diameters. A pipette was then mounted onto Eppendorf InjectMan NI2 (Eppendorf, Westbury, NY) and connected to a 30 mL plastic syringe. The micropipette was placed at the center of the camera's field of view, approximately one micron above the substrate. This position was stored in the micromanipulator; after a cell was captured in suspension, the position was recalled to automatically place the cell in contact with the substrate. CCD images were captured before cell contact and the final contact area was established within 50 ms, clearly defining an initial time point. In some instances, cells ruptured on contact; these were discarded.
Fluorescence microscopy
Objective-based TIRF microscopy was carried out on an Olympus IX 71 (Olympus America, Center Valley, PA) inverted microscope with a 60× or 150× 1.45 NA oil objective using a 488 nm laser (Coherent, Santa Clara, CA) with a 50 nm (1/e) calculated penetration depth of the evanescent wave. A 1.6× tube lens was also used for the 60× objective. Interference filters (Semrock, Rochester, NY) were used to block excitation. The EMCCD (iXon 887; Andor Technologies, Belfast, Northern Ireland) was cooled to −100°C (CCD cooler: Thermo Neslab, Waltham, MA); gain = 100. Sample temperatures were maintained at 37°C for POPC and cross-linked surfaces and at 32°C for DPPC surfaces with an objective heater (Cell MicroControls, Norfolk, VA). Images were collected with in-house software implemented in MATLAB (The MathWorks, Natick, MA).
Cluster tracking
Fluorescent anti-DNP IgE clusters were tracked with ImageJ (NIH, Bethesda, MD) SpotTracker plug-in (36) for up to 90 s in 1-s intervals. The mean-square displacement (MSD) was calculated for each time difference Δt in the track record. As the MSD was generally not linear in time, the tracks were fit to a drift + diffusion model. Raw tracks were fit by choosing a drift vx, vy so as to best linearize the residual MSD (estimated by χ2). For fitting, each MSD was weighted by the number of independent time intervals sampled minus one.
Image processing and camera calibration
The spatial and temporal correlation analysis, explicit noise removal, and masking was implemented in DIPimage (Delft University of Technology, Delft, The Netherlands) a MATLAB toolbox for scientific image processing. Each image was corrected by subtracting a dark image (average of 900) and then normalizing to the TIRF illumination profile. To measure the typical size of receptor heterogeneities the images were preprocessed by masking; the RBL cell exterior was filled with the mean of the cell interior to remove cell-size artifacts. Cell outlines were determined via a DIPimage thresholding function that uses Isodata algorithm (37). The correlation image was then rotationally averaged and the typical cluster size was extracted from the full width at half-maximum (FWHM). For temporal correlation analyses of image series taken every second with 50 ms exposure, a square region (29 × 29 pixels) inside the cell was selected and analyzed. The spatial correlation function was calculated by Fourier transform methods, whereas the temporal correlation function was calculated directly.
Diffusion trapping model and analysis
To test whether the early increase in spot fluorescence was consistent with diffusion-mediated trapping, a finite-element diffusion simulation was done using MATLAB. At each time iteration receptors in traps were removed; traps were taken to be the clusters of receptors on each cell. Traps were obtained by low-pass spatial filtering of cell images (removing the 10 lowest frequency components) and thresholding at 5% of the maximum intensity. As receptor clusters are dim at the very earliest time points, the trap mask generated at 0.5 s was used for earlier times as well. This method produced stationary receptor cluster traps for immobile bilayers and slowly moving cluster traps on mobile bilayers. In modeling diffusional trapping, periodic boundary conditions were used on a square lattice with an area equal to the total cell surface area. (For the times modeled (0–3 s), the boundary conditions are irrelevant, as the total pool of receptors is not depleted significantly.)
Results and Discussion
When RBL cells loaded with fluorescent IgE were either pipette-pressed or allowed to settle onto surfaces (10), an initial heterogeneous fluorescence distribution was always observed, even on bare glass (Fig. 1, inset). To characterize the heterogeneous distribution, the rotationally averaged image correlation function was computed; the width of this distribution is a measure of the spatial scale of heterogeneities (convolved with the point spread function of the optical system). Fig. 1 (Top) shows a typical image and correlation function of a RBL cell loaded with fluorescent IgE contacting a mobile bilayer containing DNP ligands. To facilitate comparison between different surfaces, the FWHM of the correlation function was used as described in Materials and Methods.
Figure 1.
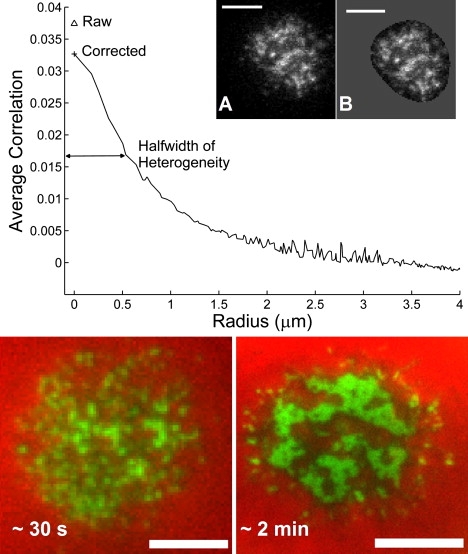
ICS was carried out to determine the sizes of fluorescence heterogeneities on single cells pipette-pressed against ligand-bearing or ligand-free surfaces. The rotationally averaged correlation shows explicit noise removal from the variance (g(0)) and the resultant halfwidth at half-maximum. Inset (A) is a TIRF image of a pipette-pressed RBL cell on a mobile bilayer with 1 mol % DNP-Cap PE and (B) same image masked with mean of cell interior, as was done in the analyses to eliminate the cell-size artifact from the autocorrelation. (Bottom) Two-color TIRF images of FcɛRI receptors (green) and a soluble buffer marker (red). Close contacts between the cell and the coverslip exclude the soluble fluorophore, and essentially every close contact contains IgE. (Left) Pipette-pressed RBL cell after ∼30 s (90×). (Right) Settled RBL cell after ∼2 min (150×) on mobile bilayer with 25 mol % DNP-Cap PE lipid. Scale bar = 5 μm.
The exponential decay of the evanescent field can result in heterogeneous fluorescence intensity even on membranes with uniform receptor distributions, if the membrane itself has regions that are apposed to the substrate more closely. On the length scale of optical microscopy, IgE-loaded FcɛRI are distributed uniformly on the RBL surface, when not bound to antigens. Thus, we interpret the heterogeneities on bare glass surfaces as points of close contact between the cell and the surface. This interpretation is supported by previous studies using scanning electron microscopy (38) and transmission electron microscopy in cross section (39).
Using pipette manipulation to precisely fix the time of first contact with the surface, the early time evolution of the fluorescence heterogeneities can be studied. On bare glass surfaces the heterogeneities do not change significantly in brightness or size after initial contact for the first few seconds (data not shown). On all ligand-coated surfaces, however, the heterogeneities brighten substantially in the first few seconds. If the ligand is immobile, the spatial scale and positions of the clusters do not change over time. With mobile ligands clusters do move and at longer times (∼2 min) they coalesce into a large central patch reminiscent of the IS formed by T cells (40). This large scale reorganization and localization of signaling molecules has been more fully characterized elsewhere (10).
Additional support for the hypothesis that receptor clustering initially occurs at close contacts was obtained by adding a water soluble fluorescent dye (20 nM Alexa Fluor 647-R-phycoerythrin streptavidin; Invitrogen) to the buffer (41) (Fig. 1 Bottom). At both early (left panel) and late (right panel) time points, the receptor clusters and patches are found only where the soluble dye is excluded. Furthermore, every contact zone (as indicated by dark regions in the red channel) contains clustered receptors. As a consequence, there are no dark regions in the composite image: all close contacts appear green, owing to the presence of IgE.
Qualitatively, there are no apparent differences in the size or distribution of the heterogeneities formed immediately after contact, regardless of the nature of the contacted surface, Fig. 1 inset and Fig. S2 in the Supporting Material. Quantitatively, the spatial scale of the heterogeneities, as assessed by the FWHM of the correlation function, showed no statistically significant differences between surfaces, Fig. 2. The average size of heterogeneities on all surfaces (30 cells, 3 on each surface) is 1.2 ± 0.2 (SD) μm. Measured fluorescent heterogeneities are significantly larger than the FWHM of 0.34 μm computed for the theoretical microscope point spread function (Fig. 2, vertical black line) and 0.63 μm for measured 100 nm fluorescent beads (Fig. 2, vertical gray line). All analyzed data was obtained within 5 s of initial contact, during which time no coalescence of clusters was observed. The qualitative and quantitative similarities between heterogeneities observed on different surfaces, including surfaces that display no ligands for IgE, strongly support the contention that clusters of IgE receptors observed on contacting ligand-presenting surfaces are formed by diffusion-mediated trapping at points of close contact between the cell and the surface.
Figure 2.
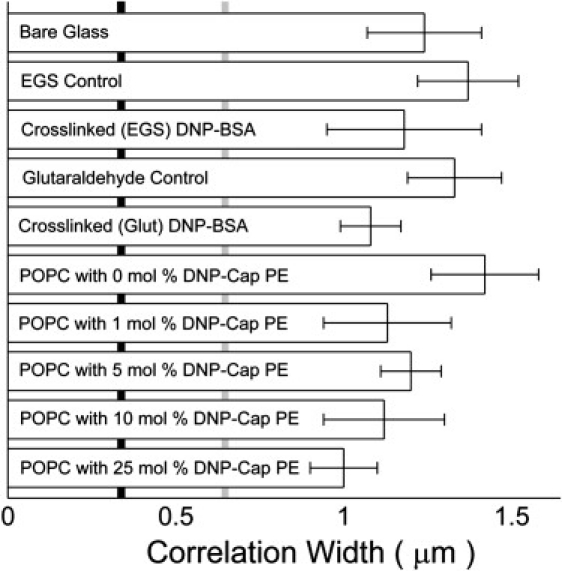
Size of FcɛRI receptor fluorescence heterogeneities on different surfaces, measured by the FWHM of the rotationally averaged intensity autocorrelation. Fluorescence heterogeneities may arise from clustering of receptors, or from variations in the proximity of the cell membrane to the substrate. The size of heterogeneities is independent of the presence/absence of ligand or the ligand mobility. Three pipette-pressed cells were analyzed for each surface. Error bars are the standard deviation. The vertical solid gray line shows the measured size of 100 nm fluorescent beads and the vertical black line is the size of the theoretical point spread function (Airy function) with effective NA = 1.33 and a wavelength of 550 nm.
To further test this hypothesis, we fit the rapidly increasing fluorescence in clusters to a simple finite-element diffusion model in MATLAB. The initial, brighter heterogeneities were treated as diffusion traps. The initial receptor distribution was taken to be uniform at t = 0, then evolved in time via diffusion, with receptors in traps removed from the diffusing pool at each time step. Traps were taken as the actual cluster locations, determined by thresholding cell images. Accumulation in the traps was then fit to the measured accumulation (i.e., fluorescence increase) with two adjustable parameters: the diffusion coefficient and the total fluorescence (Fig. 3). Only data from the first three seconds of accumulation (brightening) was analyzed because photobleaching causes significant dimming at long times. Very good fits were found for both immobile and mobile ligands. From measurements on three cells, the mean receptor diffusion coefficient was 0.30 ± 0.08 (SD) μm2/s (immobile ligands) and 0.24 ± 0.07 (SD) μm2/s (mobile ligands), consistent with reported IgE receptor diffusivity (42,43). To test the sensitivity of the fit diffusion coefficient to the trap sizes, the intensity threshold was increased from 5% to 10% and the data was refit; this gave the same diffusion coefficients, within experimental uncertainty.
Figure 3.
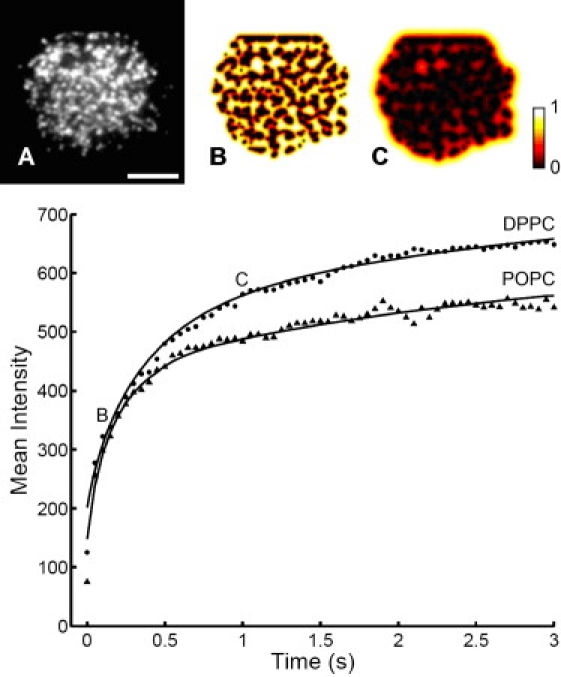
Mean receptor cluster intensity is used as a measure of number of receptors trapped in cell protrusions by immobile (DPPC) or mobile (POPC) ligands. The data was fit to a numerical 2D diffusion trapping model. The extracted diffusion coefficients for cells shown here are 0.16 μm2/s and 0.26 μm2/s for DPPC and POPC respectively. (The differences in overall intensities likely reflect cell to cell receptor number variations, and were not consistently different for mobile versus immobile bilayers.) Top images depict (A) analyzed cell on DPPC bilayer and evolved free receptor concentration after (B) 0.16 s and (C) 1 s. Scale bar = 5 μm.
It has been shown previously that mobile bilayers with 5, 10, and 25 mol % DNP-Cap PE can cause downstream signaling and degranulation (10). To confirm that the observed IgE receptor clusters on immobile bilayers with 25 mol % DNP-Cap PE can also cause signaling, an exocytosis assay was conducted to determine the percentage of cellular β-hexosaminidase released (44). To ensure immobility of the bilayer, the assay was conducted at 32°C. Consistent with previous results for mobile bilayers with similar mol % DNP-Cap PE, the mean percentage of β-hexosaminidase release from two independent experiments with immobile bilayers was 7.3 ± 2.5 (SD) %; small but still significantly higher than the spontaneous release of 0.5 ± 0.4 (SD) %. Degranulation with 25 mol % mobile ligands has been found to be 10.1 ± 1 (SD) % (10).
Cluster distributions and dynamics
At early times, the distribution of heterogeneities (clusters) on immobile substrates appeared random, using two tests. First, the radial distribution of clusters was examined, by histogram analysis of cluster distances from the center of the contact area (Fig. 4 A). The number of clusters at a radial distance (r) was proportional to r, as expected for a uniform density of randomly positioned clusters. This radially uniform distribution was found on all immobile substrates, and on mobile substrates at early time points after cell contact. Thus, at early times, clusters are no more likely to be found in the center of the contact zone than in the periphery. It is thus likely that points of close contact are formed by cell protrusions (such as microvilli) that are distributed randomly on the cell surface before the cell is brought into contact with the planar substrate. Similar, apparently random distributions of receptor clusters have been observed in RBL cells (10) and in T cells (12) that are allowed to settle by gravity onto ligand-bearing membrane substrates.
Figure 4.
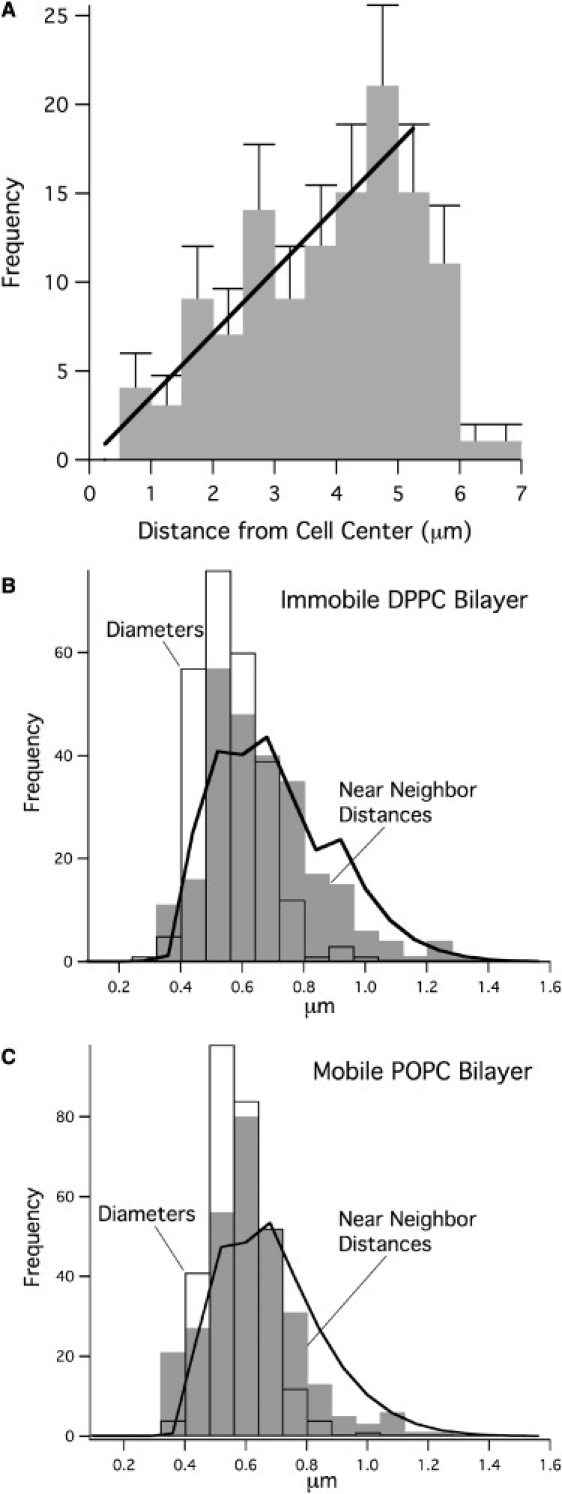
(A) The number of clusters at a radial distance r is proportional to the distance from the center of a cell (Fig. 3A) as expected for a uniform density of randomly positioned clusters. Distributions of cluster diameters and nearest-neighbor distances on immobile (B) and mobile (C) ligand substrates. The area of each bar is the fraction of clusters with nearest-neighbor distances (gray bars) or diameters (white bars) in the x-range of the bar. The solid lines are the nearest-neighbor distances expected from a random spot model P(r) ∝ r exp(−ρπr2), where ρ is the measured density of clusters, weighted with the measured distribution of cluster diameters (see Mathematical Background). Cluster density = 1.139 clusters/μm2 on DPPC (255 clusters on 3 cells), and 1.145 clusters/μm2 on POPC (296 clusters on 3 cells). On immobile DPPC (B), the nearest-neighbor distribution predicted for random clusters fits the observed distribution well. On mobile POPC (C), the fit is slightly poorer, with clusters appearing closer together than predicted for a perfectly random distribution. This may reflect the initial stages of membrane reorganization that lead to the formation of the large patch or synapse on mobile substrates.
A further test of cluster randomness was carried out by measuring nearest-neighbor distance distributions for receptor clusters from three cells on immobile (Fig. 4 B) and on mobile bilayers (Fig. 4 C). Identical, randomly positioned disks give a Rayleigh distribution (Eq. 7), but with a sharp cutoff for distances less than the disk diameter. The width of the Gaussian is determined solely by the density of clusters, ρ. As the actual clusters have a range of sizes, we plotted the random disk model, weighted with the measured distribution of cluster diameters (P′(h) in Eq. 8). Using the measured cluster density ρ and the total number of receptor clusters, the theoretical nearest-neighbor distribution (Fig. 4 B, solid line) then has zero free parameters. This theoretical distribution fits the measured distribution on immobile bilayers well, supporting the contention that clusters are randomly distributed. On mobile bilayers (Fig. 4 C), clusters are slightly closer together than predicted for a perfect random distribution at the measured density. This result may indicate that, even at early times, there is some change in the cluster distribution on mobile ligands that may be a precursor to later coalescence.
Receptor dynamics differed depending on the mobility of the ligand. On immobile ligand substrates, the clusters brightened but did not move appreciably over the observation time of >1 min. On mobile ligand substrates, clusters of receptors on pipette-pressed cells (and on gravity-settled cells) showed directed, weakly correlated motion (vide infra), and coalesced to form a large central patch of receptors within 2 min, whereas small isolated clusters remained at the periphery of the contact zone. Similar receptor patching has been observed in T cells and has been termed a synapse. The similarity in receptor organization has led to the designation of this large patch as a mast cell synapse (10).
Before the onset of big patch formation ∼2 min after contact, the receptor clusters do not grow measurably in size, nor do they coalesce. Thus, individual receptor clusters may be tracked to phenomenologically characterize their motion, to compare the dynamics with other immunoreceptor clusters. MSD plots for 20 individual clusters were obtained from extracted coordinates. All plots had upward curvature; such MSD curves can be best fit with a combination of diffusion and drift velocity. A typical MSD plot is shown in Fig. 5. Fig. 5 inset shows the root mean-square diffusional spread (parabolas) and drift velocity (arrows) of six receptor trajectories during 55 s of initial contact. The cluster drift velocity was 37 ± 5 (mean ± SE.) nm/s and did not seem to change over time; in contrast, TCR clusters have slightly slower velocities when initially formed, but faster, strongly centripetal velocities during synapse formation (45). For IgE receptor clusters, there was only a very weak correlation among track directions: over a 10-s interval, roughly twice as many tracks had an inward component of drift as had an outward component. The computed mean diffusion coefficient of clusters was 5.1 ± 0.7 × 10−3 (mean ± SE) μm2/s, which is comparable to that of TCR clusters when their centripetal motion is inhibited by pharmacological or physical mechanisms (12).
Figure 5.
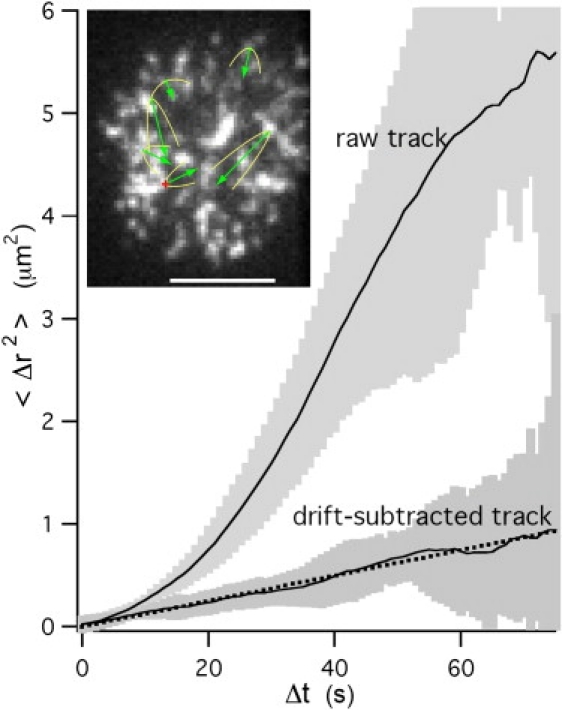
Raw and drift-subtracted MSD for cluster track indicated with + (red online) in inset. Shaded area represents the standard error of the mean from multiple measurements. (Inset) Tracked receptor clusters after ∼55 s of initial contact of POPC bilayer with 25 mol % DNP-Cap PE lipid. Parabolas (yellow online) show the root mean-square diffusional spread of the cluster. Arrows (green online) are proportional to the drift velocity and show how far each cluster would drift in 66 s. Scale bar = 5 μm.
Receptor clusters on ligand-bearing membranes undergo active (directed) transport. Interestingly, cells on ligand-free surfaces also exhibit fluorescence fluctuations in TIRF imaging (Movie S1). These fluctuations are much too large in intensity and in spatial extent to be caused by stochastic receptor density fluctuations—they most likely reflect fluctuations in the proximity of various regions of the cell surface to the substrate. To further explore the possible relationship between cell fluctuations on unliganded surfaces and cluster motion on ligand-containing membranes, the temporal autocorrelation was computed. The decay of the temporal autocorrelation function depends on the persistence of the intensity variations between images in a time-series (32). Fig. 6 depicts the temporal autocorrelation of fluorescence images from six different cells (three on 0 mol % DNP-Cap PE and three on 25 mol % DNP-Cap PE bilayer). For intervals of a few seconds or less, the kinetics of both cluster motion and cell membrane fluctuations are similar (although one cell on ligand-free membranes was somewhat slower, as shown). For longer time intervals, the autocorrelation on ligand-free membranes showed greater persistence; this may be caused by the fact that bright patches on cells on ligand-free membranes are somewhat larger than receptor clusters, and they grow in time as the cells spread. (Fig. 6, images). The similarities in the kinetics of cell membrane fluctuations and cluster motions suggest that similar biological mechanisms may be responsible for both. It is possible that as the clusters initially formed at contact zones move, the contact zones themselves move, via (for example) cytoskeletal dynamics. In this context, it is interesting that motion of microvilli over the cell surface has been reported in A6 toad kidney epithelium cells, using scanning ion conductance microscopy (46).
Figure 6.
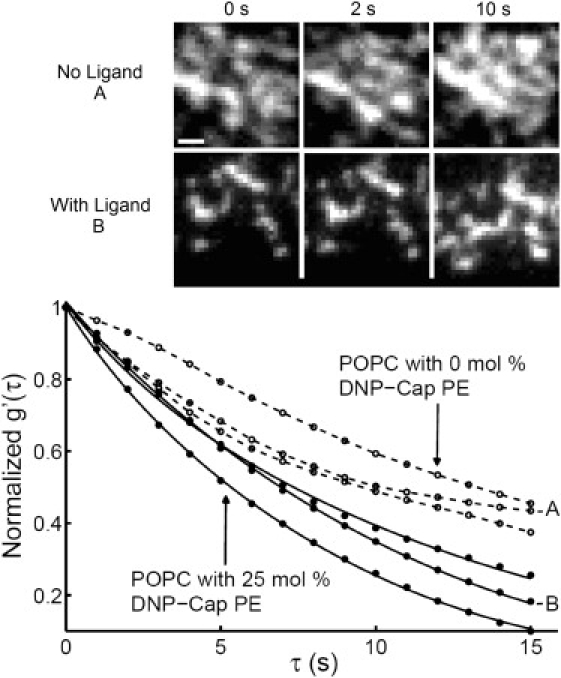
Temporal autocorrelation functions of three cell images on mobile bilayers with 25 mol % DNP-Cap PE (solid circles) and 0 mol % DNP-Cap PE (open circles). Data obtained from 25 mol % bilayers was fit to the flow + diffusion model (solid lines) outlined in Mathematical Background. The extracted mean cluster diffusion coefficient and flow speed were 7.4 ± 1.2 × 10−3 (SD) μm2/s and 30 ± 7 (SD) nm/s respectively, which is consistent with results from tracking single clusters. Fluorescence fluctuation dynamics are quite similar for short lag times regardless of the presence of ligand. Images depict cell regions analyzed here for POPC bilayer with 0 mol % DNP-Cap PE (top row, A in bottom plot) and with 25 mol % DNP-Cap PE (bottom row, B in bottom plot) at 0 s, 2 s, and 10 s. Scale bar = 1 μm.
The temporal autocorrelation on ligand-bearing membranes was also checked for consistency with the results from individually tracked clusters. A diffusion + flow model was fit to the autocorrelation (Eq. 11). A very good fit was obtained (Fig. 6, solid lines) and the extracted mean diffusion coefficient and flow speed were 7.4 ± 1.2 × 10−3 (SD) μm2/s and 30 ± 7 (SD) nm/s respectively, consistent with numbers obtained from IgE receptor tracks.
Conclusions
This study presents what we believe to be the first quantitative evidence that anti-DNP IgE-FcɛRI complexes form microclusters at RBL cell protrusions through diffusion mediated trapping at initial contact with monovalent ligands in SLBs. To quantitatively measure early IgE receptor dynamics, the time of cell-surface contact was fixed by micropipette cell manipulation, with an onset precision of ±50 ms. At early time points, the typical size of the fluorescence heterogeneities was the same for all surfaces, regardless of the presence or absence of ligand, or the ligand mobility. On ligand-free surfaces, these heterogeneities most likely reflect the variation in the separation between the cell and the substrate, as the exponential decay in the TIRF field will cause close contacts to appear brighter. The hypothesis that receptor clusters develop at close contacts was supported by simultaneous TIRF imaging of a water-soluble fluorescent marker dye.
The cluster brightness increases substantially in the first few seconds of initial contact with monovalent ligands incorporated in either mobile or immobile SLBs, and this increase is well modeled by diffusional trapping. The initial cluster locations seemed to be random, as quantified by both a radial density analysis and a near-neighbor distance analysis. At later times (>10 s), liganded IgE receptor clusters on mobile surfaces undergo a combination of directed and diffusive motion, indicating involvement of active cellular processes. Whether the receptor clusters remain associated with or restricted to cellular protrusions remains to be determined.
Supporting Material
Two figures and one movie are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00527-8.
Supporting material
Acknowledgments
The authors thank Dr. V. M. Kenkre for discussions on receptor diffusion and trapping, Dr. Fernando C. Valenzuela for helping with pulling micropipettes, and Dr. George Bachand for loaning the micromanipulator.
This work was supported in part by the Laboratory Directed Research and Development program at Sandia National Laboratories (to J.A.T.), by the Army Research Office Grant W911NF0510464, and by the Program in Interdisciplinary Biological and Biomedical Sciences funded by the University of New Mexico (to K.S.). Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy's National Nuclear Security Administration under Contract DE-AC04-94AL85000.
References
- 1.Thomas J.L., Feder T.J., Webb W.W. Effects of protein concentration on IgE receptor mobility in rat basophilic leukemia cell plasma membranes. Biophys. J. 1992;61:1402–1412. doi: 10.1016/S0006-3495(92)81946-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas J.L., Holowka D., Webb W.W. Large-scale co-aggregation of fluorescent lipid probes with cell surface proteins. J. Cell Biol. 1994;125:795–802. doi: 10.1083/jcb.125.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Posner R.G., Subramanian K., Baird B. Simultaneous cross-linking by two nontriggering bivalent ligands causes synergistic signaling of IgE Fc epsilon RI complexes. J. Immunol. 1995;155:3601–3609. [PubMed] [Google Scholar]
- 4.Metzger H. Transmembrane signaling: the joy of aggregation. J. Immunol. 1992;149:1477–1487. [PubMed] [Google Scholar]
- 5.Galli S.J., Tsai M., Piliponsky A.M. The development of allergic inflammation. Nature. 2008;454:445–454. doi: 10.1038/nature07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaizuka Y., Douglass A.D., Vale R.D. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sohn H.W., Tolar P., Pierce S.K. Membrane heterogeneities in the formation of B cell receptor-Lyn kinase microclusters and the immune synapse. J. Cell Biol. 2008;182:367–379. doi: 10.1083/jcb.200802007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monks C.R., Freiberg B.A., Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 9.Weis R.M., Balakrishnan K., McConnell H.M. Stimulation of fluorescence in a small contact region between rat basophil leukemia cells and planar lipid membrane targets by coherent evanescent radiation. J. Biol. Chem. 1982;257:6440–6445. [PubMed] [Google Scholar]
- 10.Carroll-Portillo A., Spendier K., Timlin J.A. Formation of a mast cell synapse: FcɛRI membrane dynamics upon binding mobile or immobilized ligands on surfaces. J. Immunol. 2010;184:1328–1338. doi: 10.4049/jimmunol.0903071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tempelman L.A., Hammer D.A. Receptor-mediated binding of IgE-sensitized rat basophilic leukemia cells to antigen-coated substrates under hydrodynamic flow. Biophys. J. 1994;66:1231–1243. doi: 10.1016/S0006-3495(94)80907-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMond A.L., Mossman K.D., Groves J.T. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J. 2008;94:3286–3292. doi: 10.1529/biophysj.107.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolar P., Sohn H.W., Pierce S.K. Viewing the antigen-induced initiation of B-cell activation in living cells. Immunol. Rev. 2008;221:64–76. doi: 10.1111/j.1600-065X.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- 14.Ma Z., Sharp K.A., Finkel T.H. Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 2008;6:328–342. doi: 10.1371/journal.pbio.0060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Z., Janmey P.A., Finkel T.H. The receptor deformation model of TCR triggering. FASEB J. 2008;22:1002–1008. doi: 10.1096/fj.07-9331hyp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman M.A., Shoag J., Koretzky G.A. Disruption of SLP-76 interaction with Gads inhibits dynamic clustering of SLP-76 and FcɛRI signaling in mast cells. Mol. Cell. Biol. 2006;26:1826–1838. doi: 10.1128/MCB.26.5.1826-1838.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waugh R., Evans E.A. Thermoelasticity of red blood cell membrane. Biophys. J. 1979;26:115–131. doi: 10.1016/S0006-3495(79)85239-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans E., Needham D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Phys. Chem. 1987;91:4219–4228. [Google Scholar]
- 19.Tözeren A., Sung K.-L.P., Chien S. Micromanipulation of adhesion of a Jurkat cell to a planar bilayer membrane containing lymphocyte function-associated antigen 3 molecules. J. Cell Biol. 1992;116:997–1006. doi: 10.1083/jcb.116.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chesla S.E., Selvaraj P., Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCloskey M.A., Poo M.M. Contact-induced redistribution of specific membrane components: local accumulation and development of adhesion. J. Cell Biol. 1986;102:2185–2196. doi: 10.1083/jcb.102.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patrick S.M., Kim S., Leonard E.F. Controlled cell deformation produces defined areas of contact between cells and ligand-coated surfaces. Ann. Biomed. Eng. 2001;29:1–8. doi: 10.1114/1.1332081. [DOI] [PubMed] [Google Scholar]
- 23.Snijder-Van As M.I., Rieger B., Kanger J.S. A hybrid total internal reflection fluorescence and optical tweezers microscope to study cell adhesion and membrane protein dynamics of single living cells. J. Microsc. 2009;233:84–92. doi: 10.1111/j.1365-2818.2008.03099.x. [DOI] [PubMed] [Google Scholar]
- 24.Petersen N.O., Höddelius P.L., Magnusson K.E. Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys. J. 1993;65:1135–1146. doi: 10.1016/S0006-3495(93)81173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiseman P.W., Petersen N.O. Image correlation spectroscopy. II. Optimization for ultrasensitive detection of preexisting platelet-derived growth factor-β receptor oligomers on intact cells. Biophys. J. 1999;76:963–977. doi: 10.1016/S0006-3495(99)77260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershko A.Y., Rivera J. Mast cell and T cell communication; amplification and control of adaptive immunity. Immunol. Lett. 2010;128:98–104. doi: 10.1016/j.imlet.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudenzio N., Espagnolle N., Espinosa E. Cell-cell cooperation at the T helper cell/mast cell immunological synapse. Blood. 2009;114:4979–4988. doi: 10.1182/blood-2009-02-202648. [DOI] [PubMed] [Google Scholar]
- 28.Unruh J.R., Gratton E. Analysis of molecular concentration and brightness from fluorescence fluctuation data with an electron multiplied CCD camera. Biophys. J. 2008;95:5385–5398. doi: 10.1529/biophysj.108.130310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Digman M.A., Dalal R., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lidke K.A., Rieger B., Jovin T.M. The role of photon statistics in fluorescence anisotropy imaging. IEEE Trans. Image Process. 2005;14:1237–1245. doi: 10.1109/tip.2005.852458. [DOI] [PubMed] [Google Scholar]
- 31.Torquato S., Lu B., Rubinstein J. Nearest-neighbor distribution function for systems of interacting particles. J. Phys. Math. Gen. 1990;23:L103–L107. doi: 10.1103/physreva.41.2059. [DOI] [PubMed] [Google Scholar]
- 32.Kolin D.L., Wiseman P.W. Advances in image correlation spectroscopy: measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 2007;49:141–164. doi: 10.1007/s12013-007-9000-5. [DOI] [PubMed] [Google Scholar]
- 33.Liu F.T., Bohn J.W., Katz D.H. Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J. Immunol. 1980;124:2728–2737. [PubMed] [Google Scholar]
- 34.Seagrave J., Pfeiffer J.R., Oliver J.M. Relationship of IgE receptor topography to secretion in RBL-2H3 mast cells. J. Cell. Physiol. 1991;148:139–151. doi: 10.1002/jcp.1041480117. [DOI] [PubMed] [Google Scholar]
- 35.Werner J.H., Montaño G.A., Shreve A.P. Formation and dynamics of supported phospholipid membranes on a periodic nanotextured substrate. Langmuir. 2009;25:2986–2993. doi: 10.1021/la802249f. [DOI] [PubMed] [Google Scholar]
- 36.Sage D., Neumann F.R., Unser M. Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans. Image Process. 2005;14:1372–1383. doi: 10.1109/tip.2005.852787. [DOI] [PubMed] [Google Scholar]
- 37.Ridler T.W., Calvard S. Picture thresholding using an iterative selection method. IEEE Trans. Syst. Man Cybern. 1978;8:630–632. [Google Scholar]
- 38.Hamawy M.M., Oliver C., Siraganian R.P. Inhibition of IgE binding to RBL-2H3 cells by a monoclonal antibody (BD6) to a surface protein other than the high affinity IgE receptor. J. Immunol. 1992;148:524–531. [PubMed] [Google Scholar]
- 39.Oliver J.M., Seagrave J., Deanin G.G. Signal transduction and cellular response in RBL-2H3 mast cells. Prog. Allergy. 1988;42:185–245. [PubMed] [Google Scholar]
- 40.Grakoui A., Bromley S.K., Dustin M.L. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 41.Andrews N.L., Lidke K.A., Lidke D.S. Actin restricts FcɛRI diffusion and facilitates antigen-induced receptor immobilization. Nat. Cell Biol. 2008;10:955–963. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larson D.R., Gosse J.A., Webb W.W. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell Biol. 2005;171:527–536. doi: 10.1083/jcb.200503110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menon A.K., Holowka D., Baird B. Cross-linking of receptor-bound IgE to aggregates larger than dimers leads to rapid immobilization. J. Cell Biol. 1986;102:541–550. doi: 10.1083/jcb.102.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith A.J., Pfeiffer J.R., Wilson B.S. Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic. 2003;4:302–312. doi: 10.1034/j.1600-0854.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 45.Yokosuka T., Sakata-Sogawa K., Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 46.Gorelik J., Shevchuk A.I., Korchev Y.E. Dynamic assembly of surface structures in living cells. Proc. Natl. Acad. Sci. USA. 2003;100:5819–5822. doi: 10.1073/pnas.1030502100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.