

Abstract
We demonstrate that an external constant electric field is able to modify the secondary structure of a protein and induce a transition from a β-sheet into a helix-like conformation. This dramatic change is driven by a global rearrangement of the dipole moments at the amide planes. We also predict electric-field-induced modifications of the intermediate states of the protein.
Introduction
Misfolding of a protein occurs when it becomes trapped in a local minimum of the potential energy surface (PES) where the conformation differs from the native-state structure. If the local minimum is stable enough, serious diseases may be caused, especially if the secondary structure of the misfolded conformation differs from the native one. Prions, a type of protein, provide an example of this behavior. Whereas, in normal prions, the secondary structure is dominated by α-helices, the conformation of abnormal, disease-producing prions exhibits a considerable fraction of β-sheets. Prions are responsible for mad cow and Creutzfeldt-Jakob diseases, among other transmissible spongiform encephalopathies (1–3). Moreover, evidences have been recently presented that prions could also be involved in Alzheimer's disease (4). Therefore, it is of fundamental interest to investigate whether and how abnormal prions and, in general, misfolded proteins can be healed.
In this work, we demonstrate that an external constant electric field can directly induce a dramatic conformational change in the secondary structure of proteins. The external electric field couples to the dipoles in the peptide units, which are parallel to the direction of the and bonds in the amide planes (see Fig. 1) and have a magnitude equal to 1.1 × 10−29 Cm (5), i.e., 20 times larger than the dipole moment of a water molecule. This interaction can lead to structural changes. During the past few years the alignment of hydrated proteins and small molecules in the gas phase was achieved experimentally using polarized light (6,7). The alignment of the tertiary structure of large macromolecules under static and oscillating electric fields has also been described by molecular dynamics simulations (8,9).
Figure 1.
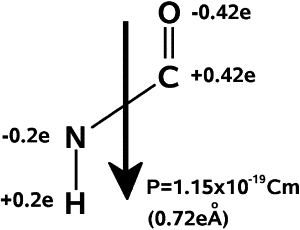
The dipoles of NH and OC in the amide plane give rise to a total dipole moment for each amino acid, which has the value 1.1 × 10−29 Cm.
Here, we show that the folding dynamics in the presence of an electric field can be analyzed in a similar way as classical spin systems under magnetic fields. For that we generalize and implement a Monte Carlo approach used for spin glasses to be able to describe structural changes of macromolecules in real space.
Note that the superposition of the individual dipoles in a protein gives rise to a total dipole moment
(N ≡ number of amino acids) when all the dipoles are aligned ferroelectrically as in the case of the α-helix (5,10). In contrast, the total dipole of a β-sheet should vanish, because in this case the individual dipoles corresponding to the ith and i + 1th amino acids in the sequence are oriented antiparallel to each other.
Now, it is known for polymers in general that a rotation of local dipoles can occur without any significant change in bond length and bonding angle (11). Therefore, a protein under an external electric field decreases its potential energy through dipole alignment. On the other hand, the dipole-dipole interactions lead to an interplay between the conformation and the dipole arrangement in the peptide. Very high field strengths will force the protein-structure to be aligned. However, for field intensities corresponding to a coupling strength comparable to the energy of hydrogen bonds, we expect a more complex and physically more interesting potential energy surface.
Theory
The model
To analyze the direct influence of external fields on the secondary structure of proteins, we study in this work the small peptide V3-loop, Protein DataBank ID 1NJ0, which consists of a β-sheet structure in its native state. This peptide conforms the V3-loop of the exterior membrane glycoprotein (GP120) of the Human Immunodeficiency Virus type 1 (HIV-1). We describe the protein by using an unbiased off-lattice model recently developed by Chen et al. (12). This coarse-grained model contains the most important ingredients needed to describe folding. In particular, local hydrophobic interactions between the residues i and i + 1 and dipole-dipole interactions are treated on the same footing.
In the following we briefly describe the model (for more details, see (12)). Each amino acid is represented by a unit that contains the atoms N, Cα, C′, O, and H. The residues are modeled as spherical beads R attached to the Cα values. The only remaining degrees of freedom are the Ramachandran angles ψ and ϕ. Thus, the force field is given by
| (1) |
where VSt represents hard-core potentials to avoid unphysical overlaps, VHB accounts for the hydrogen bonding and VDD for the dipole-dipole interaction. VMJ is a distance-dependent version of the Miyazawa-Jernigan (MJ) matrix (13), which describes the interaction between residues. VL–HP represents the local hydrophobic effect. The role of the presence of water molecules is taken into account by the terms VMJ and VL–HP. Notice that VMJ partially includes the effect of water polarization (14). The dipole-dipole interactions VDD are divided into local and nonlocal terms. The latter account for the interactions between dipoles belonging to amino acids which are not nearest neighbors, and are described by the term
| (2) |
where μ and ν refer to OC or NH and piμ is the corresponding OC or NH dipole moment in the ith amino acid of the sequence. The dipole-dipole interactions between amino acids which are nearest neighbors (local terms) are approximated as
where Pi = piCO + piNH refer to the total dipole moment of the ith amino acid. The values εnlDD and εlDD are coupling constants. Note that the nonlocal interaction has a dependence on the distance between dipoles while the local one is roughly independent of it, because the dipoles are localized in the center of the amide plane and the distance between nearest neighbors remains unchanged upon conformational transformations.
The term describing the coupling of the protein to the external electric field E reads
The value ε is a dimensionless quantity, it measures the field strength with respect to a reference field intensity E0 = 5.16108 V/m. It should be noted that for field magnitudes of ∼108 V/m, no chemical reactions occur (15–17) and therefore the information provided by our model is enough to describe the thermodynamics of proteins.
Note also that our approach does not involve an all-atom simulation and therefore our results do not have the accuracy of an ab initio approach. More-sophisticated potentials should be used to confirm our predictions. Our model yields a good description of the secondary structure of small proteins, and the field intensities considered are small, in order to produce changes on the bond character or electronic configuration of the individual atoms and molecules. Therefore, we do not expect major differences regarding the physical processes described in our work if an atomistic model is used.
Simulations
We have determined the thermodynamic properties of the V3-loop under an external electric field using the Wang-Landau algorithm (18). As the joint density of states g(E,Q) for biomolecules is difficult to obtain with the classical Wang-Landau algorithm, we implemented here the 1/t convergence criterion (recently proposed by Belardinelli and Pereyra (19)) for the avoidance of saturation errors, which is essential for the appropriate treatment of complex PES. To our knowledge, this modified algorithm has not been reported before for proteins.
Details of the implementation of the Wang-Landau algorithm can be found in the literature (18–20). We calculated the joint density of states g(E,Q) as a function of the configurational energy E (in Kcal/mol) and of the end-to-end distance of the protein Q (in Å). With the help of the Wang-Landau simulation, we explored the volume [–160 < E < –90] × [4 < Q < 50] in the reduced phase space defined by E and Q. At each Monte Carlo step, we changed the Ramachandran angles ϕ and ψ. After 1 × 1010 Monte Carlo steps, we obtained the unnormalized density of states (using the convergence criterion ffinal ∼ exp(10−8)).
Results and Discussion
From the computed g(E, Q), we obtained the free energy surface as a function of E and Q for different values of ε, running from 0 to 2 Kcal/mol, which is shown in Fig. 2. The temperature used for this calculation was T = Tf = 321 K, which is the optimal folding temperature of the V3-loop peptide. In the absence of an external field (ε = 0.0), the free energy shows the typical funnel-like form around the native state N1. In addition to the native- and the unfolded states, two characteristic local minima, I1 and I2, can be distinguished, which correspond to intermediates (20). In the presence of an external field, the whole free energy landscape is modified. For ε = 0.4, no considerable changes occur. However, for ε = 0.8, the native state N1, which is strongly by the field, is no longer the global minimum of the free energy; it becomes degenerate with the intermediates. The protein exhibits no global minimum. Therefore, we also consider N1 as a metastable state and label it as I3 (see Fig. 2 c).
Figure 2.
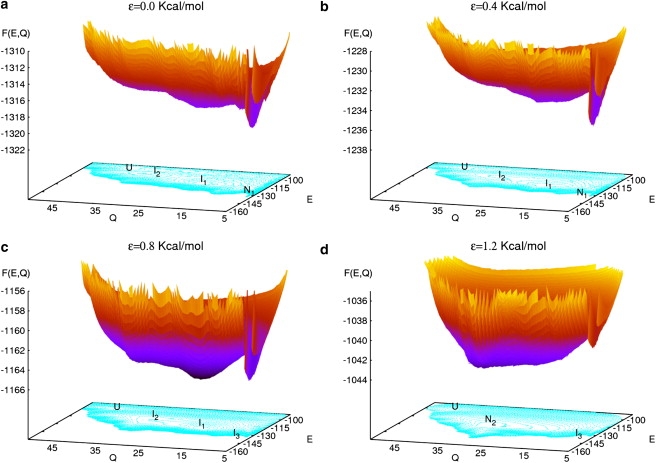
(Color online) Free energy surface of the V3-loop as a function of the configurational energy E and the end-to-end distance Q for different strengths of the external electric field εE0: ε = 0.0, 0.4, 0.8, and 1.2. Local minima labeled as I1 and I2 correspond to intermediates. N1 refers to the native state in absence of field, which becomes metastable (I3) for ε = 0.8. Note the formation of a new global minimum N2 for the field strength ε = 1.2. U corresponds to the unfolded states. The temperature in all cases is T = Tf = 321 K.
For larger field strengths, dramatic qualitative changes in the potential landscape occur, giving rise to a new phenomenon—namely, a transition from a β-sheet to an α-helixlike secondary structure. This effect can be observed, directly, in Fig. 2 d: for ε = 1.2, a conformation which does not correspond to any local minima in the absence of the field becomes the new native state (N2). The state N2 is characterized by the coordinates (E = −145, Q = 30). Note, for comparison, the native state in the absence of the field N1 is located at the point (−135,5) in the plane (E, Q).
This result suggests that, by increasing the magnitude of the external field, one should be able to control the conformation of the V3-loop and, in general, the secondary structure of proteins.
To visualize the field-induced transition from the native state N1 to the state N2, we looked for the structure that yields the largest contribution to the partition function
for each value of the temperature T and the strength ε. Such conformation constitutes the observable structure, i.e., the one having the highest probability to be present. In Fig. 3 , we show the coordinates (E,Q) of the observable conformations for ε = 0, 0.4, 0.8, and 1.2 and for temperatures running from 10 K up to 600 K. In the absence of the field and for T < Tf the native state N1 (E = −135, Q = 5) contributes most to . At very high temperatures, only the unfolded state can be observed. The intermediates I1 and I2, although present as local minima in the PES (see Fig. 2), do not provide the dominant contribution at any temperatures. New features appear when the external field is switched on. For ε = 0.4, the intermediate state I2 (E = –110, Q = 35) becomes observable for temperatures above Tf. At very high temperatures again the unfolded states dominate. A peculiar situation occurs for ε = 0.8, where both intermediates I1 and I2 can be observed and yield the dominant contribution on a wide range of temperatures. Interestingly, I1 (E = –120, Q = 20) can be observed even below Tf. The state N1 is only dominant at low temperatures.
Figure 3.
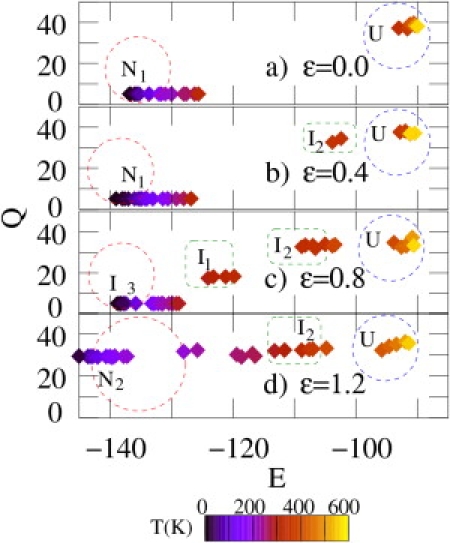
(Color online) Coordinates in the (E, Q)-plane of the conformations yielding the maximal contribution to the partition function for ε = 0.0, 0.4, 0.8, and 1.2 and at different temperatures. The field strength is given by εE0 (see text). Note that for ε = 0.0 the observable structures lie around the point (E = − 135, Q = 5) (β-sheet) while for ε = 1.2 they are located near the point (E = –150, Q = 30) (helix). Dark (blue) and light (yellow) diamonds refer to low and high temperatures, respectively (see temperature scale).
Finally, the conformation that yields the largest contribution to Z for ε = 1.2 and T < Tf is the helixlike structure with coordinates (E = –150, Q = 30), corresponding to the new native state N2. As the temperature is further increased, the intermediate I2 starts to be the most probable structure. As in the case of smaller field strengths, at high temperatures (T > 400 K) the unfolded state U yields the largest contribution to the partition function.
A more graphical description of the transition and particularly of the structure of the field-induced new native state N2 can be gained from the Ramachandran plots. In Fig. 4, we show the Ramachandran plots for ε = 0.0, 0.4, 0.8 and 1.2 and at the folding temperature T = Tf. In the absence of the external field, most of the bond-angles of the peptide lie in the upper-left part of the Ramachandran plot, inside the region which characterizes the β-sheet of the state N1 (see the crosses in Fig. 4). There are, of course, angles that lie outside this region. This dispersion is due to the bonds at the ends and in the turn of the β-sheet.
Figure 4.
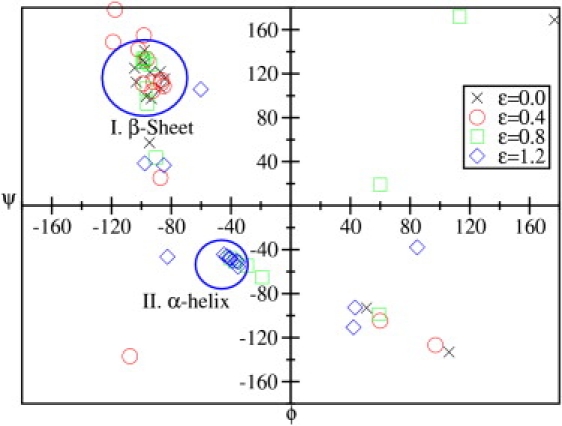
(Color online) Ramachandran plot of the V3-loop for different strengths of the external electric field represented by the dimensionless factor ε (see text) at T = Tf = 321 K. The regions corresponding to helices and β-sheets are indicated.
As the magnitude of the field is increased, the local dipole moments of the amino acids start to change their orientation. This leads to rearrangements in the structure and, in particular, in the bond angles. As a consequence, for ε = 0.4 and 0.8, the dispersion of the points in the Ramachandran plot increases. For ε = 0.8, a considerable fraction of the angles lies not only outside the β-sheet region, but they are distributed over the four panels of the plot. Now, if ε = 1.2 again a qualitative change can be observed. A considerable fraction of the angles is concentrated inside and around the region which characterizes α- and a 310-helices. This happens because for large fields all dipoles in the peptide tend to be aligned parallel to the applied field in order to minimize the interaction energy (note that ε > 0). The resulting helixlike structure has a nonzero total dipole moment of ∼17.6 × 10−29 Cm.
The energy of the native state N1 undergoes slight changes with increasing ε with respect to the case without field (E(N1)|ε =0 = –137.5 Kcal/mol). In particular, ΔE/|E| ∼ –0.009 for ε = 0.4 and ΔE/|E| ∼ –0.015 for ε = 0.8. The energy of the native state N2 for ε = 1.2 is equal to −150.0 Kcal/mol. The energy difference of ∼12.5 Kcal/mol between the native states ε = 0 and ε = 1.2 results from the balance between the field-peptide interaction energy, i.e., the energy gained from the alignment of the dipole moments, and the different number of hydrogen bonds in the configurations N1 and N2.
From our calculations, it is clear that an external electric field forces the peptide to undergo a conformational change to a structure in which the dipoles are aligned parallel to each other and parallel to the field. For ε = 1.2, this structure is a helixlike one. If the field strength is further increased, the native state displays more features related to helices, and for a sufficiently high value of ε (ε ≥ 2.5), a perfect α-helix is formed. A picture of the native state N2 for ε = 1.2 is displayed in Fig. 5 showing a helixlike structure; note that three of the 16 residues in the tail of the structure did not form an α-helix for ε = 1.2. Note that the situation is analogous to that of a classical spin system interacting with a magnetic field. The paramagnetic state would represent the unfolded protein structure, in which dipoles are oriented randomly in the absence of a field; the antiferromagnetic state would be the analog of a β-sheet, where neighboring dipoles are antiparallel to each other, and the ferromagnetic state would correspond to the α-helix. However, and in addition to the case of spin systems, the protein is free to move in space and the Hamiltonian consists of different competing many-body interaction terms.
Figure 5.
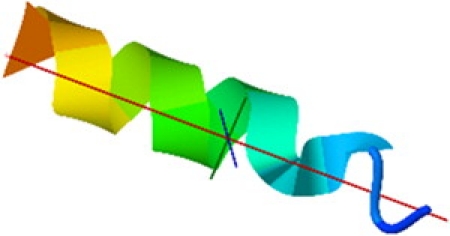
The α-helix structure of the native state for a field strength of εE0 with εE0 = 5.16 × 108.
At this stage, it is important to mention that recent molecular dynamics simulations performed to study the effect of static and oscillating electric fields on insulin (8) and an β-Amyloid peptide (9) yield a destabilization of α-helices and a change to a β-sheet or to a random-coil structure. Note, however, for both cases, the studied proteins are rather long and exhibit a tertiary structure that consists of subunits forming different secondary structures, and that the most important effect of the field was the alignment of the tertiary structure. Therefore, the destabilization of a helix is only part of this global alignment process and does not contradict our results, because the field does not interact directly with the secondary structure, as is the case in our study.
Moreover, from the results of our calculations and the above discussion, we can propose the following alternative picture for externally assisted folding: a transition from a β-sheet to an α-helix within a large protein can be induced by an electric field which directly interacts with the secondary structure.
Finally, it is important to mention that a much more efficient field-induced transition from a β-sheet to an α-helix should occur if both conformations are already present as minima in the free energy in absence of the field. Research in this direction is in progress.
Acknowledgments
P.O.-M. is grateful to the German Service of Academic Exchange for its support for the accomplishment of his PhD.
Footnotes
Dr. Ojeda-May's present address is Institute for Computational Physics, Stuttgart University, Pfaffenwaldring 27, 70569 Stuttgart, Germany.
Contributor Information
Pedro Ojeda-May, Email: pojeda@physik.uni-kassel.de.
Martin E. Garcia, Email: garcia@physik.uni-kassel.de.
References
- 1.Etienne M.A., Aucoin J.P., Hammer R.P. Stoichiometric inhibition of amyloid β-protein aggregation with peptides containing alternating α,α-disubstituted amino acids. J. Am. Chem. Soc. 2006;128:3522–3523. doi: 10.1021/ja0600678. [DOI] [PubMed] [Google Scholar]
- 2.Kelly J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 3.Lynn D.G., Meredith S.C. Review: Model peptides and the physicochemical approach to β-amyloids. J. Struct. Biol. 2000;130:153–173. doi: 10.1006/jsbi.2000.4287. [DOI] [PubMed] [Google Scholar]
- 4.Laurén J., Gimbel D.A., Strittmatter S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wada A. The α-helix as an electric macro-dipole. Adv. Biophys. 1976;9:1–63. [PubMed] [Google Scholar]
- 6.Spence J., Schmidt K., Fromme P. Diffraction and imaging from a beam of laser-aligned proteins: resolution limits. Acta Cryst. A. 2005;61:237–245. doi: 10.1107/S0108767305002710. [DOI] [PubMed] [Google Scholar]
- 7.Reckenthaeler P., Centurion M., Fill E.E. Time-resolved electron diffraction from selectively aligned molecules. Phys. Rev. Lett. 2009;102:213001. doi: 10.1103/PhysRevLett.102.213001. [DOI] [PubMed] [Google Scholar]
- 8.Legge F.S., Budi A., Yarovsky I. Protein flexibility: multiple molecular dynamics simulations of insulin chain B. Biophys. Chem. 2006;119:146–157. doi: 10.1016/j.bpc.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Toschi F., Lugli F., Zerbetto F. Effects of electric field stress on a β-amyloid peptide. J. Phys. Chem. B. 2009;113:369–376. doi: 10.1021/jp807896g. [DOI] [PubMed] [Google Scholar]
- 10.Hol W.G. The role of the α-helix dipole in protein function and structure. Prog. Biophys. Mol. Biol. 1985;45:149–195. doi: 10.1016/0079-6107(85)90001-x. [DOI] [PubMed] [Google Scholar]
- 11.Cai L., Wang X., Dowben P.A. Energetics of the dipole flip-flop motion in a ferroelectric polymer chain. J. Chem. Phys. 2007;126:124908. doi: 10.1063/1.2646731. [DOI] [PubMed] [Google Scholar]
- 12.Chen N.Y., Su Z.-Y., Mou C.-Y. Effective potentials for folding proteins. Phys. Rev. Lett. 2006;96:078103. doi: 10.1103/PhysRevLett.96.078103. [DOI] [PubMed] [Google Scholar]
- 13.Miyazawa S., Jernigan R.L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z.H., Lee H.C. Origin of the native driving force for protein folding. Phys. Rev. Lett. 2000;84:574–577. doi: 10.1103/PhysRevLett.84.574. [DOI] [PubMed] [Google Scholar]
- 15.Pompa P.P., Bramanti A., Rinaldi R. Retention of nativelike conformation by proteins embedded in high external electric fields. J. Chem. Phys. 2005;122:181102. doi: 10.1063/1.1902903. [DOI] [PubMed] [Google Scholar]
- 16.Callis P., Burgess B. Tryptophan fluorescence shifts in proteins from hybrid simulations: an electrostatic approach. J. Phys. Chem. B. 1997;101:9429–9432. [Google Scholar]
- 17.Pellegrino M., Apollonio F. Molecular simulations of biochemical processes in presence of a MW signal. IEEE Ant. Prop. Soc. Int. Symp. 2008 1–9: 2990–2993. [Google Scholar]
- 18.Wang F., Landau D.P. Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 2001;86:2050–2053. doi: 10.1103/PhysRevLett.86.2050. [DOI] [PubMed] [Google Scholar]
- 19.Belardinelli R.E., Pereyra V.D. Fast algorithm to calculate density of states. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007;75:046701. doi: 10.1103/PhysRevE.75.046701. [DOI] [PubMed] [Google Scholar]
- 20.Ojeda P.A., Garcia M.E., Chen N.-Y. Monte Carlo simulations of proteins in cages: influence of confinement on the stability of intermediate states. Biophys. J. 2009;96:1076–1082. doi: 10.1529/biophysj.107.125369. [DOI] [PMC free article] [PubMed] [Google Scholar]