

Abstract
Background
Thalamic deep brain stimulation (DBS) is proven therapy for essential tremor, Parkinson's disease, and Tourette's Syndrome. We tested the hypothesis that high-frequency electrical stimulation results in local thalamic glutamate release.
Methods
Enzyme-linked glutamate amperometric biosensors were implanted in anesthetized rat thalamus adjacent to the stimulating electrode. Electrical stimulation was delivered to investigate the effect of frequency, pulse width, voltage-controlled or current-controlled stimulation, and charge balancing.
Results
Monophasic electrical stimulation-induced glutamate release was linearly dependent on stimulation frequency, intensity and pulse width. Prolonged stimulation evoked glutamate release to a plateau that subsequently decayed back to baseline after stimulation. Glutamate release was less pronounced with voltage-controlled stimulation and not present with charge balanced current-controlled stimulation.
Conclusions
Using fixed potential amperometry in combination with a glutamate bioprobe and adjacent microstimulating electrode, the present study has shown that monophasic current-controlled stimulation of the thalamus in the anesthetized rat evoked linear increases in local extracellular glutamate concentrations that were dependent on stimulation duration, frequency, intensity, and pulse width. However, the efficacy of monophasic voltage-controlled stimulation, in terms of evoking glutamate release in the thalamus, was substantially lower compared to monophasic current-controlled stimulation and entirely absent with biphasic (charge balanced) current-controlled stimulation. It remains to be determined whether similar glutamate release occurs with human DBS electrodes and similar charge balanced stimulation. As such, the present results indicate the importance of evaluating local neurotransmitter dynamics in studying the mechanism of action of DBS.
Keywords: deep brain stimulation, essential tremor, glutamate, electrochemistry, amperometry
Introduction
Deep brain stimulation (DBS) of the thalamus is an effective treatment for epilepsy (1), essential tremor (2-6), and Tourette syndrome (7). Despite similar therapeutic effect of tissue ablation and DBS of the thalamus, recent studies have reported physiological effects of DBS differ from those expected by lesioning, such as distal and local release of neurotransmitters (8). Mimicking parameters used in clinical DBS, in vitro intracellular electrode recordings in thalamus and subthalamus (9,10) have shown the presence of both inhibitory and excitatory postsynaptic potentials as a consequence of high frequency stimulation (HFS), but the precise neurochemical species released are only now being elucidated. For example, using microdialysis techniques, Windels et al. (11) and Zhang et al. (12) reported that subthalamic nucleus high-frequency stimulation (STN HFS) was able to alter neurotransmitter concentrations in the efferent target areas of the STN. These included increases in extracellular glutamate which were detected in the globus pallidus, while both glutamate and γ-aminobutyric acid (GABA) were increased in substantia nigra pars reticulata. In addition, Bruet et al. (13) reported unilateral STN HFS evoked a bilateral increase in glutamate and GABA release in the striatum of both intact and hemiparkinsonian rats.
Release of a various neurotransmitters has also been reported in the region of the simulating electrode. For example, an enhancement in local glutamate release was found in the STN of rats in vivo during HFS using an enzyme-linked glutamate biosensor (14,15). Recently, Bekar et al. (16) reported an increase in adenosine release with thalamic stimulation, hypothesizing that adenosine is responsible for the anti-tremor effects of thalamic DBS. In contrast, Hiller et al. (17) reported a selective increase of GABA without a corresponding increase in glutamate release at the site of electrical stimulation in the caudate of the awake rat. Thus, the specific effects that DBS can have on neurochemical extracellular concentrations near the stimulating electrode in various sites of the brain remain poorly understood.
Here, we employed in vivo fixed potential amperometry coupled with a glutamate oxidase-based enzyme-linked biosensor to evaluate glutamate release at the site of HFS of the thalamic nucleus in the anesthetized rat. Using an adjacent concentric bipolar stimulating electrode we demonstrate a stimulation frequency-, intensity- and pulse width-dependent increase in glutamate release during thalamic monophasic current-controlled stimulations. Interestingly glutamate release was not observed when stimulation parameters more similar to clinical DBS were used. Under the present recording conditions only relatively high voltages were able to induce a measurable response. Moreover, glutamate release was not observed with current-controlled stimulation when charge balancing of the pulses was employed, supporting the notion that DBS utilizing monophasic electrical stimulation could potentially result in neurotoxic extracellular glutamate concentrations at the site of stimulation.
Methods
Rat Surgery
All experiments were approved by a Mayo Clinic Institutional Animal Care and Use Committee in accordance with National Institute of Health guidelines for use of animals in teaching and research. Male Sprague Dawley rats weighing 240-450g were used throughout. Before surgery, each rat was anesthetized with an intraperitoneal (i.p.) injection of ketamine and xylazine (100 and 10 mg/kg, respectively). Supplementary administrations of these anesthetics were given as necessary to maintain anesthesia. Rats were secured in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) and body temperature was maintained at 37°C with a temperature regulating heating pad (Peco Services Ltd, Brough, UK). A 1.5-2.0 cm incision of the scalp was made to expose the cranial landmarks (i.e., bregma and lambda). A trephine burr hole was drilled to allow placement of the stimulating electrode and glutamate biosensor into the left thalamus.
The stimulating electrode and the glutamate biosensor were mounted on a single holder within 0.2 mm of each other with the biosensor more medial and the tip of the stimulating electrode in the middle of the biosensor sensing cavity as shown in figure 1A. Stereotaxic coordinates for placement of the electrodes were in mm AP -2.4 from bregma, ML 2.2 from midline, and DV 6 from dura in accordance with Paxinos and Watson (18). Concentric bipolar platinum/iridium (Pt/Ir) electrodes with an outer and inner diameter of 200 μm and 50 μm, respectively (style CB-BFE75, FHC, Inc.), were used to stimulate thalamic tissue. Since glutamate is not an electro-active analyte, its electrochemical measurement is possible only with the use of enzyme-linked biosensors (Pinnacle Technology Inc., Lawrence, KS). These biosensors consist of a Pt wire (0.18 mm diam.) coated with a selectively permeable membrane to block interfering electroactive compounds and a coating containing glutamate oxidase. Glutamate is converted by the enzyme to α-ketoglutaric acid and hydrogen peroxide at the active surface (∼0.5 mm length). Hydrogen peroxide diffuses through the selectively permeable coating and is reduced at the Pt wire surface producing an amperometric signal whose magnitude is directly proportional to the concentration of glutamate.
Figure 1.
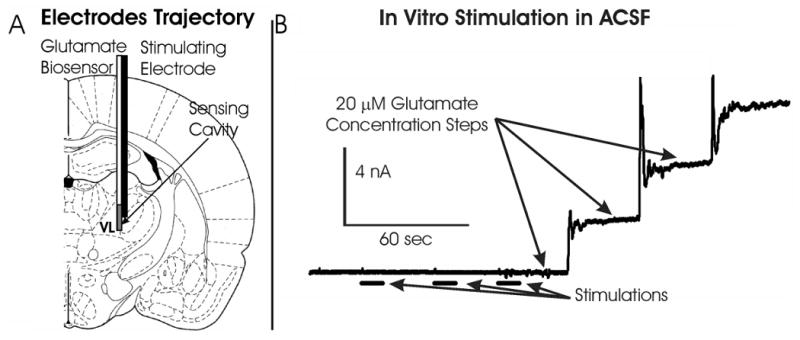
(A):Illustration showing the configuration of the electrical bipolar stimulating electrode and glutamate biosensor implanted in the rat thalamus to evoke and record glutamate release, respectively. (B): Oxidation current measured by the glutamate biosensor in artificial cerebrospinal fluid during 10 seconds monophasic stimulations (1 mA, 100 Hz, 100 μsec pulse width) and after 20 μM step increases in glutamate concentration.
To eliminate the possibility that concentrations of hydrogen peroxide produced by electrical stimulation at the stimulating electrode surface-tissue interface may have contributed significantly to the electrochemical signal recorded at the glutamate biosensor in the thalamus in vitro experiments in artificial CSF were performed with the stimulating and recording electrode in the same configuration as the in vivo experiments. In all cases of application of similar stimulation parameters (monophasic or biphasic) used in vivo, electrical stimulation adjacent to the recording electrode failed to alter the electrochemical baseline response as shown in figure 1B.
Electrical Stimulation
HFS was applied for 10 seconds using an optically isolated pulse generator (Iso-Flex/Master-8, AMPI, Jerusalem, Israel) with frequencies between 10 and 300 Hz, intensities between 0.1-1.3 mA, and a monophasic pulse width between 50 and 500 μsec to evaluate the dependence of the amperometric signal on stimulation parameters. To investigate the dynamics of longer stimulations, we performed stimulations up to 10 minutes duration. Clinically employed human DBS systems (Medtronic, inc.) are currently equipped to deliver voltage-controlled stimulations only, while investigational devices designed to deliver constant-current stimulations are presently undergoing clinical trials for safety and therapeutic efficacy. Therefore, we performed a series of 10 sec voltage-controlled stimulations in the thalamus with intensities between 1 and 90 V at a fixed frequency and pulse width (100 Hz, 100 μsec). Given that clinically applied DBS stimulation is always performed by balancing the charge injected in the tissue, we evaluated the effect of balanced and unbalanced biphasic pulses on glutamate release. To evaluate the potential source of the stimulation-evoked glutamate release we also performed a set of “paired-pulse” stimulations consisting of a 100 Hz train of paired pulses (100 Hz, 0.1 mA, and 100 μsec) with an interpulse delay between 100 μsec and 5 msec.
Amperometry and Data Analysis
Fixed potential amperometric recordings in a two-electrode configuration were acquired using a commercial potentiostat connected to an analog to digital converter (e-DAQ Picostat and E-corder, Colorado Springs, CO) and stored on a PC for off-line analysis. Once the glutamate biosensor was in position, a fixed potential of +0.6 Volts was applied between the working and the integrated Ag/AgCl reference electrode. After the oxidation current reached relatively steady-state baseline levels, electrical stimulations were performed allowing on-line current monitoring during each stimulation period. Changes in oxidation current elicited by stimulations had a relatively slow time course and was low-pass filtered at 1 Hz. Pre-stimulation baseline to peak change in oxidation current was determined for every stimulation. Oxidation currents were converted to glutamate concentrations with a post-operative calibration performed with step-wise increases of glutamate (Sigma-Aldrich, St. Louis, MI) concentration in a stirred beaker.
Measurement of voltage and currents delivered during stimulation
The voltage across electrode contacts was measured by connecting an analog to digital converter (10 MΩ input resistance) to the stimulator poles. In separate experiments, the current passing through the electrode was evaluated by measuring the voltage across a 100 Ω resistor inserted in series with the electrode. To avoid interactions with stimulation-evoked measurements, voltage and current measurements were not performed during the measurement of stimulation-evoked glutamate release.
Histology
At the end of each experiment, rats were sacrificed by decapitation. The brains were removed and placed in 10% formalin solution. Once fixated, the brains were rinsed with saline and sliced with a vibratome (Leica microsystems, Bannockburn, IL) at a thickness of 200 μm. Localization of the electrodes was visually confirmed and compared to the rat brain atlas of Paxinos and Watson (18).
Results
Glutamate release increases linearly with increasing stimulation duration
As shown in figure 2A and B, the magnitude of thalamic glutamate release evoked by 100 Hz stimulation (0.2 mA, 100 μsec pulse width) increased linearly with respect to increasing stimulation duration (1-15 sec). A typical response to stimulation consisted of a delayed increase in extracellular glutamate concentration, reaching a peak many seconds after stimulation had been discontinued and returning back to baseline within minutes. When a longer duration of stimulation (10 min of 100 Hz at 0.2 mA, 100 μsec pulse width) was applied at varying intensities (30, 50, and 100 μA) the extracellular concentration of glutamate in the thalamus increased to a relatively steady-state plateau with a slow return to baseline following termination of stimulation (figure 2C). As shown in figure 2D, thalamic extracellular glutamate concentration recorded during the plateau (∼8 min post-stimulation) increased in a linear fashion with respect to increasing stimulation current intensity.
Figure 2.

Stimulation Duration Dependency. (A): Evoked glutamate release in the thalamus of one rat at different monophasic stimulation durations (current intensity and pulse width held constant: 0.2 mA, 100 Hz, 100 μs). (B): Peak magnitudes in thalamic glutamate extracellular concentrations obtained at varying monophasic stimulation durations (n=3 rats). Different symbols represent measurements obtained from separate animals. Black lines represent best fit for a given animal. (C): Thalamic glutamate release obtained from one rat during 10 min of monophasic stimulation at a fixed frequency and pulse width (100 Hz, 100 μsec). The black line above the x-axis represents the 10 min period of stimulation. (D): Peak magnitudes of thalamic glutamate extracellular concentrations obtained with 10 min monophasic stimulation (100 Hz, 100 μsec) and varying stimulation intensity in three rats. Different symbols represent measurements obtained from separate animals. Black lines represent best fit for a given animal.
Glutamate release increases linearly with increasing frequency, intensity, and pulse width
To explore further the relationship between thalamic glutamate release and varying stimulation parameters, 10 seconds of current-controlled stimulation was applied while systematically varying one parameter (frequency, intensity, and pulse width) at a time (figure 3). Examples of glutamate stimulation-evoked responses in the thalamus for varying frequency (10-300 Hz), intensity (0.1-1.3 mA), and pulse width (50-500 μsec) are shown in figures 3 A, C, and E, respectively. Peak magnitudes of extracellular glutamate concentrations increased linearly with systematic increases in each of the specific stimulation parameters tested. Correlation coefficients for frequency, intensity, and pulse width were R2=0.907 (n=5 rats), 0.97 (n=7 rats), and 0.926 (n=3 rats), (figure 3B, D, and F, respectively).
Figure 3.

Stimulation Parameter Dependence. (A): Evoked glutamate release in the thalamus of one rat at different monophasic stimulation frequencies (10-300 Hz) with current intensity and pulse width held constant (0.5 mA, 100 μsec). The inset shows the initial phase of the response around the 10 sec (black bar) stimulation period. (B): Peak magnitudes in thalamic glutamate extracellular concentrations elicited at each test stimulation frequency (n=5 rats). The dashed line is the average and solid line the linear regression (r2 =0.907). (C): Evoked glutamate release in the thalamus of one rat at different monophasic stimulation intensities (0.1-1.3 mA) with frequency and pulse width held constant (100 Hz and 100 μsec). The inset shows the initial phase of the response around the 10 sec (black bar) stimulation period. (D): Peak magnitudes in thalamic glutamate extracellular concentrations elicited at each test stimulation current intensity (n=7 rats). The dashed line is the average and solid line the linear regression (r2 =0.97). (E) Evoked glutamate release in the thalamus of one rat at different monophasic stimulation pulse widths (50-500 μsec) with frequency and current intensity held constant (100 Hz, 0.2 mA). The inset shows the initial phase of the response around the 10 sec (black bar) stimulation period. (F) Peak magnitudes in thalamic glutamate extracellular concentrations elicited at each test stimulation pulse width (n=3 rats).
Comparison of glutamate release evoked by voltage-controlled and current-controlled stimulation
As shown in figure 4A, relatively high voltage-controlled intensities (e.g., 90 V) were required to obtain a nearly equivalent peak increase in extracellular glutamate observed in the same animal with 0.5 mA current-controlled stimulation (100 Hz and 0.5 msec pulse duration held constant). Current-controlled stimulation for 10 sec evoked robust increases in thalamic glutamate release with as little as 0.2 mA of current (22±8 μM, n=7 rats). As shown in table 1, voltage-controlled stimulations of 70 V were required to evoke comparable peak amplitude increases in extracellular glutamate concentration (19.9±4.5 μM, n=5 rats).
Figure 4.

Voltage-controlled Stimulation. (A): Glutamate release in the thalamus in one rat evoked by monophasic voltage-controlled stimulation (60-90 V) compared to 0.5 mA current-controlled stimulation with stimulation frequency and pulse width held constant (100 Hz, 0.5 msec). (B): Comparison of current-controlled and voltage stimulations. Each line represents the linear regression of peak glutamate extracellular concentrations attained with 10 seconds of current-controlled stimulations for four rats. Symbols correspond to peak glutamate extracellular concentrations obtained in these animals. (C): Comparison of stimulation voltages measured across electrode contact during 0.1 mA current-controlled stimulation (left) and a comparable 4V voltage-controlled stimulation (right). (D): The first pulse of both stimulations showing differences in the interpulse decay for current-controlled (solid line) and voltage-controlled (dashed line) stimulations. (E): Voltages measured across electrode contacts during current-controlled stimulations with increasing current intensities (0.05-0.3 mA). (F): Voltages measured across electrode contacts during voltage-controlled stimulations of increasing voltage intensities (1-9 V). (G): Overlap of the last pulse of 1 sec long stimulation train delivered with voltage-controlled stimulation showing the interpulse decay is minimal with respect to a relatively higher stimulation voltage of 90 V. (H): Peak current measured at different applied voltage-controlled stimulations (1-90 V).
As shown in figure 4B, regression analysis of peak glutamate release (in terms of peak oxidation current) as a function of applied current-controlled intensity (0.1-0.7 mA) showed a highly linear relationship in four test animals. In these same animals, peak oxidation current values corresponding to peak increases in glutamate release (in terms of peak oxidation current) evoked by voltage-controlled stimulation (50-90 V) were determined and superimposed on these current intensity-oxidation current plots. As can be seen in figure 4B, the peak increases in glutamate release evoked by these relatively high voltage stimulations across four test animals were always within the range of peak increases in glutamate release evoked by current-controlled stimulations recorded in the same animals.
As can be seen in figure 4B, oxidation current values obtained with these relatively high voltage stimulations exhibited regression slopes that did not differ significantly from regression slopes calculated for current-controlled evoked responses.
To investigate why relatively high voltage-controlled intensities, compared to current-controlled intensities, were required to evoke comparable increases in thalamic glutamate release, the voltages across the stimulating electrode contacts or the currents passing through the stimulating electrode were determined during current-controlled and voltage-controlled stimulations, respectively. As shown in figure 4C (left panel) the voltage measured across the electrode contacts during current-controlled stimulation at 0.1 mA achieved a peak value of ∼4 V. However, these voltages can be seen to be riding on an elevated baseline over the course of current-controlled stimulation, compared to voltages measured during voltage-controlled stimulation at 4 V (right panel). As shown in figure 4D, expanding the time scale to examine the voltage decay during the interpulse period of current-controlled stimulation revealed that the elevation in baseline voltage was due to a relatively slow rate of decay. As can be seen in figure 4E, this “baseline shift” in voltage measured across the electrode contacts was proportional to the charge injected during current-controlled stimulation, while it was not significantly present during constant (1-9 V) voltage stimulations (figure 4F) until a relatively high voltage (90 V, figure 4G) was applied. This was despite the fact that during voltage-controlled stimulations measurements of current passing through the electrode indicated that relatively large currents (1-9 mA) were being delivered (figure 4H) with an impedance of ∼10-11 kOhms.
Biphasic charge balanced stimulations
Two sets of current-controlled stimulations balancing the charge delivered during cathodic pulses with a second anodic pulse were performed to evaluate the correlation between the voltage baseline shift across the stimulating electrode contacts and the release of glutamate in the thalamus. The first set of current-controlled stimulations (referred to as biphasic) consisted of a 0.2 mA cathodic pulse (100 μsec width) that was followed immediately by an anodic pulse of similar intensity applied at 100 Hz for 10 sec. Thereafter, the anodic pulse was systematically reduced in amplitude by 75, 50, 25 and 0% of the original 0.2 mA (figure 5A). The second set of stimulations (referred to as charge balanced stimulation) consisted of a 0.2 mA cathodic pulse (100 μsec width) that was followed immediately by an anodic pulse with a tenth of the cathodic current and 10 times longer (1000 μs) pulse applied at 100 Hz for 10 sec. Thereafter, the anodic pulse was systematically reduced in duration by 75, 50, 25 and 0% of the original 1000 μs (figure 5E).
Figure 5.

Biphasic to Monophasic Stimulation. (A): Current pulse waveform delivered during current-controlled biphasic stimulation from biphasic (left) to monophasic (right). (B): Voltage measured across electrode contacts showing progressive deviation from baseline with increasing charge unbalance. (C): Thalamic glutamate release measured during 10 sec (0.2 mA, 100 Hz, 100 μs pulse width), progressively unbalanced, stimulations shown in (A). (D): Peak glutamate responses elicited by progressively unbalanced biphasic stimulations relative to the peak glutamate response (set at 1) obtained from monophasic stimulation (n=3 rats). (E): Current pulse waveform delivered during current-controlled charge balanced stimulation from balanced (left) to monophasic (right). (F): Voltage measured across electrode contacts showing progressive deviation from baseline with increasing charge unbalance. (G): Thalamic glutamate release measured during 10 sec (0.2 mA, 100 Hz, 100 μs cathodic and 0-1ms anodic pulse width), progressively charge unbalanced stimulations as shown in (E). (H): Peak glutamate response elicited by progressively unbalanced biphasic stimulations relative to the peak glutamate response (set at 1) obtained from monophasic stimulation (n=3 rats). Black bar below each evoked response corresponds to the period of high frequency stimulation (HFS).
As shown in figure 5B and F, balanced stimulation did not produced notable deviations of the voltage measured across the stimulating electrode contacts and induced no or barely detectible glutamate release in the thalamus (figure 5C and G). As stimulations become progressively more unbalanced (i.e., lower amplitude or duration anodic pulses), the shift of voltage above baseline became more pronounced with corresponding increases in stimulation-evoked glutamate release. As shown in figure 5D and H for three separate animals, glutamate release (in terms of oxidation current) increased in a linear and progressive fashion as charge balance approached zero (pure monophasic cathodic pulse condition).
Paired-pulse stimulations
Two interleaved current-controlled cathodic pulses (100 Hz, 0.1 mA, 100μs pulse width) separated by a time delay (ΔT) varying between 100 μs and 5 msec (when the two stimulations are equivalent to a 200 Hz stimulation of equal intensity and pulse width) were applied to evaluate the possibility of non-neuronal (e.g., astrocyte) origin of stimulation-evoked thalamic glutamate release. A schematic representation of the applied cathodic current pulses is shown in figure 6A. To assess the origin of the stimulation-evoked glutamate release in the thalamus (i.e., neuronal vs. astrocytic), paired-pulse stimulations were compared to single cathodic pulse 100 Hz (0.1 mA, 100μs pulse width) stimulation.
Figure 6.
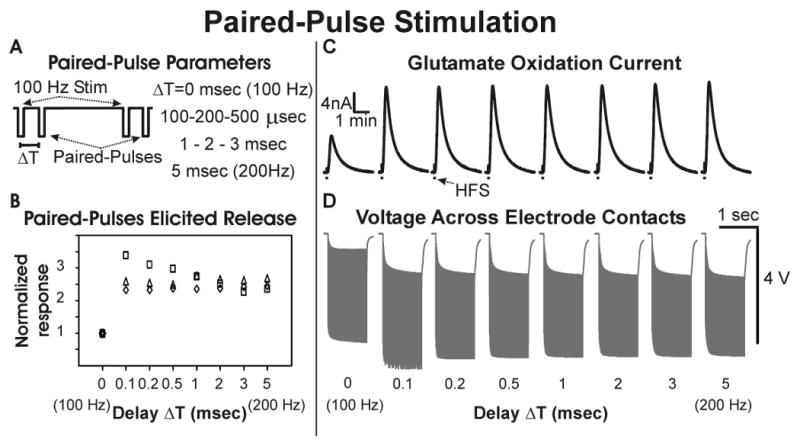
Paired-Pulse Stimulation. (A): Schematic representation of paired-pulse stimulation with a 100 Hz stimulation interleaved with paired pulses at an interpulse delay of ΔT (0.1-5 msec). (B): Peak thalamic glutamate response elicited with different interpulse delays relative to the the peak glutamate response (set at 1) obtained with 100 Hz stimulations at ΔT=0 (n=3 rats). (C): Thalamic glutamate release elicited at different interpulse delays in one rat (0.1 mA, 100 μs pulse width). (D): Voltage measured across electrode contacts during paired-pulse stimulations.
Under these conditions, the assumption was that the second “paired” pulse should not elicit a significant enhancement in stimulation-evoked glutamate release until ΔT attained a delay that was equal to or greater than the typical absolute refractory period (∼0.5-1 msec) for recovery of axonal sodium channel activation. As shown in figure 6B and C, the magnitude of thalamic glutamate release evoked by paired-pulse current-controlled stimulation was independent of the ΔT delay and comparable to the responses obtained with 200 Hz stimulation (i.e., a ΔT delay of 5 msec). Interestingly, the baseline shift of voltage measured across the stimulating electrode contacts was similar across all ΔT delays (0.1 to 3 msec), including the voltages measured at 200 Hz stimulation (figure 6D).
Discussion
Using fixed potential amperometry in combination with a glutamate bioprobe, the present study has shown that monophasic current-controlled stimulation of the thalamus in the anesthetized rat evoked linear increases in local extracellular glutamate concentrations that were dependent on stimulation duration, frequency, intensity, and pulse width. A comparison of current-controlled with voltage-controlled stimulation revealed that the efficacy of voltage-controlled stimulation, in terms of evoking glutamate release in the thalamus, was substantially lower than current-controlled stimulation. Indeed, relatively high monophasic voltages (50-90 V), well above the range (1-10 V) that would be considered therapeutically effective in thalamic voltage-controlled human DBS (e.g., 1-6), were required to evoke comparable peak increases in extracellular glutamate concentrations utilizing relatively low current intensities (0.1-0.5 mA), similar to those published in animal studies (e.g., 11,14,17), including human thalamic DBS using monophasic current-controlled stimulation (0.1 mA, 333 Hz, 0.2 msec pulse width) (19).
The discrepancy in efficacy between current-controlled and voltage stimulation did not appear to be related to the impedance in the stimulator-electrode circuit since relatively large currents were delivered with voltage-controlled stimulations. For example, with 90 V voltage-controlled stimulation a peak current value of 8.9 mA was measured passing across a 100 Ω resistor positioned in series with the stimulating electrode. This stimulation voltage evoked an average increase (∼40 μM) in glutamate concentration, equivalent to the increase in glutamate release seen with 0.3 mA current-controlled stimulation (see table 1). The difference between current-controlled and voltage stimulation may be due to the disparate interpulse decay rates (see figure 4D) resulting from the electronic configurations of the stimulator typically utilized for current-controlled versus voltage stimulation (20). For example, in the case of monophasic voltage-controlled stimulation, an internal 10 KΩ resistor across the plus/minus poles of the stimulus isolator allows the charge accumulated at the electrode surface at the end of each pulse to discharge rapidly. In contrast, the current applied to the electrode during each monophasic current-controlled pulse has no comparable fast discharge path for current, as the external stimulator circuit is opened between pulses. As described by Merrill et al. (20), at the end of a current-controlled pulse, the charge that has accumulated can only dissipate via redistribution of charged chemical species at the electrode surface that do not transfer electrons between the electrode and electrolyte (non-faradaic current) and electron transfer between the electrode and electrolyte as a result of oxidation or reduction of electroactive compounds (faradaic current). With the circuit open between current-controlled pulses, at the end of the pulse the charge on the double layer capacitance continues to discharge through faradaic reactions resulting in an exponential discharge of the electrode. Thus, the relatively slow discharge kinetics due to these processes, compared to current discharge to ground through a resistor, likely account for the comparatively slower discharge rates with current-controlled versus voltage stimulation.
Table 1.
Comparison of mean thalamic glutamate extracellular concentrations obtained at different intensities of current-controlled (n=7) versus voltage-controlled stimulation (n=5) with fixed frequency and pulse width (100 Hz and 100 μsec, respectively).
| Current (mA) | 0.1 | 0.2 | 0.3 | 0.5 | 0.9 | 1.3 |
| Concentration (μM) | 7±3 | 22±8 | 38±13 | 71±25 | 123±42 | 146±50 |
| Voltage (V) | 10 | 20 | 30 | 50 | 70 | 90 |
| Concentration (μM) | 0.6±0.3 | 0.8±0.9 | 2.9±2.2 | 7.1±2 | 19.9±4.5 | 40.7±19.8 |
Support for this notion was provided by the observation that application of an anodic pulse of equivalent magnitude or duration to the cathodic pulse (biphasic or charge balanced stimulation; see figure 5A and E, leftmost trace) resulted in negligible or no increases in glutamate release (see figure 5C and G, leftmost trace). Thus, a systematic decrease in charge balance at the electrode surface resulted in progressive and linearly-related increase in stimulation-evoked thalamic glutamate release, as well as an increase in baseline voltage measured across the electrode contacts (see figure 5D and H). It is worth noting that providing a current discharge path during current-controlled stimulation resulted in no delayed voltage decay and corresponding baseline elevation. This was accomplished by connecting a second stimulus isolator in parallel with the main stimulus isolator delivering current-controlled stimulations to the bipolar stimulating electrode implanted in the thalamus. The second stimulus isolator was turned “off” and set on voltage-controlled stimulation thus presenting an internal 10 kOhm resistance path between the stimulator poles of the second stimulus isolator. In this configuration roughly half of the delivered current is shunted through this internal 10 kOhm resistor in parallel with the 10-11 kOhm electrode-tissue path, thus providing a means for rapid discharge of any charge accumulated at the electrode surface during stimulation. Under these conditions, no detectable glutamate release was observed (data not shown) suggesting that if the charge accumulated at the electrode surface at the end of each pulse is allowed to discharge rapidly, glutamate release does not occur.
Source of stimulation-evoked glutamate release in the thalamus
A potential source of glutamate recorded in the thalamus in response to local electrical stimulation could arise from depolarization of cortical glutamatergic afferents to the thalamus (21). All neurons exhibit an axonal refractory period for voltage-gated sodium channel-dependent impulse activation. Thus, paired-pulse stimulation where the interpulse interval (ΔT) was less than the absolute refractory period (∼0.5-1 msec) for sodium channel activation would be expected to evoke a significantly smaller increase in glutamate release compared to paired-pulses applied at a higher interpulse interval. However, the glutamate responses recorded with paired-pulse stimulations suggest a non-neuronal origin of glutamate as stimulation-evoked glutamate release did not appear to be dependent on the refractory period. Paired-pulse stimulation with a ΔT within 1 msec produced the same increase in glutamate release compared to release evoked with a delay greater than 1 msec (see figure 6B and C). Together, these data strongly suggest that, under these conditions, the principle source of thalamic glutamate release evoked by current-controlled local stimulation was likely from a non-neuronal source.
Astrocytes contain a readily releasable pool of glutamate and thus may provide an alternate source of stimulation-evoked glutamate release in the thalamus (22-26). Glutamate release from astrocytes has been shown to operate through a Ca2+ dependent mechanism with internal Ca2+ stores being the principal source of the Ca2+ required to elicit glutamate release (27, 28). As electrical stimulation interferes with voltage-sensitive ion channels, it is conceivable that the semi-constant field measured as a shift of baseline voltage present between the contacts of the electrode during monophasic current-controlled stimulation could affect Ca2+ voltage-activated channels leading to Ca2+ entry into astrocytes. This, in turn, could activate Ca2+ sensitive ryanodin receptors on the endoplasmic reticulum triggering a larger Ca2+ release from the internal stores, compared to that elicited by voltage-controlled stimulation. Such a non-physiological Ca2+ dynamic could easily trigger many nonspecific events in astrocytes including exocytosis of glutamate stored in astrocytic vesicles.
Entry of sufficient extracellular Ca2+ to trigger a “second messenger-like” cascade in astrocytes, as well as the cascade itself, may explain the delay between stimulation onset and the observed increase in oxidation current. Such a mechanism could also account for the further observation that with 10 sec duration stimulations the majority of glutamate release occurred well after stimulation was discontinued. Similarly, since astrocytes are non-excitable cells, they are more likely to produce a linear response to electrical stimulation, where the overall power provided by the stimulation is more important than the individual parameters necessary to deliver it. Astrocytes release glutamate in the extracellular space to influence activity in the surrounding region and are therefore more likely to produce large extracellular glutamate concentrations similar to those observed in the present study. Thus, it is possible that the source of glutamate release evoked by local electrical stimulation in the thalamus may, at least in part, be from astrocytes. Indeed, we have recently demonstrated that electrical stimulation of purified primary astrocytic cultures was able to evoke intracellular calcium transients and glutamate release, and bath application of a calcium chelator inhibited glutamate release (29). Taken together, these results suggest that vesicular astrocytic glutamate release may be an important mechanism by which DBS is able to achieve clinical benefits, and warrants further investigation.
Methodological considerations
There are a number of methodological issues that are worth considering. For example, there was a high degree of inter-animal variability to stimulation-evoked release of glutamate. This may have been due to differences in the distance between stimulating electrode tip and the biosensor sensing cavity. An additional consideration relates to the post-operative glutamate calibration procedure. It is safe to assume that during implantation the sensitivity of the biosensor diminishes to some extent due to interactions of the sensing cavity with endogenous proteins. This would lead to an overestimation of glutamate concentrations. As well, glutamate calibrations were performed at room temperature while measurements were at body temperature. Enzymes are more active at body temperature and, as a consequence, this too would lead to overestimations. It is also important to note that although the bioprobe utilized in the present study incorporates the anion exchange resin Nafion to provide a high degree of selectivity, it is not completely insensitive to other electroactive interferents, such as ascorbate and catecholamines. Thus, it is not inconceivable that at least a part of the measured response could be due to interfering compounds. A strategy to avoid contamination of the amperometric signal from these substances involves simultaneous recordings from a sentinel (self-referencing) biosensor that is made from the same materials as the glutamate biosensor, but lacks the enzyme glutamate oxidase and/or Nafion. Lastly, clinical DBS is typically performed using charge balanced voltage-controlled pulses in the range of 1-10 V. Although glutamate release evoked by 1-10 V voltage-controlled stimulation could not be readily detected with the present sensing techniques, it remains possible that even with clinical stimulations small amounts of glutamate may be released within the region of stimulation, particularly considering the larger size Pt/Ir ring stimulating electrodes used for DBS. Further studies are needed to address these issues.
Conclusion
The major findings of this study are: 1) In the thalamus of rats, local glutamate release occurs during the application of high frequency electrical stimulation in a parameter and waveform dependent fashion, 2) Stimulation-evoked glutamate release was observed only under conditions wherein the stimulation produced a shift of voltage baseline across the stimulating electrode contacts. More studies are needed to evaluate this phenomenon. Given that clinical human DBS currently utilizes only charge balanced voltage-controlled simulation, the present results suggest that such potentially neurotoxic local glutamate release is likely absent in the clinical setting.
Acknowledgments
This work was supported by: NIH (K08 NS 52232 award to KHL), Mayo Foundation (2008-2010 Research Early Career Development Award for Clinician Scientists award to KHL.
Abbreviations
- DBS
deep brain stimulation
- GABA
γ-aminobutyric acid
- HFS
high frequency stimulation
References
- 1.Hodaie M, Wennberg RA, Dostrovsky JO, Lozano AM. Chronic anterior thalamus stimulation for intractable epilepsy. Epilepsia. 2002;43:603–608. doi: 10.1046/j.1528-1157.2002.26001.x. [DOI] [PubMed] [Google Scholar]
- 2.Benabid AL, Pollak P, Gao D, Hoffmann D, Limousin P, Gay E. Chronic electrical stimulation of the ventralis intermedius nucleus of the thalamus as a treatment of movement disorders. J Neurosurg. 1996;84:203–214. doi: 10.3171/jns.1996.84.2.0203. [DOI] [PubMed] [Google Scholar]
- 3.Koller WC, Pahwa PR, Lyons KE, Wilkinson SB. Deep brain stimulation of the Vim nucleus of the thalamus for the treatment of tremor. Neurology. 2000;55:S29–33. [PubMed] [Google Scholar]
- 4.Lozano AM. Vim thalamic stimulation for tremor. Arch Med Res. 2000;31:266–269. doi: 10.1016/s0188-4409(00)00081-3. [DOI] [PubMed] [Google Scholar]
- 5.Hariz GM, Lindberg M, Bergenheim AT. Impact of thalamic deep brain stimulation on disability and health-related quality of life in patients with essential tremor. J Neurol Neurosurg Psychiatry. 2002;72:47–52. doi: 10.1136/jnnp.72.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berk C, Carr J, Sinden M, Martzke J, Honey CR. Thalamic deep brain stimulation for the treatment of tremor due to multiple sclerosis: a prospective study of tremor and quality of life. J Neurosurg. 2002;97:815–820. doi: 10.3171/jns.2002.97.4.0815. [DOI] [PubMed] [Google Scholar]
- 7.Maciunas RJ, Maddux BN, Riley DE, Whitney CM, Schoenberg MR, Ogrocki PJ, Albert JM, Gould DJ. Prospective randomized double-blind trial of bilateral thalamic deep brain stimulation in adults with Tourette syndrome. J Neurosurg. 2007;107:1004–1014. doi: 10.3171/JNS-07/11/1004. [DOI] [PubMed] [Google Scholar]
- 8.Lee KH, Blaha CD, Garris PA, Mohseni P, Horne AE, Bennet KE, Agnesi F, Bledsoe JM, Lester DB, Kimble C, Min HK, Kim YB, Cho ZH. Evolution of Deep Brain Stimulation: Human Electrometer and Smart Devices Supporting the Next Generation of Therapy. Neuromodulation. 2009;12:85–103. doi: 10.1111/j.1525-1403.2009.00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson T, Hu B, Pittman Q, Kiss ZH. Mechanisms of deep brain stimulation: an intracellular study in rat thalamus. J Physiol. 2004;559:301–313. doi: 10.1113/jphysiol.2004.064998. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KH, Chang SY, Roberts DW, Kim U. Neurotransmitter release from high-frequency stimulation of the subthalamic nucleus. J Neurosurg. 2004;101:511–517. doi: 10.3171/jns.2004.101.3.0511. [DOI] [PubMed] [Google Scholar]
- 11.Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M. Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res. 2003;72:259–267. doi: 10.1002/jnr.10577. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B, Chu J, Zhang J, Ma Y. Change of extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in epileptic rats. Stereotact Funct Neurosurg. 2008;86:208–215. doi: 10.1159/000131657. [DOI] [PubMed] [Google Scholar]
- 13.Bruet N, Windels F, Bertrand A, Feuerstein C, Poupard A, Savasta M. High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J Neuropathol Exp Neurol. 2001;60:15–24. doi: 10.1093/jnen/60.1.15. [DOI] [PubMed] [Google Scholar]
- 14.Lee KH, Kristic K, van Hoff R, Hitti FL, Blaha C, Harris B, et al. High-frequency stimulation of the subthalamic nucleus increases glutamate in the subthalamic nucleus of rats as demonstrated by in vivo enzyme-linked glutamate sensor. Brain Res. 2007;1162:121–129. doi: 10.1016/j.brainres.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Behrend CE, Cassim SM, Pallone MJ, Daubenspeck JA, Hartov A, Roberts DW, et al. Toward feedback controlled deep brain stimulation: dynamics of glutamate release in the subthalamic nucleus in rats. J Neurosci Methods. 2009;180:278–289. doi: 10.1016/j.jneumeth.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Bekar L, Libionka W, Tian GF, Xu Q, Torres A, Wang X, et al. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008;14:75–80. doi: 10.1038/nm1693. [DOI] [PubMed] [Google Scholar]
- 17.Hiller A, Loeffler S, Haupt C, Litza M, Hofmann U, Moser A. Electrical high frequency stimulation of the caudate nucleus induces local GABA outflow in freely moving rats. J Neurosci Methods. 2007;159:286–290. doi: 10.1016/j.jneumeth.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 18.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1998. [Google Scholar]
- 19.Anderson TR, Hu B, Iremonger K, Kiss ZH. Selective attenuation of afferent synaptic transmission as a mechanism of thalamic deep brain stimulation-induced tremor arrest. J Neurosci. 2006;26:841–850. doi: 10.1523/JNEUROSCI.3523-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merrill DR, Bikson M, Jefferys JG. Electrical stimulation of excitable tissue: design of efficacious and safe protocols. J Neurosci Methods. 2005;141:171–198. doi: 10.1016/j.jneumeth.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Ray JP, Russchen FT, Fuller TA, Price JL. Sources of presumptive glutamatergic/aspartatergic afferents to the mediodorsal nucleus of the thalamus in the rat. J Comp Neurol. 1992;320:435–456. doi: 10.1002/cne.903200403. [DOI] [PubMed] [Google Scholar]
- 22.Bezzi P, Vesce S, Panzarasa P, Volterra A. Astrocytes as active participants of glutamatergic function and regulators of its homeostasis. Adv Exp Med Biol. 1999;468:69–80. doi: 10.1007/978-1-4615-4685-6_6. [DOI] [PubMed] [Google Scholar]
- 23.Bezzi P, Volterra A. A neuron–glia signaling network in the active brain. Curr Opin Neurobiol. 2001;11:387–394. doi: 10.1016/s0959-4388(00)00223-3. [DOI] [PubMed] [Google Scholar]
- 24.Bezzi P, Gunderson V, Galbete JL, Seifert G, Steinhäuser C, Pilati E, et al. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- 25.Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. J Neurosci. 2000;20:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anlauf E, Derouiche A. Astrocytic exocytosis vesicles and glutamate: a high-resolution immunofluorescence study. Glia. 2004;49:96–106. doi: 10.1002/glia.20094. [DOI] [PubMed] [Google Scholar]
- 27.Parpura V, Scemes E, Spray DC. Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytic release. Neurochem Int. 2004;45:259–264. doi: 10.1016/j.neuint.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Hua X, Malarkey EB, Sunjara V, Rosenwald SE, Li W, Parpura V. Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J Neurosci Res. 2004;76:86–97. doi: 10.1002/jnr.20061. [DOI] [PubMed] [Google Scholar]
- 29.Tawfik V, Chang SY, Hitti FL, Roberts DW, Leiter JC, Jovanovic S, Lee KL. Deep Brain Stimulation results in local glutamate and adenosine release: Investigation into the role of astrocytes. Neurosurgery. doi: 10.1227/01.NEU.0000371988.73620.4C. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]