

Abstract
A facile synthetic approach for the preparation of well-defined (co)polymers bearing pendent alkene functionalities was established by selective reversible addition-fragmentation chain transfer (RAFT) (co)polymerization. A divinyl monomer 4-(3′-buten-1′-oxy)-2,3,5,6-tetrafluorostyrene (1) with a styrenyl group and a pendent alkene group was synthesized. Due to a very high reactivity of the styrenyl group relative to the alkene group in 1, functional fluoro(co)polymers with both well-defined structures and pendent alkene groups were prepared by RAFT polymerizations of 1 and copolymerization of 1 with pentafluorostyrene (PFS). Alkene-functionalized diblock copolymers were also prepared by RAFT copolymerization of 1 with PFS or styrene, extending from a poly(styrene-alt-maleic anhydride) macro-chain transfer agent. Hydrolysis and ammonolysis of these copolymers resulted in amphiphilic diblock fluorocopolymers with alkene-functionalized hydrophobic segments, which were shown to form internally-functionalized micelles in THF-water.
Introduction
The preparation of polymers with accurately controlled structures has become a research theme of modern polymer chemistry. Because properties and performances of polymers depend greatly on the kind and number of functional groups, the development of synthetic strategies that exert excellent control over functionalities of polymers has attracted increasing interests.1–8 Due to the broad applications of vinyl groups in polymerization,9 crosslinking,10–17 radical coupling, 18–22 post-modification and transformations into other useful functionalities,23 well-defined polymers with pendent vinyl groups have received significant attention.20, 24–35
Similar to most types of polymers with functional side groups, those having multiple pendent vinyl groups have been synthesized by either post-polymerization functionalization or polymerization of vinyl-functionalized monomers. With high selectivity towards substituted vinyl groups, anionic polymerization of non-symmetric divinyl monomers has provided a variety of well-defined vinyl-functionalized polymers. For example, as reported by Zhang and Ruckenstein,25, 26 anionic polymerizations of 4-(vinylphenyl)-1-butene and trans,trans-1-methacryloyloxy-2,4-hexadiene showed high selectivity, whereby only styrenyl and methacrylic groups underwent polymerization and well-defined alkene-functionalized polystyrenes and polymethacrylates were afforded. The vinyl groups also exhibit no or little reactivity under the reaction conditions of most ring-opening polymerizations (except ring-opening radical polymerization). Ring-opening polymerizations,20, 28, 29, 36, 37 including ring-opening metathesis polymerization,38 of vinyl-functionalized cyclic monomers have yielded polymers with maintenance of side chain vinylic groups. As illustrated by Coates and co-workers,28 ring-opening polymerization using vinylcyclohexene oxide with other comonomers gave vinyl-functionalized polycarbonates, which were further converted into unimolecular nanoparticles through intramolecular crosslinking.
Throughout the past decade, controlled radical polymerization (CRP) has become one of the most powerful synthetic tools for the preparation of well-defined polymers. Three major CRP techniques have been developed, including nitroxide-mediated polymerization (NMP),39 atom transfer radical polymerization (ATRP),40 and reversible addition-fragmentation chain transfer (RAFT) polymerization.41–47 With good functional group tolerance, CRP of functional monomers has readily provided a broad variety of polymers with terminal or side-chain functional groups. For instance, the syntheses of norbornenyl-based α-alkene macromonomers by ATRP or RAFT of vinylic monomers, having high reactivity toward radical polymerization, have been demonstrated using norbornene-functionalized initiators.48, 49 In such selective polymerizations, it is important to balance the relative reactivities of the multiple alkenyl groups in the system. The significant difference in reactivity between the norbornenyl units and styrenyl- or acrylate-based monomers provided for good control, however, there are situations in which the outcomes have not been as positive. Although, theoretically, methacrylate vinyl groups are more reactive than allyl groups toward ATRP, their difference is not sufficient. ATRP of allyl methacrylate (AMA) experienced crosslinking at early stages of polymerization, indicating that the selectivity between its two vinyl groups was not sufficient to afford well-defined allyl-functionalized polymers.31–33
Because CRP can provide polymers with C-C backbones under less stringent reaction conditions than typical anionic polymerization or ring-opening polymerization, we are interested in establishing selective CRP of non-symmetrical divinyl monomers as an approach for the preparation of well-defined polymers with alkene functionalities. In this paper, we report our recent research on selective RAFT polymerization of 4-(3′-buten-1′-oxy)-2,3,5,6-tetrafluorostyrene. As one of the major living polymerization techniques, RAFT polymerization provides homogeneous and metal-free living polymerization systems applicable for a wide variety of monomers under relatively mild temperature, and therefore, was chosen in this study.
Experimental section
Materials
2,3,4,5,6-Pentafluorostyrene (PFS) and styrene were purified by passing through a column packed with neutral alumina gel and then were stored under argon at 4 °C. The chain transfer agent S-1-dodecyl-S′-(α,α′-dimethyl-α″-acetic acid)trithiocarbonate (DDMAT) was prepared following the published procedure.50 All other chemicals and reagents were purchased from Aldrich Chemical Co. and used as received, unless otherwise noted.
Characterization Methods
1H NMR spectra were recorded at 300 MHz in CDCl3 or acetone-d6 or DMSO-d6 on a Varian Mercury 300 spectrometer, with the solvent proton signal as standard. 13C NMR spectra were recorded at 150.8 MHz in CDCl3 on a Varian Unity 600 spectrometer with the solvent carbon signal as standard. 19F NMR spectra were recorded at 282.2 MHz in CDCl3 on a Varian Mercury 300 spectrometer with external CFCl3 as standard. Infrared spectra were obtained on a Perkin-Elmer Spectrum BX FT-IR system using diffuse reflectance sampling accessories, and were analyzed using FT-IR Spectrum v2.00 software (Perkin-Elmer Corp., Beaconsfield, Bucks, England).
Size exclusion chromatography (SEC) was conducted on a Waters 1515 HPLC (Waters Chromatography, Inc.), equipped with a Waters 2414 differential refractometer, a PD2026 dual-angle (15 and 90°) light scattering detector (Precision detectors, Inc.), and a three-column series PL gel 5 μm Mixed C, 500 Å, and 104 Å, 300×7.5 mm columns (Polymer Laboratories, Inc.). The system was equilibrated at 35 °C in THF, which served as the polymer solvent and eluent with a flow rate of 1.0 mL/min. Polymer solutions were prepared at a known concentration (ca. 3 mg/mL), and an injection volume of 200 μL was used. Data collection and analysis were performed, respectively, with Precision Acquire software and Discovery 32 software (Precision Detectors, Inc.). The interdetector delay volume and the light-scattering detector calibration constant were determined by calibration using a nearly monodispersed polystyrene standard (Pressure Chemical Co., Mp) 90 kDa, Mw/Mn < 1.04). The differential refractometer was calibrated with standard polystyrene reference material (SRM 706 NIST), of known specific refractive index increment dn/dc (0.184 mL/g). The dn/dc values of the analyzed polymers were then determined from the differential refractometer response.
Samples for transmission electron microscopy (TEM) measurements were diluted with 1 wt% of phosphotungstic acid (PTA) stain solution (v/v, 1:1). Carbon grids were exposed to oxygen plasma treatment to increase the surface hydrophilicity. Micrographs were collected at 100,000× magnification and calibrated using a 41 nm polyacrylamide bead from NIST. The number average particle diameters (Dav) and standard deviations were generated from the analysis of a minimum of 150 particles from at least three different micrographs.
Hydrodynamic diameters (Dh) and size distributions for the micelles in aqueous solutions were determined by dynamic light scattering (DLS). The DLS instrumentation consisted of a Brookhaven Instruments Limited (Worcestershire, U.K.) system, including a model BI-200SM goniometer, a model BI-9000AT digital correlator, a model EMI-9865 photomultiplier, and a model 95-2 Ar ion laser (Lexel, Corp.; Farmindale, NY) operated at 514.5 nm. Measurements were made at 20 ± 1 °C. Prior to analysis, solutions were filtered through a 0.22 μm Millex®-GV PVDF membrane filter (Millipore Corp., Medford, MA) and then centrifuged in a model 5414 microfuge (Brinkman Instruments, Inc.; Westbury, NY) for 10 minutes to remove dust particles. Scattered light was collected at a fixed angle of 90°. The digital correlator was operated with 522 ratio spaced channels, and initial delay of 5 μs, a final delay of 100 ms, and a duration of 10 minutes. A photomulitplier aperture of 400 μm was used, and the incident laser intensity was adjusted to obtain a photon counting of between 200 and 300 kcps. Only measurements in which the measured and calculated baselines of the intensity autocorrelation function agreed to within 0.1 % were used to calculate particle size. The calculations of the particle size distributions and distribution averages were performed with the ISDA software package (Brookhaven Instruments Company), which employed single-exponential fitting, cumulants analysis, non-negatively constrained least-squares (NNLS) and CONTIN particle size distribution analysis routines. All determinations were made in triplicate.
Synthesis of S-1-dodecyl-S′-(α,α′-dimethyl-α″-methyl acetate)trithiocarbonate (DDMMAT)
DDMAT (2.92 g, 8.0 mmol), CH3OH (0.79 g, 25 mmol), dicyclohexylcarbodiimide (DCC, 1.72 g, 8.4 mmol), 4-dimethylaminopyridine (DMAP, 0.10 g, 0.80 mmol) were loaded into a 250 mL round-bottom flask containing 100 mL of dry CH2Cl2. The reaction mixture was allowed to stir at room temperature under nitrogen atmosphere for 40 h. The mixture was filtered and the filtrate was concentrated in vacuo, and separated by flash chromatography using CH2Cl2/hexane (v/v, 1/4) as eluent to yield 2.85 g (94%) DDMMAT as a yellow oil. IR (NaCl, cm−1): 2924, 2852, 1740, 1465, 1382, 1363, 1263, 1193, 1154, 1128, 1067, 817. 1H NMR (300 MHz, CDCl3, ppm): δ 0.88 (t, J = 6.5 Hz, 3H, -(CH2)11CH3), 1.26 (br, 18H, -SCH2CH2(CH2)9CH3), 1.66 (m, 2H, -SCH2CH2-), 1.70 (s, 6H, -SC(CH3)2COOCH3), 3.28 (t, J = 7.5 Hz, 2H, -SCH2CH2-), 3.71 (s, 3H, -OCH3). 13C NMR (150.8 MHz, CDCl3, ppm): δ 14.1, 22.8, 25.1, 29.4, 31.9, 35.8, 52.5, 55.0, 174.5, 222.1. MS (ESI, mol. ion.): [C18H34O2S3 + H+], calcd: 379.1793, found: 379.1784.
Synthesis of 4-(3′-buten-1′-oxy)-2,3,5,6-tetrafluorostyrene, 1
3-Buten-1-ol (6.53 g, 90.5 mmol) was diluted with dry THF (60 mL) and deprotonated by addition of NaH (2.71 g, 113 mmol) over 20 min at 0 °C. PFS (14.1 g, 72.4 mmol) in dry THF (30 mL) was then added dropwise. The reaction mixture was heated at reflux for 3 h, cooled to room temperature and concentrated under reduced pressure. Saturated NH4Cl solution (100 mL) was added slowly, and the organic products were extracted into CH2Cl2 (70 mL × 3), which was then washed with water (70 mL), and dried over MgSO4. The organic phase was concentrated to give the crude product, which was purified by flash chromatography using CH2Cl2/hexane (v/v, 1/19) as eluent to yield 11.8 g (66%) of product as a colorless liquid. IR (NaCl, cm−1): 3080-2960, 1660-1620, 1530-1400, 1353, 1291, 1252, 1180-1060, 980-962, 944, 895, 847, 667. 1H NMR (300 MHz, CDCl3, ppm): δ 2.55 (td, J = 6.8 and 6.6 Hz, 2H, -OCH2CH2CH=CH2), 4.28 (t, J = 6.8 Hz, 2H, -OCH2CH2CH=CH2), 5.15 (dd, 1H, J = 18.0 Hz and 2.8 Hz, cis OCH2CH2CH=CHH), 5.11 (dd, J = 11.8 Hz and 2.8 Hz, 1H, trans OCH2CH2CH=CHH), 5.63 (d, J = 12.0 Hz, 1H, cis CHH=CHAr), 5.89 (m, 1H, -OCH2CH2CH=CH2), 6.04 (d, J = 17.6 Hz, 1H, trans CHH=CHAr), 6.63 (dd, J1 = 17.6 and 12.0 Hz, 1H, CH2=CHAr). 13C NMR (150.8 MHz, CDCl3, ppm): δ 34.4, 74.4, 118.5, 122.2, 134.4, 136.2, 139.9, 143.3, 144.1, 147.2. 19F NMR (282.2 MHz, CDCl3, ppm): δ −158.7 (m, 2F, meta-F), −145.6 (m, 2F, ortho-F). MS (ESI, mol. ion.): [C12H10OF4 + H+], calcd: 247.0741, found: 247.0690.
Synthesis of poly(1), 2
A 10 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with 1 (1.24 g, 5.02 mmol), DDMMAT (37.9 mg, 0.10 mmol) and 2,2′-azobisisobutyronitrile (AIBN, 1.6 mg, 0.010 mmol) and 0.60 mL of 2-butanone as the co-solvent. After three cycles of freeze-pump–thaw to degas the reaction mixture, the flask was then placed into an oil bath at 69 °C to allow for polymerization under N2. During polymerization, small aliquots (0.1 mL) of polymerization solution were withdrawn with a syringe and were analyzed by 1H NMR spectroscopy for the determination of conversions of 1 and by SEC for the determination of molecular weights and polydispersities. Finally, after 12 h, the flask was immersed in liquid N2 to quench the polymerization. The polymer solution was precipitated twice into 200 mL of methanol. The product was collected and dried in vacuo for 24 h at room temperature to afford 2 as a yellow solid. Yield: 0.35 g (53% based on the 63% conversion of 1). MnNMR = 8.2 kDa, MnGPC = 8.9 kDa, PDI = 1.35, averaged degree of polymerization (DP)ave = 32. IR (NaCl, cm−1): 3090-2900, 2360, 2343, 1649, 1540, 1493, 1459, 1384, 1144, 1098, 960, 921, 667. 1H NMR (300 MHz, CDCl3, 7.27 ppm): δ 0.87 (br, -CH3 of DDMMAT), 1.25 (br, alkyl protons of DDMMAT), 2.51 (br, -OCH2CH2CH=CH2 on polymer side chain), 2.80-1.40 (br, protons on polymer backbone), 3.39 (br, -SCH2- of DDMMAT), 4.18 (br, -OCH2CH2-), 5.14 (br, -CH2CH=CH2), 5.87 (br, -CH2CH=CH2). 13C NMR (150.8 MHz, CDCl3, 77.2 ppm): δ 19.2, 22.9, 29.1–32.3, 37.7, 74.4, 114.0, 115.2, 118.1, 135.2–144.1, 147.3. 19F NMR (282.2 MHz, CDCl3): δ −158.9 (br, 2F, meta-F), −145.7 to −142.5 (br, 2F, ortho-F).
Synthesis of poly(10.32-co-PFS0.68)n, 3
A 10 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with PFS (1.45 g, 7.5 mmol), 1 (0.62 g, 2.5 mmol), DDMMAT (37.0 mg, 0.10 mmol), AIBN (1.6 mg, 0.010 mmol) and 3 mL of 2-butanone as the co-solvent. After three cycles of freeze-pump–thaw, the mixture was allowed to stir for 10 min at room temperature to ensure that the mixture became homogeneous. The flask was then placed into an oil bath at 68 °C to allow for polymerization under N2. During polymerization, small aliquots (0.1 mL) of polymerization solution were withdrawn with a syringe and were analyzed by 1H NMR spectroscopy for the determination of conversion of each comonomer. Finally, after 20 h, the flask was immersed in liquid N2 to quench the polymerization. The polymer solution was precipitated twice into 200 mL of methanol. The product was collected and dried in vacuo to afford 3 as a slightly green-yellow copolymer. Isolated yield: 1.02 g (83% based on 51% conversion of PFS and 71% conversion of 1). MnNMR = 12.0 kDa, MnGPC = 13.2 kDa, PDI = 1.13, (DP)ave = 57. IR (NaCl, cm−1): 3080-2850, 2358, 2332, 1646, 1540, 1493, 1455, 1381, 1224, 1136, 1082, 957, 922, 760, 702, 666. 1H NMR (300 MHz, CDCl3, ppm): δ 0.88 (br, -CH3 of DDMMAT), 1.26 (br, alkyl protons of DDMMAT), 2.50 (br, -OCH2CH2CH=CH2 on polymer side chain), 3.00-1.40 (br, protons on polymer backbone), 3.32 (br, -SCH2- of DDMMAT), 4.17 (br, -OCH2CH2-), 5.14 (br, -CH2CH=CH2), 5.89 (br, -CH2CH=CH2). 13C NMR (150.8 MHz, CDCl3, ppm): δ 22.4, 29.3–33.0, 38.2, 74.5, 115.3, 118.1, 136.2, 139.4–145.2, 147.1. 19F NMR (282.2 MHz, CDCl3, ppm): δ −161.6 (br, meta-F of PFS units), −157.2 (br, meta-F of 1 units), -154.8 (br, para-F of PFS units), −147.0 (br, ortho-F of 1 units and PFS units).
Synthesis of poly(St-alt-MAn)n, 4
A 50 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with styrene (5.31 g, 51.0 mmol), maleic anhydride (5.00 g, 51.0 mmol), DDMMAT (387 mg, 10.2 mmol) and AIBN (16.5 mg, 1.00 mmol) and 10 mL of 1,4-dioxane as the co-solvent. The reaction mixture was degassed by three cycles of freeze-pump-thaw and then heated with an oil bath at 54 °C under N2. The polymerization was quenched, by being allowed to cool to room temperature, and then opening of the flask to air and dilution of the reaction mixture by addition of THF, at a polymerization time of 3 h, with estimated conversions of both comonomers of 70%. The polymer solution was precipitated twice into 500 mL of diethyl ether and dried in vacuo to yield 4 as a paled yellow powder. Isolated yield: 6.89 g (93% based on the 70% conversions of the comonomers). MnNMR = 7.8 kDa, MnGPC = 8.1 kDa, PDI = 1.16, (DP)ave = 38. IR (NaCl, cm−1): 3090-2860, 2360, 2341, 1856, 1779, 1720, 1540-1410, 1371, 1224, 1081, 956, 924, 703, 667. 1H NMR (300 MHz, acetone-d6, ppm): δ 0.85 (br, -CH3 of DDMMAT), 1.25 (br, alkyl protons of DDMMAT), 1.30–3.00 (br, backbone protons of styrene units), 3.00–3.90 (br, backbone protons of MAn units), 6.00–7.60 (br, Ar-H). 13C NMR (150.8 MHz, CDCl3, ppm): δ 31.0–38.5, 40.2–46.4, 52.5, 127.0–131.2, 136.5, 140.2, 171.1–174.9.
Synthesis of poly(St-alt-MAn)n-b-P(St0.55-co-10.45)m, 5
A 10 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with St (0.79 g, 7.6 mmol), 1 (0.94 g, 3.8 mmol), macro-chain transfer agent 4 (0.81 g, 0.10 mmol) and AIBN (3.2 mg, 0.020 mmol) and 3 mL of 2-butanone as the co-solvent. The reaction mixture was degassed by three cycles of freeze-pump-thaw and then heated with an oil bath at 70 °C for 5 h under N2. The polymer solution was precipitated three times into 100 mL of pentane and the product was collected and dried in vacuo to give 5 as a yellow powder. Isolated yield: 1.35 g (95% based on based on the 26% conversion of St and 43% conversion of 1). MnNMR = 14.1 kDa, MnGPC = 13.6 kDa, PDI = 1.18, (DP)ave, n = 38, (DP)ave, m = 25,. IR (NaCl, cm−1): 3100-2900, 1858, 1782, 1735, 1648, 1492, 1454, 1382, 1332, 1225, 1136, 1082, 957, 923, 760, 702, 670. 1H NMR (300 MHz, CDCl3, ppm): δ 0.87 (br, -CH3 of DDMMAT), 1.26 (br, alkyl protons of DDMMAT), 2.51 (br, -OCH2CH2CH=CH2 on polymer side chain), 1.30–3.00 (br, protons on polymer backbone except MAn units), 3.00–4.00 (br, backbone protons of MAn units), 4.15 (br, -OCH2CH2-), 5.13 (br, -CH2CH=CH2), 5.86 (br, -CH2CH=CH2), 6.00–7.80 (m, all aromatic protons). 13C NMR (150.8 MHz, CDCl3, ppm): δ 22.5, 29.2–33.1, 34.5–48.9, 51.3, 52.2, 53.5, 74.1, 115.0 118.1, 124.3, 137.9–145.0, 147.2, 171.0–175.0 19F NMR (282.2 MHz, CDCl3, ppm): δ −159.1 (br, 2F, meta-F), −145.0 to −141.9 (br, 2F, ortho-F).
Synthesis of poly(St-alt-MAn)n-b-P(FSt0.60-co-10.40)n, 6
A 10 mL-Schlenk flask with a stir bar and sealed by a rubber septum was charged with PFS (1.48 g, 7.63 mmol), 1 (0.94 g, 3.8 mmol), macro-chain transfer agent 4 (0.81 g, 0.10 mmol), AIBN (3.2 mg, 0.020 mmol) and 3 mL of 2-butanone as the co-solvent. The reaction mixture was degassed by three cycles of freeze-pump-thaw and then heated with an oil bath at 70 °C for 5 h under N2. The polymer solution was precipitated three times into 100 mL of pentane and the product was collected and dried in vacuo to give 6 as a yellow powder. Isolated yield: 1.73 g (97% based on the 36% conversion of PFS and 48% conversion of 1). MnNMR = 17.8 kDa, MnGPC = 18.5 kDa, PDI = 1.12, (DP)ave, n = 38, (DP)ave, m = 49. IR (NaCl, cm−1): 3000-2850, 1859, 1782, 1735, 1653, 1523, 1501, 1457, 1419, 1383, 1303, 1225, 1142, 1088, 981, 959, 922, 870, 764, 703. 1H NMR (300 MHz, acetone-d6, ppm): δ 0.88 (br, -CH3 of DDMMAT), 1.26 (br, alkyl protons of DDMMAT), 2.51 (br, -OCH2CH2CH=CH2 on polymer side chain), 1.30–3.00 (br, backbone protons of styrene units), 3.00–3.90 (br, backbone protons of MAn units), 4.18 (br, -OCH2CH2-), 5.15 (br, -CH2CH=CH2), 5.88 (br, -CH2CH=CH2), 6.00–7.70 (br, all aromatic protons). 13C NMR (150.8 MHz, CDCl3, ppm): δ 22.9, 29.5–32.1, 33.8–47.1, 50.5–53.4, 74.3, 115.4, 118.2, 122.5, 136.3, 140.0–145.0, 147.2, 171.5–174.1. 19F NMR (282.2 MHz, CDCl3, ppm): δ −161.6 (br, meta-F of PFS units), −157.3 (br, meta-F of 1 units), −154.7 (br, para-F of PFS units), −147.0 and −141.0 (br, ortho-F of 1 units and PFS units).
Synthesis of 7
Copolymer 5 (71 mg, 5.8×10−3 mmol, 0.22 mmol of maleic anhydride units) was dissolved in 15 mL of THF and the solution was stirred for 1 h at room temperature. KOH solution (1 mL, 30 mg/mL, 0.5 mmol) was added and the mixture was stirred overnight. The solution was then adjusted with 1 N HCl solution to pH 4 and dialyzed against water (MWCO = 3,500 Da) for 3 days. Water was removed by evaporation and the product was then dried in vacuo for 24 h to yield 69 mg (93%) of 7 as a yellow solid. IR (KBr, cm−1): 3550-2500, 1719, 1654, 1492, 1458, 1419, 1384, 1247, 1173, 1150, 1118, 960, 860, 810, 660, 604. 1H NMR (300 MHz, DMSO-d6, ppm): δ 0.82 (br, -CH3 of DDMMAT), 1.18 (br, alkyl protons of DDMMAT), 3.80-1.50 (br, protons on polymer backbone), 4.13 (br, -OCH2CH2-), 5.12 (br, -CH2CH=CH2), 5.82 (br, -CH2CH=CH2), 6.18–7.60 (m, all aromatic protons), 12.0 (br, -COOH).
Synthesis of 8
Copolymer 6 (152 mg, 8.22×10−3 mmol, 0.312 mmol of maleic anhydride units) was dissolved in 20 mL of THF and stirred for 30 min. An aqueous solution of NH4OH (0.50 mL, 2 M) was added and the reaction was allowed to stir for 30 h at room temperature. The mixture was allowed to dialyze against water (MWCO = 3,500 Da) for 3 days. Water was removed by evaporation and the product was dried in vacuo for 24 h to yield in 125 mg (78%) of 8 as a yellow solid. IR (KBr, cm−1): 3700-2500, 1718, 1656, 1560, 1493, 1457, 1398, 1216, 1148, 1076, 962, 861, 763, 702, 660. 1H NMR (300 MHz, DMSO-d6, ppm): δ 0.83 (br, -CH3 of DDMMAT), 1.20 (br, alkyl protons of DDMMAT), 4.00-1.20 (br, protons on polymer backbone), 4.08 (br, -OCH2CH2-), 5.03 (br, -CH2CH=CH2), 5.76 (br, -CH2CH=CH2), 6.00–7.50 (m, all aromatic protons).
Preparation of micelle solution from copolymer 7
Copolymer 7 (10 mg) was dissolved in 10 mL of THF, and the solution was stirred for 1 h at room temperature. DI water (10 mL) was added dropwise into the copolymer solution at a rate of 20 mL/h. The mixture was transferred into dialysis tubing (MWCO = 3,500 Da) and allowed to dialyze against DI water for 3 days to result in 37 mL of micelle solution. The concentration of the final micelle solution was 0.30 mg/mL. (Dh)n (DLS) = 8 ± 1 nm; (Dh)v (DLS) = 13 ± 1 nm; (Dh)i (DLS) = 137 ± 19 nm (bimodal with populations at 13 and 228 nm); Dav (TEM) = 10 ± 1 nm.
Preparation of micelle solution from copolymer 8
A similar procedure for the preparation of micelle solution 7 was followed. The final micelle solution of copolymer 8 (24 mL) was obtained at a concentration of 0.21 mg/mL. (Dh)n (DLS) = 11 ± 1 nm; (Dh)v (DLS) = 14 ± 1 nm; (Dh)i (DLS) = 155 ± 24 nm (bimodal with populations at 21 and 387 nm); Dav (TEM) = 10 ± 2 nm.
Results and discussion
Monomer Design and Synthesis
Initially, a variety of potential divinyl monomers was considered from both radical polymerization and organic chemistry aspects (Chart 1). As shown in Table 1, each divinyl monomer can be viewed as a copolymerization system of two vinylic monomer units.51 Based on the (Q, e) values52 from Alfrey-Price theory,52, 53, 54 the reactivity ratios (r1 and r2) of each pair of vinyl groups within each divinyl monomer were calculated (Table 1). For this treatment, vinylic unit 1 was designated as the one having a higher tendency toward polymerization, whereas vinylic unit 2 was expected to be retained. A divinyl monomer that is capable of undergoing polymerization with good selectivity between the two vinylic bonds should have a significant difference between r1 and r2. (i.e., r1 with a large value, r2 with a value close to 0, and a high ratio of r1/r2).
Chart 1.

Various divinyl monomers designed for selective radical polymerization.
Table 1.
Calculated reactivity ratios for divinyl monomers.a
| Divinyl monomersb | (Q1, e1) | (Q2, e2) | r1 | r2 | r1/r2 |
|---|---|---|---|---|---|
| A: MA vs. BE | (0.45, 0.64) | (0.007, −0.06) | 41 | 0.015 | 2,800 |
| B: MMA vs. VAc | (0.78, 0.40) | (0.026, −0.88) | 18 | 0.011 | 1,700 |
| C; MMA vs. AAc | (0.78, 0.40) | (0.24, −1.07) | 1.8 | 0.06 | 28 |
| D: MMA vs. BE | (0.78, 0.40) | (0.007, −0.06) | 89 | 0.009 | 10,200 |
| E/F: MSt vs. BEc | (1.10, −0.60) | (0.007, −0.06) | 110 | 0.007 | 17,000 |
| G: MOSt vs. BE | (1.53, −1.40) | (0.007, −0.06) | 33 | 0.005 | 6,800 |
| 1: PFS vs. BEd | (0.86, 0.75) | (0.007, −0.06) | 68 | 0.008 | 8,800 |
(Q, e) values are cited from Polymer Handbook,52 and reactivity ratios were calculated based on Alfrey-Price equations: r1=(Q1/Q2)·exp[−e1(e1−e2)]; r2=(Q2/Q1)·exp[−e2(e2−e1)].
Individual vinylic functionalities within the divinyl monomer structures. MA: methyl acrylate; BE: 1-butene; MMA: methyl methacrylate; VAc: vinyl acetate; AAc: allyl acetate; MSt: p-methyl styrene; MOSt: p-methoxy styrene.
Both 4-(3-buten-1-yl)-styrene (E) and (3′-buten-1′-oxy) 4-vinylbenzyl ether (F) were approximated to the same copolymerization system as 4-methoxy styrene vs. 1-butene.
4-(3′-Buten-1′-oxy)-2,3,5,6-tetrafluorostyrene (1) was approximated to a copolymer pair of PFS with BE. The (Q, e) of PFS is cited from the literature.55
The acrylate-based divinyl monomer (A), due to the low intrinsic reactivity of the acrylate unit (Q = 0.45), is expected to have poor selectivity between the two vinyl groups, and, therefore, monomer A might be potentially used for the synthesis of branched polymers or crosslinked networks under radical polymerization conditions. Although the methacrylate moieties in vinyl methacrylate (VMA, B) and allyl methacrylate (AMA, C) are highly reactive (Q = 0.78), the vinyl acetate (VAc) moiety in B and allyl acetate (AAc) moiety in C also have noticeable reactivities (Q(VAc) = 0.026, Q(AAc) = 0.24), which could be reflected by the relatively low r1 values (18 and 1.8 for B and C, respectively). Moreover, the allyl protons of C have appreciable tendency to undergo chain transfer reactions in radical polymerization because the resulting radical can be stabilized through resonances with both allyl and ester groups. Therefore, both B and C, although commercially available, were not considered to be ideal candidates for selective radical polymerization. Each of the monomers D, E, F, G and 1 exhibit characteristic reactivity ratios that suggest they would allow for the controlled radical polymerization-based preparation of well-defined linear polymers bearing side chain vinylic groups. 4-(3′-Buten-1′-oxy)-2,3,5,6-tetrafluorostyrene (1) was selected as the non-symmetrical divinyl monomer for these initial studies of selective RAFT polymerization (Figure 1), due to the expected high contrast of radical polymerization reactivity of its two vinyl groups and its synthetic feasibility. In other work,56 a monomer that at first glance appears to be similar to 1, yet has a non-fluorinated styrenyl ring and, significantly, a benzylic ether linkage to the butenyl unit, was studied for the preparation of branched polystyrenes via RAFT at high temperatures. In contrast to that work and our earlier exploitation of non-reactive 1-alkenes,9 our current interest is in linear polymers that are prepared via selective RAFT polymerization that does not involve the side chain vinylic groups.
Figure 1.
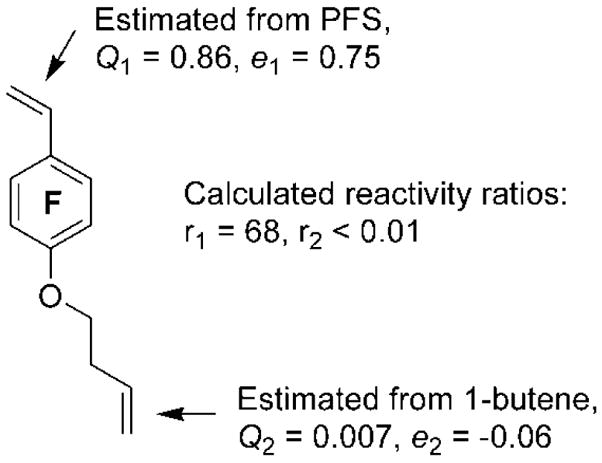
Structural design of nonsymmetrical divinyl monomer 1 for selective RAFT polymerization.
Based on structural similarity, the reactivity of the substituted styrenyl group and alkene group in 1 can be estimated through PFS (Q1 = 0.86, e1 = 0.75) and 1-butene (Q2 = 0.007, e2 = −0.06), respectively. The calculated reactivity ratios, r1 of 68 and r2 of less than 0.01, indicate that the styrenyl group has significantly higher reactivity than does the alkene group, and further suggest that the polymerization of 1 might be conducted as a homopolymerization of its styrenyl group, until a high molar ratio of alkene group to styrenyl group is reached.
The non-symmetric divinyl monomer 1 was synthesized by the nucleophilic aromatic substitution57 of PFS with the commercially available 3-butenyl-1-ol. The reaction was conducted with a slight excess of the alcohol (1.25 eq. relative to PFS, to drive the complete consumption of PFS) in dry THF heated at reflux for 3 h to afford 1 in 66% yield, after chromatographic purification (Scheme 1). The chemical structure of 1 was verified by IR, 1H, 13C, and 19F NMR spectroscopies. As shown in Figure 2a, the three styrenyl protons resonated at 6.63, 6.04 and 5.63 ppm, while the three protons of the other vinyl group protons resonated at 5.89, 5.15 and 5.11 ppm. Their resonance intensity ratios were in excellent agreement with their proton number ratios. The signal for the methyleneoxy protons (-OCH2-) at 4.28 ppm confirmed the covalent connection between the two vinyl group-containing units. Further, the 19F NMR spectra of PFS and 1 allowed for confirmation that the nucleophilic substitution occurred in the para position, as the para-F signal of PFS (−157.4 ppm) disappeared coincident with the introduction of two new ortho- (−145.6 ppm for 1 vs. −144.5 ppm for PFS) and meta-F (−158.7 ppm for 1 vs. −164.4 ppm for PFS) resonances upon formation of 1 (Figure 3).
Scheme 1.
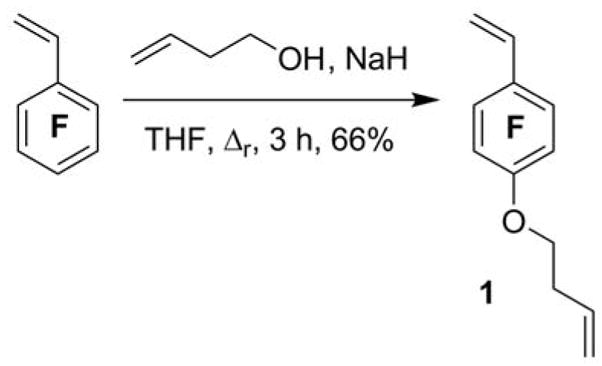
Synthesis of 4-(3′-buten-1′-oxy)-2,3,5,6-tetrafluorostyrene (1)
Figure 2.

1H NMR spectra of the divinyl monomer 1 (a) and copolymer 3 (b). (300 MHz, CDCl3)
Figure 3.

19F NMR spectra of PFS (a), divinyl monomer 1 (b), polymer 2 (c) and copolymer 3 (d). (282 MHz, CDCl3)
RAFT Homopolymerization
With two different types of vinyl groups on monomer 1, selective RAFT homopolymerization of 1 was investigated using 2,2′-azobisisobutyronitrile (AIBN) as the thermal initiator, S-1-dodecyl-S′-(α,α′-dimethyl-α″-methyl acetate) trithiocarbonate (DDMMAT), as the RAFT agent, in 2-butanone (~50 vol %) at 68 °C ([1]0:[AIBN]0:[DDMMAT]0 = 50:0.1:1). DDMMAT was prepared by the esterification reaction of S-1-dodecyl-S′-(α,α′-dimethyl-α″-acetic acid) trithiocarbonate (DDMAT),50 a widely used RAFT agent, with methanol. At time intervals during polymerization, small aliquots were withdrawn from the polymerization solution and analyzed by 1H NMR spectroscopy for the determination of monomer conversions and by SEC for the determination of molecular weights and polydispersities of the resulting polymers.
Comparison of the resonance intensities of the styrenyl protons at 6.63, 6.04 and 5.63 ppm with the resonance intensities of methylene protons (-OCH2-) at 4.20–4.35 ppm gave 7, 32, 48 and 63% conversions of the styrenyl groups of 1 at 1.2, 5, 8, and 12 h, respectively. The resonance intensities of the protons of the pendent alkene groups remained consistent during the polymerization, indicating that essentially only the styrenyl groups were polymerized under the reaction conditions. SEC monitoring showed that, with the increase of polymerization times and monomer conversions, the molecular weights (MW) of the resulting polymers increased (Figure 4). Narrow MW distributions (PDI < 1.20) were observed for the polymers, at monomer conversions below ca. 50%. The final polymer 2, obtained with 63% monomer conversion after 12 h of polymerization, had a number-average molecular weight (Mn) of 8.87 kDa and a PDI of 1.35. The experimental Mn value of 2 by SEC was in agreement with the theoretical value of 8.2 kDa. However, the broadened molecular weight distribution of 2 suggested a slight occurrence of reactions of the pendent alkene functionalities at the later polymerization stage. Although not detectable by 1H NMR spectroscopy, consumption of a small fraction of the side-chain vinylic groups could occur as the molar ratio of the alkene group increased, relative to the remaining styrenyl group at higher conversions, resulting in a broadening of the MW distribution.
Figure 4.

A composite of SEC curves, collected during the RAFT polymerization of 1 (left), and a plot showing the dependency of Mncalc, MnSEC and PDI of the resulting polymers on the monomer conversions (right).
A conventional radical polymerization of 1 was performed under the similar conditions of RAFT polymerization, for which apparent crosslinking was observed when the conversion of styrenyl groups reached 43% after 5 h of polymerization. After precipitating the reaction mixture into methanol, a material that was insoluble in organic solvents was obtained.58 These results further support the controlled characteristics of the RAFT polymerization process.
RAFT Copolymerization
To improve the conversions of 1, while maintaining suppression of the reactions of the pendent alkene groups, copolymerization of 1 with PFS was studied under similar polymerization conditions to those employed for the homopolymerizations ([PFS]0:[1]0:[AIBN]0:[DDMMAT]0 = 75:25:0.1:1; 60 vol% of 2-butanone; 68 °C). With PFS as comonomer, the initial molar ratio of reactive styrenyl group to the pendent alkene group for this copolymerization system was 4:1, much higher than the ratio of 1:1 for the RAFT homopolymerization of 1. As detected by 1H NMR spectroscopic analysis of polymerization solutions, based on the intensities of the resonances of unreacted styrenyl protons at 5.72 ppm for PFS and at 5.63 ppm for 1, the conversions of the comonomers increased with the copolymerization time, and finally reached 51% and 71%, respectively, for PFS and 1 after 20 h. SEC analysis showed that the resulting copolymer 3 had a Mn of 13.2 kDa, which agreed with the calculated Mn (12.2 kDa), and a narrow MW distribution (PDI = 1.19) was obtained. As shown in the 1H NMR spectrum (Figure 2b) of 3 (poly(1-co-PFS)), the resonances of the pendent vinyl protons at ~5.8 and ~5.2 ppm had the same intensities as the methylene protons (-CH2O-) at 4.25 ppm (Figure 2), indicating maintenance of the pendent alkene functionality of 3. The well-defined structure of 3 could also be verified by the methyl protons at 0.84 ppm and methylene protons (-SCH2-) at 3.31 ppm of the RAFT agent chain end functionality. The unit ratio of PFS and 1 in copolymer 3 was calculated to be 0.68:0.32 based on the comonomer conversions, indicating that above 30 mol % of 1, bearing the pendent alkenyl functionalities, was incorporated into the copolymer.
The 19F NMR spectra (Figure 3c) for fluorocopolymer 3 showed four broad resonance peaks in the area from −140 to −165 ppm, which, according to the literature,47 were assigned to the ortho-fluorines of 1- and PFS-based repeat units (−144.0 ppm), the para-fluorines of PFS units (−154.8 ppm), the meta-fluorines of repeat units from 1 (−157.2 ppm), and the meta-fluorines of PFS units (−161.6 ppm), respectively. The ratio of 19F NMR resonance intensities from the peaks at −154.8 ppm and −157.2 ppm were calculated to be 1:0.89, indicating the molar ratio of the PFS : 1 repeat units to be 69:31 (2.00:0.89), which is in good agreement with the theoretical ratio, based upon the feed ratio and the comonomer conversions.
RAFT Block Copolymerization
With the importance of amphiphilic block copolymers and their ability to afford well-defined nanoscale objects, our interest has been directed toward the use of amphiphilic block copolymers for the construction of functional nanoscale materials. Therefore, our final goal in this current work was to investigate the potential of this selective RAFT polymerization methodology for the preparation of alkene-functionalized amphiphilic block copolymers, extending from an initial macro-RAFT agent as a hydrophilic precursor. The alternating copolymer poly(St-alt-MAn)38 (4, Mn = 8.1 kDa, PDI = 1.16) was prepared by RAFT copolymerization of styrene with equal amount of MAn at 54 °C using DDMMAT as the CTA following a literature method.59 Starting from the resulting poly(St-alt-MAn)n-based macro-CTA, two types of alkene-functionalized diblock copolymers 5 and 6 were successfully synthesized (Scheme 3). RAFT copolymerization of styrene and 1 ([St]0:[1]0:[AIBN]0:[4]0=76:38:0.2:1) was conducted at 70 °C in 50 vol% of 2-butanone. The polymerization was allowed to proceed for 5 h, and as a result, 26% conversion of styrene and 43 % conversion of 1 were obtained (the intensities of 1H NMR resonances of the unreacted styrenyl protons at 5.22 ppm for styrene and at 5.63 ppm for 1 were used for conversion determination). Under the same reaction conditions, RAFT copolymerization of PFS and 1 ([PFS]0:[1]0:[AIBN]0:[4]0=76:38:0.2:1) was found to reach conversions of 36% and 48% for PFS and 1, respectively (the intensities of 1H NMR resonances of the unreacted styrenyl protons at 6.09 ppm for PFS and at 5.63 ppm for 1 were used for conversion determination). The formations of diblock copolymers poly(St-alt-MAn)n-b-poly(St0.55-co-10.45)m (5, MnSEC = 13.6 kDa, PDI = 1.18, nave = 38, mave = 25) and poly(St-alt-MAn)n-b-poly(FSt0.60-co-10.40)m (6, MnSEC = 18.2 kDa, PDI = 1.12, nave = 38, mave = 49) by chain extension from the poly(St-alt-MAn)n macro-CTA were verified by SEC analysis. Moreover, the good agreement between the experimental and calculated molecular weights (Mncalcd = 14.0 kDa for 5 and Mncalcd = 17.9 kDa for 6, respectively) and the mono-modal molecular weight distributions of the diblock copolymers illustrates the quantitative chain transfer efficiency of the macro-CTA.
Scheme 3.

Syntheses of well-defined amphiphilic diblock copolymers with pendent alkene functionalities.
The block ratios of the diblock copolymers 5 and 6 were also analyzed by 1H NMR spectroscopy (Figure 7), based upon comparisons of the intensities of characteristic resonances of MAn units (for protons j′ and j″ at 3.05–4.00 ppm), styrene units (for aromatic protons at 6.00–7.75 ppm), and repeat units of 1 (for protons f′ and f″ at 5.02–5.34 ppm). The molar ratio of repeat units of MAn:St:1 = 2.90:3.94:1.00 was determined for copolymer 5, indicating that the ratio of the degree of polymerization for the block segments of poly(St-alt-MAn)n and poly(St-co-1)m was 1.45:1, which agrees with the theoretical value of 1.52:1. A molar fraction of 0.49 for 1 was further obtained for the poly(St-co-1)m block of 5, and it agrees with the theoretical value of 0.45 calculated from conversions and the molar feed ratio of comonomers. A molar ratio of repeat units of MAn:St:1 = 1.88:1.91:1.00 was obtained for copolymer 6. This ratio, coupled with the theoretical molar fraction of 0.40 for 1 within the poly(PFS-co-1)m block segment, indicates that the ratio of the degree of polymerization for block segments of poly(St-alt-MAn)n and poly(PFS-co-1)m is 0.76:1.00, which is close to the theoretical value of 0.78:1.00. Additionally, the molar fraction of 1 in the poly(PFS-co-1)m block segment of 6 was also determined by 19F NMR spectroscopy. In terms of the intensities of resonances of para-fluorines of PFS centered at -154.7 ppm and meta-fluorines of 1 at -157.3 ppm, the experimental value of 0.42 was obtained, in excellent agreement with the theoretical value of 0.40.
Figure 7.

1H NMR spectra of copolymer 4 (a), copolymer 5 (b) and copolymer 6 (c). (300 MHz, acetone-d6).
Amphiphilic Block Copolymers
By transformations of the MAn units in the poly(St-alt-MAn) blocks into hydrophilic acid and amide units, 5 and 6 could be further converted into amphiphilic block copolymers 7 and 8, respectively. Hydrolysis of 5 was first attempted at neutral conditions in 10% water-THF, but no reaction was observed by 1H NMR spectroscopy, after stirring overnight. However, hydrolysis proceeded readily at room temperature under basic conditions using 1 M KOH(aq) solution to promote the reaction.59 Aminolysis-hydrolysis was carried out by treating 6 with an excess amount of 2 M NH4OH(aq) solution for 6 h at room temperature.60 1H NMR and FT-IR spectroscopic characterizations were used to verify the chemical structures of 7 and 8. Resonances of carboxylic protons centered at 12.0 ppm were observed by 1H NMR measurement of 6 in DMSO-d6, indicating the formation of carboxylic acids by hydrolysis of 5. According to the consistent intensities of the resonances of the pendent alkene protons at ~5.8 and ~5.1 ppm for these polymers before and after the chemical transformations, the pendent alkene groups remained intact. Both precursors 5 and 6 showed two strong C=O stretching absorptions at 1855 and 1772 cm−1 for their cyclic anhydride groups, with absences of any O-H or N-H stretching absorptions. By comparison with 5, polymer 7 had only one C=O stretching absorption at 1719 cm−1 and a broad O-H stretching absorption at 3500-2500 cm−1, indicating functional group transformation from the anhydrides to carboxylic acid groups. Copolymer 8 had N-H stretching absorptions at 3700–3400 cm−1 with a C=O stretching mode at 1656 cm−1, verifying the formation of amides.
Amphiphilic copolymers 7 and 8 could undergo self assembly in THF-water mixtures. Upon the slow addition of nanopure water to a dilute solution of 7 or 8 in THF (ca. 1 mg/mL), followed by dialysis against nanopure water, water-dispersible micelles were formed. As detected by dynamic light scattering (DLS) measurements, the micelles of 7 had a number-average hydrodynamic diameter (Dh)n of 8 ± 1 nm and a volume-average hydrodynamic diameter (Dh)v of 13 ± 1 nm, and the micelles of 8 had a (Dh)n of 11 ± 1 nm and a (Dh)v of 14 ± 1 nm. In each case, only the intensity-weighted DLS data indicated a population of larger aggregates, together with the well-defined micellar assemblies. The micelles were further characterized by transmission electron microscopy (TEM). As shown in Figure 8, both micelles from 7 and 8 had average diameters of 10 nm and relatively narrow size distributions. These particles are expected to have a “core-shell” structure, potentially with an ability to adopt unusual multi-compartment morphologies,61 with the core domain(s) functionalized with alkene groups. Further study on the post-modification of these domain-specific alkene functionalities are in progress.
Figure 8.

TEM images of the self-assembled micelles of diblock copolymers 7 (a) and 8 (b) in aqueous media. Scale bars = 100 nm.
Conclusions
In summary, a facile synthetic approach for the preparation of well-defined (co)polymers with pendent alkene functionalities has been established by selective RAFT polymerization. Well-defined fluoro(co)polymers bearing vinylic side chain functionalities were prepared by RAFT homopolymerization of a divinyl monomer 1 and by its copolymerization with PFS. Alkene-functionalized diblock fluorocopolymers were prepared by RAFT copolymerization of 1 together with styrene or PFS, extending from a poly(styrene-alt-maleic anhydride) macro-chain transfer agent. Hydrolysis and aminolysis of these copolymers resulted in amphiphilic fluorocopolymers with alkene-functionalized hydrophobic block segments, which generated functional micelles upon supramolecular assembly of the amphiphilic block copolymers in aqueous solution. These functionalized polymers are expected to have significant applications for the preparation of advanced polymer architectures by crosslinking, grafting, or other chemical transformations via the pendent alkene groups.
Figure 5.

A composite of SEC traces collected during the RAFT copolymerization of 1 with PFS (left), and plots of molecular weights of polymers and conversions of comonomers vs. polymerization time (right).
Figure 6.
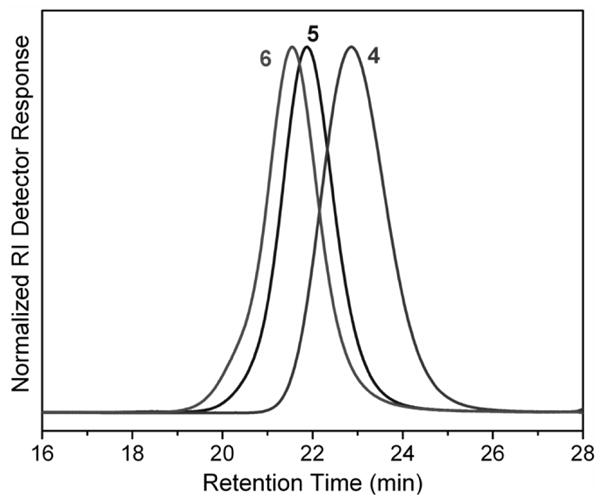
SEC traces of copolymers poly(St-alt-MAn)n (4), poly(St-alt-MAn)n-b-P(St0.55-co-10.45)m (5) and poly(St-alt-MAn)n-b-P(FSt0.60-co-10.40)m (6).
Scheme 2.

RAFT homopolymerization of 1, and RAFT copolymerization of 1 and PFS.
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant No. 0451490 and by the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HL080729). The authors thank Mr. G. M. Veith for the kind assistance with acquisition of TEM images.
References and notes
- 1.Takemoto K, Ottenbrite RM, Kamachi M, editors. Functional Monomers and Polymers. 2. Marcel Dekker; New York: 1997. [Google Scholar]
- 2.Coessens V, Pintauer T, Matyjaszewski K. Prog Polym Sci. 2001;26:337–377. [Google Scholar]
- 3.Braunecker WA, Matyjaszewski K. Prog Polym Sci. 2007;32:93–146. [Google Scholar]
- 4.Jeong JH, Kim SW, Park TG. Prog Polym Sci. 2007;32:1239–1274. [Google Scholar]
- 5.Ladmiral V, Mantovani G, Clarkson GJ, Cauet S, Irwin JL, Haddleton DM. J Am Chem Soc. 2006;128:4823–4830. doi: 10.1021/ja058364k. [DOI] [PubMed] [Google Scholar]
- 6.Yanjarappa MJ, Sivaram S. Prog Polym Sci. 2002;27:1347–1398. [Google Scholar]
- 7.Li YS, Yang J, Benicewicz BC. J Polym Sci, Part A: Polym Chem. 2007;45:4300–4308. [Google Scholar]
- 8.Wang R, Lowe AB. J Polym Sci, Part A: Polym Chem. 2007;45:2468–2483. [Google Scholar]
- 9.Ma J, Cheng C, Sun G, Wooley KL. J Polym Sci, Part A: Polym Chem. 2008;46:3488–3498. [Google Scholar]
- 10.Doura M, Naka Y, Aota H, Matsumoto A. Macromolecules. 2005;38:5955–5963. [Google Scholar]
- 11.Ishizu K, Tsubaki K, Mori A, Uchida S. Prog Polym Sci. 2003;28:27–54. [Google Scholar]
- 12.Antony P, De SK, van Duin M. Rubber Chem Tech. 2001;74:376–408. [Google Scholar]
- 13.Wendland MS, Zimmerman SC. J Am Chem Soc. 1999;121:1389–1390. [Google Scholar]
- 14.Hustad PD, Coates GW. Macromolecules. 2002;124:11578–11579. doi: 10.1021/ja0273748. [DOI] [PubMed] [Google Scholar]
- 15.Beil JB, Zimmerman SC. Macromolecules. 2004;37:778–787. [Google Scholar]
- 16.Ornelas C, Mery D, Cloutet E, Aranzaes JR, Astruc D. J Am Chem Soc. 2008;130:1495–1506. doi: 10.1021/ja077392v. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerman SC, Zharov I, Wendland MS, Rakow NA, Suslick KS. J Am Chem Soc. 2003;125:13504–13518. doi: 10.1021/ja0357240. [DOI] [PubMed] [Google Scholar]
- 18.David RLA, Kornfield JA. Macromolecules. 2008;41:1151–1161. [Google Scholar]
- 19.Campos LM, Meinel I, Guino RG, Schierhorn M, Gupta N, Stucky GD, Hawker CJ. Adv Mater. 2008;20:3728–3733. [Google Scholar]
- 20.Gress A, Vo1lkel A, Schlaad H. Macromolecules. 2007;40:7928–7933. [Google Scholar]
- 21.Carioscia JA, Schneidewind L, O’Brien C, Ely R, Feeser C, Cramer N, Bowman CN. J Polym Sci, Part A: Polym Chem. 2007;45:5686–5696. [Google Scholar]
- 22.Podesva J, Hruby M, Spevacek J, Hrdlickova M, Netopilik M. J Polym Sci, Part A: Polym Chem. 2008;46:3919–3925. [Google Scholar]
- 23.Larock RC. Comprehensive Organic Transformations: A Guide to Functional Group Transformations. 2. Wiley-VCH; New York: 1999. [Google Scholar]
- 24.Ruckenstein E, Zhang HM. 1999;32:6082–6087. [Google Scholar]
- 25.Zhang H, Ruckenstein E. Macromolecules. 1999;32:5495–5500. [Google Scholar]
- 26.Zhang HM, Ruckenstein E. Macromolecules. 2001;34:3587–3593. [Google Scholar]
- 27.Liu F, Liu G. Macromolecules. 2001;34:1302–1307. [Google Scholar]
- 28.Cherian AE, Sun FC, Sheiko SS, Coates GW. J Am Chem Soc. 2007;129:11350–11351. doi: 10.1021/ja074301l. [DOI] [PubMed] [Google Scholar]
- 29.Hu X, Chen X, Xie Z, Liu S, Jing X. J Polym Sci, Part A: Polym Chem. 2007;45:5518–5528. [Google Scholar]
- 30.Lin Y, Liu XH, Li XR, Zhan J, Li YS. J Polym Sci, Part A: Polym Chem. 2007;45:26–40. [Google Scholar]
- 31.Paris R, De la Fuente JL. J Polym Sci, Part A: Polym Chem. 2005;43:2395–2406. [Google Scholar]
- 32.Paris R, De La Fuente JL. J Polym Sci, Part A: Polym Chem. 2005;43:6247–6261. [Google Scholar]
- 33.Paris R, de la Fuente JL. Euro Polym J. 2008;44:1403–1413. [Google Scholar]
- 34.Sugiyama F, Satoh K, Kamigaito M. Macromolecules. 2008;41:3042–3048. [Google Scholar]
- 35.Venkatesh R, Vergouwen F, Klumperman B. J Polym Sci, Part A: Polym Chem. 2004;42:3271–3284. [Google Scholar]
- 36.Chojnowski J, Cypryk M, Fortuniak W, Scibiorek M, Rozga-Wijas K. Macromolecules. 2003;36:3890–3897. [Google Scholar]
- 37.Nagai A, Ochiai B, Endo T. Macromolecules. 2004;37:4417–4421. [Google Scholar]
- 38.Cheng C, Yang NL. Macrom Rapid Commun. 2005;26:1395–1399. [Google Scholar]
- 39.Hawker CJ, Bosman AW, Harth E. Chem Rev. 2001;101:3689–3745. doi: 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- 40.Matyjaszewski K, Xia J. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 41.Lowe AB, McCormick CL. Prog Polym Sci. 2007;32:283–351. [Google Scholar]
- 42.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2005;58:379 – 410. [Google Scholar]
- 43.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2005;59:669–692. [Google Scholar]
- 44.Moad G, Rizzardo E, Thang SH. Polymer. 2008;49:1079–1131. [Google Scholar]
- 45.Perrier S, Takolpuckdee P. J Polym Sci, Part A: Polym Chem. 2005;43:5347–5397. [Google Scholar]
- 46.Rizzardo E, Chiefari J, Mayadunne RTA, Moad G, Thang SH. Controlled/Living Radical Polymerization. In: Matyjaszewski K, editor. Progress in ATRP, NMP, and RAFT. Vol. 768. American Chemical Society; Washington, DC: 2000. pp. 278–296. [Google Scholar]
- 47.Barner-Kowollik C, Perrier S. J Polym Sci, Part A: Polym Chem. 2008;46:5715–5723. [Google Scholar]
- 48.Patton DL, Advincula RC. Macromolecules. 2006;39:8674–8683. [Google Scholar]
- 49.Cheng C, Khoshdel E, Wooley KL. Macromolecules. 2005;38:9455–9465. [Google Scholar]
- 50.Lai JT, Filla D, Shea R. Macromolecules. 2002;35:6754–6756. [Google Scholar]
- 51.For example, 3-butenoxy-methacrylate (D) could be approximately viewed as a comonomer pair of methyl methacrylate (MMA) and 1-butene.
- 52.Greenley RZ. In: Polymer Handbook. 3. Immergut EH, editor. John Wiley & Sons; 1989. pp. 267–274. [Google Scholar]
- 53.Odian G. Principles of polymerization. 4. Wiley; Hoboken, N.J: 2004. pp. 490–505. [Google Scholar]
- 54.Alfrey-Price equations were shown in table 1, footnote a.
- 55.Pryor WA, Huang TL. Macromolecules. 1969;2:70–77. [Google Scholar]
- 56.Dong ZM, Liu XH, Lin Y, Li YS. J Polym Sci, Part A: Polym Chem. 2008;46:6023–6034. [Google Scholar]
- 57.Powell KT, Cheng C, Wooley KL. Macromolecules. 2007;40:4509–4515. doi: 10.1021/ma0628937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.The obtained crosslinked materials could not dissolve in CH2Cl2, CHCl3, THF, DMF and DMSO at room temperature.
- 59.Cheng C, Khoshdel E, Wooley KL. Macromolecules. 2007;40:2289–2292. [Google Scholar]
- 60.Henry SM, El-Sayed MEH, Pirie CM, Hoffman AS, Stayton PS. Biomacromolecules. 2006;7:2407–2414.1. doi: 10.1021/bm060143z. [DOI] [PubMed] [Google Scholar]
- 61.Harrisson S, Wooley KL. Chem Commun. 2005:3259–3261. doi: 10.1039/b504313a. [DOI] [PubMed] [Google Scholar]