

Abstract
The time course of inactivation of voltage-activated potassium (Kv) channels is an important determinant of the firing rate of neurons. In many Kv channels highly unsaturated lipids as arachidonic acid, docosahexaenoic acid and anandamide can induce fast inactivation. We found that these lipids interact with hydrophobic residues lining the inner cavity of the pore. We analysed the effects of these lipids on Kv1.1 current kinetics and their competition with intracellular tetraethylammonium and Kvβ subunits. Our data suggest that inactivation most likely represents occlusion of the permeation pathway, similar to drugs that produce ‘open-channel block'. Open-channel block by drugs and lipids was strongly reduced in Kv1.1 channels whose amino acid sequence was altered by RNA editing in the pore cavity, and in Kv1.x heteromeric channels containing edited Kv1.1 subunits. We show that differential editing of Kv1.1 channels in different regions of the brain can profoundly alter the pharmacology of Kv1.x channels. Our findings provide a mechanistic understanding of lipid-induced inactivation and establish RNA editing as a mechanism to induce drug and lipid resistance in Kv channels.
Keywords: anandamide, arachidonic acid, inactivation, ion channel, potassium
Introduction
Voltage-activated potassium channels (Kv channels) are opened by membrane depolarization and control the repolarization of the action potential in neurons and in other excitable cells. Some of the Kv channels close rapidly during continued membrane depolarization, a process called N-type inactivation, whereas other Kv channels show little or no inactivation. The rate of inactivation of potassium channels after depolarization is an important determinant of the firing rate of neurons and thus of the transmission of information in the nervous system (Trimmer and Rhodes, 2004). Rapid inactivation is mediated by plugging of the pore by an ‘inactivation particle' attached to the N-terminus of the channels or by β-subunits carrying a similar inactivation particle (Rettig et al, 1994). It has been reported that highly unsaturated fatty acids as arachidonic acid (AA) can convert non-inactivating Kv channels to rapidly inactivating channels (Honore et al, 1994; Oliver et al, 2004), but the mechanism underlying this ‘conversion' and especially the binding site of AA have remained unclear. Amphiphilic substances such as AA could modulate the function of the channel protein (i) by changing the physical characteristics of the lipid bilayer, (ii) by changing the hydrophobic interaction between the membrane protein and the lipid bilayer or (iii) by binding to a specific site that is important for the gating or the permeation properties of the channel (Oliver et al, 2004; Boland and Drzewiecki, 2008; Lundbaek, 2008; Meves, 2008). In this study, we have examined the binding site and the mechanism of action of AA and obtained evidence that, contrary to expectations, the fatty acid can bind to hydrophobic residues lining the inner cavity of potassium channels of the Kv1 subfamily, provided the channels have been opened earlier by depolarization.
Inactivation of Kv channels is also observed during application of AA, docosahexaenoic acid (DHA) and anandamide (N-arachidonylethanolamide, AEA). The latter molecules resemble AA in possessing highly unsaturated C20–C22 hydrocarbon tails. In neurons, various signal transduction pathways result in the release of AA, DHA and AEA from the cell membrane into the cytosol. AEA is the most studied member of the endocannabinoids, a family of neuromodulatory lipids. AEA liberated from cell membranes to the cytosol is released into the extracellular space through a putative endocannabinoid transporter and has a major role in the regulation of synaptic transmission in the brain (Katona and Freund, 2008). All three endogenous lipids mentioned here, AA, DHA and AEA, have been implicated in neurological disorders, including stroke, Alzheimer's disease and depression (Marcheselli et al, 2003; Calon et al, 2004; Bisogno and Di Marzo, 2007). Previous experimental results suggest that these important signalling lipids, in addition to their indirect effects through metabolites or receptors, directly modulate Kv channels (Honore et al, 1994; Oliver et al, 2004). We show here that this direct modulation of Kv channels is achieved through an open-channel block mechanism.
Kv1.1 belongs to the increasing number of proteins whose amino acid sequence has been found to be altered by RNA editing (Hoopengardner et al, 2003). In the edited channels an isoleucine residue facing the inner cavity of the pore is replaced by valine (Figure 1A). The extent of RNA editing varies within different regions of the brain, with highest levels in medulla, spinal cord and thalamus (Hoopengardner et al, 2003). We show here that open-channel block by AA, DHA or AEA is strongly decreased in ‘edited' Kv1.1 channels. In addition, we found that RNA editing at a single subunit of the Kv1.1 tetramer is sufficient to alter the sensitivity of heteromeric Kv1.x channels to various drugs known to cause open-channel block. Furthermore, we show that RNA editing in neurons of the lateral geniculate nucleus (LGN) of the thalamus markedly alters the sensitivity of Kv1 channels to pharmacological inhibition. Taken together, our results suggest that (i) AA, DHA and AEA cause open-channel block of Kv1.x channels, and (ii) the sensitivity of Kv1.x channels to these endogenous lipids is modulated by differential RNA editing of Kv1.1 subunits in different regions of the brain. The combination of these two effects may make an important contribution to the fine-tuning of neuronal information processing.
Figure 1.

Editing prevents block of Kv1.1 channels by HUFAs. (A) Localization of the residue I400 in the pore of Kv1.1 channels. (B, C) Kv1.1 (B) and Kv1.1I400V (C) currents in Xenopus oocytes, activated by voltage steps to +40 mV, before and after application of AA. (D) Percentage of ‘inactivation' at +40 mV induced by HUFAs for Kv1.1 and Kv1.1I400V; ** indicates P<0.01. (E, F) Application of AA (E) or AEA (F) to the intracellular side of patches expressing either Kv1.1 or Kv1.1I400V. Insets show the dose–response curves of AA (E) and AEA (F) for Kv1.1 (blue) and Kv1.1I400V (red). (G) Application of AA to the intracellular side of patches expressing either Kv3.1 or Kv3.1I428V; currents were evoked by voltage steps from −80 to +40 mV. (H) Time course of Kv1.1 block by different HUFAs applied through the bath solution in inside–out patch-clamp experiments (top) or whole-cell recordings from oocytes (bottom). (I) Time course of Kv1.1 and Kv1.1I400V inhibition induced by 10 μM AA applied from the intracellular side of inside–out patches. (J) Binding rates (kon) of AA inhibition for Kv1.1 and Kv1.1I400V calculated assuming a bimolecular reaction mechanism kon=(1/τ) × (1−Iss/Ipeak). (K) Recovery kinetics of AA-induced ‘inactivation' for Kv1.1 and Kv1.1I400V channels recorded with test pulses to +40 mV. Cells were held at −80 mV during the interpulse intervals. (L) Plot of normalized peak current amplitude of the test pulse versus the interpulse interval. Blue Kv1.1 and red Kv1.1I400V lines represent mono-exponential fits for the time constant of recovery from ‘inactivation'.
Results
Highly unsaturated fatty acids cause open-channel block of Kv1 channels
We have analysed the mechanism by which the highly unsaturated lipids AA (C20:4n-6), DHA (C22:6n-3) and anandamide (AEA, N-arachidonylethanolamide; C20:4n-6) influence the function of Kv channels. The starting point of our study was an observation in Xenopus oocytes expressing Kv channels (Figure 1B and D). Expression of wild-type (WT) Kv1.1 channels gave rise to non-inactivating potassium currents; application of 10 μM AA for 8 min, while the cells were held at −80 mV, did not change the initial current but produced a marked time-dependent ‘inactivation' of the current (Figure 1B). The isoleucine residue at position 400 of the potassium channel Kv1.1 (I400), which is located in the middle of the S6 segment and faces the inner cavity of the pore (Figure 1A), can be changed to a valine by RNA editing (Bhalla et al, 2004). Expression of Kv1.1I400V channels in oocytes showed that the ‘edited' channels were insensitive to inhibition by AA. In fact, application of 10 μM AA slightly increased the initial outward current but did not induce inactivation (Figure 1C). Similar results were obtained for drosophila Shaker and Shab channels that undergo an I-to-V editing at the equivalent position. Like Kv1.1I400V, the ‘edited' drosophila channels were insensitive to inhibition by AA (data not shown). Inactivation of Kv1.1 channels, but not of Kv1.1I400V, could also be induced by the highly unsaturated fatty acids DHA and eicosatetraynoic acid (ETYA) (Figure 1D).
The observation that, before the onset of inactivation, Kv1.1 currents were fully activated even after prolonged exposure to AA (Figure 1B) is consistent with the view that lipid-induced ‘inactivation' occurs exclusively from the open state. The finding that a single amino acid exchange in the inner cavity abolished lipid-induced ‘inactivation' (Figure 1C) suggests that AA, DHA and ETYA bind in the pore. These initial experiments led us to the following working hypothesis: AA and other highly unsaturated lipids enter the inner cavity of Kv1.1 channels through the open activation gate, bind to a site in the inner cavity (which includes the isoleucine residue at position 400) and cause occlusion of the permeation pathway. The hypothesis of ‘open-channel block' by AA (and other closely related lipids) from the intracellular side of the membrane was then subjected to a series of experimental tests.
First, we compared application of AA to the intracellular side of the membrane in macropatches with application to the outside of intact Xenopus oocytes. Intracellular application of AA in macropatches caused an even stronger ‘inactivation' than extracellular application in whole-cell recordings. In inside–out patches, 2 μM AA produced a strong reduction in the steady-state Kv1.1 outward current but had virtually no effect on Kv1.1I400V channels (Figure 1E). Application of 2 μM AEA to inside–out patches produced an even stronger ‘inactivation' of Kv1.1 channels (81.6±2.8%; n=5) and had almost no effect on edited Kv1.1 channels (Figure 1F). The IC50 of Kv1.1 for AA was shifted 4.3-fold, from 1.5±0.1 μM (Hill coefficient, H=2.0) to 6.5±0.4 μM (H=1.7) (Figure 1E, inset). The IC50 of Kv1.1 for AEA was shifted 3.9-fold, from 1.3±0.1 μM (H=1.8) to 5.1±0.9 μM (H=1.5) (Figure 1F, inset). The results obtained with the I400V mutant are consistent with the idea that AA binds in the pore of the channel. To correlate our results with a previous study on the effects of AA on Kv3.1 channels (Oliver et al, 2004), in which the AA-binding site had not been identified, we mutated the isoleucine residue at the corresponding position (I428) in the pore cavity of Kv3.1 to valine. We found that the inactivation induced by intracellular application of AA in macropatches containing Kv3.1 was strongly reduced in the Kv3.1I428V mutant (Figure 1G). Thus, the mechanism of action of AA appears to be similar in Kv1.1 and Kv3.1.
In inside–out patches, the time course of the effects of AA and AEA was by an order of magnitude faster than in whole-cell recordings. Application of AA or AEA to the inside of the patches produced ‘inactivation' within ∼30 s (Figure 1H, top), whereas application of these lipids to the outside of intact oocytes changed the rate of ‘inactivation' very slowly, reaching a steady state after >10 min (Figure 1H, bottom). We then tested the effects of another lipid, VDM11 (N-(4-hydroxy-2-methylphenyl) arachidonylamide), which is closely related to AEA and has been used as a blocker of AEA uptake (De Petrocellis et al, 2000). VDM11 also induced strong ‘inactivation' in inside–out patches and had no measurable effects within 10 min when applied to intact oocytes (Figure 1H, bottom). These findings support the hypothesis that the observed lipid-induced ‘inactivation' in fact represents open-channel block from the intracellular side by AA and related lipids.
Biophysical tests for open-channel block by AA
Next, we studied the kinetics of block by 10 μM AA in inside–out macropatches from Xenopus oocytes expressing either WT or ‘edited' Kv1.1 channels. Although the extent of block by intracellular AA was markedly reduced in Kv1.1I400V, the time course of inhibition was similar for both in Kv1.1 and in Kv1.1I400V (Figure 1I). Indeed, the binding rate (kon) calculated from the time constant and steady state of AA inhibition was not significantly different for WT and ‘edited' Kv1.1 (Figure 1J). In contrast, recovery from block by AA was strongly enhanced by the I400V exchange. The time constant of recovery from block (unbinding of AA) was 385±35 ms (n=8) for WT Kv1.1 channels and 49±8 ms (n=7) for Kv1.1I400V (Figure 1K and L), corresponding to rate constants (koff) of 2.6 and 20.4 s−1, respectively. These findings suggest that the rate of unbinding of AA is greatly increased by the I400V exchange, consistent with a reduction in binding affinity. A similar effect on the recovery from block by AA was observed for Kv3.1I428V channels (Supplementary Figure 1).
The classical test for an open-channel blocker acting from the inside is the simultaneous application of the blocker with intracellular tetraethylammonium (TEA), which is known to enter the pore. If intracellular TEA competes with the blocker (Figure 2A), open-channel block is the most likely mechanism (Armstrong, 1971; Yellen, 1998). Indeed, the time course of AA block of the residual current in the presence of TEA was found to be much slower than the time course of AA block under control conditions (Figure 2B). Figure 2C illustrates the open-channel block caused by 20 μM AA in the presence of different TEA concentrations; the current traces were normalized to the initial peak current. At all TEA concentrations, the time course of the current changes measured after addition of AA could be adequately described by a single time constant. Figure 2D shows that the time constants (•) increased with increasing TEA concentration. These experimental data were compared to the predictions derived from a simple kinetic scheme of competitive open-channel block by TEA and AA:
Figure 2.

Open-channel block of Kv1.1 channels by HUFAs. (A) Cartoon illustrating the competition of TEA and AA for pore binding. (B) Block of Kv1.1 by AA alone (τ=17 ms, black fit), by TEA (grey) and by AA in the presence of TEA (τ=132 ms, black fit). (C) Normalized Kv1.1 currents recorded in the presence of different TEA concentrations. The IC50 for TEA was 0.67±0.06 mM (n=3, not illustrated). (D) Time constants of Kv1.1 block by AA in the presence of different TEA concentrations. The filled circles represent the measured time constants of block and the continuous blue line the calculated time constants as a function of the TEA concentration. (E) Block of Kv1.1 by AA at +40 mV. The deactivation tails recorded at −40 mV show a clear cross over. (F–M) Data derived from cell-attached recordings in HEK cells transfected with Kv1.1. (F) Single-channel recordings before and (G) after application of AA. (H) Superposition of the ensemble currents in the absence and presence of AA (average of 12 traces). (I) Amplitude histogram before and after application of AA. (J) Closed-time histogram and (K) histogram of open times within a burst before (black) and after (blue) application of AA. (L) Changes in burst duration and (M) in nPo induced by AA.
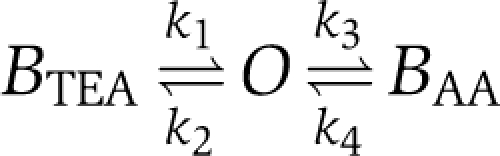 |
O is the open state, BTEA is the blocked state produced by TEA, BAA is the blocked state produced by AA. A similar kinetic scheme has been used previously to describe the competition between TEA and the inactivation particle in Shaker K+ channels (Choi et al, 1991). Figure 2D illustrates that the measured time constants (•) agreed well with the predictions derived from the kinetic scheme (continuous line). The values for the rate constants k3 and k4 obtained in this way were 88.9 and 3.54 s−1, respectively (see Supplementary data); these values are in good agreement with the values for kon and koff determined in the absence of TEA (Figure 1J–L). The finding that the effects of TEA and AA on Kv1.1 currents could be described adequately by competitive inhibition provides strong support for the hypothesis that AA causes open-channel block of Kv1.1.
Another hallmark of open-channel block is the so-called ‘foot-in-the-door' phenomenon (Armstrong, 1971). This term refers to the observation that deactivation of the current is slowed after application of certain blockers because the activation gate of the channel cannot close until the blocker is cleared from the pore. As a result, blocked channels deactivate more slowly than channels that are not blocked, and the superposition of the two deactivation curves measured at the same potential ‘cross over'. Consistent with the ‘foot-in-the-door' phenomenon, the two deactivation curves of Kv1.1, recorded in the absence and in the presence of AA, showed a typical cross over (Figure 2E), i.e. in the presence of AA deactivation was much slower. In conclusion, the kinetics of current deactivation were found to be consistent with the idea that the apparent inactivation of Kv1.1 channels observed in the presence of AA was caused by open-channel block.
Next, we set out to study open-channel block by AA at single-channel level using a mammalian cell line (HEK293 cells) transiently transfected with Kv1.1 channels. Whole-cell experiments showed that in HEK293 cells the effects of AA on the time course of ‘inactivation' were similar to those observed in Xenopus oocytes (Supplementary Figure 2). The membrane potential of the patch was estimated using the average membrane potential of the cells determined in a separate series of experiments (−47±1 mV; n=52). In most patches, several channels opened on depolarization from (approximately) −77 to +53 mV (Figure 2F). In agreement with previous studies (Stuhmer et al, 1988; Koren et al, 1990), we found that the mean single-channel conductance was 10.9±1.2 pS (5 mM Kout+, n=9) and that the openings of Kv1.1 channels occurred in bursts, i.e. the openings within a burst were interrupted by brief closures (<2 ms) and bursts were separated by long closures (>10 ms). The mean burst duration (τb) under control conditions, calculated using a variant of the algorithm described by Fenwick et al (1982) (Supplementary Figure 3), was 293.2±22.5 ms. In the presence of 10 μM AA the number of openings decreased dramatically within the first 300 ms of the depolarizing voltage step (Figure 2G). The ensemble current, obtained by adding 12 individual current traces, showed the kinetics of AA block (Figure 2H). The unitary current amplitude (Figure 2I) and the kinetics of the short closures within the burst were not changed by application of AA (Figure 2J and K). However, the mean burst duration decreased from 293.2±22.5 to 61.0±21.9 ms (Figure 2L; see Supplementary Figure 3 for details) and the overall open probability nPo decreased by a factor of ∼5 (Figure 2M). These observations are consistent with the hypothesis of open-channel block: we propose that in the presence of AA the bursts were terminated prematurely by the entry of AA into the inner cavity of the channel.
The binding site for open-channel block
The data described above suggest that AA, DHA and AEA interact with the isoleucine at position 400 of Kv1.1. This residue is conserved in all Kv channels and is located in the middle of the S6 segment (which forms the wall of the central cavity). We tried to characterize the lipid-binding site in the inner cavity more completely by carrying out a systematic mutagenesis of the S6 segment of the closely related channel Kv1.5, as described earlier (Decher et al, 2004). The Kv1.5 mutants are suitable for analysing the binding of AA because the inner cavity is identical in all Kv1.x channels; as expected, the effects of AA on the time course of Kv1.5 were very similar to the effects on Kv1.1 described above.
Kv1.5 channels with single mutations to alanine (or alanine to valine) were expressed in Xenopus oocytes and block by 10 μM AA was measured (Figure 3A and B). The rationale was that mutations that remove block should be part of the AA-binding site. We found that mutants with non-pore-facing S6 segment mutations (blue) were blocked by AA essentially similar to WT Kv1.5 channels, whereas channels with pore-facing S6 segment mutations (red) showed virtually no block (Figure 3A), leaving the increase in peak amplitude by AA intact. Mutation of the isoleucine residue at position 508 in Kv1.5, equivalent to the isoleucine at position 400 in Kv1.1, resulted in a small increase in peak current without subsequent block (Figure 3A, box), as observed for the I400V editing at the homologous site of Kv1.1. The alanine scan showed a helical pattern for the AA-binding site. Substitutions by alanine, which disturbed block, were located in distances of about 3.5 amino acids and were facing the inner cavity (Figure 3B and D). These data suggest that the fatty acid binds to the pore-facing side of the S6 segment. Unlike other pore blockers such as AVE0118, S0100176 (Decher et al, 2004, 2006), AA did not appear to interact with the threonine residues (T479, T480) of the pore signature sequence (Figure 3B). Taken together, these findings suggest that AA can bind to the central cavity and induces apparent inactivation by occluding the channel and that the homologous (and edited) isoleucine residue at position 400 of Kv1.1 is an important part of the AA-binding site (Figure 3B, red bar).
Figure 3.

The binding site for HUFAs in Kv1 channels. (A) Currents measured during voltage steps to +40 mV in Xenopus oocytes expressing WT or mutant Kv1.5 channels. Blue traces: WT and non-pore-facing mutants in the presence of AA. Red traces: pore-facing mutants in the presence of AA. The mutation at site I508 (homologous to I400 in Kv1.1) is shown in a red box. (B) Ala-scanning mutagenesis of the pore domain of Kv1.5. Relative current (%) remaining after addition of AA, determined at the end of 1.5 s pulses to +40 mV (ne, non-expressing); * indicates P<0.05, ** indicates P<0.01. (C) Relative inhibition of Kv1.1 by AA, measured with (○) or without (•) co-expression of Kvβ1.1. No significant voltage dependence of block was observed in the voltage range of 0 to +70 mV for both, Kv1.1 alone or Kv1.1+Kvβ1.1; * indicates P<0.05, ** indicates P<0.01. (D) Stereo-view of the channel shown with the lipid-binding site identified by the Ala-scan. For a better view into the central cavity only three subunits are depicted. The inset illustrates from which orientation of the homology model the close-up was made. Residue I508 is shown in red, valines in yellow, proline and isoleucines in white. (E–H) The open channel viewed from the cytosolic side. The solvent-accessible surface area of the protein is shown in blue. The PVP motif of the channel is highlighted in yellow. Residue T480 near the selectivity filter is indicated in red and white. (F) Closer view of the empty open-channel cavity shown in (E). (G) Space-filling model of AEA (grey) in the cavity of the channel. (H) AEA (grey) and a potassium ion (purple) with its square-antiprismatic solvation shell in the entrance of the blocked channel.
As the AA-binding site resembles the Kvβ1.1- and Kvβ1.3-binding site (Zhou et al, 2001; Decher et al, 2005, 2008), we speculated that Kvβ1 and AA might compete for the same binding domain in the inner cavity. To test this hypothesis, we studied the block of Kv1.1 by AA in the absence and in the presence of Kvβ1.1. When the Kv1.1 channels were co-expressed with Kvβ1.1, the fractional block of the steady-state current by AA was strongly reduced (Figure 3C). These data are consistent with the idea that Kvβ1.1 and AA compete for the same or overlapping binding sites located in the inner pore cavity. Thus, the molecular mechanism of inactivation by AA might be analogous to N-type inactivation by Kvβ1 subunits.
AA can physically occlude the pore
A pore homology model that was generated on the basis of the crystal structure of the open channel (Decher et al, 2008) shows that the residues identified by our Ala-scan face into the central cavity (Figure 3D). To test the plausibility of our hypothesis that AA, DHA and AEA can enter the inner cavity of Kv1 channels, we docked AEA into this pore homology model. In most cases, AEA penetrated into the cavity, placing the ethanolamide group next to the threonine residues at the entrance of the selectivity filter and forming extended contacts with non-polar residues lining the wall of the channel cavity. The docking program GOLD found multiple solutions that differed in the orientation of the C20 tail of the ligand, suggesting that there is not one distinct binding orientation for the probe ligand. Some of the most prominent solutions are shown in Supplementary Figure 4. These relatively crude docking considerations are consistent with the idea that the pore is wide enough to fully accommodate a molecule such as AEA (Figure 3E–G) and that the binding of the ligand (Figure 3G) will significantly narrow the pore width so that its remaining diameter is too small to allow a potassium ion together with its square-antiprismatic solvation shell (K (H2O)8+) to pass through the channel (Figure 3H).
Editing of Kv channel in different human tissues
Editing of Kv1.1 channels may represent a physiologically relevant mechanism because it may modulate the effects of AA, DHA and AEA in different regions of the brain. Therefore, we studied the extent of editing in various regions of the brain. In agreement with previous results (Hoopengardner et al, 2003), we found that Kv1.1 has high editing rates in medulla, thalamus and spinal cord (Figure 4A). In addition, we observed editing rates of 80 and 65% in human brain stem and striatum, respectively (Figure 4B). We found no editing of Kv1.1 in heart and kidney. We also tested whether other Kv channels undergo a homologous I400V RNA editing. In human medulla, thalamus, spinal cord and heart none of the other Kv channels studied was edited in the S6 segment (Figure 4A).
Figure 4.

Kv channel editing in different human tissues. (A) I–V editing of different Kv channels in human tissues. (B) Percentage of I–V editing of Kv1.1 in different human tissues.
Edited Kv1.1 channels have a lower drug affinity
As RNA editing appeared to modulate open-channel block by AA we asked the question whether editing might also alter the effect of synthetic compounds known to act as open-channel blockers. We studied the effects of four different K+ channel blockers on Kv1.1 and on Kv1.1I400V channels. Three of these drugs (S0100176, AVE0118, S020951) block all members of the Kv channel family (Decher et al, 2004, 2006), the fourth one, Psora-4, is a potent Kv1.x selective channel blocker (IC50 for Kv1.1=62 nM) (Vennekamp et al, 2004). Figure 5A–D (left) show current traces of Kv1.1 and Kv1.1I400V before and after addition of the different drugs. With all compounds the dose–response curve for the Kv1.1I400V mutant was shifted to the right as compared with the curve for ‘non-edited' Kv1.1 (Figure 5A–D, right). The IC50 for S0100176, AVE0118 and S020951 was increased 6.5-, 3.8- and 5.5-fold, respectively (Figure 5A–D, right). For Psora-4, the effect was particularly striking, the edited channel was resistant to application of 500 nM Psora-4 (Figure 5D, left) and the IC50 was increased 70-fold.
Figure 5.

Editing reduces drug affinity of Kv1.1 channels. (A–F) The effects of the drugs (A) S020951, (B) AVE0118, (C) S0100176, (D) Psora-4, (E) 4-aminopyridine and (F) the scorpion toxin rHongotoxin-1 (rHgTX1) on Kv1.1 and Kv1.1I400V channels. Left-hand panels show Kv1.1 or Kv1.1I400V currents activated by voltage steps to +40 mV before and after application of the same concentration of blocker (black, controls; blue, Kv1.1 plus drug; red, Kv1.1I400V channels plus drug). The right-hand panels show the dose–response curves for Kv1.1 (blue) and Kv1.1I400V (red).
We also examined the effects of Kv1.1 editing on the action of 4-aminopyridine (4-AP) and the scorpion toxin rHongotoxin-1 (HgTX1) (Figure 5E and F). 4-AP is a non-specific Kv channel blocker, which is frequently used to characterize or identify native Kv1 potassium channels because Kv1 channels are more sensitive to 4-AP than the other members of the Kv family. The sensitivity of Kv1.1 to block by 4-AP was strongly reduced by the I400V exchange (Figure 5E); RNA editing caused a 68-fold increase in the IC50 of 4-AP (from 98 μM to 6.7 mM) and fully prevented block by 500 μM 4-AP. In contrast, I400V editing did not affect inhibition of Kv1.1 by HgTX1 (Figure 5F), as expected because the toxin binds to the outer vestibule of the channel.
The pharmacology of heteromeric Kv1 channels
I400V editing is tissue dependent and rarely reaches a level of 100%. To further clarify the physiological significance of our findings, we analysed whether heteromers of I400V subunits with WT Kv1.1 subunits also show altered drug sensitivity and fatty acid sensitivity. We mixed cRNA of (unedited) Kv1.1 and Kv1.1I400V at different ratios before injecting a constant amount of the cRNA mixture into Xenopus oocytes. When we injected 25% Kv1.1I400V plus 75% non-edited Kv1.1 cRNA, block by 500 nM Psora-4 (Figure 6A) and by 10 μM AA (Figure 6B) were reduced by 65 and 60%, respectively. These results are consistent with random assembly and an alteration of channel properties by a single subunit in the tetrameric channel. For these conditions, a binomial distribution of assembled subunits predicts a reduction of block by 68.4%, as 68.4% of the channels would contain at least one I400V subunit in the tetramer. The co-expression with 50 or 75% of I400V cRNA did not perfectly follow a binomial distribution for the block of Psora-4 (grey lines and squares in Figure 6A and B). Nevertheless, our data suggest that a single edited I400V subunit in the channel tetramer is sufficient to suppress block of the channel by drugs and by fatty acids (Figure 6A and B). Thus, a small percentage of RNA editing, as for example found in the aorta (Figure 4B), can significantly alter the ‘inactivation' properties and the pharmacological profile of Kv1.1 channels.
Figure 6.

Analysis of heteromeric Kv1.x channels containing Kv1.1I400V subunits. (A, B) The effects of (A) Psora-4 and (B) AA measured after co-expression of Kv1.1 and Kv1.1I400V channels at different ratios in Xenopus oocytes. The left-hand panels show typical currents measured at +40 mV. The right-hand panels show the percentage of inhibition by (A) Psora-4 and (B) AA, observed after co-expression of different ratios of WT and Kv1.1I400V subunits. Grey lines and squares indicate the inhibition expected for a binomial distribution; ** indicates P<0.01. (C–E) Currents measured after expression of heteromeric channels in oocytes; equal amounts of cRNA of Kv1.1I400V and Kv1.x were co-injected. (C) The panel shows typical currents measured at +40 mV for Kv1.5 channels alone or co-expressed with Kv1.1 or Kv1.1I400V subunits. (D) The effects of Psora-4 on homomeric Kv1.x channels and on heteromeric Kv1.x channels containing either WT Kv1.1 or Kv1.1I400V. The concentration of Psora-4 was 2 μM for Kv1.3 and Kv1.4 and 500 nM for all other Kv1.x channels; ** indicates P<0.01. (E) AA sensitivity of homomeric Kv1.x channels and heteromeric Kv1.x channels containing edited Kv1.1I400V subunits, analysed at +40 mV; ** indicates P<0.01.
Editing modulates drug sensitivity and lipid sensitivity of heteromeric Kv1.x channels
As Kv1.x subunits can form heteromeric channels, we tested whether Kv1.1I400V subunits can heteromerize with other Kv1.x subfamily members and whether Kv1.1I400V modulates their properties. Homomeric Kv1.1, Kv1.2, Kv1.5 and Kv1.6 channels were all blocked by 500 nM Psora-4 to a similar extent. Kv1.3 and Kv1.4 had a reduced Psora-4 affinity and were therefore tested with 2 μM Psora-4. Co-injection of Kv1.5 cRNA with an equal amount of Kv1.1 mRNA in Xenopus oocytes generated Kv currents sensitive to block by Psora-4 (Figure 6C, top). In contrast, co-expression of Kv1.1I400V with Kv1.5 produced currents resistant to block by 500 nM Psora-4 (Figure 6C, bottom). The change in drug sensitivity caused by heteromerization with edited Kv1.1 channels was not restricted to Kv1.5 channels. Co-expression of non-edited Kv1.1 channels with other Kv1.x channels did not change the sensitivity of the channels to Psora-4 (Figure 6D). In contrast, co-expression of Kv1.x channel members with equal amounts of Kv1.1I400V cRNA drastically reduced the affinity of Psora-4 and AA for these channels (Figure 6D and E). These observations suggest that the effects of Kv1.1 editing on drug sensitivity and lipid sensitivity can be transferred to heteromeric Kv1.1/Kv1.x channels.
Editing reduces drug sensitivity of Kv1.x channels in rat LGN neurons
We then set out to analyse to what extent editing alters the pharmacology of Kv1.x channels in intact neurons. We chose to study relay neurons acutely isolated from the LGN of the thalamus, which possess a slow transient outward current with low 4-AP sensitivity (Budde et al, 1992). First, we quantified the extent of I400V RNA editing in these neurons. Sequencing of the PCR-amplified Kv1.1 S6 segment indicated that ∼50% of the transcripts undergo I400V editing (Figure 7A, arrow). Quantification of the editing with MfeI restriction digests of cloned Kv1.1 S6 segments (see Supplementary data for details) gave an editing ratio of 52% (52 out of 100 clones). Using RT–PCR, we found expression of Kv1.1, Kv1.2, Kv1.4, Kv1.6, and to a lesser extent Kv1.3, and Kv1.5 (Figure 7A, right).
Figure 7.

Analysis of RNA editing and Psora-4-resistant Kv currents in native TCR neurons. (A) Sequencing of PCR amplified Kv1.1 S6 segments and RT–PCR of Kv1.x channels from rat LGN neurons. (B) Voltage-activated outward currents recorded under control conditions. Currents were evoked by 2 s test pulses from a 2 s conditioning potential of −112 mV to potentials between −82 and +38 mV. (C) Currents from the same cell after application of Psora-4. During the wash in of Psora-4 cells were depolarized every 13 s from −112 to +38 mV to monitor the drug effects and the use dependence of block. (D) Currents after application of Psora-4 plus recombinant HgTX1. (E) Psora-4-sensitive current component, as difference between the currents measured before and after application of Psora-4 (B minus C). (F) Psora-4-resistant current component, as difference between the currents measured before and after additional application of HgTX1 (C minus D). (G) I–V plots derived from the currents measured at the end of the test pulse. (H) Normalized total voltage-activated current from (G) analysed at +38 mV. (I) Pharmacological dissection of the components of Kv1.x current in LGN neurons. (J–L) Analysis of the components of Kv1.x current in hippocampal CA1 pyramidal neurons. Data analysis was performed as described in (G–I). For all experiments * indicates P<0.05 and ** indicates P<0.01.
Our co-expression experiments with different Kv subunits in Xenopus oocytes (Figure 6) show that heteromeric Kv1.x channels containing Kv1.1I400V have a reduced affinity to Psora-4. In contrast, the affinity of HgTX1 was not altered by the I400V exchange, because it binds to the external vestibule of the channel (Figure 5F). The rationale of the experiments in rat thalamocortical relay (TCR) neurons was to determine the component of the Kv current that could be blocked by HgTX1 but was resistant to Psora-4. This current component should give an indication of the effects of editing on the pharmacology in these native neurons. For these experiments, we used 100 nM Psora-4, a concentration sufficient to completely block all members of the Kv1 family (Vennekamp et al, 2004). We confirmed in HEK293 cells that 100 nM Psora-4 almost completely blocked all Kv1.x channels (except for Kv1.4, data not shown), but produced only a negligibly small block of Kv1.1I400V (IC50, 1.93 μM).
Our voltage protocol evoked the typical voltage-dependent K+ currents (Figure 7B) described earlier in TCR neurons (Budde et al, 1992). Application of 100 nM Psora-4 inhibited all components of the outward current to a similar extent (Figure 7C and G); the late outward current measured at +38 mV was reduced to 79.7±3.9% of control (Figure 7C, G and H). HgTX1 specifically blocks Kv1.1, Kv1.2, Kv1.3 and Kv1.6 channels (Koschak et al, 1998), which are also blocked by Psora-4. Thus, in the absence of Kv1.1 editing, additional application of HgTx1 in the presence of Psora-4 should not result in further inhibition. However, we found that additional application of 100 nM HgTX1 resulted in a further significant reduction in the late outward current to 54.8±7.7% of control (Figure 7D, G and H). The Psora-4-sensitive and Psora-4-resistant components of a single cell are shown in Figure 7E and F. We infer from the relative size of these two components (Figure 7I) that in TCR neurons about two-thirds (63±8%) of the total Kv1.x current was Psora-4 resistant. The latter component most likely represents Kv1.x channels containing at least one edited Kv1.1 subunit and (non-edited) Kv1.1, Kv.1.2, Kv1.3 or Kv1.6 subunits (which are sensitive to HgTX1). We conclude from these results that in TCR neurons of the LGN a major fraction of the voltage-activated K+ current shows substantially altered pharmacological properties as a result of a Kv1.1 editing.
To confirm the validity of our pharmacological approach to determine the extent of editing we repeated these experiments in neurons that are known to have very low Kv1.1I400V editing. Consistent with a previous report (Hoopengardner et al, 2003), we found that in cells from the CA1–CA3 region of the rat hippocampus only 7% of the Kv1.1 transcripts undergo I400V editing (7 out of 100 clones), much less than the 50% editing found in the LGN neurons. Our patch-clamp measurements of the Psora-4-sensitive and Psora-4-resistant Kv currents in rat hippocampal CA1 pyramidal neurons are illustrated in Supplementary Figure 5. In contrast to the experiments in TCR neurons, additional application of 100 nM HgTX1 resulted in only a minor reduction of the late outward currents (Figure 7J and K). The Psora-4-resistant component of Kv1.x currents was very small (Figure 7L). This is consistent with the low Kv1.1 editing ratio found in hippocampal neurons and supports our observation that RNA editing in LGN neurons causes drug resistance.
Discussion
Open-channel block of Kv1.1 channels by highly unsaturated fatty acids
Our results suggest that lipid-induced inhibition of Kv1.1 channels occurs exclusively from the open state and that AA and other highly unsaturated lipids enter the inner cavity of Kv1.1 channels, causing physical occlusion of the permeation pathway. The experimental evidence supporting the idea that endogenous lipids cause open-channel block of Kv1.1 channels can be summarized as follows: (i) when applied from the extracellular side in Xenopus oocytes, the effects of AA, DHA and ETYA on ‘inactivation' developed slowly (within minutes), consistent with a relatively slow increase in the cytosolic concentration of the fatty acids; AEA and VDM11 had little or no effect when applied to the extracellular side. When applied to inside–out patches, the effect of the endogenous lipids on ‘inactivation' developed much faster (within seconds). (ii) The ‘inactivation' induced by AA was antagonized by intracellular TEA ions in inside–out patches. Competition with intracellular TEA is a classical criterion for open-channel block (Yellen, 1998). (iii) The deactivation of Kv1 channels was retarded after inhibition of the channels with AA, consistent with a ‘foot-in-the-door' mechanism (Armstrong, 1971; Yellen, 1998). (iv) The mean burst duration of single Kv1.1 channels was reduced in the presence of AA, consistent with a termination of the burst by AA entering the pore. (v) Our systematic Ala-mutagenesis screen identified several hydrophobic residues facing the inner cavity of the channel as binding sites of AA; mutation of any one of these residues to alanine strongly attenuated the ‘inactivation' induced by AA. (vi) The fractional block of Kv1.1 by AA was reduced in the presence of Kvβ1.1 subunits that are known to bind to the inner cavity of the pore, consistent with competition for an overlapping binding site.
We interpret these findings to indicate that AA, similar to Kvβ, binds to the inner pore cavity and that the bound fatty acid causes open-channel block. An alternative interpretation is that binding of AA to the inner cavity allosterically induces pore closure by altering the conformation of the cavity, which, in turn, might lead to a change in the conformation of the selectivity filter (Panyi and Deutsch, 2007). At present, we cannot rule out this alternative explanation, although we favour the view that binding of AA physically occludes the pore. Rapid decay of Kv1.1 currents in inside–out patches was induced not only by AA but also by DHA, AEA and VDM11. These findings suggest that the endogenous lipids DHA and AEA also cause open-channel block. Using a pore homology model of Kv channels we found that the inner cavity of the channel is wide enough to fully accommodate an AEA molecule (and also AA and DHA, not illustrated) and narrow enough to prevent potassium ions with their hydration shells from passing through the channels in the presence of the lipid. Thus, even if the binding of the lipid would cause an allosteric change, it is likely that it would still occlude the pore.
It is noteworthy that the AA-binding site in Kv1.1 overlaps very well with the previously reported Kvβ1.1-binding site (Zhou et al, 2001). Zhou et al found that Kvβ1.1 binds to the pore-facing residues V551, I554, V558 and V562 of Kv1.4, which are homologous to V505, I508, V512 and V516 of Kv1.5. These residues are lining the inner cavity and consistently they interact with both Kvβ1.1 and AA. In addition, block by AA was perturbed by I502 and P513, although these residues are not perfectly facing into the pore. However, as the alkyl chains of AA and AEA are highly flexible an additional interaction with these residues seems plausible.
In neurons, various signal transduction pathways result in the liberation of AA, DHA and AEA from the cell membrane via PLA2, PLD, NAPE-PLD and other pathways (Farooqui and Horrocks, 2006; Okamoto et al, 2007). Unsaturated fatty acids have intrinsically high rates of dissociation from membranes and can diffuse rapidly to local sites of action (Brash, 2001; Balsinde et al, 2002). The free cytosolic concentrations of AA, DHA and AEA are probably <10 μM under normal conditions but local phospholipase action is likely to result in a high concentration (>50 μM) of these amphiphaths in the submembrane zone (Brash, 2001; Farooqui and Horrocks, 2006; Darios et al, 2007). Our analysis of the effects of AA in inside–out patches (Figure 1I and J) suggests that AA can diffuse into the pore cavity of Kv1.1 and Kv1.1I400V with a similar rate constant. In contrast, the recovery of the Kv1.1 current from block by AA was changed drastically by the I400V exchange (Figure 1K and L). These data suggest that the dissociation rate was dependent on the interaction of the fatty acid with the hydrophobic side chains lining the inner cavity of the channel, similar as described earlier for Kvβ molecules (Bhalla et al, 2004).
The binding of lipids to the inner cavity of ion channels has not been reported earlier. In the seminal study of Oliver et al (2004) the inner pore cavity has not been probed systematically by mutagenesis and the binding site of AA in Kv channels has not been identified. The authors mention that a double mutation introduced in the lower S6 helix of Kv3.1 does not affect AA-induced inactivation. However, this double mutation, corresponding to a V512I/V514I double mutation in Kv1.5, is conservative and includes a non-pore-facing mutation (V514I). In contrast to our findings on Kv1.1, Oliver et al (2004) found that lipid-induced ‘inactivation' of Kv3.1 channels was unaffected by the presence of TEA at the extracellular or intracellular side of the channel. This experimental finding may be explained by the fact that Kv3.1 channels have a much lower TEA affinity than Kv1.1 channels.
Functional consequences of Kv1.x editing
The binding site of AA in the inner cavity of Kv1.1 channels includes the isoleucine residue at position 400, which is altered to valine by RNA editing. We found that in the edited Kv1.1I400V channel low concentrations of AA, DHA and AEA no longer induced open-channel block. On the contrary, application of 10 μM AA slightly increased the peak and steady-state Kv1.1I400V currents. Editing of Kv1 channels is selective for Kv1.1, as we found no editing in other members of the Kv1.x family. However, Kv1.x channels show heteromerization, although the extent of which in native tissues is still unknown. Heteromeric channels are likely to predominate in neurons expressing multiple subtypes of Kv channels. We have found that heteromerization with Kv1.1 confers sensitivity to editing to the heteromeric channels. Our co-expression analysis suggests that open-channel block by AA, DHA and AEA was strongly decreased in heteromeric Kv1.x channels containing at least one Kv1.1I400V subunit. Thus, it appears that editing of one of the four channel subunits is sufficient to drastically alter the properties of the channel. In other words, the lipids may interact with channels containing a valine residue at position 400 with a lower affinity than with unedited channels containing a ring of four isoleucine residues. Consistent with the idea that editing of heteromeric Kv1.1 channels occurs in vivo, our patch-clamp measurements in TCR neurons of the LGN suggest that about ⅔ of the voltage-activated K+ current is carried by (Psora-4 insensitive) Kv1.x channels containing at least one edited Kv1.1 subunit.
In drosophila Shaker (Kv1), Shab (Kv2) and squid sqKv2 channels, the position corresponding to I400 in Kv1.1 is also an I-to-V editing site, which represents a clear case of convergent evolution (Patton et al, 1997; Bhalla et al, 2004; Ryan et al, 2008). In addition to the previously reported functional consequences for editing at this paralogous position (Patton et al, 1997; Ryan et al, 2008), we found that Shaker and Shab channels have also a reduced HUFA sensitivity when they undergo this conserved I-to-V editing.
Kv1 channels are novel drug targets against multiple sclerosis, autoimmune diseases, neuroinflammatory diseases and atrial fibrillation (Schwid et al, 1997; Vennekamp et al, 2004; Beraud et al, 2006; Decher et al, 2006). As the drug-binding site in the inner cavity of Kv channels is highly conserved (Decher et al, 2004) it is difficult to develop Kv subtype specific drugs. Our results show that the characterization of drug effects in animal models (for example, inferring involvement of Kv channels from sensitivity to 4-AP) can give erroneous results if Kv1.1 editing is not considered. Depending on the fraction of edited Kv1.1 mRNA in a cell, the affinity and selectivity of drugs can be altered drastically. Thus, it may be possible to design drugs that take into account the differential editing of Kv1.1 channels in different regions of the brain and in neurological disease states. In conclusion, we have obtained evidence that the endogenous lipids AA, DHA and AEA can produce open-channel block of Kv channels, mimicking an increase in the rate of inactivation. These direct effects of the lipids, as well as the effects of pharmacological open-channel blockers, can be antagonized by RNA editing of Kv1.1 channels. The presence of one edited Kv1.1 subunit in heteromeric Kv1.x channels appears to be sufficient to substantially decrease the effects of lipids and drugs on Kv1.x currents. It has long been known that the time course of inactivation of Kv channels is an important determinant of the firing pattern and of the input/output relation of neurons (Connor and Stevens, 1971; Daut, 1973; Boland and Drzewiecki, 2008). Our results suggest that the combined effects of open-channel block of Kv1 channels by highly unsaturated lipids together with differential RNA editing may contribute to the fine-tuning of neuronal information processing in different regions of the brain.
Materials and methods
Two-electrode voltage clamp
Isolation of Xenopus laevis oocytes and synthesis of capped cRNA were performed as described (Decher et al, 2004). Kv channels and the rat Kvβ1.1 subunit were cloned into different expression vectors, including pSGEM, ALMV, pXOOM and pGEM. Oocytes were injected with 0.05–10 ng cRNA encoding the different channels. Voltage-clamp recordings were performed at room temperature (21–22°C) with an Axoclamp 900A amplifier, a Digidata 1440A and pClamp10 software (Axon Instruments). Macroscopic currents were recorded 2–4 days after injection. The holding potential was always −80 mV. The cycle time for all pulse protocols was 10 s or slower.
Inside–out macropatches from Xenopus oocytes
Inside–out macropatches were excised from oocytes and currents were measured at room temperature 4 days after mRNA injection. Pipettes made from thick-walled borosilicate glass with resistances of 0.3–0.9 MΩ (tip diameter of 5–15 μm) were filled with 4 mM KCl, 116 mM N-methylglucamine, 10 mM HEPES, 1.8 mM CaCl2; pH 7.2. Currents were recorded with an EPC9 amplifier (HEKA electronics) and sampled at 10 kHz with analog filter set to 3 kHz. To characterize the Kv currents, double pulses were applied from −80 mV (holding potential) to +40 mV with an interpulse interval of 7 s. Solutions were applied to the cytoplasmic side of excised patches through a multi-barrel pipette and had the following composition: 120 mM KCl, 10 mM HEPES, 2 mM K2EGTA; pH 7.2.
Preparation of LGN neurons
All animal procedures were approved by local authorities. Rats (P14–P22) were deeply anaesthetized with halothane and decapitated. Brains were removed and placed in iced, oxygenated artificial cerebrospinal fluid containing 210 mM sucrose, 20 mM PIPES, 2.4 mM KCl, 10 mM MgCl2, 0.5 mM CaCl2, 10 mM dextrose; pH 7.25. Thalamic slices (thickness, 500 μm) were obtained from coronal vibratome sections (Ted Pella) and dLGN tissue was transferred to a spinner flask and incubated for 25–30 min at 30°C in an oxygenated solution containing trypsin (0.5–1 mg/ml; Sigma) and 120 mM NaCl, 5 mM KCl, 3 mM MgCl2, 1 mM CaCl2, 20 mM PIPES, 25 mM dextrose; pH 7.35. Single neurons were obtained by trituration.
Electrophysiology of LGN neurons
Whole-cell recordings were performed on identified TCR neurons of the dorsal part of the dLGN at room temperature. Borosilicate glass pipettes with a resistance of 3–5 MΩ were used; typical access resistance was in the range of 3–8 MΩ. Series resistance compensation of >40% was routinely applied. Voltage-clamp experiments with an EPC-7 amplifier (E.S.F. electronics) were controlled by pClamp software operating through a Digidata 1200 interface (Axon Instruments). The extracellular solution contained 140 mM NaCl, 2 mM KCl, 10 mM HEPES, 10 mM dextrose, 3 mM MgCl2, 1 mM CaCl2, 0.001 mM TTX; pH 7.35, osmolarity 305 mOsm. The intracellular solution contained 85 mM K-gluconate, 10 mM K3citrate, 10 mM NaCl, 10 mM KCl, 3 mM K-BAPTA, 0.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 3 mM MgATP, 0.5 mM Na2GTP, 15 mM phosphocreatine; pH 7.25 and 295 mOsm. Data were corrected for the liquid junction potential of 21.5±1.6 mV (n=3). All results are presented as mean±s.e.m. Statistical significance was tested using a Student's t-test; * indicates P<0.05, **P<0.01 and ***P<0.001.
Drugs and IC50 values
Recombinant HgTX1 (Alomone) and 5-(4-phenylbutoxy)psoralen (Psora-4) were dissolved in water or DMSO, respectively, aliquoted, stored at −20°C and added to the external solution on the day of experimentation. AA and AEA were stored as stock solutions (10 mM) at −80°C. The IC50 was determined from Hill plots using 2–5 concentrations for each mutant or construct.
Supplementary Material
Acknowledgments
We thank Elke Nass for LGN patch-clamp experiments and Klaus Steinmeyer for supporting the study. This work was supported by grants of the Deutsche Forschungsgemeinschaft (DFG) to ND (DE-1482/2-1, DE-1482/3-1), to JD (SFB593 TPA4) and to TB (BU-1019/8-1/9-1), and by Research Grants of the University Medical Center Giessen and Marburg to ND and RD, the IMF Münster to TB (BU 120803), the OCC Münster to PE, the Medizin Stiftung and the PE Kempkes Stiftung 09/05, 11/06 and 12/07 to ND.
Footnotes
The authors declare that they have no conflict of interest.
References
- Armstrong CM (1971) Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol 58: 413–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde J, Winstead MV, Dennis EA (2002) Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett 531: 2–6 [DOI] [PubMed] [Google Scholar]
- Beraud E, Viola A, Regaya I, Confort-Gouny S, Siaud P, Ibarrola D, Le Fur Y, Barbaria J, Pellissier JF, Sabatier JM, Medina I, Cozzone PJ (2006) Block of neural Kv1.1 potassium channels for neuroinflammatory disease therapy. Ann Neurol 60: 586–596 [DOI] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJ, Holmgren M, Reenan R (2004) Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol 11: 950–956 [DOI] [PubMed] [Google Scholar]
- Bisogno T, Di Marzo V (2007) Short- and long-term plasticity of the endocannabinoid system in neuropsychiatric and neurological disorders. Pharmacol Res 56: 428–442 [DOI] [PubMed] [Google Scholar]
- Boland LM, Drzewiecki MM (2008) Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem Biophys 52: 59–84 [DOI] [PubMed] [Google Scholar]
- Brash AR (2001) Arachidonic acid as a bioactive molecule. J Clin Invest 107: 1339–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Mager R, Pape HC (1992) Different types of potassium outward current in relay neurons acutely isolated from the rat lateral geniculate nucleus. Eur J Neurosci 4: 708–722 [DOI] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N Jr, Ashe KH, Frautschy SA, Cole GM (2004) Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron 43: 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G (1991) Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci USA 88: 5092–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Stevens CF (1971) Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol 213: 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Connell E, Davletov B (2007) Phospholipases and fatty acid signalling in exocytosis. J Physiol 585: 699–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daut J (1973) Modulation of the excitatory synaptic response by fast transient K+ current in snail neurones. Nat New Biol 246: 193–196 [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V (2000) Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 483: 52–56 [DOI] [PubMed] [Google Scholar]
- Decher N, Gonzalez T, Streit AK, Sachse FB, Renigunta V, Soom M, Heinemann SH, Daut J, Sanguinetti MC (2008) Structural determinants of Kvbeta1.3-induced channel inactivation: a hairpin modulated by PIP2. EMBO J 27: 3164–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Pirard B, Sanguinetti MC (2006) Binding site of a novel Kv1.5 blocker: a ‘foot in the door' against atrial fibrillation. Mol Pharmacol 70: 1204–1211 [DOI] [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Renigunta V, Sanguinetti MC (2005) Structural basis for competition between drug binding and Kvβ1.3 accessory subunit-induced N-type inactivation of Kv1.5 channels. Mol Pharmacol 68: 995–1005 [DOI] [PubMed] [Google Scholar]
- Decher N, Pirard B, Bundis F, Peukert S, Baringhaus KH, Busch AE, Steinmeyer K, Sanguinetti MC (2004) Molecular basis for Kv1.5 channel block: conservation of drug binding sites among voltage-gated K+ channels. J Biol Chem 279: 394–400 [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA (2006) Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist 12: 245–260 [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E (1982) Sodium and calcium channels in bovine chromaffin cells. J Physiol 331: 599–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore E, Barhanin J, Attali B, Lesage F, Lazdunski M (1994) External blockade of the major cardiac delayed-rectifier K+ channel (Kv1.5) by polyunsaturated fatty acids. Proc Natl Acad Sci USA 91: 1937–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R (2003) Nervous system targets of RNA editing identified by comparative genomics. Science 301: 832–836 [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF (2008) Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14: 923–930 [DOI] [PubMed] [Google Scholar]
- Koren G, Liman ER, Logothetis DE, Nadal-Ginard B, Hess P (1990) Gating mechanism of a cloned potassium channel expressed in frog oocytes and mammalian cells. Neuron 4: 39–51 [DOI] [PubMed] [Google Scholar]
- Koschak A, Bugianesi RM, Mitterdorfer J, Kaczorowski GJ, Garcia ML, Knaus HG (1998) Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J Biol Chem 273: 2639–2644 [DOI] [PubMed] [Google Scholar]
- Lundbaek JA (2008) Lipid bilayer-mediated regulation of ion channel function by amphiphilic drugs. J Gen Physiol 131: 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG (2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278: 43807–43817 [DOI] [PubMed] [Google Scholar]
- Meves H (2008) Arachidonic acid and ion channels: an update. Br J Pharmacol 155: 4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Wang J, Morishita J, Ueda N (2007) Biosynthetic pathways of the endocannabinoid anandamide. Chem Biodivers 4: 1842–1857 [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B (2004) Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304: 265–270 [DOI] [PubMed] [Google Scholar]
- Panyi G, Deutsch C (2007) Probing the cavity of the slow inactivated conformation of shaker potassium channels. J Gen Physiol 129: 403–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DE, Silva T, Bezanilla F (1997) RNA editing generates a diverse array of transcripts encoding squid Kv2 K+ channels with altered functional properties. Neuron 19: 711–722 [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O (1994) Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 369: 289–294 [DOI] [PubMed] [Google Scholar]
- Ryan MY, Maloney R, Reenan R, Horn R (2008) Characterization of five RNA editing sites in Shab potassium channels. Channels (Austin) 2: 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwid SR, Petrie MD, McDermott MP, Tierney DS, Mason DH, Goodman AD (1997) Quantitative assessment of sustained-release 4-aminopyridine for symptomatic treatment of multiple sclerosis. Neurology 48: 817–821 [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Stocker M, Sakmann B, Seeburg P, Baumann A, Grupe A, Pongs O (1988) Potassium channels expressed from rat brain cDNA have delayed rectifier properties. FEBS Lett 242: 199–206 [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ (2004) Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol 66: 477–519 [DOI] [PubMed] [Google Scholar]
- Vennekamp J, Wulff H, Beeton C, Calabresi PA, Grissmer S, Hansel W, Chandy KG (2004) Kv1.3-blocking 5-phenylalkoxypsoralens: a new class of immunomodulators. Mol Pharmacol 65: 1364–1374 [DOI] [PubMed] [Google Scholar]
- Yellen G (1998) The moving parts of voltage-gated ion channels. Q Rev Biophys 31: 239–295 [DOI] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R (2001) Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411: 657–661 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.